Cellular Plasticity Cascades in the Pathophysiology and Treatment of Bipolar Disorder (original) (raw)
INTRODUCTION
Bipolar disorder (BPD) is a common, chronic, recurrent mental illness that affects the lives and functioning of millions of individuals worldwide. A growing number of recent studies indicate that for a majority of these individuals, outcome is quite poor. High rates of relapse, chronicity, lingering residual symptoms, subsyndromes, cognitive and functional impairment, psychosocial disability, and diminished well-being are unfortunately common occurrences in BPD (Belmaker, 2004). Furthermore, BPD is a systemic disease that is frequently associated with a wide range of physiological perturbations and medical problems, including cardiovascular disease, diabetes mellitus, obesity, and thyroid disease (Kupfer, 2005). Neurobiological studies of mood disorders over the past 40 years have primarily focused on abnormalities of the monoaminergic neurotransmitter systems, on characterizing alterations of individual neurotransmitters in disease states, and on assessing response to mood stabilizer and antidepressant medications. The monoaminergic systems are extensively distributed throughout the network of limbic, striatal, and prefrontal cortical neuronal circuits thought to support the behavioral and visceral manifestations of mood disorders (Drevets, 2000). Studies of cerebrospinal fluid chemistry, neuroendocrine responses to pharmacological challenge, and neuroreceptor and transporter binding have demonstrated a number of abnormalities in monoaminergic neurotransmitter and neuropeptide systems in mood disorders (Goodwin and Jamison, 2007).
Unfortunately, these observations have not yet greatly advanced our understanding of the underlying biology of recurrent mood disorders, which must include an explanation for the predilection to episodic and often profound mood disturbance that can become progressive over time. BPD likely arises from the complex interaction of multiple susceptibility (and protective) genes and environmental factors, and the phenotypic expression of the disease includes not only mood disturbance, but also a constellation of cognitive, motor, autonomic, endocrine, and sleep/wake abnormalities. Furthermore, while most antidepressants exert their initial effects by increasing intrasynaptic levels of serotonin and/or norepinephrine, their clinical antidepressant effects are observed only after chronic administration (over days to weeks), suggesting that a cascade of downstream events is ultimately responsible for their therapeutic effects. These observations have led to the idea that while dysfunction within the monoaminergic neurotransmitter systems is likely to play an important role in mediating some facets of the pathophysiology of BPD, it likely represents the downstream effects of other, more primary abnormalities in signaling pathways (Table 1) (Goodwin and Jamison, 2007) (Figure 1a).
Table 1 Putative Roles for Signaling Pathways in Mood Disorders
Figure 1
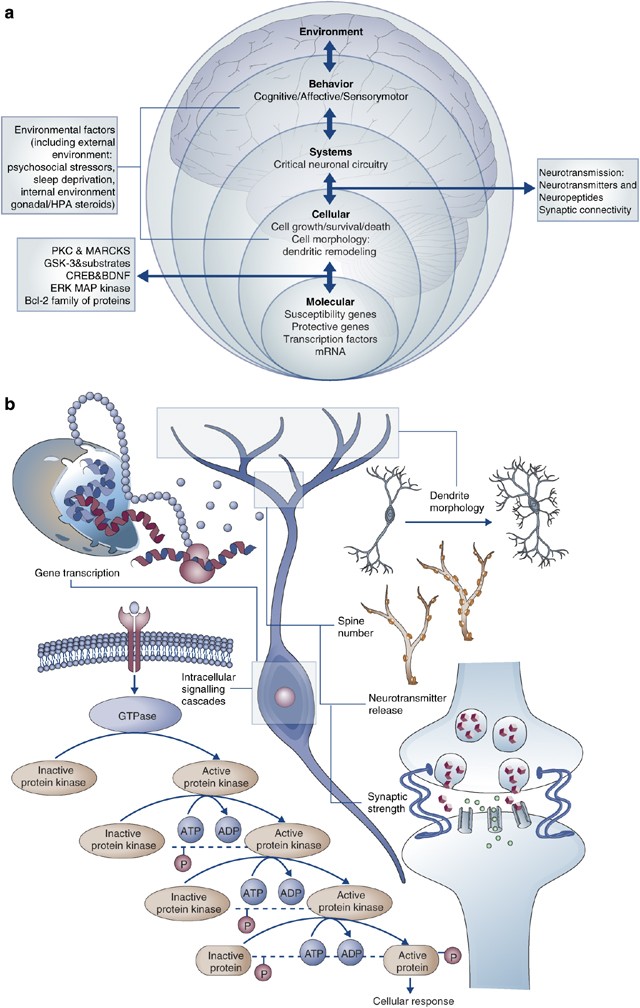
(a) A true understanding of the pathophysiology of BPD must encompass different systems on different physiological levels at which the disease manifests: molecular, cellular, and behavioral. (b) Biological mechanisms underlying neuroplasticity. The remarkable plasticity of neuronal circuits is achieved through different biological means including alterations in gene transcription and intracellular signaling cascades. These changes modify diverse neuronal properties such as neurotransmitter release, synaptic function and even morphological characteristics of neurons.
Why Consider Plasticity Cascades as Playing a Central Role in the Pathophysiology and Treatment of BPD?
Plasticity, the ability to undergo and sustain change, is essential for the proper functioning of our nervous system. This capacity for change allows organisms to adapt to complex alterations in both their internal and external environments, a feature fundamentally important for survival and reproduction. Besides the evident need for significant adaptation mechanisms in learning and memory as well as physiological homeostasis, all complex behavioral phenomena—including mood and emotion—are dynamic processes that rely on plastic neural circuitry. The biological basis of this capacity to adapt encompasses a diverse set of cellular and molecular mechanisms that fall under the broad term ‘neuroplasticity’. In this paper, we make the distinction between synaptic plasticity and neuroplasticity.
Synaptic plasticity refers to the cellular process that results in lasting changes in the efficacy of neurotransmission. More specifically, the term synaptic plasticity refers to the variability of the strength of a signal transmitted through a synapse. The regulation of transmission at the synapse may be mediated by changes in neurotransmitter levels, receptor subunit phosphorylation, surface/cellular levels or receptors, and conductance changes, among others.
Neuroplasticity is a broader term that encapsulates changes in intracellular signaling cascades and gene regulation (see McClung and Nestler, 2008 in this issue), modifications of synaptic number and strength, variations in neurotransmitter release, modeling of axonal and dendritic architecture and, in some areas of the CNS, the generation of new neurons. Modifications arising from neuroplastic mechanisms can be of short duration or long lasting, and this is determined by the qualitative, quantitative, and temporal characteristics of the precipitating stimuli. For instance, compared to acute or single stimuli, chronic and repeated stimuli often lead to qualitatively different, and often times long-lasting alterations (Hyman and Nestler, 1996); furthermore, substantial life events that occur during development of the organism often have a greater impact than they would later in life.
In recent years, research has linked mood disorders with structural and functional impairments related to neuroplasticity in various regions of the CNS. In addition, psychotropic drugs commonly used to treat these conditions target molecules and signaling cascades implicated in the control of neuroplasticity. Research on the biological underpinnings of mood disorders has therefore moved away from focusing on absolute changes in neurochemicals such as monoamines and neuropeptides, and instead has begun highlighting the role of neural circuits and synapses, and the plastic processes controlling their function. Thus, these illnesses can best be conceptualized as genetically influenced disorders of synapses and circuits rather than simply as deficits or excesses in individual neurotransmitters. The integration of knowledge derived from different physiological and phenomenological levels continues to help move us toward a more conceptual understanding of the etiology and pathophysiology of BPD. As we review in this paper, a growing body of data supports the contention that BPD arises from abnormalities in cellular plasticity cascades, leading to aberrant information processing in synapses and circuits mediating affective, cognitive, motoric, and neurovegetative functions (Catapano et al, in press; Post, 2007; Young, 2007). Indeed, in a recent whole-genome association study of BPD, all of the highly significant associations implicated signaling cascades (Baum et al, 2007).
The role of cellular signaling cascades has the potential to explain much of the complex neurobiology of BPD (Goodwin and Jamison, 2007). Cellular signaling cascades regulate the multiple neurotransmitter and neuropeptide systems implicated in the disorder, and are targets for the most effective treatments. Signaling pathways are also targets for hormones that have been implicated in the pathophysiology of BPD. The highly integrated monoamine and prominent neuropeptide pathways are known to originate and project heavily to limbic-related regions such as the hippocampus, hypothalamus, and brain stem, which are likely associated with neurovegetative symptoms. Abnormalities in cellular signaling cascades that regulate diverse physiologic functions also likely explain the tremendous medical comorbidity associated with BPD. Furthermore, many of these pathways play critical roles not only in synaptic (and therefore behavioral) plasticity, but also in long-term atrophic processes (see below) (Figure 1b).
Targeting these cascades in the treatment of mood disorders may stabilize the underlying disease process by reducing the frequency and severity of the profound mood cycling that contributes to morbidity and mortality.
In this paper, we focus upon the role of plasticity cascades in the pathophysiology and treatment of BPD. The role of neurotransmitter and neuropeptide systems has recently been extensively covered elsewhere (Goodwin and Jamison, 2007; Soares and Young, 2007), and is not addressed here. There have also been tremendous advances in our understanding of the fundamental processes underlying synaptic and neural plasticity; many of these advances are well covered in accompanying papers (Bear et al, 2008; Martinowich and Lu, 2008; Citri and Malenka, 2008; McClung and Nestler, 2008), and are not discussed in detail here. Finally, the interested reader is referred to the chapter by Pittenger and Duman for an excellent, extensive discussion of the role of plasticity cascades in depression, and as targets for antidepressants (Pittenger and Duman, 2008).
CLINICAL EVIDENCE SUPPORTING THE CONTENTION THAT ABNORMALITIES IN SYNAPTIC AND CELLULAR PLASTICITY PLAY AN IMPORTANT ROLE IN BPD
Structural Neuroimaging Findings
As discussed in the introduction, considerable data now show that many forms of neural plasticity involve structural brain changes (Figure 2). Additionally, evidence has accumulated supporting the persistence of ‘structural plasticity’ in the adult human brain. Thus, investigators have used neuroimaging and post-mortem human brain studies to investigate potential alterations in structural plasticity in mood disorders.
Figure 2
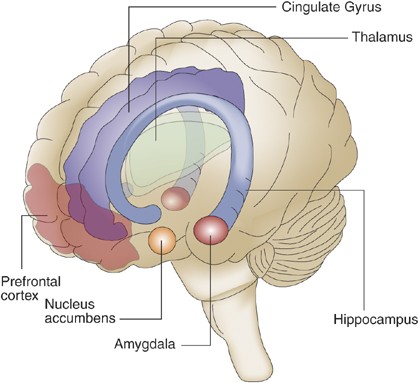
Neuroanatomical regions implicated in affective processes. Neuroimaging studies, observations on patients with selective CNS lesions, and data from animal behavioral experiments have elucidated several brain regions implicated in the perception and control of mood states and emotions. These include the PFC, amygdala, insula, hippocampus, anterior cingulate cortex, and ventral striatum. Studies on BPD using neuroimaging and post-mortem pathological techniques have revealed several functional and structural abnormalities in these regions.
Computed tomography and magnetic resonance imaging (MRI) have revealed structural abnormalities in the brains of patients with mood disorders. Overall, gray matter volumes do not significantly differ in patients with BPD as compared to healthy individuals (Brambilla et al, 2001; Schlaepfer et al, 1994; Zipursky et al, 1997); however, several studies have found region-specific reductions. The major region-specific structural imaging findings include increased ventricular size and decreased frontal cortical area volumes. Several studies have reported that BPD patients have enlarged ventricles (Kato et al, 1994; Nasrallah et al, 1982; Pearlson et al, 1984; Pearlson and Veroff, 1981; Strakowski et al, 2002), which suggests that decreases in brain tissue volume may exist. Indeed, studies have revealed decreases in specific cortical areas including the left subgenual region 24 (SG24), a structure in the anterior gyrus ventral to the genu of the corpus callosum (Drevets et al, 1997). Other studies indicate reductions of gray matter volume in the left dorsolateral prefrontal cortex (DLPC) (Brambilla et al, 2002), the ventral prefrontal cortex (PFC), and the orbital PFC (Frangou et al, 2002). Temporal lobe structures such as the hippocampus and the amygdala have not been as thoroughly researched as the frontal lobes.
In contrast to findings in major depressive disorder (MDD), significant BPD-related volumetric differences have not been consistently reported in the hippocampus; as we discuss later, the possibility that mood stabilizers exert a mitigating neurotrophic effect is an important potential confound. One study did, however, find that volumetric reductions were more prevalent in the right hippocampus of the affected individual in monozygotic twin sets discordant for BPD (Noga et al, 2001).
Periventricular and deep hyperintensities in the subcortical white matter—white matter hyperintensities (WMH)—have consistently been identified with MRI in the brains of elderly depressed patients and patients with BPD (Altshuler et al, 1995; Bearden et al, 2001; Lenox et al, 2002; Stoll et al, 2000). Although the pathophysiological and functional meanings associated with WMH still need to be elucidated, they may have multiple causes including cerebrovascular accidents, ischemia, demyelination, loss of axons, dilated perivascular space, minute brain cysts, and necrosis. To assess the cellular pathophysiology of these lesions, a histopathological assessment was correlated with neuroimaging conducted in vitro using brain slices from elderly depressed subjects and elderly controls. Deep WMH were found to be ischemic in the depressed group when compared against the control samples, and were localized mainly in the DLPC, supporting the contention that the lesions were of vascular origin in these elderly depressed subjects (Thomas et al, 2002).
Intriguingly, a growing body of data suggests that a significant percentage of young bipolar patients (including children) exhibit WMH (Lyoo et al, 2002; Pillai et al, 2002). In fact, these lesions have also been found to be increased in children with psychiatric disorders, though highest among those with BPD when compared to controls, particularly in the frontal lobes (Lyoo et al, 2002) and also early in the course of BPD in adolescents (Pillai et al, 2002). They appear to be associated with poor treatment response in patients with affective disorders (Lenox et al, 2002), particularly when they are located in subcortical rather than periventricular areas (Moore et al, 2001). Together, these results support the contention that WMH damage the structure of brain tissue, and likely disrupt the neuronal connectivity necessary for normal affective functioning.
Because these young BPD patients have no overt cerebrovascular risk factors, the findings are most consistent with the hypothesis that patients with BPD have endogenous impairments of cellular resilience, leading to hypoxic-like changes even in the face of normal cerebrovascular flow. Indeed, the relationship between reduced intracellular pH and increased WMH in BPD supports this possibility (Kato et al, 1998).
Magnetic Resonance Spectroscopy Findings Pertaining to Mitochondrial Function and Cell Energetics
It is clear that BPD is not a classical mitochondrial disorder. However, emerging data suggest that many upstream abnormalities (likely nuclear genome coded) converge to regulate mitochondrial function, and that mitochondrial function is implicated in acute abnormalities of both synaptic and neural plasticity (Goodwin and Jamison, 2007). Thus, in addition to the well-known function of energy production via oxidative phosphorylation, neuronal mitochondria also play an important role in apoptosis and in the regulation of intracellular calcium; increasing evidence suggests that the latter action may be critically important in regulating the release of, and response to, neurotransmitters. Furthermore, mounting evidence suggests that activation of mitochondrial-apoptotic cascades may lead to a process of ‘synaptic apoptosis’, in which apoptotic processes are activated in a highly localized manner (Mattson, 2007).
A growing body of evidence suggests that mitochondria may be integrally involved in the general processes of synaptic plasticity (Mattson, 2007; Yang et al, 2003). In addition, increased synaptic activity has been shown to induce the expression of mitochondrial-encoded genes, suggesting that a long-lasting up-regulation of energy production may be triggered by synaptic activity, playing a role in the long-term regulation of synaptic strength (Williams et al, 1998). Thus, mitochondrial dysfunction exerts major effects on ‘here and now’ neurotransmitter function, in addition to its better appreciated long-term cytoprotective effects.
Magnetic resonance spectroscopy (MRS) has increasingly been utilized in the study of neuropsychiatric disorders. _N_-acetyl-aspartate (NAA) is a predominant neurochemical compound that can be quantitatively assessed via MRS in the normal adult human brain. Pertinent to the present discussion is the fact that NAA is localized to mature neurons and synthesized within mitochondria.
Interestingly, inhibitors of the mitochondrial respiratory chain decrease NAA concentrations, effects that correlate with reductions in adenosine triphosphate (ATP) and oxygen consumption. High-resolution 1H-MRS imaging studies have found decreased levels of NAA in limbic and limbic-related areas of the brain as well as decreased levels in the hippocampus, independent of mood state (Bertolino et al, 2003), in euthymic and medicated familial bipolar I patients (Diecken et al, 2003); in the DLPC in euthymic and unmedicated adult bipolar I and II patients (Winsberg et al, 2000); in the orbitofrontal cortex in manic/mixed patients (Cecil et al, 2002); and in the DLPC in juvenile BPD patients (Chang et al, 2003). In addition, decreased levels of NAA were also found in the basal ganglia in both the depressive and euthymic states (Hamakawa et al, 1998). As discussed above, these findings may be the expression of underlying changes in ATP spent and availability, in oxygen consumption, or in glutamatergic activity in BPD.
In addition to the NAA findings, studies that used phosphorous-31 MRS (31P MRS), which permits the determination of abnormally high brain energy phosphate metabolism, showed a decrease in phosphocreatine (PCr) and/or ATP levels in mood disorder patients (Diecken et al, 1995; Kato et al, 1995; Volz et al, 1998). The most extensive series of studies investigating possible abnormalities in brain energy regulation in mood disorders have been conducted by Kato and colleagues. Consistent with the decreased PCr and ATP levels discussed above, this research group also found low pH levels (measured indirectly via 31P MRS) in mood disorder patients compared to normal controls, in basal ganglia and the whole brain (Hamakawa et al, 2004); these observations originally led to the notion that BPD may be associated with mitochondrial dysfunction (Hamakawa et al, 2004; Kato et al, 1998).
It is not currently known whether these neuroimaging results are developmental abnormalities that confer vulnerability to severe mood episodes, compensatory changes to other pathogenic processes, or the sequelae of recurrent affective episodes (Carlson et al, 2006). Indeed, data suggest that multiple factors may be operative. The reduced gray matter volumes, WMH, and reduced NAA levels described above often affect first-onset patients or children with BPD (Frazier et al, 2005; Sassi et al, 2005). While these studies do not demonstrate that the changes precede illness onset, they certainly suggest that these changes do not represent the toxic sequelae of decades of illness. Consistent with such a contention, a meta-analysis of imaging studies concluded that volumetric abnormalities in the subgenual PFC, striatum, hippocampus, and amygdala are seen in first-episode bipolar subjects, children with BPD, and unaffected siblings, raising the possibility that this endophenotype may constitute a heritable vulnerability factor in these patients (Hajek et al, 2005).
However, some of the brain changes may be associated with duration of illness and the consequences of affective episodes per se. Sheline et al (2003, 1996) measured hippocampal volumes of subjects with a history of major depressive episodes and found that the degree of hippocampal volume reduction correlated with total duration of MDD, and with duration of untreated depressive episodes. Another study found hippocampal volume reduction in subjects with multiple depressive episodes, but not in first-episode subjects (MacQueen et al, 2003). It is noteworthy that similar changes have not been reported in patients with BPD—this difference may reflect distinct pathophysiologies, or the neuroprotective effects of mood stabilizers (see below).
Post-Mortem Brain Findings
Neurons
In addition to accumulating neuroimaging findings, studies of post-mortem tissue from BPD patients have revealed several abnormalities (Figure 3). Most studies have focused on areas highlighted by structural and functional imaging findings such as PFC, amygdala, hippocampus, and striatum. In recent years, progress in techniques such as unbiased stereology, histopathology, and microscopy has helped to advance efforts to provide a neuropathological description of BPD. Post-mortem observations have further corroborated the concept that impaired cellular resilience and function play a role in the pathophysiology of BPD (Table 2).
Figure 3
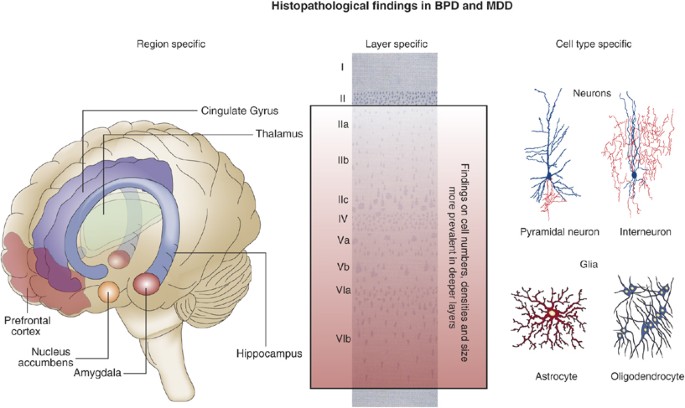
Histopathological findings in BPD research. Research on post-mortem brain tissue of BPD patients and cell biological findings in animal models have revealed several findings in areas implicated in emotion perception and control including hippocampus, amygdala, and prefrontal and cingulate cortices.
Table 2 Post-mortem Morphometric Brain Studies in Mood Disorders Demonstrating Cellular Atrophy and/or Loss
Studies have found reduced subcortical nuclei volumes in patients with recurrent mood disorders, with especially striking results in the BPD group (Baumann et al, 1999a, 1999b; Bielau et al, 2005). Other studies have shown differential neuron density and morphology, which appear to be layer- and cell-type specific. In a DLPC analysis using Nissl-stained sections from BPD and MDD brains, significant reductions in the density and soma size of some neurons in specific layers were observed. Decreased soma size could be correlated with less extensive or less active axodendritic trees, which could not be observed using standard Nissl staining procedures. The density of neurons with large cell somas (most likely corresponding to glutamatergic pyramidal neurons) was significantly decreased in layers III and V (Rajkowska, 2000, 2002), and the size of neurons in layer V and VI was found to be reduced (Cotter et al, 2002b). In different anterior cingulate subregions of BPD individuals, reduced cell densities were found in layers III–VI, nonpyramidal cell density was decreased in layer II, and neuron size was increased in layer V and in layer II nonpyramidal cells (discussed in Goodwin and Jamison, 2007). In the hippocampus, decreased pyramidal cell soma size was found in the CA1 region in BPD (Liu et al, 2007).
GABAergic interneurons can be classified by their immunoreactivity for the calcium-binding proteins calbindin, parvalbumin, and calretinin. Immunohistochemical studies in BPD patients using these markers found decreased levels of calbindin- and parvalbumin-positive cells in the anterior cingulate cortex (Benes and Berretta, 2001; Cotter et al, 2002a), the hippocampus (Benes et al, 2001), and reductions of parvalbumin-positive cells in the entorhinal cortex (Pantazopoulos et al, 2007). Clustering of parvalbumin-positive neurons was increased in the anterior cingulate cortex.
Most recently, Bezchlibnyk et al (2007) assessed the size and density of both neuronal and glial cells in discrete amygdalar nuclei in post-mortem sections from subjects with MDD, BPD, schizophrenia, and from nonpsychiatric control subjects. They found significantly decreased neuron somal size in the lateral amygdalar nucleus (LAN) and the accessory basal parvocellular nucleus in subjects with BPD relative to control subjects. These changes in cellular morphology were most prominent in the LAN in sections obtained from the left hemisphere (Bezchlibnyk et al, 2007).
Glial cells
In addition to the aforementioned abnormalities observed in neuronal populations, prominent glial cell abnormalities have been identified in post-mortem BPD brains. Glial cell density appears decreased in frontal cortical areas (Ongur et al, 1998; Rajkowska, 2000, 2002; Rajkowska et al, 2001), and is accompanied by increased nucleus size. Increased glial cell nuclei size has also been observed in reactive gliosis occurring over the course of some classic neurodegenerative diseases, but in these cases the actual number of astrocytes markedly increases. This glial pathology might be more apparent in BPD individuals with a strong genetic component, because one study only found striking reductions in the subgenual PFC (41%) in patients with a clear family history of BPD (Ongur et al, 1998). In the amygdala, glial cell numbers were decreased in MDD, but not in BPD. However, the only two BPD patients in this study that had not been treated with mood stabilizers did show decreases in glial number, indicating that treatment with mood stabilizers had a possible protective effect (Bowley et al, 2002).
Glial cells, including astrocytes, oligodendrocytes, and microglia, constitute a diverse group of nonneuronal cells each with different functions. Astrocytes play important roles in neurotransmitter catabolism, but have also been implicated in synapse formation and as providers of trophic support to neurons. The main function of oligodendrocytes is axon myelination in the CNS. Microglia are highly mobile cells constituting the major immune cells of the brain. The type of glial cells most affected in BPD remains unclear. Findings in MDD have suggested that astrocytes may be uniquely affected; however, a different morphological type of glial cell may be targeted. A proteomic study on MDD and BPD brains found disease-specific alterations in levels of glial-fibrillary acidic protein, a protein that is abundantly expressed in astrocytes (Johnston-Wilson et al, 2000). Other studies have found reductions in oligodendrocyte number (Uranova et al, 2004), as well as in the expression of key oligodendrocyte and myelin-related genes in the DLPC in individuals with BPD (Tkachev et al, 2003).
Overall, cell loss and atrophy (both neurons and glia) likely represent etiologic factors as well as the consequence of disease progression in BPD. There is almost no doubt that these atrophic brain changes contribute to illness pathophysiology by disrupting the circuits that mediate normal affective, cognitive, motoric, and neurovegetative functioning. Furthermore, these findings suggest that the neurotrophic effects of mood stabilizers (see below) may be very relevant to the treatment of BPD.
Post-mortem gene expression of key mitochondrial proteins in BPD
Interestingly, additional evidence of dysregulated mitochondrial processes in BPD comes from an elegant series of recent post-mortem brain microarray studies. Konradi et al (2004), using gene arrays that analyzed mRNA expression in hippocampus, studied 12 558 nuclear genes analyzed in three different groups (healthy controls, BPD, and schizophrenia patients). Using stringent statistical analysis, they found that the expression of only 43 genes—42% of which coded for mitochondrial proteins—decreased in BPD compared with schizophrenia. Notably, this gene expression was involved in regulating oxidative phosphorylation in the mitochondrial inner membrane, which included subunits of complexes I and the ATP-dependent process of proteasome degradation ((NADH dehydrogenase in one gene), IV (cytochrome c oxidase in one gene), and V (ATP synthase in five genes)). Based on these findings, and additionally on the decreased expression of the enzymes glutamic acid decarboxylase 67 and somatostatin, the authors suggested that a subset of hippocampal interneurons is abnormal in BPD, particularly those that affect mitochondrial energy metabolism (Heckers et al, 2002; Konradi et al, 2004). Most recently, Buttner et al (2007) investigated whether or not apoptosis is associated with GABAergic interneurons in the anterior cingulate cortex in schizophrenia and BPD. A double-labeling technique using the Klenow method of in situ end-labeling of single-stranded DNA breaks was combined with an in situ hybridization localization of mRNA for the 67 kDa isoform of glutamate decarboxylase (GAD67); an increase in Klenow-positive, GAD67-negative nuclei was observed in layer V/VI of patients with BPD, but not schizophrenics (Buttner et al, 2007). These findings suggest that there is more DNA fragmentation in cells showing no detectable GAD67 mRNA in patients with BPD than in patients with schizophrenia or controls, and further, that non-GABAergic cells may be selectively vulnerable to oxidative stress in patients with BPD.
Nevertheless, interpreting the results of post-mortem brain studies requires caution due to the numerous potentially confounding factors (including ante-mortem medication history and/or substance abuse, post-mortem interval, and cause of death). Notably, a study using lymphoblastoid cell lines from BPD patients and healthy controls reported a decreased expression level of NDUFV2 gene (a nuclear-encoded mitochondrial complex I subunit gene) in patients with bipolar I disorder (Washizuka et al, 2003). Finally, a whole-genome association study from the Wellcome Trust in the United Kingdom (Wellcome Trust Case Control Consortium, 2007) investigated 2000 bipolar subjects and 3000 controls. This study found a highly significant association between complex I and BPD; together with the data demonstrating that mood stabilizers enhance mitochondrial function (see below), these data suggest that mitochondrially mediated plasticity may be integral to BPD. Indeed, Stork and Renshaw (2005) have also posited that the many facets of the complex neurobiology of BPD can be fit into a more cohesive bioenergetic and neurochemical model. Specifically, they propose that the existence of mitochondrial dysfunction in BPD involves impaired oxidative phosphorylation, a resultant shift toward glycolytic energy production, a decrease in total energy production and/or substrate availability, and altered phospholipid metabolism (Stork and Renshaw, 2005).
Subcellular markers for neuroplasticity
Further studies have examined protein and mRNA levels of molecules implicated in neuronal and glial cell function. Several indicator proteins for synaptic number and function, such as GAP-43 and the synapsins, have been studied in the post-mortem brains of BPD patients. The neuronal plasticity marker GAP-43 is highly expressed in axonal growth cones during development (Strittmatter, 1992) and is implicated in the regulation of axonal morphology and synaptic plasticity in the mature brain (Benowitz et al, 1990). Expression of GAP-43 increases after in vitro antidepressant treatment (Chen et al, 2003) and decreased levels of this protein have been associated with depression and suicide (Hrdina et al, 1998). In BPD, GAP-43 protein levels are reduced in cingulate cortex (Eastwood and Harrison, 2001) and hippocampus (Tian et al, 2007).
Several proteins implicated in the machinery regulating synaptic vesicle and neurotransmitter release have been studied. Synapsins are a family of proteins that bind synaptic vesicles to the cytoskeleton, thereby preventing their transport to the presynaptic membrane and subsequent neurotransmitter release (Hilfiker et al, 1999). Synaptic vesicle docking and actual neurotransmitter release are regulated by a complex of proteins that includes SNAP-25, syntaxin, and synaptobrevin. Synaptophysin is another protein of unknown function that might interact with synaptobrevin during neurotransmitter release. In BPD post-mortem brain samples, reductions of synapsins (Vawter et al, 2002) in the hippocampus, and increases in SNARE complex proteins in the DLPC have been found (Scarr et al, 2006). Another study showed decreases of synaptobrevin and synaptophysin in visual association cortex (Beasley et al, 2005). Finally, mRNA levels of netrins—a class of proteins implicated in axon guidance—were found to be reduced in the CA3 region of the hippocampus and the entorhinal cortex of individuals with BPD (Eastwood and Harrison, 2007).
EVIDENCE THAT CELLULAR PLASTICITY CASCADES ARE THE TARGETS OF MOOD-STABILIZING AGENTS
Despite substantial advances in mood disorder pharmacotherapeutics in the past 20 years, lithium remains the most effective therapy for BPD. However, the direct targets and mechanisms of its action remain elusive. Several direct targets of lithium have been identified and extensively studied (reviewed in Gould and Manji, 2005; Gurvich and Klein, 2002; Li et al, 2002). These include inositol monophosphatase (IMPase) and structurally related phosphomonoesterases, phosphoglucomutase, and glycogen synthase kinase-3 (GSK-3). Given the broad range of biological functions for pathways regulated by phosphomonoesterases and GSK-3, even this limited set of direct lithium targets is likely to regulate numerous and diverse downstream effectors. Here, we discuss these two best-characterized primary direct targets, as well as those downstream targets (ie those consistent with a clinical temporal profile) that have been demonstrated to exert major effects on neural plasticity.
Inositol Monophosphatase
Inositol monophosphatase dephosphorylates inositol monophosphate, an intermediate in the turnover of the second messenger inositol 1,4,5-tris-phosphate (IP3) to inositol, and is inhibited by lithium at concentrations similar to those achieved clinically in the treatment of BPD (Berridge et al, 1989; Hallcher and Sherman, 1980). The inositol depletion hypothesis (Figure 4) proposes that lithium interferes with the regeneration of inositol and, under conditions where inositol limits phosphatidylinositol (PI) synthesis, depletes the cell of PI (Berridge et al, 1989). Because PI is an obligate precursor for phosphatidylinositol bisphosphate (PIP2), the hypothesis posits that inhibition of IMPase could disrupt PIP2/IP3-mediated signaling.
Figure 4
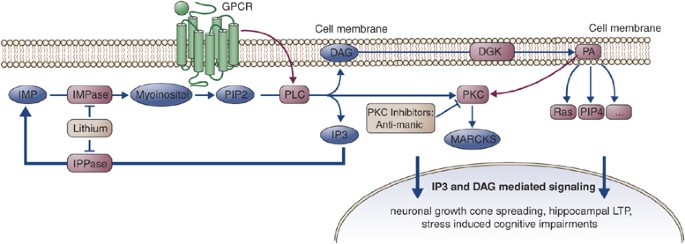
Phosphatidylinositol signaling pathway. Therapeutic levels of lithium directly inhibit several key enzymes that regulate recycling of inositol-l,4,5-trisphosphate (IP3). Inositol-monophosphatase (IMPase) is the final, common step for conversion of monophosphorylated inositols into myo-inositol, and therefore inhibition of IMPase can reduce the level of myo-inositol. The inositol depletion hypothesis proposes that inhibition of this step may interfere with the synthesis of phosphatidylinositol (PI), although this has not been demonstrated in vivo. The PI signaling cascade starts with surface receptor-mediated activation of phospholipase C (PLC). Activated PLC catalyzes the hydrolysis of PIP2 to diacylglycerol (DAG) and IP3. DAG activates protein kinase C (PKC) that, among many other functions, activates myristoylated aknine-rich C kinase substrate (MARCKS). Lithium and valproate both decrease levels of phosphorylated and total MARCKS.
Several other enzymes involved in inositol turnover, including the inositol polyphosphate 1-phosphatase (IPPase) and 3′ (2′) phosphoadenosine-5′-phosphate (PAP) phosphatase (which also hydrolyzes inositol-l,4-bisphosphate (Lopez-Coronado et al, 1999; Spiegelberg et al, 1999)), have been postulated as targets for the therapeutic action of lithium in BPD. IMPase (encoded by IMPA1 and IMPA2), IPPase (encoded by INPP1), and PAP phosphatase are all inhibited by lithium at or below therapeutically relevant concentrations. Loss of function mutations in the genes encoding IPPase (ipp in Drosophila) and IMPase (ttx in C. elegans) demonstrate synaptic phenotypes that are phenocopied by lithium treatment, strongly supporting these inositol phosphatases as targets of lithium in these settings (Acharya et al, 1998; Tanizawa et al, 2006).
Lithium has been shown to reduce inositol levels (Maslanski et al, 1992; O'Donnell et al, 2000; Shaldubina et al, 2006) and to inhibit IMPase in vivo (Hedgepeth et al, 1997), although the magnitude of inhibition is generally modest. Furthermore, the effects of lithium in several experimental systems can be reversed by adding millimolar concentrations of exogenous myoinositol, which has been interpreted to indicate reversal of inositol depletion. For example, lithium and other drugs used to treat BPD, including valproate (VPA) and carbamazepine, can stabilize growth cones, thereby increasing growth cone spreading in sensory neurons in culture (Williams et al, 2002). These effects were reversed by the addition of 1 mM inositol, suggesting that these drugs may act through inositol depletion (although inositol levels were not actually measured). Exogenous inositol also corrects the thermotaxis defect observed in ttx/IMPase mutants in C. elegans (Tanizawa et al, 2006).
Although the inositol depletion hypothesis provides an elegant potential mechanism to explain lithium action, a few important issues remain unresolved. First, therapeutic lithium does not deplete PIP2 in vivo, an essential component of the hypothesis, nor does it reduce IP3 in vivo (Dixon et al, 1994). Second, although lithium inhibits IMPase in vivo (Hedgepeth et al, 1997; Maslanski et al, 1992; O'Donnell et al, 2000; Shaldubina et al, 2006), the inhibition is incomplete, and it is not clear that the modest reduction in inositol is sufficient to impair PI or PIP2 synthesis; indeed, marked reduction in inositol through other means does not reduce PI levels in vivo in rodents (Berry et al, 2004), and potent alternative IMPase inhibitors do not mimic lithium action (Klein and Melton, 1996). Thus, in mice lacking the sodium myoinositol cotransporter 1 gene (SMIT1), a 92% reduction of intracellular inositol in fetal brain had no effect on PI levels (Berry et al, 2004). This is likely because basal intracellular inositol concentrations in the brain are in the 4–8 mM range, which is orders of magnitude above the limiting concentrations that may be found in cultured cells and neuronal slices. The 25% reduction in inositol caused by lithium in vivo therefore is unlikely to affect global PIP2 or IP3 levels.
An interesting hypothesis that has not been extensively explored posits that lithium may affect functions of inositol or inositol phosphates that are independent of PI and PIP2 (Lee et al, 2007; Odom et al, 2000; Seeds et al, 2005; Shaldubina et al, 2002). In addition, specific regions of the brain or compartments within the cell could have uniquely low basal inositol concentrations in vivo, rendering these regions exquisitely sensitive to small reductions in inositol. Alternatively, inhibition of other lithium-sensitive phosphomonoesterases may be responsible for the therapeutic response in BPD, although these potential targets have not yet been as thoroughly explored as IMPase and IPPase. In spite of these controversies, IMPase and structurally related molecules remain important potential targets for lithium action in BPD.
Notably, a recent whole-genome association study of BPD has further implicated the overall phosphoinositide/protein kinase C (PKC) pathway. This study identified a molecule in the diacylglycerol (DAG) limb of the phosphoinositide cascade as a putative BPD susceptibility gene. This study, utilizing North American bipolar pedigrees as a test sample, and German bipolars as the replication sample found a statistically highly significant association (_p_∼10−8) of diacylglycerol kinase η (DGKH) with BPD (Baum et al, in press). DAG is the major activator of PKC, and these genetic findings receive indirect support from the preclinical data suggesting that PKC is a target for antimanic agents (reviewed in Goodwin and Jamison, 2007), and the preliminary clinical efficacy of nonslective PKC inhibitors in the treatment of mania (Bebchuk et al, 2000; Zarate et al, 2007).
Glycogen Synthase Kinase-3
Glycogen synthase kinase-3 is also inhibited by clinically relevant concentrations of lithium, unlike most other protein kinases (Klein and Melton, 1996; Davies et al, 2000) (Figure 5). First identified in mammals as an inhibitor of glycogen synthase, GSK-3 is a ubiquitous, constitutively active, multisubstrate serine/threonine kinase encoded by two closely related genes, Gsk-3α (51 kDa) and Gsk-3β (46 kDa) (Woodgett, 1990). As a key component of many signaling pathways including insulin, neurotrophin, and Wnt pathways, GSK-3 plays a critical role in multiple cellular processes, including metabolism, proliferation, differentiation, axonogenesis and synaptogenesis, development, and apoptosis (Cohen and Frame, 2001; Doble and Woodgett, 2003; Gould et al, 2006; Gurvich and Klein, 2002; Hall et al, 2000; Huang and Klein, 2006; Jope and Johnson, 2004; Kim and Kimmel, 2000; Kim et al, 2006; Salinas, 1999; Shaltiel et al, 2007).
Figure 5
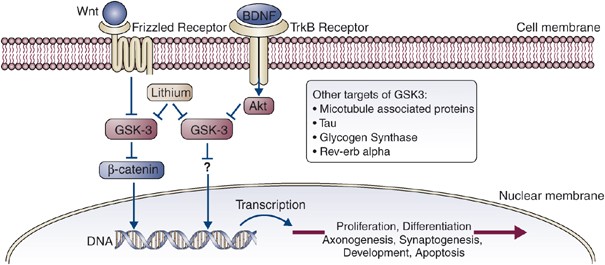
Wnt pathway and GSK-3. MSDs inhibit glycogen synthase kinase 3 (GSK-3). In the Wnt signaling pathway, Wnt glycoproteins interact with the frizzled family of receptors to stimulate the disheveled-mediated inactivation of GSK-3. Inhibition of GSK-3 prevents _β_-catenin phosphorylation and thereby inhibits degradation of this molecule that can act as a transcription factor binding to lef/tcf. Wnt proteins are implicated in the regulation of neuron morphology, neurotransmission, and synaptogenesis.
As a general rule, GSK-3 antagonizes canonical signaling pathways, including the insulin and Wnt pathways. For example, GSK-3 phosphorylates and inhibits glycogen synthase, a downstream effector of insulin action; furthermore, GSK-3-mediated phosphorylation of _β_-catenin, a downstream component of the canonical Wnt pathway, causes rapid degradation of _β_-catenin. Thus, to activate these downstream components, the pathways must inhibit GSK-3. This explains why lithium can activate glycogen synthesis, Wnt/_β_-catenin-dependent transcription, and other pathways normally inhibited by GSK-3.
The best-characterized mode of GSK-3 inhibition is mediated by N-terminal phosphorylation of GSK-3. Insulin activates the protein kinase Akt/PKB, which directly phosphorylates GSK-3 (at serine-21 in GSK-3_α_ and serine-9 in GSK-3_β_) converting the N-terminal sequence of GSK-3 into a pseudosubstrate that autoinhibits the enzyme (Frame et al, 2001; Doble and Woodgett, 2003). Akt activation via other receptor tyrosine kinases (such as neurotrophin receptors) or through G-protein-coupled receptors (such as serotonin receptors) also causes phosphorylation and inhibition of GSK-3. Several other protein kinases (including PKA, PKC, p70S6kinase, and p90rsk) can also phosphorylate GSK-3 in vitro and may inhibit GSK-3 in vivo (Doble and Woodgett, 2003).
Wnt signaling also inhibits GSK-3 enzymatic activity (Cook et al, 1996; Ruel et al, 1999), preventing _β_-catenin degradation, which in turn activates transcription of Wnt-dependent target genes. Wnt-mediated inhibition of GSK-3 is independent of serine-21/9 phosphorylation, as mice lacking serine-21 and -9 have no defects in Wnt signaling (McManus et al, 2005). Instead, the pool of GSK-3 involved in Wnt signaling is associated with the Axin/APC complex and is inaccessible to N-terminal protein kinases (Ding et al, 2000). This has been proposed as a mechanism to insulate Wnt signaling from crosstalk with other GSK-3-regulated signaling pathways.
Lithium directly inhibits GSK-3 at therapeutically relevant concentrations in vitro (Klein and Melton, 1996) and in vivo in diverse cell types, including cultured neurons and rodent brain (Gould et al, 2004a; Hedgepeth et al, 1997; Hong et al, 1997; Lovestone et al, 1999; Munoz-Montano et al, 1997; Noble et al, 2005; O'Brien et al, 2004; Stambolic et al, 1996). Many of the known effects of lithium can be explained in terms of GSK-3 inhibition based on parallels to other physiological (ie insulin- or Wnt-induced effects), genetic (Gsk-3 loss of function), or pharmacological (ie small molecule inhibitors of GSK-3) modes of GSK-3 inhibition, including the effects on glycogen synthesis, early development (Klein and Melton, 1996), neurogenesis, neuronal survival (Chalecka-Franaszek and Chuang, 1999; Chuang, 2004; Gould et al, 2006; Li et al, 2002), and behavior (Beaulieu et al, 2004; Gould et al, 2004b; Kaidanovich-Beilin et al, 2004; O'Brien et al, 2004).
For example, lithium increases neuronal growth cone area (Goold et al, 1999; Hall et al, 2000; Lucas et al, 1998; Williams et al, 2002), alters synaptogenesis (Hall et al, 2000; Salinas, 1999), and stimulates hippocampal neurogenesis (Chen et al, 2000); all of these effects mimic Wnt signaling (Burden, 2000; Lie et al, 2005; Salinas, 1999). Furthermore, N-terminal-phosphorylated GSK-3_β_ localizes to growth cones, and agents that induce growth cone collapse, such as sema-3A and lysophosphatidic acid, also induce dephosphorylation of GSK-3 (Eickholt et al, 2002; Sayas et al, 1999).
GSK-3 also regulates neuronal polarity in developing hippocampal neurons. Local GSK-3 inactivation specifies axon site formation, and global GSK-3 inhibition induces multiple axons (Jiang et al, 2005; Kim et al, 2006; Yoshimura et al, 2005). The effects of lithium on growth cone stability and on neuronal polarity may be mediated via changes in phosphorylation of microtubule-associated proteins that are known targets of GSK-3, including MAP-1B, APC, and CRMP-1 (Cole et al, 2004; Goold et al, 1999; Lucas et al, 1998; Yoshimura et al, 2005; Zhou et al, 2004, 2005). Taken together, these observations indicate that inhibition of GSK-3 by lithium may regulate multiple aspects of axonal morphogenesis.
However, inhibition of GSK-3 by lithium (IC50 ∼1–2 mM in vitro) is achieved at the higher end of its therapeutic range (0.5–1.5 mEq/l), raising some questions about whether this level of inhibition is sufficient to have significant biological effects. On the other hand, IC50 is strongly affected by in vitro assay conditions; lithium inhibits GSK-3 competitively with respect to magnesium and therefore inhibition is reversed by raising the [Mg2+] to superphysiological levels (Gurvich and Klein, 2002; Ryves and Harwood, 2001). Intracellular [Mg2+] is significantly lower than that generally used for in vitro kinase assays, and when GSK-3 is assayed at physiological [Mg2+], the observed IC50 is 0.8–1.0 mM. Nevertheless, this degree of inhibition might still be insufficient to explain the mood-stabilizing effects of lithium, as clinical efficacy can be observed at the low end of the response window.
To address this issue, Jope and colleagues have proposed that a secondary mode of inhibition enhances the direct inhibition by GSK-3 (De Sarno et al, 2002). Thus, in addition to directly inhibiting GSK-3, lithium may also induce N-terminal phosphorylation, enhancing its more direct inhibitory effects (as discussed above) (Bhat et al, 2000; Chalecka-Franaszek and Chuang, 1999; De Sarno et al, 2002; Hall et al, 2002; Lochhead et al, 2006; Noble et al, 2005; Roh et al, 2005; Song et al, 2002; Zhang et al, 2003). Proposed mechanisms of secondary inhibition (Figure 6) include inhibition of the phosphatase that dephosphorylates GSK-3 (which is itself regulated by GSK-3) (Zhang et al, 2003), and activation of Akt through an unknown mechanism (Chalecka-Franaszek and Chuang, 1999). In either case this indirect inhibition would arise through direct inhibition of GSK-3 itself, shown with alternative GSK-3 inhibitors and genetic disruption of Gsk-3β, revealing a positive autoregulatory loop for GSK-3 (Zhang et al, 2003) (Figure 6). In support of this indirect mode of inhibition, evidence for increased N-terminal phosphorylation of GSK-3 in peripheral blood mononuclear cells has been reported in patients treated with lithium (Li et al, 2007). In addition, lithium prevents an activating autophosphorylation on tyrosine of newly translated GSK-3, suggesting an additional mode of inhibition that might show a delayed effect, as it would only affect protein translated in the presence of lithium and therefore would not be manifested until existing GSK-3 had turned over sufficiently (Lochhead et al, 2006).
Figure 6
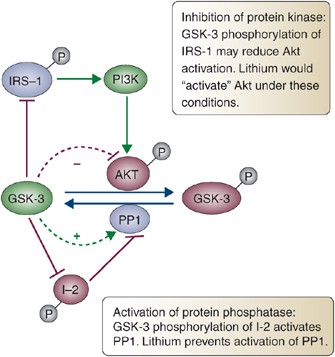
Glycogen synthase kinase 3 (GSK-3) autoregulatory loop. In addition to directly inhibiting GSK-3, lithium (as well as other inhibitors) induces N-terminal phosphorylation of GSK-3. This inhibitory phosphorylation may enhance the effect of direct inhibitors and appears to be achieved both through activation of Akt and through inhibition of protein phosphatase 1 (PP1). These observations reveal an autoregulatory loop in which GSK-3 maintains itself in an active state, which is then interrupted by direct inhibition of GSK-3 (‘P’ indicates phosphorylation; IRS-1, insulin receptor substrate-1; PI3K, phosphatidylinositol-3 kinase; PP1, protein phosphatase 1; I-2, PP1-specific inhibitor-2; - -∣ indicates inhibitory step; and - -> indicates activating step).
Behavioral Effects of VPA and Lithium that may be Mediated by the GSK-3 and/or IMPase Cascades
Preclinical work has found that chronic lithium treatment in mice reduces immobility time in the forced swim test (FST) and reduces exploratory behavior in the hole-board apparatus without affecting overall activity in the open field test (OFT) or other general measures of the state of the animal (O'Brien et al, 2004). Similarly, mice treated with VPA for 5–10 days display reduced immobility in the FST (Semba et al, 1989) and reduced exploratory behavior without an accompanying reduction in locomotor activity or rearing (File and Aranko, 1988; Rao et al, 1991). Lithium and VPA also attenuate amphetamine- and chlordiazepoxide-induced hyperlocomotion (Cao and Peng, 1993; Murphy, 1977). The hypothesis that these behavioral effects could be mediated by direct or indirect inhibition of GSK-3 is supported by pharmacological and genetic evidence. For instance, two structurally distinct GSK-3 inhibitors—AR-A014418 (Gould et al, 2004b) and the peptide L803-mts (Kaidanovich-Beilin et al, 2004)—reduce immobility in the FST and attenuate amphetamine-induced hyperactivity. In addition, GSK-3 overexpression reportedly increases activity (although this transgenic line also displayed compensatory changes in Akt signaling that may complicate analysis of their behavior (Prickaerts et al, 2006)). The specific behavioral responses to lithium and other GSK-3 inhibitors are observed in animals lacking one copy of the Gsk-3β gene (Beaulieu et al, 2004; O'Brien et al, 2004), establishing strong genetic and pharmacological support that these behavioral effects of lithium are mediated by inhibition of GSK-3.
In contrast, genetic perturbations that reduce inositol in rodent brain do not affect lithium-sensitive behaviors. As discussed above, lithium and VPA partially reduce in vivo brain inositol levels, and in invertebrate model systems, mutations that interfere with inositol turnover cause clear defects in synaptic function, including at the neuromuscular junction in Drosophila and in synaptic function regulating thermotaxis behavior in C. elegans. However, in adult mice heterozygous for the inositol transporter SMIT1, inositol levels in the brain are reduced to an even greater extent (33–37% reduction) than in lithium-treated wild-type siblings (22–25%); nevertheless, this reduction in inositol has no effect on the FST (Shaldubina et al, 2006) or other lithium-sensitive behaviors (Shaldubina et al, 2007), indicating that inositol reduction is not responsible for the behavioral effects of lithium in mice. Knockout of the IMPA2 gene in mice reportedly also did not yield lithium-like behaviors, but apparently these mice also did not show a reduction in brain inositol, perhaps because of redundancy with IMPA1 (Cryns et al, 2007).
Reduction of GSK-3 activity by lithium is predicted to increase _β_-catenin signaling and activation of Wnt-dependent gene expression. This was confirmed by examining a transgenic mouse line expressing a Wnt reporter (BAT-Gal); when these reporter mice were treated with lithium (1 mEq/l) or VPA, Wnt/_β_-catenin reporter activity enhanced specifically in the dentate gyrus (DG), medial amygdala, and, weakly, in the hypothalamus (O'Brien et al, 2004; Freidman, O'Brien, and Klein, unpublished data). Furthermore, overexpression of a stabilized form of _β_-catenin leads to reduced immobility in the FST (Gould et al, 2007), paralleling the effects of lithium, other GSK-3 inhibitors, and Gsk-3β+/− mice.
Another interesting observation concerns circadian period lengths, which are shortened in patients with BPD and lengthened with lithium treatment. This behavioral effect is paralleled by changes in GSK-3 phosphorylation in both mice and cultured cells. In Drosophila, both lithium and VPA can prolong the circadian period (Dokucu et al, 2005; Kaladchibachi et al, 2007; Padiath et al, 2004) (reviewed in Gould and Manji, 2005; Martinek et al, 2001; McClung, 2007). Moreover, a dominant-negative mutation in the mouse CLOCK gene results in hyperactivity, decreased sleep, increased mobility in the FST, and heightened reward value for cocaine or sucrose. These effects have been interpreted to indicate a manic state, and interestingly, these alterations can be reversed by chronic lithium treatment (Roybal et al, 2007). The Bmal1/CLOCK complex regulates expression of the nuclear hormone receptor rev-erbα; the rev-erb_α_ protein is in turn stabilized by GSK-3 phosphorylation and represses Bmal1 expression. Lithium therefore destabilizes rev-erb_α_ and results in activation of Bmal1 (Yin et al, 2006), which could play a central role in the circadian response to lithium.
Because both inositol phosphates and GSK-3 affect multiple downstream pathways, inhibition of IMPase and GSK-3 could affect overlapping targets, thereby yielding similar cellular or morphological readouts in specific settings, such as growth cone spreading/collapse or neuronal survival. In support of this idea, interesting recent work from Greenberg and colleagues showed that GSK-3 is required for optimal myoinositol-3 phosphate synthase activity and de novo inositol biosynthesis (Azab et al, 2007) in budding yeast. If similar regulation is present in mammals, this could provide an additional mechanism for inositol reduction caused by lithium, and, more generally, suggest interplay between GSK-3 and inositol pathways.
Cellular Signaling Cascades Converge to Regulate Synaptic Plasticity and Behavioral Plasticity
As discussed previously, it is now clear that certain intracellular signaling cascades play important roles in the pathophysiology and treatment of severe mood disorders. An important consideration to address at this point is how can changes in intracellular molecules bring about complex behavioral changes? These signaling cascades undoubtedly converge to regulate synaptic plasticity, and thereby information processing in critical circuits mediating the affective, cognitive, motoric, and somatic manifestations of mood disorders.
In this context, it is now clear that modification of the levels of synaptic AMPA receptors, in particular by receptor subunit trafficking, insertion, and internalization, is a critically important mechanism for regulating various forms of synaptic plasticity (Malinow and Malenka, 2002). Thus, through phosphorylation of specific sites on AMPA receptor subunit GluR1, GluR1 trafficking is regulated by protein kinase A (PKA), Ca2+/calmodulin-dependent protein kinase II, and PKC (Du et al, 2004b, 2006). Phosphorylation/dephosphorylation of the receptor subunits regulates both the intrinsic channel properties of the receptor and the interaction of the receptor with associated proteins that modulate the membrane trafficking and synaptic targeting of the receptor. _N_-methyl-D-aspartate (NMDA) receptor subunits are regulated by similar mechanisms. It was therefore postulated that the effects of mood stabilizers/antidepressants ultimately converge to regulate AMPA and NMDA synaptic transmission. The regulation of transmission at the synapse may be mediated by changes in neurotransmitter levels, receptor subunit phosphorylation, surface/cellular levels or receptors, and conductance changes, among others. For the purposes of general discussion, we use the term ‘throughput,’ as we discuss the studies that identified AMPA and NMDA receptors as targets for the actions of mood stabilizers and antidepressants.
Chronic lithium and VPA regulate surface/synaptic GluR1 and GluR2 levels
A series of studies was undertaken to test the hypothesis that lithium and VPA's effects on intracellular signaling cascades converge to regulate surface and/or synaptic AMPA receptors (Du et al, 2003, 2004a, 2004b). Using three independent assays, it was demonstrated that chronic treatment of rats with therapeutically relevant concentrations of lithium or VPA reduced hippocampal synaptosomal GluR1 and GluR2 levels via a reduction of surface GluR1 and GluR2 distribution onto the neuronal membrane (Du et al, 2003, 2004a, 2004b). In addition, both agents induced a decrease in GluR1 phosphorylation at a specific PKA site (GluR1p845) known to facilitate AMPA receptor insertion and opening of the sodium channel. Notably, in striking contrast to the effects observed with lithium and VPA, antidepressants, psychostimulants, dopamine agonists, and sleep deprivation have all been shown to increase phosphorylation and/or synaptic levels of GluR1 receptors (Du et al, 2004a, 2007). Complementary behavioral studies suggest that attenuating hippocampal AMPA throughput attenuates manic-like behaviors (Du et al, submitted).
Anticonvulsants with a predominantly antidepressant profile enhance surface/synaptic GluR1 and GluR2 receptors
Anticonvulsants are being increasingly used in the treatment of BPD. They were initially thought to exert predominantly antimanic effects by suppressing neuronal excitability; however, recent data suggest that some anticonvulsants—notably lamotrigine and riluzole—may have an antidepressant profile (Zarate et al, 2005; Zarate et al, 2006b). It was found that lamotrigine and riluzole significantly enhanced the surface expression of GluR1 and GluR2 in a time and dose-dependent manner in cultured hippocampal neurons; by contrast, the antimanic anticonvulsant VPA significantly reduced surface expression of GluR1 and GluR2 (Du et al, 2007). Concomitant with the GluR1 and GluR2 changes, the peak value of depolarized membrane potential evoked by AMPA was significantly higher in lamotrigine- and riluzole-treated neurons, supporting the surface receptor changes. Phosphorylation of GluR1 at the PKA site (S845) was enhanced in both lamotrigine- and riluzole-treated hippocampal neurons, but reduced in VPA-treated neurons. In addition, lamotrigine and riluzole, as well as the traditional antidepressant imipramine, increased GluR1 phosphorylation at GluR1 (S845) in the hippocampus after chronic in vivo treatment (Du et al, 2007).
Ketamine's antidepressant effects involve NMDA/AMPA receptor interactions
Building upon these preclinical data, recent clinical trials have investigated the clinical effects of glutamatergic agents in subjects with mood disorders. These studies have demonstrated effective and rapid antidepressant action of glutamatergic agents, including ketamine, an NMDA receptor antagonist (Zarate et al, 2006a, 2006b). It was found that a single intravenous dose of the noncompetitive NMDA antagonist ketamine produced a rapid (within 2 h), robust (70% response on day 1) and relatively sustained (approximately 1 week) antidepressant effect in patients with treatment-resistant major depression (Zarate et al, 2006a). Preclinical studies have revealed that in addition to its antidepressant effects in humans ketamine produces antidepressant-like behavioral effects in rodent models. A single administration of ketamine facilitated animal recovery from behavioral despair within 24 h in the learned helplessness paradigm. A single administration of ketamine also reduced animal immobility in the FST, with effects lasting for at least 2 weeks. A selective NR2B antagonist also exerted antidepressant-like effects (Maeng et al, 2007). Pretreatment with NBQX, an AMPA receptor antagonist, attenuated both ketamine-induced antidepressant-like behavioral alterations and regulation of hippocampal phosphorylations of GluR1 AMPA receptors at serine 845 (Maeng et al, 2007). These and other data have led to the hypothesis that alterations in neural plasticity in critical limbic and reward circuits, mediated by increasing the postsynaptic AMPA to NMDA throughput, may represent a convergent mechanism for antidepressant action (Zarate et al, 2006b). This line of research holds considerable promise for developing new treatments for both MDD and BPD.
Indirect, Long-Term Targets of Mood-Stabilizing Agents Involved in Neuroplastic Processes
Several clinical observations of patients undergoing treatment for BPD are noteworthy when considering alterations in neuroplastic processes as an underlying mechanism of mood stabilizer action. These include the time course of onset for antimanic and antidepressant effects of mood stabilizers, the efficiency of mood stabilizer maintenance treatment in episode prevention, and observations on the time to recurrence after treatment withdrawal (Goodwin and Jamison, 2007). The mood stabilizers discussed in this article, namely lithium and the anticonvulsants, do not appear to have rapid effects (within minutes or hours), but instead require days to weeks to achieve their maximal response (Goodwin and Jamison, 2007). Further support for long-term neuroplastic adaptations comes from observations regarding episode relapse after lithium cessation. Particularly when done rapidly, discontinuation of lithium maintenance is clearly associated with increased rates of relapse (Davis et al, 1999; Suppes et al, 1991). Interestingly, and again consistent with longer-term neuroplastic changes, new episodes are not experienced immediately—as would be expected for an acute withdrawal mechanism—but, rather, are seen approximately 3–9 months after medication withdrawal. The recurrence of episodes during the course of BPD is a defining feature and occurs in a nonrandom fashion. Therefore, treatments for BPD must not only address symptoms of mania and depression during acute episodes, but also the underlying vulnerability predisposing to their recurrence. Indeed, maintenance treatment with mood stabilizers seems capable of preventing the onset of new episodes and attenuating the symptoms experienced upon eventual relapse (Baastrup and Schou, 1967; Gyulai et al, 2003).
REGULATION OF CELL SURVIVAL AND RESILIENCE PATHWAYS BY MOOD STABILIZERS
Lithium and VPA Activate the ERK Signaling Cascade
In view of the important role that the extracellular receptor-coupled kinase (ERK) signaling cascade plays in mediating long-term neuroplastic events (Figure 7), a series of studies were undertaken to investigate the effects of lithium and VPA on this signaling cascade (Chen and Manji, 2006; Yuan et al, 2001). These studies showed that lithium and VPA, at therapeutically relevant concentrations, robustly activate the ERK MAPK cascade in human neuroblastoma SH-SY5Y cells (Chen and Manji, 2006; Yuan et al, 2001). Recent follow-up studies showed that similar to the effects observed in neuroblastoma cells in vitro chronic lithium and VPA also robustly increase the levels of activated ERK in areas of the brain that have been implicated in the pathophysiology and treatment of BPD—the anterior cingulate cortex and hippocampus (Chen and Manji, 2006).
Figure 7
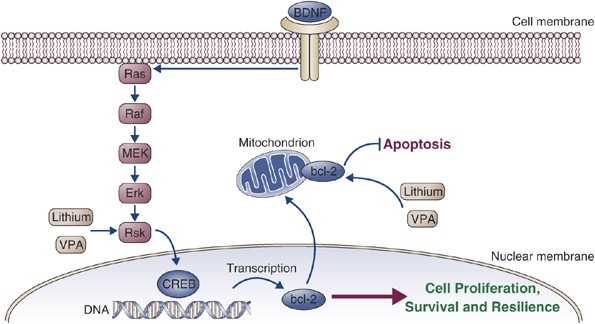
Erk pathway and bcl-2. Lithium and valproate (VPA), at therapeutically relevant concentrations, robustly activate the extracellular receptor-coupled kinase (ERK) MAPK cascade. One of the major downstream targets of the ERK MAPK cascades is arguably one of the most important neuroprotective proteins, bcl-2. Bcl-2 is expressed in the rodent and mammalian nervous system and is localized to the outer mitochondrial membrane, endoplasmic reticulum, and nuclear membrane. It is now clear that bcl-2 is a protein that inhibits both apoptotic and necrotic cell death induced by diverse stimuli. Bcl-2 attenuates apoptosis by sequestering proforms of caspases, preventing the release of mitochondrial apoptogenic factors such as calcium and cytochrome c, and by enhancing mitochondrial calcium uptake.
Animal behavioral studies have shown that chemical inhibition of brain ERK pathway in rats reduces immobility in the FST and increased locomotive/explorative activity in the large OFT. These studies also show that ERK1 (one of two ERK subtypes) knockout mice have a brain region-specific functional deficit of the ERK pathway and exhibit reduced immobility in the FST, increased activity in the OFT, persistently increased home-cage wheel running activity for at least 30 days, and enhanced response to psychostimulants (reviewed in Chen and Manji, 2006). Very recent studies have therefore examined the role of the ERK pathway as a behavioral modulator in the left anterior cingulate cortex, one of the brain regions being implicated in the pathophysiology of mood disorders by human brain imaging and post-mortem studies; these pharmacological and genetic studies have shown that attenuating ERK activity in the ACC may be associated with manic-like behaviors (Chen and Manji, 2006).
However, it would undoubtedly be an oversimplication to suggest that ERK inhibition in the brain always results in manic-like behavior. Recent data from several laboratories clearly show that certain molecules with functions related to neuronal plasticity (eg BDNF, CREB, and ERK) can have divergent effects on rodent behavior, depending on the brain region manipulated (Berton et al, 2006; Duman et al, 2007; Eisch et al, 2003). Notably, recent studies from the Duman (Duman et al, 2007) and Manji laboratories (Shaltiel et al, 2007) raise the possibility that ERK may exert regionally selective effects on affective-like behaviors. Moreover, emerging evidence suggests that BDNF plays different and perhaps opposing roles in the brain stress system including the hippocampus and hypothalamus–pituitary–adrenocortical axis, and the brain reward system including the nucleus accumbens and the ventral tegmental area (Berton et al, 2006; Eisch et al, 2003). It is clear that specific circuits and brain regions are undoubtedly involved in the pathophysiology of mood disorders; defining the effects of altering-specific signaling pathways, in specific brain regions of rodents, will allow for translational studies of similar brain regions/functions in humans (ie studies of human endophenotypes).
Bcl-2 is a Therapeutic Target for the Actions of Lithium and VPA
One of the major downstream targets of the ERK MAPK cascades is arguably one of the most important neuroprotective proteins, bcl-2. Bcl-2 is expressed in the rodent and mammalian nervous system and is localized to the outer mitochondrial membrane, endoplasmic reticulum, and nuclear membrane. It is now clear that bcl-2 is a protein that inhibits both apoptotic and necrotic cell death induced by diverse stimuli (Adams and Cory, 1998; Bruckheimer et al, 1998; Merry and Korsmeyer, 1997, and references therein). Several cellular mechanisms are likely involved in mediating bcl-2's protective effects, including sequestering the proforms of caspases, inhibiting the effects of caspase activation, antioxidant effects, enhancing mitochondrial calcium uptake, and attenuating the release of calcium and cytochrome c from mitochondria (reviewed in Adams and Cory, 1998; Bruckheimer et al, 1998; Li and Yuan, 1999; Sadoul, 1998). The role for bcl-2 in protecting neurons from cell death is now supported by abundant evidence; bcl-2 has been shown to protect neurons from a variety of insults in vitro including growth factor deprivation, glucocorticoids, ionizing radiation, and oxidant stressors such as hydrogen peroxide, _tert_-butylhydroperoxide, reactive oxygen species, and buthionine sulfoxamine (Adams and Cory, 1998; Bruckheimer et al, 1998). In addition to these potent in vitro effects, bcl-2 also prevents cell death in numerous studies in vivo.
In the absence of pharmacological means of increasing CNS bcl-2 expression (until recently), all the studies have hitherto utilized transgenic mouse models or viral vector-mediated delivery of the bcl-2 gene into the CNS. In these models, bcl-2 overexpression has been shown to prevent motor neuron death induced by facial nerve axotomy and sciatic nerve axotomy, to save retinal ganglion cells from axotomy-induced death, to protect cells from the deleterious effects of MPTP or focal ischemia, and to protect photoreceptor cells from two forms of inherited retinal degeneration; interestingly, neurons that survive ischemic lesions or traumatic brain injury in vivo show upregulation of bcl-2 (Bonfanti et al, 1996; Chen et al, 1997; Lawrence et al, 1996; Merry and Korsmeyer, 1997; Raghupathi et al, 1998; Sadoul, 1998; Yang et al, 1998, and references therein).
Overexpression of bcl-2 has also recently been shown to prolong survival and attenuate motor neuron degeneration in a transgenic animal model of amyotrophic lateral sclerosis (Kostic et al, 1997). Not only does bcl-2 overexpression protect against apoptotic and necrotic cell death, it can also promote regeneration of axons in the mammalian CNS, leading to the intriguing postulate that bcl-2 acts as a major regulatory switch for a genetic program that controls the growth of CNS axons (Chen et al, 1997). Because bcl-2 has also recently been shown to promote neurite sprouting, increasing CNS bcl-2 levels may represent a very effective therapeutic strategy for the treatment of many neurodegenerative diseases (Chen et al, 1997).
Lithium and VPA Robustly Increase the Expression of bcl-2
Chronic treatment of rats with therapeutic doses of lithium and VPA doubled bcl-2 levels in the frontal cortex, an effect primarily due to a marked increase in the number of bcl-2 immunoreactive cells in layers II and III of the anterior cingulated cortex (Chen et al, 1999; Manji et al, 1999, 2000). Interestingly, the importance of neurons in the anterior cingulate has recently been emphasized in neuroimaging studies of BPD, particularly because these areas provide connections with other cortical regions and are targets for subcortical input (Rajkowska, 2000). Chronic lithium was also found to markedly increase the number of bcl-2 immunoreactive cells in the DG and striatum (Manji et al, 1999). Subsequent to these findings, lithium was shown to increase bcl-2 levels in C57BL/6 mice (Chen et al, 1999), in human neuroblastoma SH-SY5Y cells in vitro (Manji et al, 2000), and in rat cerebellar granule cells in vitro (Chen et al, 1999).
Overall, the data clearly show that chronic lithium robustly increases levels of the neuroprotective protein bcl-2 in areas of rodent frontal cortex, hippocampus, and striatum in vivo, and in cultured cells of both rodent and human neuronal origin in vitro. Furthermore, at least in cultured cell systems, lithium reduces levels of the proapoptotic protein p53. As demonstrated recently, repeated electroconvulsive shocks also significantly increase precursor cell proliferation in the DG of the adult monkey, an effect that appears to be due to increased expression of bcl-2 (Perera et al, 2007). These results suggest that stimulation of neurogenesis and enhanced expression of bcl-2 may contribute to the therapeutic actions of ECT.
Behavioral studies have also been undertaken to determine if bcl-2 plays a role in the pathogenesis and treatment of depression (Yuan et al, unpublished data). Bcl-2+/− mice and wild-type littermates were also treated with the antidepressant citalopram (10 mg/kg/day) acutely and chronically, and responses in the tail suspension and learned helplessness tests were examined. In the learned helplessness test, bcl-2+/− mice demonstrated a significantly higher rate of escape failures in the bcl-2+/− mice. Furthermore, while chronic citalopram decreased escape failures in the wild-type mice, it did not affect the bcl-2+/− mice. Similarly, citalopram was effective in wild-type mice in the tail suspension test, but did not affect the bcl-2+/− mice. These data demonstrate that Bcl-2+/− mice are insensitive to the SSRI antidepressant citalopram in two animal models of depression, suggesting that some of the therapeutic effects of antidepressants may be mediated through the actions of bcl-2. In total, these observations suggest that regulation of bcl-2-mediated plasticity is likely to play an important role in regulating synaptic throughput in the circuitry-mediating complex behaviors (Yuan et al, unpublished data).
BAG1 (bcl-2-Associated Athanogene) is also a Long-Term Target for Mood Stabilizers
Recent microarray studies with stringent validating criteria identified bcl-2-associated athanogene (BAG1) as a target for the actions of mood stabilizers (Zhou et al, 2005). BAG1 interacts with glucocorticoid receptors, Bcl-2, Hsp70, and Raf, thereby regulating cell survival pathways and glucocorticoid function. The potential role of BAG1 in regulating affective-like behavior was investigated in mice selectively overexpressing BAG1 in the brain. BAG1-overexpressing mice displayed less anxiety on the anxiety-related tests. The mice did not differ from controls on measures of immobility in the FST or helplessness in the learned helplessness paradigm; however, the BAG1 mice showed higher spontaneous recovery rates from the helplessness behavior. On mania-related tests, BAG1-overexpressing mice recovered much faster in the amphetamine-induced hyperlocomotion test, and displayed a clear resistance to cocaine-induced behavioral sensitization. BAG1-overexpressing mice had specific hippocampal neurochemical alterations including increased Hsp70 and decreased FKBP51 levels. These data suggest that BAG1, a novel target for the actions of mood stabilizers, plays an important role in affective resilience (Maeng et al, 2007).
Lithium Exerts Robust Neuroprotective Effects in Preclinical Paradigms
In view of its major effects on GSK-3, bcl-2, and BAG1, it is not surprising that recent studies have investigated lithium's potential neuroprotective effects in a variety of preclinical paradigms. Lithium demonstrated robust neuroprotective properties against a variety of insults (reviewed in Bachmann et al, 2005; Chuang and Priller, 2006; Manji et al, 2000) (Table 3). Notably, lithium pretreatment protected cerebral and cerebellar neurons in primary culture from glutamate-induced, NMDA receptor-mediated apoptosis (reviewed in Chuang and Priller, 2006). Excessive NMDA throughput is likely involved in stress-induced hippocampal atrophy, and has been implicated in the pathogenesis of a variety of neurodegenerative diseases such as stroke, Huntington's disease, ALS, spinal cord injury, brain trauma, and cerebellar degeneration. In cultured neurons, lithium-induced neuroprotection against glutamate excitotoxicity occurred within the therapeutic concentration range of this drug and required 5–6 days pretreatment for maximal effects. Lithium neuroprotection involved BDNF induction and activation of its receptor, TrkB, and was associated with upregulation of the antiapoptotic protein bcl-2, downregulation of the proapoptotic proteins p53 and Bax, and inhibition of caspase-3. Treatment of cultured neurons with other GSK-3 inhibitors or transfection with GSK-3 siRNA mimicked the neuroprotective effects of lithium (Liang and Chuang, 2007), again suggesting a critical role for GSK-3 in mediating neuroprotection.
Table 3 Neurotrophic and Neuroprotective Effects of Lithium
Lithium has also showed beneficial effects in a number of animal models of neurodegenerative diseases. For example, pre- or post-insult treatment with lithium suppressed cerebral ischemia-induced brain infarction, caspase-3 activation, and neurological deficits in rats, and these neuroprotective effects were associated with induction of heat shock protein 70 and decreased expression of Bax (Ren et al, 2003; Xu et al, 2003). Several independent studies demonstrated that lithium has neuroprotective effects in animal and cellular models of Alzheimer's, Huntington's, and Parkinson's diseases, retinal degeneration, spinal cord injury, and HIV infection (reviewed in Chuang and Priller, 2006). Notably, therapeutic concentrations of lithium, by acting on GSK-3, block the production of A_β_ peptides by interfering with amyloid precursor protein (APP) cleavage at the γ_-secretase step (Phiel et al, 2003; Rockenstein et al, 2007; Ryder et al, 2003; Su et al, 2004). Importantly, lithium also blocks the accumulation of A_β peptides in the brains of mice that overproduce APP (Phiel et al, 2003; Rockenstein et al, 2007; Ryder et al, 2003; Su et al, 2004). Similarly, lithium administration has been shown to significantly lower levels of phosphorylation at several epitopes of τ known to be hyperphosphorylated in Alzheimer's disease and to significantly reduce levels of aggregated, insoluble τ (Munoz-Montano et al, 1997; Noble et al, 2005). Furthermore, levels of aggregated τ correlate strongly with the degree of axonal degeneration, and lithium-treated mice showed less degeneration if administration was started during early stages of tangle development.
Lithium is also neuroprotective in APP transgenic mice (Rockenstein et al, 2007). Thus, mice treated with lithium displayed improved performance in the water maze, preservation of the dendritic structure in the frontal cortex and hippocampus, and decreased τ phosphorylation (Rockenstein et al, 2007). Chronic lithium treatment also protected against neurodegeneration and improved spatial learning deficits in rats perfused with A_β_ fibrils (De Ferrari et al, 2003). Interestingly, some preliminary retrospective human studies show that long-term lithium treatment may have protective effects against Alzheimer's disease and/or cognitive deficits (Angst et al, 2007; Nunes et al, 2007; Terao et al, 2006).
Mood Stabilizers Increase Neurogenesis
Neurogenesis persists in the mammalian brain into adulthood and is stimulated by several mood stabilizers. Seminal work by the Duman laboratory demonstrated that various classes of antidepressants enhance adult hippocampal neurogenesis (Pittenger and Duman, 2008). Lithium stimulates progenitor cell proliferation in cultured neurons (Hashimoto et al, 2003) and increases BrdU-positive cells in the adult rat hippocampus by approximately 25% (Chen et al, 2000; Son et al, 2003). VPA has similar effects on neurogenesis, specifically increasing the proliferation of rat cerebral cortical cells in culture and the number of BrdU-positive neurons in the mouse DG (Hao et al, 2004). Interestingly, the Wnt ligands Wnt3a, Wnt7a, and Wnt7b are expressed in the subgranular zone of the DG, and canonical Wnt signaling appears to be critical for postnatal hippocampal neurogenesis (Lie et al, 2005; Maekawa et al, 2005). In addition, a Wnt-dependent transcription reporter is active in the DG (Lie et al, 2005; Maekawa et al, 2005) and is further enhanced in vivo by lithium treatment (O'Brien et al, 2004). Moreover, recent findings suggest canonical Wnt signaling in general, and GSK-3 specifically, are major regulators of stem cell self-renewal and proliferation (Reya and Clevers, 2005; Trowbridge et al, 2006).
HUMAN EVIDENCE FOR THE NEUROTROPHIC EFFECTS OF LITHIUM
While the body of preclinical data demonstrating neurotrophic and neuroprotective effects of lithium is striking, considerable caution must clearly be exercised in extrapolating to the clinical situation with humans. In view of lithium's robust effects on the levels of the cytoprotective protein bcl-2 in the anterior cingulate, Drevets and colleagues re-analyzed older data demonstrating ∼40% reductions in subgenual PFCPFC volumes in familial mood disorder subjects (Drevets, 2001). Consistent with neurotrophic/neuroprotective effects of lithium, they found that patients treated with chronic lithium or VPA exhibited subgenual PFC volumes that were significantly higher than those in non-lithium- or non-VPA-treated patients, and not significantly different from controls. To investigate the potential neurotrophic effects of lithium in humans more definitively, Moore et al (2000a) used proton MRS and demonstrated that treatment of bipolar patients with lithium for 4 weeks increased the level of NAA, a marker of neuronal viability, in the cerebral cortex. A follow-up volumetric MRI study demonstrated that 4 weeks of lithium treatment also significantly increased total gray matter content in the human brain (Moore et al, 2000b), suggesting the possibility of an increase in the volume of the neuropil (the moss-like layer comprised of axonal and dendritic fibers that occupies much of the cortex gray matter volume). A subsequent study by Sassi et al (2002) confirmed a similar increase by lithium in gray matter volume, compared to the brains of untreated patients and healthy subjects.
Another study of familial pediatric BPD revealed that subjects with BPD with past exposure to lithium or VPA tended to have greater amygdala gray matter volume than subjects with BPD without such exposure (Chang et al, 2005). Yucel et al (2007) compared the volume of the hippocampus, hippocampal head (Hh), and body/tail in three groups with no history of medication use before entry into the study: (1) a group of patients treated with lithium for 1–8 weeks and then scanned, (2) a group comprising patients who were unmedicated at the time of scan, and (3) a group of patients treated with either VPA or lamotrigine for 1–8 weeks. They observed a bilateral increase in hippocampal and Hh volumes in the lithium-treated group compared to the unmedicated group, an effect that was apparent even over a brief treatment period (Yucel et al, 2007).
Another study used high-resolution MRI and cortical pattern matching methods to map gray matter differences in 28 BPD patients, 20 of whom were lithium treated, and 28 healthy were controls (Bearden et al, 2007). Their results showed that gray matter density was significantly greater in diffused cortical regions in bipolar patients than in healthy subjects; the differences were most pronounced in the bilateral cingulated and paralimbic cortices, which are areas used in attention, motivation, and emotion. In addition, their data revealed greater gray matter density in the right anterior cingulate in lithium-treated patients relative to the bipolar subjects not taking lithium. Their lithium-treated sample included subjects who were on the drug for varying time durations and their dosages were not uniform. The lack of difference in the gray mater density between the untreated patients and healthy controls, as well as the growing evidence that lithium exerts major effects on a number of cellular proteins and pathways (see above) known to regulate cell atrophy/death lend support to the view that the gray matter enlargement is mediated through the trophic actions of lithium in the brain (Chuang and Manji, 2007).
FUTURE RESEARCH DIRECTIONS
As we have reviewed here, a considerable body of evidence supports abnormalities in the regulation of cellular plasticity cascades as integral to the underlying neurobiology of BPD. Many of these pathways play critical roles not only in ‘here and now’ synaptic plasticity (and therefore profound changes in mood), but also in long-term cell growth/atrophy and cell survival/cell death. Indeed, the atrophic changes observed in multiple cell types (neurons and glia), as well as the reversibility of the changes with treatment support a role for intracellular plasticity cascades. It is likely that the major defect is in the ability to regulate neuroplastic adaptations to perturbations (both physiological and pathophysiological)—an inability to handle ‘normal loads’ (neurochemical, hormonal, stress-induced, pharmacologically induced, etc) without failing or invoking compensatory adaptations that overshoot and predispose to oscillations. Indeed, the allostatic load would thus contribute to long-term disease progression (and potentially to cycle acceleration). Many of the very same cascades involved in regulating synaptic plasticity also play a critical role in cell atrophy and cellular resilience. These observations serve to explain the atrophic aspect of the illness in some patients, as well as the presence of stigmata normally associated with ischemic/hypoxic insults, such as WMH.
In the past decade several studies have questioned the common belief that BPD is associated with a favorable prognosis and good long-term outcome; this has led both clinicians and researchers to focus on the early stages of the illness to study psychopathological prodromes and biological developmental abnormalities that could guide new treatment algorithms in high-risk populations. Neuroimaging studies have shown widespread cortical and subcortical involvement in BPD even in patients experiencing their first episode; prospective studies have also suggested that after onset, new brain pathological remodeling takes place in areas involved in BPD pathophysiology, with gray matter loss, white matter abnormalities, and new functional and cognitive deficits. Evidence also suggests that somewhat akin to the treatment of conditions such as hypertension and diabetes, early and sustained treatment may be necessary to adequately prevent the deleterious long-term sequelae associated with mood disorders (Leverich et al, 2007; Post, 2007). However, careful selection of the subjects who might benefit from early intervention and appropriate study designs for a correct evaluation of the outcome are needed; prospective large-scale studies from high-risk populations with appropriate biological markers could help identify ‘real’ high-risk subjects and develop new treatment algorithms.
Unfortunately, there has been little progress in developing truly novel medications specifically for the treatment of BPD, and most recent additions to the pharmacopeia are brain penetrant drugs developed for the treatment of epilepsy or schizophrenia. This era may now be over as there are a number of pharmacological ‘plasticity-enhancing’ strategies that may be of considerable utility in the treatment of BPD. Indeed, these next-generation drugs, in addition to treating the core symptoms of BPD, might be able to target other important aspects of the illness. These include enhancing cognition independent of any improvement in mood symptoms, and preventing or reversing epigenetic factors that may have long-term negative impacts on the course of the illness.
We are optimistic that the advances outlined here will ultimately lead to the discovery of new approaches for the prevention and treatment of some of mankind's most devastating and least understood illnesses. The development of novel therapeutics holds much promise for the long-term treatment of severe mood disorders and for improving the lives of the many who suffer from them.
References
- Acharya JK, Labarca P, Delgado R, Jalink K, Zuker CS (1998). Synaptic defects and compensatory regulation of inositol metabolism in inositol polyphosphate 1-phosphatase mutants. Neuron 20: 1219–1229.
Article CAS PubMed Google Scholar - Adams JM, Cory S (1998). The Bcl-2 protein family: arbiters of cell survival. Science 281: 1322–1326.
Article CAS PubMed Google Scholar - Altshuler LL, Curran JG, Hauser P, Mintz J, Denicoff K, Post RM (1995). T2 hyperintensities in bipolar disorder: magnetic resonance imaging comparison and literature meta-analysis. Am J Psychiatry 152: 1139–1144.
Article CAS PubMed Google Scholar - Angst J, Gamma A, Gerber-Werder R, Zarate CA, Manji HK (2007). Does long-term medication with lithium prevent or attenuate dementia in bipolar and depressed patients? Int J Psychiatry Clin Pract 11: 2–8.
Article CAS PubMed Google Scholar - Azab AN, He Q, Ju S, Li G, Greenberg ML (2007). Glycogen synthase kinase-3 is required for optimal de novo synthesis of inositol. Mol Microbiol 63: 1248–1258.
Article CAS PubMed Google Scholar - Baastrup PC, Schou M (1967). Lithium as a prophylactic agents. Its effect against recurrent depressions and manic-depressive psychosis. Arch Gen Psychiatry 16: 162–172.
Article CAS PubMed Google Scholar - Bachmann RF, Schloesser RJ, Gould TD, Manji HK (2005). Mood stabilizers target cellular plasticity and resilience cascades: implications for the development of novel therapeutics. Mol Neurobiol 32: 173–202.
Article CAS PubMed Google Scholar - Baum A, Akula N, Cabenero M, Cardona I, Corona W, Klemens B et al (2007). A genome-wide association study implicates diacylglycerol kinase eta (DGKH) and several other genes in the etiology of bipolar disorder. Mol Psychiatry; advance online publication 8 May 2007; doi:101038/sjmp4002012.
- Baumann B, Danos P, Krell D, Diekmann S, Leschinger A, Stauch R et al (1999a). Reduced volume of limbic system-affiliated basal ganglia in mood disorders: preliminary data from a postmortem study. J Neuropsychiatry Clin Neurosci 11: 71–78.
Article CAS PubMed Google Scholar - Baumann B, Danos P, Krell D, Diekmann S, Wurthmann C, Bielau H et al (1999b). Unipolar-bipolar dichotomy of mood disorders is supported by noradrenergic brainstem system morphology. J Affect Disord 54: 217–224.
Article CAS PubMed Google Scholar - Bear M, Dolen G, Osterweil E, Nagarajan N (2008). Fragile X: translation in action. Neuropsychopharmacology Rev 33: 84–87.
Article CAS Google Scholar - Bearden CE, Hoffman KM, Cannon TD (2001). The neuropsychology and neuroanatomy of bipolar affective disorder: a critical review. Bipolar Disord 3: 106–150.
Article CAS PubMed Google Scholar - Bearden CE, Thompson PM, Dalwani M, Hayashi KM, Lee AD, Nicoletti M et al (2007). Greater cortical gray matter density in lithium-treated patients with bipolar disorder. Biol Psychiatry 62: 7–16.
Article CAS PubMed PubMed Central Google Scholar - Beasley CL, Honer WG, Bergmann K, Falkai P, Lutjohann D, Bayer TA (2005). Reductions in cholesterol and synaptic markers in association cortex in mood disorders. Bipolar Disord 7: 449–455.
Article CAS PubMed Google Scholar - Beaulieu JM, Sotnikova TD, Yao WD, Kockeritz L, Woodgett JR, Gainetdinov RR et al (2004). Lithium antagonizes dopamine-dependent behaviors mediated by an AKT/glycogen synthase kinase 3 signaling cascade. Proc Natl Acad Sci USA 101: 5099–5104.
Article CAS PubMed PubMed Central Google Scholar - Bebchuk JM, Arfken CL, Dolan-Manji S, Murphy J, Hasanat K, Manji HK (2000). A preliminary investigation of a protein kinase C inhibitor in the treatment of acute mania. Arch Gen Psychiatry 57: 95–97.
Article CAS PubMed Google Scholar - Belmaker RH (2004). Bipolar disorder. N Engl J Med 351: 476–486.
Article CAS PubMed Google Scholar - Benes FM, Berretta S (2001). GABAergic interneurons: implications for understanding schizophrenia and bipolar disorder. Neuropsychopharmacology 25: 1–27.
Article CAS PubMed Google Scholar - Benes FM, Vincent SL, Todtenkopf M (2001). The density of pyramidal and nonpyramidal neurons in anterior cingulate cortex of schizophrenic and bipolar subjects. Biol Psychiatry 50: 395–406.
Article CAS PubMed Google Scholar - Benowitz LI, Perrone-Bizzozero NI, Neve RL, Rodriguez W (1990). GAP-43 as a marker for structural plasticity in the mature CNS. Prog Brain Res 86: 309–320.
Article CAS PubMed Google Scholar - Berridge MJ, Downes CP, Hanley MR (1989). Neural and developmental actions of lithium: a unifying hypothesis. Cell 59: 411–419.
Article CAS PubMed Google Scholar - Berry GT, Buccafusca R, Greer JJ, Eccleston E (2004). Phosphoinositide deficiency due to inositol depletion is not a mechanism of lithium action in brain. Mol Genet Metabolism 82: 87–92.
Article CAS Google Scholar - Bertolino A, Frye MA, Callicott JH, Mattay VS, Rakow R, Shelton-Repella J et al (2003). Neuronal pathology in the hippocampal area of patients with bipolar disorder: a study with proton magnetic resonance spectroscopic imaging. Biol Psychiatry 53: 906–913.
Article PubMed Google Scholar - Berton O, McClung CA, Dileone RJ, Krishnan V, Renthal W, Russo SJ et al (2006). Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science 311: 864–888.
Article CAS PubMed Google Scholar - Bezchlibnyk YB, Sun X, Wang J, MacQueen GM, McEwen BS, Young LT (2007). Neuron somal size is decreased in the lateral amygdalar nucleus of subjects with bipolar disorder. J Psychiatry Neurosci 32: 203–210.
PubMed PubMed Central Google Scholar - Bhat RV, Shanley J, Correll MP, Fieles WE, Keith RA, Scott CW et al (2000). Regulation and localization of tyrosine216 phosphorylation of glycogen synthase kinase-3beta in cellular and animal models of neuronal degeneration. Proc Natl Acad Sci USA 97: 11074–11079.
Article CAS PubMed PubMed Central Google Scholar - Bielau H, Trubner K, Krell D, Agelink MW, Bernstein HG, Stauch R et al (2005). Volume deficits of subcortical nuclei in mood disorders: a postmortem study. Eur Arch Psychiatry Clin Neurosci 255: 401–412.
Article PubMed Google Scholar - Bonfanti L, Strettoi E, Chierzi S, Cenni MC, Liu XH, Martinou J-C et al (1996). Protection of retinal ganglion cells from natural and axotomy-induced cell death in neonatal transgenic mice overexpressing bcl-2. J Neurosci 16: 4186–4194.
Article CAS PubMed PubMed Central Google Scholar - Bowley MP, Drevets WC, Ongur D, Price JL (2002). Low glial numbers in the amygdala in major depressive disorder. Biol Psychiatry 52: 404–412.
Article PubMed Google Scholar - Brambilla P, Harenski K, Nicoletti MA, Mallinger AG, Frank E, Kupfer DJ et al (2001). Differential effects of age on brain gray matter in bipolar patients and healthy individuals. Neuropsychopharmacology 43: 242–247.
CAS Google Scholar - Brambilla P, Nicoletti MA, Harenski K, Sassi RB, Mallinger AG, Frank E et al (2002). Anatomical MRI study of subgenual prefrontal cortex in bipolar and unipolar subjects. Neuropsychopharmacology 27: 792–799.
Article PubMed Google Scholar - Bruckheimer EM, Cho SH, Sarkiss M, Hermann J, McDonnell TJ (1998). The Bcl-2 gene family and apoptosis. Adv Biochem Eng Biotechnol 62: 75–105.
CAS PubMed Google Scholar - Burden SJ (2000). Wnts as retrograde signals for axon and growth cone differentiation. Cell 100: 495–497.
Article CAS PubMed Google Scholar - Buttner N, Bhattacharyya S, Walsh J, Benes FM (2007). DNA fragmentation is increased in non-GABAergic neurons in bipolar disorder but not in schizophrenia. Schizophr Res 93: 33–41.
Article PubMed PubMed Central Google Scholar - Cao BJ, Peng NA (1993). Magnesium valproate attenuates hyperactivity induced by dexamphetamine-chlordiazepoxide mixture in rodents. Eur J Pharmacol 237: 177–181.
Article CAS PubMed Google Scholar - Carlson PJ, Singh JB, Zarate CA, Drevets WC, Manji HK (2006). Neural circuitry and neuroplasticity in mood disorders: insights for novel therapeutic targets. NeuroRx 3: 22–41.
Article CAS PubMed PubMed Central Google Scholar - Catapano L, Chen G, Du J, Zarate CA, Manji HK (in press). Cellular plasticity cascades: genes to behavior pathways in the pathophysiology and treatment of bipolar disorder. In: Charney DS, Nestler EJ (eds). Neurobiology of Mental Illness. Oxford University Press: New York.
- Cecil KM, DelBello MP, Morey R, Strakowski SM (2002). Frontal lobe differences in bipolar disorder as determined by proton MR spectroscopy. Bipolar Disord 4: 357–365.
Article CAS PubMed Google Scholar - Chalecka-Franaszek E, Chuang DM (1999). Lithium activates the serine/threonine kinase Akt-1 and suppresses glutamate-induced inhibition of Akt-1 activity in neurons. Proc Natl Acad Sci USA 96: 8745–8750.
Article CAS PubMed PubMed Central Google Scholar - Chang K, Adleman N, Dienes KA, Barnea-Goraly N, Reiss A, Ketter TA (2003). Decreased _N_-acetylaspartate in children with familial bipolar disorder. Biol Psychiatry 53: 1059–1065.
Article CAS PubMed Google Scholar - Chang K, Barnea-Goraly N, Karchemskiy A, Simeonova DI, Barnes P, Ketter TA et al (2005). Cortical magnetic resonance imaging findings in familial pediatric bipolar disorder. Biol Psychiatry 58: 197–203.
Article PubMed Google Scholar - Chen B, Wang JF, Sun X, Young LT (2003). Regulation of GAP-43 expression by chronic desipramine treatment in rat cultured hippocampal cells. Biol Psychiatry 53: 530–537.
Article CAS PubMed Google Scholar - Chen DF, Schneider GE, Martinou JC, Tonewaga S (1997). Bcl-2 promotes regeneration of severed axons in mammalian CNS. Nature 385: 434–439.
Article CAS PubMed Google Scholar - Chen G, Manji HK (2006). The extracellular signal-regulated kinase pathway: an emerging promising target for mood stabilizers. Curr Opin Psychiatry 19: 313–323.
Article PubMed Google Scholar - Chen G, Rajkowska G, Du F, Seraji-Bozorgzad N, Manji HK (2000). Enhancement of hippocampal neurogenesis by lithium. J Neurochem 75: 1729–1734.
Article CAS PubMed Google Scholar - Chen G, Zeng WZ, Yuan PX, Huang LD, Jiang YM, Zhao ZH et al (1999). The mood-stabilizing agents lithium and valproate robustly increase the levels of the neuroprotective protein bcl-2 in the CNS. J Neurochem 72: 879–882.
Article CAS PubMed Google Scholar - Chuang DM (2004). Neuroprotective and neurotrophic actions of the mood stabilizer lithium: can it be used to treat neurodegenerative diseases? Crit Rev Neurobiol 16: 83–90.
Article CAS PubMed Google Scholar - Chuang DM, Manji HK (2007). In search of the holy grail for the treatment of neurodegenerative disorders: has a simple cation been overlooked? Biol Psychiatry 62: 4–6.
Article PubMed PubMed Central Google Scholar - Chuang DM, Priller J (2006). Potential use of lithium in neurodegenerative disorders. In: Bauer M, Grof P, Muler-Oerlingausen B (eds). Lithium in Neuropsychiatry: The Comprehensive Guide. Taylor & Francis: London. pp 381–397.
Chapter Google Scholar - Citri A, Malenka RC (2008). Mechanisms of plasticity in excitatory synapses. Neuropsychopharmacology Rev 1 (in press).
- Cohen P, Frame S (2001). The renaissance of GSK3. Nat Rev Mol Cell Biol 2: 769–776.
Article CAS PubMed Google Scholar - Cole AR, Knebel A, Morrice NA, Robertson LA, Irving AJ, Connolly CN et al (2004). GSK-3 phosphorylation of the Alzheimer epitope within collapsin response mediator proteins regulates axon elongation in primary neurons. J Biol Chem 279: 50176–50180.
Article CAS PubMed Google Scholar - Cook D, Fry MJ, Hughes K, Sumathipala R, Woodgett JR, Dale TC (1996). Wingless inactivates glycogen synthase kinase-3 via an intracellular signalling pathway which involves a protein kinase C. EMBO J 15: 4526–4536.
Article CAS PubMed PubMed Central Google Scholar - Cotter D, Landau S, Beasley C, Stevenson R, Chana G, MacMillan L et al (2002a). The density and spatial distribution of GABAergic neurons, labelled using calcium binding proteins, in the anterior cingulate cortex in major depressive disorder, bipolar disorder, and schizophrenia. Biol Psychiatry 51: 377–386.
Article CAS PubMed Google Scholar - Cotter D, Mackay D, Chana G, Beasley C, Landau S, Everall IP (2002b). Reduced neuronal size and glial cell density in area 9 of the dorsolateral prefrontal cortex in subjects with major depressive disorder. Cereb Cortex 12: 386–394.
Article PubMed Google Scholar - Cryns K, Shamir A, Shapiro J, Daneels G, Goris I, Van Craenendonck H et al (2007). Lack of lithium-like behavioral and molecular effects in IMPA2 knockout mice. Neuropsychopharmacology 32: 881–891.
Article CAS PubMed Google Scholar - Davies SP, Reddy H, Caivano M, Cohen P (2000). Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J 351: 95–105.
Article CAS PubMed PubMed Central Google Scholar - Davis JM, Janicak PG, Hogan DM (1999). Mood stabilizers in the prevention of recurrent affective disorders: a meta-analysis. Acta Psychiatr Scand 100: 406–417.
Article CAS PubMed Google Scholar - De Ferrari GV, Chacon MA, Barria MI, Garrido JL, Godoy JA, Olivares G et al (2003). Activation of Wnt signaling rescues neurodegeneration and behavioral impairments induced by beta-amyloid fibrils. Mol Psychiatry 8: 195–208.
Article CAS PubMed Google Scholar - De Sarno P, Li X, Jope RS (2002). Regulation of Akt and glycogen synthase kinase-3beta phosphorylation by sodium valproate and lithium. Neuropharmacology 43: 1158–1164.
Article CAS PubMed Google Scholar - Diecken RF, Fein G, Weiner MW (1995). Abnormal frontal lobe phosphorous metabolism in bipolar disorder. Am J Psychiatry 152: 915–918.
Article Google Scholar - Diecken RF, Pegues MP, Anzalone S, Feiwell R, Soher B (2003). Lower concentration of hippocampal _N_-acetylaspartate in familial bipolar I disorder. Am J Psychiatry 160: 873–882.
Article Google Scholar - Ding VW, Chen RH, McCormick F (2000). Differential regulation of glycogen synthase kinase 3beta by insulin and Wnt signaling. J Biol Chem 275: 32475–32481.
Article CAS PubMed Google Scholar - Dixon JF, Los GV, Hokin LE (1994). Lithium stimulates glutamate ‘release’ and inositol 1,4,5-trisphosphate accumulation via activation of the _N_-methyl-D-aspartate receptor in monkey and mouse cerebral cortex slices. Proc Natl Acad Sci USA 91: 8358–8362.
Article CAS PubMed PubMed Central Google Scholar - Doble BW, Woodgett JR (2003). GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci 116: 1175–1186.
Article CAS PubMed Google Scholar - Dokucu ME, Yu L, Taghert PH (2005). Lithium- and valproate-induced alterations in circadian locomotor behavior in Drosophila. Neuropsychopharmacology 30: 2216–2224.
Article CAS PubMed Google Scholar - Drevets WC (2000). Neuroimaging studies of mood disorders. Biol Psychiatry 48: 813–829.
Article CAS PubMed Google Scholar - Drevets WC (2001). Neuroimaging and neuropathological studies of depression: implications for the cognitive–emotional features of mood disorders. Curr Opin Neurobiol 11: 240–249.
Article CAS PubMed Google Scholar - Drevets WC, Price JL, Simpson Jr JR, Todd RD, Reich T, Vannier M et al (1997). Subgenual prefrontal cortex abnormalities in mood disorders. Nature 386: 824–827.
Article CAS PubMed Google Scholar - Du J, Creson T, Gray NA, Falke CA, Wei Y, Wang YM et al (submitted). The role of hippocampal GluR1 and GluR2 receptors in manic-like behaviors.
- Du J, Gray NA, Falke CA, Chen W, Yuan P, Szabo ST et al (2004a). Modulation of synaptic plasticity by antimanic agents: the role of AMPA glutamate receptor subunit 1 synaptic expression. J Neurosci 24: 6578–6589.
Article CAS PubMed PubMed Central Google Scholar - Du J, Gray NA, Falke CA, Yuan PX, Szabo ST, Manji HK (2003). Structurally dissimilar antimanic agents modulate synaptic plasticity by regulating AMPA glutamate receptor subunit GluR1 synaptic expression. Ann NY Acad Sci 1003: 378–380.
Article CAS PubMed Google Scholar - Du J, Machado-Vieira R, Maeng S, Martinowich K, Manji HK, Zarate CA (2006). Enhancing AMPA to NMDA throughput as a convergent mechanism for antidepressant action. Drug Discov Today: Ther Strat 3: 519–526.
Google Scholar - Du J, Quiroz J, Gray NA, Szabo ST, Zarate Jr CA, Manji HK (2004b). Regulation of cellular plasticity and resilience by mood stabilizers: the role of AMPA receptor trafficking. Dialog Clin Neurosci 6: 143–157.
Google Scholar - Du J, Wei Y, Chen Z, Falke CA, Zarate Jr CA, Manji HK (2007). The anticonvulsants lamotrigine, riluzole and valproate differentially regulate AMPA receptor trafficking: relationship to clinical effects in mood disorders. Neuropsychopharmacology 32: 793–802.
Article CAS PubMed Google Scholar - Duman CH, Schlesinger L, Kodama M, Russell DS, Duman RS (2007). A role for MAP kinase signaling in behavioral models of depression and antidepressant treatment. Biol Psychiatry 61: 661–670.
Article CAS PubMed Google Scholar - Eastwood SL, Harrison PJ (2001). Synaptic pathology in the anterior cingulate cortex in schizophrenia and mood disorders. A review and a western blot study of synaptophysin, GAP-43 and the complexins. Brain Res Bull 55: 569–578.
Article CAS PubMed Google Scholar - Eastwood SL, Harrison PJ (2007). Decreased mRNA expression of netrin-G1 and netrin-G2 in the temporal lobe in schizophrenia and bipolar disorder. Neuropsychopharmacology; advance online publication, 16 May 2007; doi:101038/sjnpp1301457.
- Eickholt BJ, Walsh FS, Doherty P (2002). An inactive pool of GSK-3 at the leading edge of growth cones is implicated in Semaphorin 3A signaling. J Cell Biol 157: 211–217.
Article CAS PubMed PubMed Central Google Scholar - Eisch AJ, Bolanos CA, de Wit J, Simonak RD, Pudiak CM, Barrot M et al (2003). Brain-derived neurotrophic factor in the ventral midbrain–nucleus accumbens pathway: a role in depression. Biol Psychiatry 54: 994–1005.
Article CAS PubMed Google Scholar - File SE, Aranko K (1988). Sodium valproate decreases exploratory behaviour in mice: development of tolerance and cross-tolerance with chlordiazepoxide. Eur J Pharmacol 151: 293–299.
Article CAS PubMed Google Scholar - Frame S, Cohen P, Biondi RM (2001). A common phosphate binding site explains the unique substrate specificity of GSK3 and its inactivation by phosphorylation. Moi Cell 7: 1321–1327.
Article CAS Google Scholar - Frangou S, Raymont V, Bettany D (2002). The Maudsley bipolar disorder project. A survey of psychotropic prescribing patterns in bipolar I disorder. Bipolar Disord 4: 378–385.
Article PubMed Google Scholar - Frazier JA, Ahn HS, DeJong S, Bent EK, Breeze JL, Giuliano AJ (2005). Magnetic resonance imaging studies in early-onset bipolar disorder: a critical review. Harv Rev Psychiatry 13: 125–140.
Article PubMed Google Scholar - Goodwin FK, Jamison KR (2007). Manic-Depressive Illness: Bipolar Disorders and Recurrent Depression. Oxford University Press: New York.
Google Scholar - Goold RG, Owen R, Gordon-Weeks PR (1999). Glycogen synthase kinase 3beta phosphorylation of microtubule-associated protein 1B regulates the stability of microtubules in growth cones. J Cell Sci 112: 3373–3384.
CAS PubMed Google Scholar - Gould TD, Chen G, Manji HK (2004a). In vivo evidence in the brain for lithium inhibition of glycogen synthase kinase-3. Neuropsychopharmacology 29: 32–38.
Article CAS PubMed Google Scholar - Gould TD, Einat H, Bhat R, Manji HK (2004b). AR-A014418, a selective GSK-3 inhibitor, produces antidepressant-like effects in the forced swim test. Int J Neuropsychopharmacol 7: 387–390.
Article CAS PubMed Google Scholar - Gould TD, Einat H, O'Donnell KC, Picchini AM, Schloesser RJ, Manji HK (2007). Beta-catenin overexpression in the mouse brain phenocopies lithium-sensitive behaviors. Neuropsychopharmacology; advance online publication, 14 February 2007; doi:101038/sjnpp1301338.
- Gould TD, Manji HK (2005). Glycogen synthase kinase-3: a putative molecular target for lithium mimetic drugs. Neuropsychopharmacology 30: 1223–1237.
Article CAS PubMed Google Scholar - Gould TD, Picchini AM, Einat H, Manji HK (2006). Targeting glycogen synthase kinase-3 in the CNS: implications for the development of new treatments for mood disorders. Curr Drug Targets 7: 1399–1409.
Article CAS PubMed Google Scholar - Gurvich N, Klein PS (2002). Lithium and valproic acid: parallels and contrasts in diverse signaling contexts. Pharmacol Ther 96: 45–66.
Article CAS PubMed Google Scholar - Gyulai L, Bowden CL, McElroy SL, Calabrese JR, Petty F, Swann AC et al (2003). Maintenance efficacy of divalproex in the prevention of bipolar depression. Neuropsychopharmacology 28: 1374–1382.
Article CAS PubMed Google Scholar - Hajek T, Carrey N, Alda M (2005). Neuroanatomical abnormalities as risk factors for bipolar disorder. Bipolar Disord 7: 393–403.
Article PubMed Google Scholar - Hall AC, Brennan A, Goold RG, Cleverley K, Lucas FR, Gordon-Weeks PR et al (2002). Valproate regulates GSK-3-mediated axonal remodeling and synapsin I clustering in developing neurons. Mol Cell Neurosci 20: 257–270.
Article CAS PubMed Google Scholar - Hall AC, Lucas FR, Salinas PC (2000). Axonal remodeling and synaptic differentiation in the cerebellum is regulated by WNT-7a signaling. Cell 100: 525–535.
Article CAS PubMed Google Scholar - Hallcher LM, Sherman WR (1980). The effects of lithium ion and other agents on the activity of myo-inositol-1-phosphatase from bovine brain. J Biol Chem 255: 10896–10901.
CAS PubMed Google Scholar - Hamakawa H, Kato T, Murashita J, Kato N (1998). Quantitative proton magnetic resonance spectroscopy of the basal ganglia in patients with affective disorders. Eur Arch Psychiatry Clin Neurosci 248: 53–58.
Article CAS PubMed Google Scholar - Hamakawa H, Murashita J, Yamada N, Inubushi T, Kato N, Kato T (2004). Reduced intracellular pH in the basal ganglia and whole brain measured by 31P-MRS in bipolar disorder. Psychiatry Clin Neurosci 58: 82–88.
Article CAS PubMed Google Scholar - Hao Y, Creson T, Zhang L, Li P, Du F, Yuan P et al (2004). Mood stabilizer valproate promotes ERK pathway-dependent cortical neuronal growth and neurogenesis. J Neurosci 24: 6590–6599.
Article CAS PubMed PubMed Central Google Scholar - Hashimoto R, Senatorov V, Kanai H, Leeds P, Chuang DM (2003). Lithium stimulates progenitor proliferation in cultured brain neurons. Neurosci 117: 55–61.
Article CAS Google Scholar - Heckers S, Stone D, Walsh J, Shick J, Koul P, Benes FM (2002). Differential hippocampal expression of glutamic acid decarboxylase 65 and 67 messenger RNA in bipolar disorder and schizophrenia. Arch Gen Psychiatry 59: 521–529.
Article CAS PubMed Google Scholar - Hedgepeth C, Conrad L, Zhang Z, Huang H, Lee V, Klein P (1997). Activation of the Wnt signaling pathway: a molecular mechanism for lithium action. Dev Biol 185: 82–91.
Article CAS PubMed Google Scholar - Hilfiker S, Pieribone VA, Czernik AJ, Kao HT, Augustine GJ, Greengard P (1999). Synapsins as regulators of neurotransmitter release. Philos Trans R Soc Lond B Biol Sci 354: 269–279.
Article CAS PubMed PubMed Central Google Scholar - Hong M, Chen DC, Klein PS, Lee VM-Y (1997). Lithium reduces tau phosphorylation by inhibition of glycogen synthase kinase-3. J Biol Chem 272: 25326–25332.
Article CAS PubMed Google Scholar - Hrdina P, Faludi G, Li Q, Bendotti C, Tekes K, Sotonyi P et al (1998). Growth-associated protein (GAP-43), its mRNA, and protein kinase C (PKC) isoenzymes in brain regions of depressed suicides. Mol Psychiatry 3: 411–418.
Article CAS PubMed Google Scholar - Huang HC, Klein PS (2006). Multiple roles for glycogen synthase kinase-3 as a drug target in Alzheimer's disease. Curr Drug Targets 7: 1389–1397.
Article CAS PubMed Google Scholar - Hyman SE, Nestler EJ (1996). Initiation and adaptation: a paradigm for understanding psychotropic drug action. Am J Psychiatry 153: 151–162.
Article CAS PubMed Google Scholar - Jiang H, Guo W, Liang X, Rao Y (2005). Both the establishment and the maintenance of neuronal polarity require active mechanisms: critical roles of GSK-3beta and its upstream regulators. Cell 120: 123–135.
CAS PubMed Google Scholar - Johnston-Wilson NL, Sims CD, Hofmann JP, Anderson L, Shore AD, Torrey EF et al (2000). Disease-specific alterations in frontal cortex brain proteins in schizophrenia, bipolar disorder, and major depressive disorder. The Stanley Neuropathology Consortium. Mol Psychiatry 5: 142–149.
Article CAS PubMed Google Scholar - Jope RS, Johnson SL (2004). The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Science 29: 95–102.
Article CAS Google Scholar - Kaidanovich-Beilin O, Milman A, Weizman A, Pick CG, Eldar-Finkelman H (2004). Rapid antidepressive-like activity of specific glycogen synthase kinase-3 inhibitor and its effect on beta-catenin in mouse hippocampus. Biol Psychiatry 55: 781–784.
Article CAS PubMed Google Scholar - Kaladchibachi SA, Doble B, Anthopoulos N, Woodgett JR, Manoukian AS (2007). Glycogen synthase kinase 3, circadian rhythms, and bipolar disorder: a molecular link in the therapeutic action of lithium. J Circadian Rhythms 5: 3.
Article PubMed PubMed Central CAS Google Scholar - Kato T, Murashita J, Kamiya A, Shiori T, Kato N, Inubushi T (1998). Decreased brain intracellular pH measured by 31P-MRS in bipolar disorder: a confirmation in drug-free patients and correlation with white matter hyperintensity. Eur Arch Psychiatry Clin Neurosci 248: 301–306.
Article CAS PubMed Google Scholar - Kato T, Shiori T, Murashita J, Hamakawa H, Inubushi T, Takahashi JS (1994). Phosphorus-31 magnetic resonance spectroscopy and ventricular enlargement in bipolar disorder. Psychiatry Res 55: 41–50.
Article CAS PubMed Google Scholar - Kato T, Shiori T, Murashita J, Hamakawa H, Takahashi Y, Inubushi T et al (1995). Lateralized abnormality of high energy phosphate metabolism in the frontal lobes of patients with bipolar disorder detected by phase-encoded 31P-MRS. Psychol Med 25: 557–566.
Article CAS PubMed Google Scholar - Kim L, Kimmel AR (2000). Regulation of a cyclin-CDK-CDK inhibitor complex by inositol pyrophosphates. Curr Opin Genet Dev 10: 508–514.
Article CAS PubMed Google Scholar - Kim WY, Zhou FQ, Zhou J, Yokota Y, Wang YM, Yoshimura T et al (2006). Essential roles for GSK-3s and GSK-3-primed substrates in neurotrophin-induced and hippocampal axon growth. Neuron 52: 981–996.
Article CAS PubMed PubMed Central Google Scholar - Klein PS, Melton DA (1996). A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci USA 93: 8455–8459.
Article CAS PubMed PubMed Central Google Scholar - Konradi C, Eaton M, MacDonald ML, Walsh J, Benes FM, Heckers S (2004). Molecular evidence for mitochondrial dysfunction in bipolar disorder. Arch Gen Psychiatry 61: 300–308.
Article CAS PubMed Google Scholar - Kostic V, Jackson-Lewis V, de Bilbao F, Dubois-Dauphin M, Przedborski S (1997). Bcl-2: prolonging life in a transgenic mouse model of familial amyotrophic lateral sclerosis. Science 277: 559–562.
Article CAS PubMed Google Scholar - Kupfer DJ (2005). The increasing medical burden in bipolar disorder. JAMA 293: 2528–2530.
Article CAS PubMed Google Scholar - Lawrence MS, Ho DY, Sun GH, Steinberg GK, Sapolsky RM (1996). Overexpression of Bcl-2 with herpes simplex virus vectors protects CNS neurons against neurological insults in vitro and in vivo. J Neurosci 16: 486–496.
Article CAS PubMed PubMed Central Google Scholar - Lee YS, Mulugu S, York JD, O'Shea EK (2007). Regulation of a cyclin-CDK-CDK inhibitor complex by inositol pyrophosphates. Science 316: 845–846.
Article Google Scholar - Lenox RH, Gould TD, Manji HK (2002). Endophenotypes in bipolar disorder. Am J Med Genet 114: 391–406.
Article PubMed Google Scholar - Leverich GS, Post RM, Keck PE, Altshuler LL, Frye MA, Kupka RW et al (2007). The poor prognosis of childhood-onset bipolar disorder. J Pediatrics 150: 485–490.
Article Google Scholar - Li H, Yuan J (1999). Deciphering the pathways of life and death. Curr Opin Cell Biol 11: 261–266.
Article CAS PubMed Google Scholar - Li X, Bijur GN, Jope RS (2002). Glycogen synthase kinase-3beta, mood stabilizers, and neuroprotection. Bipolar Disord 4: 137–144.
Article CAS PubMed PubMed Central Google Scholar - Li X, Friedman AB, Zhu W, Wang L, Boswell S, May RS et al (2007). Lithium regulates glycogen synthase kinase-3beta in human peripheral blood mononuclear cells: implication in the treatment of bipolar disorder. Biol Psychiatry 61: 216–222.
Article CAS PubMed Google Scholar - Liang MH, Chuang DM (2007). Regulation and function of glycogen synthase kinase-3 isoforms in neuronal survival. J Biol Chem 282: 3904–3917.
Article CAS PubMed Google Scholar - Lie DC, Colamarino SA, Song HJ, Desire L, Mira H, Consiglio A et al (2005). Wnt signalling regulates adult hippocampal neurogenesis. Nature 437: 1370–1375.
Article CAS PubMed Google Scholar - Liu L, Schulz SC, Lee S, Reutiman TJ, Fatemi SH (2007). Hippocampal CA1 pyramidal cell size is reduced in bipolar disorder. Cell Mol Neurobiol 27: 351–358.
Article PubMed Google Scholar - Lochhead PA, Kinstrie R, Sibbet G, Rawjee T, Morrice N, Cleghon V (2006). A chaperone-dependent GSK3beta transitional intermediate mediates activation-loop autophosphorylation. Mol Cell 24: 627–633.
Article CAS PubMed Google Scholar - Lopez-Coronado JM, Belles JM, Lesage F, Serrano R, Rodriguez PL (1999). A novel mammalian lithium-sensitive enzyme with a dual enzymatic activity, 3′-phosphoadenosine 5′-phosphate phosphatase and inositol-polyphosphate 1-phosphatase. J Biol Chem 274: 16034–16039.
Article CAS PubMed Google Scholar - Lovestone S, Davis DR, Webster MT, Kaech S, Brion JP, Matus A et al (1999). Lithium reduces tau phosphorylation: effects in living cells and in neurons at therapeutic concentrations. Biol Psychiatry 45: 995–1003.
Article CAS PubMed Google Scholar - Lucas FR, Goold RG, Gordon-Weeks PR, Salinas PC (1998). Inhibition of GSK-3beta leading to the loss of phosphorylated MAP-1B is an early event in axonal remodelling induced by WNT-7a or lithium. J Cell Sci 111: 1351–1361.
CAS PubMed Google Scholar - Lyoo IK, Lee HK, Jung JH, Noam GG, Renshaw PF (2002). White matter hyperintensities on magnetic resonance imaging of the brain in children with psychiatric disorders. Comp Psychiatry 43: 361–368.
Article Google Scholar - MacQueen GM, Campbell S, McEwen BS, Macdonald K, Amano S, Joffe R et al (2003). Course of illness, hippocampal function, and hippocampal volume in major depression. Proc Natl Acad Sci USA 100: 1387–1392.
Article CAS PubMed PubMed Central Google Scholar - Maekawa M, Takashima N, Arai Y, Nomura T, Inokuchi K, Yuasa S et al (2005). Pax6 is required for production and maintenance of progenitor cells in postnatal hippocampal neurogenesis. Genes Cells 10: 1001–1114.
Article CAS PubMed Google Scholar - Maeng S, Zarate CA, Du J, Schloesser RJ, McCammon J, Chen G et al (2007). Cellular mechanisms underlying the antidepressant effects of ketamine: role of AMPA receptors. Biol Psychiatry [20 July 2007; E-pub ahead of print].
- Malinow R, Malenka RC (2002). AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci 25: 103–126.
Article CAS PubMed Google Scholar - Manji HK, Moore GJ, Chen G (1999). Lithium at 50: have the neuroprotective effects of this unique cation been overlooked? Biol Psychiatry 46: 929–940.
Article CAS PubMed Google Scholar - Manji HK, Moore GJ, Chen G (2000). Lithium up-regulates the cytoprotective protein Bcl-2 in the CNS in vivo: a role for neurotrophic and neuroprotective effects in manic depressive illness. J Clin Psychiatry 61 (Suppl 9): 82–96.
CAS PubMed Google Scholar - Martinek S, Inonog S, Manoukian AS, Young MW (2001). A role for the segment polarity gene shaggy/GSK-3 in the Drosophila circadian clock. Cell 105: 769–779.
Article CAS PubMed Google Scholar - Martinowich K, Lu B (2008). Interaction between BDNF and serotonin: role in mood disorders. Neuropsychopharmacology Rev 33: 73–83.
Article CAS Google Scholar - Maslanski JA, Leshko L, Busa WB (1992). Lithium-sensitive production of inositol phosphates during amphibian embryonic mesoderm induction. Science 256: 243–245.
Article CAS PubMed Google Scholar - Mattson MP (2007). Mitochondrial regulation of neuronal plasticity. Neurochem Res 32: 707–715.
Article CAS PubMed Google Scholar - McClung CA (2007). Circadian genes, rhythms and the biology of mood disorders. Pharmacol Ther 114: 222–232.
Article CAS PubMed PubMed Central Google Scholar - McClung CA, Nestler EJ (2008). Neuroplasticity mediated by altered gene expression. Neuropsychopharmacology Rev 33: 3–17.
Article CAS Google Scholar - McManus EJ, Sakamoto K, Armit LJ, Ronaldson L, Shpiro N, Marquez R et al (2005). Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knockin analysis. EMBO J 24: 1571–1583.
Article CAS PubMed PubMed Central Google Scholar - Merry DE, Korsmeyer SJ (1997). Bcl-2 gene family in the nervous system. Annu Rev Neurosci 20: 245–267.
Article CAS PubMed Google Scholar - Moore GJ, Bebchuk JM, Hasanat K, Chen G, Seraji-Bozorgzad N, Wilds IB et al (2000a). Lithium increases _N_-acetyl-aspartate in the human brain: in vivo evidence in support of bcl-2's neurotrophic effects? Biol Psychiatry 48: 1–8.
Article CAS PubMed Google Scholar - Moore GJ, Bebchuk JM, Wilds IB, Chen G, Manji HK (2000b). Lithium-induced increase in human brain grey matter. Lancet 356: 1241–1242.
Article CAS PubMed Google Scholar - Moore PB, Schepherd DJ, Eccleston D, Macmillan IC, Goswami U, McAllister VL et al (2001). Cerebral white matter lesions in bipolar affective disorder: relationship to outcome. Br J Psychiatry 178: 172–176.
Article CAS PubMed Google Scholar - Munoz-Montano JR, Moreno FJ, Avila J, Diaz-Nido J (1997). Lithium inhibits Alzheimer's disease-like tau protein phosphorylation in neurons. FEBS Lett 411: 183–188.
Article CAS PubMed Google Scholar - Murphy DL (1977). Animal models for mania. In: Hanin I, Usdin E (eds). Animal Models in Psychiatry and Neurology. Pergamon Press: Oxford. pp 211–223.
Google Scholar - Nasrallah HA, McCalley-Whitters M, Jacoby CG (1982). Cortical atrophy in schizophrenia and mania: a comparative CT study. J Clin Psychiatry 43: 439–441.
CAS PubMed Google Scholar - Noble W, Planel E, Zehr C, Olm V, Meyerson J, Suleman F et al (2005). Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc Natl Acad Sci USA 102: 6990–6995.
Article CAS PubMed PubMed Central Google Scholar - Noga JT, Vladar K, Torrey EF (2001). A volumetric magnetic resonance imaging study of monozygotic twins discordant for bipolar disorder. Psychiatry Res 106: 25–34.
Article CAS PubMed Google Scholar - Nunes PV, Forlenza OV, Gattaz WF (2007). Lithium and the risk for Alzheimer's disease in elderly patients with bipolar disorder (abstract). Br J Psychiatry 190: 359–360.
Article PubMed Google Scholar - O'Brien WT, Harper AD, Jove F, Woodgett JR, Maretto S, Piccolo S et al (2004). Glycogen synthase kinase-3beta haploinsufficiency mimics the behavioral and molecular effects of lithium. J Neurosci 24: 6791–6798.
Article CAS PubMed PubMed Central Google Scholar - O'Donnell T, Rotzinger S, Nakashima TT, Hanstock CC, Ulrich M, Silverstone PH (2000). Chronic lithium and sodium valproate both decrease the concentration of myo-inositol and increase the concentration of inositol monophosphates in rat brain. Brain Res 880: 84–91.
Article CAS PubMed Google Scholar - Odom AR, Stahlberg A, Wente SR, York JD (2000). A role for nuclear inositol 1,4,5-trisphosphate kinase in transcriptional control. Science 287: 2026–2029.
Article CAS PubMed Google Scholar - Ongur D, Drevets WC, Price JL (1998). Glial reduction in the subgenual prefrontal cortex in mood disorders. Proc Natl Acad Sci USA 95: 13290–13295.
Article CAS PubMed PubMed Central Google Scholar - Padiath QS, Paranjpe D, Jain S, Sharma VK (2004). Glycogen synthase kinase 3beta as a likely target for the action of lithium on circadian clocks. Chronobiol Int 21: 43–55.
Article CAS PubMed Google Scholar - Pantazopoulos H, Lange N, Baldessarini RJ, Berretta S (2007). Parvalbumin neurons in the entorhinal cortex of subjects diagnosed with bipolar disorder or schizophrenia. Biol Psychiatry 61: 640–652.
Article CAS PubMed Google Scholar - Pearlson GD, Garbacz DJ, Tompkins RH, Ahn HS, Gutterman DF, Veroff AE et al (1984). Clinical correlates of lateral ventricular enlargement in bipolar affective disorder. Am J Psychiatry 141: 253–256.
Article CAS PubMed Google Scholar - Pearlson GD, Veroff AE (1981). Computerised tomographic scan changes in manic-depressive illness. Lancet 2: 470.
Article CAS PubMed Google Scholar - Perera TD, Coplan JD, Lisanby SH, Lipira CM, Arif M, Carpio C et al (2007). Antidepressant-induced neurogenesis in the hippocampus of adult nonhuman primates. J Neurosci 27: 4894–4901.
Article CAS PubMed PubMed Central Google Scholar - Phiel CJ, Wilson CA, Lee VM, Klein PS (2003). GSK-3alpha regulates production of Alzheimer's disease amyloid-beta peptides. Nature 423: 435–439.
Article CAS PubMed Google Scholar - Pillai JJ, Friedman L, Stuve TA, Trinidad S, Jesberger JA, Lewin JS et al (2002). Increased presence of white matter hyperintensities in adolescent patients with bipolar disorder. Psychiatry Res 114: 51–56.
Article PubMed Google Scholar - Pittenger C, Duman R (2008). Stress, depression, and neuroplasticity: a convergence of mechanisms. Neuropsychopharmacology Rev 33: 88–109.
Article CAS Google Scholar - Post RM (2007). Kindling and sensitization as models for affective episode recurrence, cyclicity, and tolerance phenomena. Neurosci Biobehav Rev 31: 858–873.
Article PubMed Google Scholar - Prickaerts J, Moechars D, Cryns K, Lenaerts I, van Craenendonck H, Goris I et al (2006). Transgenic mice overexpressing glycogen synthase kinase 3beta: a putative model of hyperactivity and mania. J Neurosci 26: 9022–9029.
Article CAS PubMed PubMed Central Google Scholar - Raghupathi R, Fernandez SC, Murai H, Trusko SP, Scott RW, Nishioka WK et al (1998). BCL-2 overexpression attenuates cortical cell loss after traumatic brain injury in transgenic mice. J Cereb Blood Flow Metab 18: 1259–1269.
Article CAS PubMed Google Scholar - Rajkowska G (2000). Postmortem studies in mood disorders indicate altered numbers of neurons and glial cells. Biol Psychiatry 48: 766–777.
Article CAS PubMed Google Scholar - Rajkowska G (2002). Cell pathology in bipolar disorder. Bipolar Disord 4: 105–116.
Article PubMed Google Scholar - Rajkowska G, Halaris A, Selemon LD (2001). Reductions in neuronal and glial density characterize the dorsolateral prefrontal cortex in bipolar disorder. Biol Psychiatry 49: 741–752.
Article CAS PubMed Google Scholar - Rao S, Rajesh KR, Joseph T (1991). Effect of antiepileptic drugs valproic acid, carbamazepine and ethosuccimide on exploratory behaviour in mice. Ind J Exper Biol 29: 127–130.
CAS Google Scholar - Ren M, Senatorov VV, Chen RW, Chuang DM (2003). Postinsult treatment with lithium reduces brain damage and facilitates neurological recovery in a rat ischemia/reperfusion model. Proc Natl Acad Sci USA 100: 6210–6215.
Article CAS PubMed PubMed Central Google Scholar - Reya T, Clevers H (2005). Wnt signalling in stem cells and cancer. Nature 434: 843–850.
Article CAS PubMed Google Scholar - Rockenstein E, Torrance M, Adame A, Mante M, Bar-on P, Rose JB et al (2007). Neuroprotective effects of regulators of the glycogen synthase kinase-3beta signaling pathway in a transgenic model of Alzheimer's disease are associated with reduced amyloid precursor protein phosphorylation. J Neurosci 27: 1981–1991.
Article CAS PubMed PubMed Central Google Scholar - Roh MS, Eom TY, Zmijewska AA, De Sarno P, Roth KA, Jope RS (2005). Hypoxia activates glycogen synthase kinase-3 in mouse brain in vivo: protection by mood stabilizers and imipramine. Biol Psychiatry 57: 278–286.
Article CAS PubMed Google Scholar - Roybal K, Theobold D, Graham A, Dinieri JA, Russo SJ, Krishnan V et al (2007). Mania-like behavior induced by disruption of CLOCK. Proc Natl Acad Sci USA 104: 6406–6411.
Article CAS PubMed PubMed Central Google Scholar - Ruel L, Stambolic V, Ali A, Manoukian AS, Woodgett JR (1999). Regulation of the protein kinase activity of Shaggy(Zeste-white3) by components of the wingless pathway in Drosophila cells and embryos. J Biol Chem 274: 21790–21796.
Article CAS PubMed Google Scholar - Ryder J, Su Y, Liu F, Li B, Zhou Y, Ni B (2003). Divergent roles of GSK3 and CDK5 in APP processing. Biochem Biophys Res Commun 312: 922–929.
Article CAS PubMed Google Scholar - Ryves WJ, Harwood AJ (2001). Lithium inhibits glycogen synthase kinase-3 by competition for magnesium. Biochem Biophys Res Commun 280: 720–725.
Article CAS PubMed Google Scholar - Sadoul R (1998). Bcl-2 family members in the development and degenerative pathologies of the nervous system. Cell Death Differ 5: 805–815.
Article CAS PubMed Google Scholar - Salinas PC (1999). Wnt factors in axonal remodelling and synaptogenesis. Biochem Soc Symp 65: 101–109.
CAS PubMed Google Scholar - Sassi RB, Nicoletti M, Brambilla P, Mallinger AG, Frank E, Kupfer DJ et al (2002). Increased gray matter volume in lithium-treated bipolar disorder patients. Neurosci Lett 329: 243–245.
Article CAS PubMed Google Scholar - Sassi RB, Stanley JA, Axelson D, Brambilla P, Nicoletti MA, Keshavan MS et al (2005). Reduced NAA levels in the dorsolateral prefrontal cortex of young bipolar patients. Am J Psychiatry 162: 2109–2115.
Article PubMed Google Scholar - Sayas CL, Moreno-Flores MT, Avila J, Wandosell F (1999). The neurite retraction induced by lysophosphatidic acid increases Alzheimer's disease-like tau phosphorylation. J Biol Chem 274: 37046–37052.
Article CAS PubMed Google Scholar - Scarr E, Gray L, Keriakous D, Robinson PJ, Dean B (2006). Increased levels of SNAP-25 and synaptophysin in the dorsolateral prefrontal cortex in bipolar I disorder. Bipolar Disord 8: 133–143.
Article CAS PubMed Google Scholar - Schlaepfer TE, Harris GJ, Tien AY, Peng LW, Lee S, Federman EB et al (1994). Decreased regional cortical gray matter volume in schizophrenia. Am J Psychiatry 151: 842–848.
Article CAS PubMed Google Scholar - Seeds AM, Bastidas RJ, York JD (2005). Molecular definition of a novel inositol polyphosphate metabolic pathway initiated by inositol 1,4,5-trisphosphate 3-kinase activity in Saccharomyces cerevisiae. J Biol Chem 280: 27654–27661.
Article CAS PubMed Google Scholar - Semba J, Kuroda Y, Takahashi R (1989). Potential antidepressant properties of subchronic GABA transaminase inhibitors in the forced swimming test in mice. Neuropsychobiology 21: 152–156.
Article CAS PubMed Google Scholar - Shaldubina A, Buccafusca R, Johanson RA, Agam G, Belmaker RH, Berry GT et al (2007). Behavioural phenotyping of sodium-myo-inositol cotransporter heterozygous knockout mice with reduced brain inositol. Genes Brain Behav 6: 253–259.
Article CAS PubMed Google Scholar - Shaldubina A, Johanson RA, O'Brien WT, Buccafusca R, Agam G, Belmaker RH et al (2006). SMIT1 haploinsufficiency causes brain inositol deficiency without affecting lithium-sensitive behavior. Mol Genet Metab 88: 384–388.
Article CAS PubMed Google Scholar - Shaldubina A, Ju S, Vaden DL, Ding D, Belmaker RH, Greenberg ML (2002). Epi-inositol regulates expression of the yeast INO1 gene encoding inositol-1-P synthase. Mol Psychiatry 7: 174–180.
Article CAS PubMed Google Scholar - Shaltiel G, Chen G, Manji HK (2007). Neurotrophic signaling cascades in the pathophysiology and treatment of bipolar disorder. Curr Opin Pharmacol 7: 22–26.
Article CAS PubMed Google Scholar - Sheline YI, Gado MH, Kraemer HC (2003). Untreated depression and hippocampal volume loss. Am J Psychiatry 160: 1516–1518.
Article PubMed Google Scholar - Sheline YI, Wang PW, Gado MH, Csernansky JG, Vannier M (1996). Hippocampal atrophy in recurrent major depression. Proc Natl Acad Sci USA 93: 3908–3913.
Article CAS PubMed PubMed Central Google Scholar - Soares JC, Young A (2007). Bipolar Disorder: Basic Mechanisms and Therapeutic Implications. Marcel Dekker, Inc.: New York.
Book Google Scholar - Son H, Yu IT, Hwang SJ, Kim JS, Lee SH, Lee YS et al (2003). Lithium enhances long-term potentiation independently of hippocampal neurogenesis in the rat dentate gyrus. J Neurochem 85: 872–881.
Article CAS PubMed Google Scholar - Song L, De Sarno P, Jope RS (2002). Central role of glycogen synthase kinase-3beta in endoplasmic reticulum stress-induced caspase-3 activation. J Biol Chem 277: 44701–44708.
Article CAS PubMed Google Scholar - Spiegelberg BD, Xiong JP, Smith JJ, Gu RF, York JD (1999). Cloning and characterization of a mammalian lithium-sensitive bisphosphate 3′-nucleotidase inhibited by inositol 1,4-bisphosphate. J Biol Chem 274: 13619–13628.
Article CAS PubMed Google Scholar - Stambolic V, Ruel L, Woodgett J (1996). Lithium inhibits glycogen synthase kinase-3 activity and mimics wingless signalling in intact cells. Curr Biol 6: 1664–1668.
Article CAS PubMed Google Scholar - Stoll AL, Renshaw PF, Yurgelen-Todd DA, Cohen BM (2000). Neuroimaging in bipolar disorder: what have we learned? Biol Psychiatry 48: 505–517.
Article CAS PubMed Google Scholar - Stork C, Renshaw PF (2005). Mitochondrial dysfunction in bipolar disorder: evidence from magnetic resonance spectroscopy research. Mol Psychiatry 10: 900–919.
Article CAS PubMed Google Scholar - Strakowski SM, DelBello MP, Zimmerman ME, Getz GE, Mills NP, Ret J et al (2002). Ventricular and periventricular structural volumes in first- versus multiple-episode bipolar disorder. Am J Psychiatry 159: 1841–1847.
Article PubMed Google Scholar - Strittmatter SM (1992). GAP-43 as a modulator of G protein transduction in the growth cone. Perspect Dev Neurobiol 1: 13–19.
CAS PubMed Google Scholar - Su Y, Ryder J, Li B, Wu X, Fox N, Solenberg P et al (2004). Lithium, a common drug for bipolar disorder treatment, regulates amyloid-beta precursor protein processing. Biochem 43: 6899–6908.
Article CAS Google Scholar - Suppes T, Baldessarini RJ, Faedda GL, Tohen M (1991). Risk of recurrence following discontinuation of lithium treatment in bipolar disorder. Arch Gen Psychiatry 48: 1082–1088.
Article CAS PubMed Google Scholar - Tanizawa Y, Kuhara A, Inada H, Kodama E, Mizuno T, Mori I (2006). Inositol monophosphatase regulates localization of synaptic components and behavior in the mature nervous system of C. elegans. Genes Dev 20: 3296–3310.
Article CAS PubMed PubMed Central Google Scholar - Terao T, Nakano H, Inoue Y, Okamoto T, Nakamura J, Iwata N (2006). Lithium and dementia: a preliminary study. Prog Neuropsychopharmacol Biol Psychiatry 30: 1125–1128.
Article CAS PubMed Google Scholar - Thomas AJ, O'Brien JT, Davis S, Ballard C, Barber R, Kalaria RN et al (2002). Ischemic basis for deep white matter hyperintensities in major depression: a neuropathological study. Arch Gen Psychiatry 59: 785–792.
Article PubMed Google Scholar - Tian SY, Wang JF, Bezchlibnyk YB, Young LT (2007). Immunoreactivity of 43 kDa growth-associated protein is decreased in post mortem hippocampus of bipolar disorder and schizophrenia. Neurosci Lett 411: 123–127.
Article CAS PubMed Google Scholar - Tkachev D, Mimmack ML, Ryan MM, Wayland M, Freeman T, Jones PB et al (2003). Oligodendrocyte dysfunction in schizophrenia and bipolar disorder. Lancet 362: 798–805.
Article CAS PubMed Google Scholar - Trowbridge JJ, Xenocostas A, Moon RT, Bhatia M (2006). Glycogen synthase kinase-3 is an in vivo regulator of hematopoietic stem cell repopulation. Nat Med 12: 89–98.
Article CAS PubMed Google Scholar - Uranova NA, Vostrikov VM, Orlovskaya DD, Rachmanova VI (2004). Oligodendroglial density in the prefrontal cortex in schizophrenia and mood disorders: a study from the Stanley Neuropathology Consortium. Schizophr Res 67: 269–275.
Article PubMed Google Scholar - Vawter MP, Thatcher L, Usen N, Hyde TM, Kleinman JE, Freed WJ (2002). Reduction of synapsin in the hippocampus of patients with bipolar disorder and schizophrenia. Mol Psychiatry 7: 571–578.
Article CAS PubMed Google Scholar - Volz HP, Rzanny R, Riehemann S, May S, Hegewald H, Preussler B et al (1998). 31P magnetic resonance spectroscopy in the frontal lobe of major depressed patients. Eur Arch Psychiatry Clin Neurosci 248: 289–295.
Article CAS PubMed Google Scholar - Washizuka S, Kakiuchi C, Mori K, Kunugi H, Tajima O, Akiyama T et al (2003). Association of mitochondrial complex I subunit gene NDUFV2 at 18p11 with bipolar disorder. Am J Med Genet 120B: 72–78.
Article PubMed Google Scholar - Wellcome Trust Case Control Consortium (2007). Genome-wide association study of 14 000 cases of seven common diseases and 3,000 shared controls. Nature 447: 661–678.
Article CAS Google Scholar - Williams JM, Thompson VL, Mason-Parker SE, Abraham WC, Tate WP (1998). Synaptic activity-dependent modulation of mitochondrial gene expression in the rat hippocampus. Brain Res Mol Brain Res 60: 50–56.
Article CAS PubMed Google Scholar - Williams RS, Cheng L, Mudge AW, Harwood AJ (2002). A common mechanism of action for three mood-stabilizing drugs. Nature 417: 292–295.
Article CAS PubMed Google Scholar - Winsberg ME, Sachs N, Tate DL, Adalsteinsson E, Spielman D, Ketter TA (2000). Decreased dorsolateral prefrontal _N_-acetyl aspartate in bipolar disorder. Biol Psychiatry 47: 475–481.
Article CAS PubMed Google Scholar - Woodgett JR (1990). Molecular cloning and expression of glycogen synthase kinase-3/factor A. EMBO J 9: 2431–2438.
Article CAS PubMed PubMed Central Google Scholar - Xu J, Culman J, Blume A, Brecht S, Gohlke P (2003). Chronic treatment with a low dose of lithium protects the brain against ischemic injury by reducing apoptotic death. Stroke 34: 1287–1292.
Article CAS PubMed Google Scholar - Yang F, He XP, Russell J, Lu B (2003). Ca2+ influx-independent synaptic potentiation mediated by mitochondrial Na(+)-Ca2+ exchanger and protein kinase C. J Cell Biol 163: 511–523.
Article CAS PubMed PubMed Central Google Scholar - Yang L, Matthews RT, Schulz JB, Klockgether T, Liao AW, Martinou J-C et al (1998). 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyride neurotoxicity is attenuated in mice overexpressing Bcl-2. J Neurosci 18: 8145–8152.
Article CAS PubMed PubMed Central Google Scholar - Yin L, Wang J, Klein PS, Lazar MA (2006). Nuclear receptor Rev-erbalpha is a critical lithium-sensitive component of the circadian clock. Science 311: 1002–1005.
Article CAS PubMed Google Scholar - Yoshimura T, Kawano Y, Arimura N, Kawabata S, Kikuchi A, Kaibuchi K (2005). GSK-3beta regulates phosphorylation of CRMP-2 and neuronal polarity. Cell 120: 137–149.
Article CAS PubMed Google Scholar - Young LT (2007). Is bipolar disorder a mitochondrial disease? J Psychiatry Neurosci 32: 160–161.
PubMed PubMed Central Google Scholar - Yuan PX, Huang LD, Jiang YM, Gutkind JS, Manji HK, Chen G (2001). The mood stabilizer valproic acid activates mitogen-activated protein kinases and promotes neurite growth. J Biol Chem 276: 31674–31683.
Article CAS PubMed Google Scholar - Yucel K, Taylor VH, McKinnon MC, Macdonald K, Alda M, Young LT et al (2007). Bilateral hippocampal volume increase in patients with bipolar disorder and short-term lithium treatment. Neuropsychopharmacology [E-pub ahead of print; doi:10.1038/sj.npp.1301405].
- Zarate Jr CA, Quiroz JA, Singh JB, Denicoff KD, De Jesus G, Luckenbaugh DA et al (2005). An open-label trial of the glutamate-modulating agent riluzole in combination with lithium for the treatment of bipolar depression. Biol Psychiatry 57: 430–432.
Article CAS PubMed Google Scholar - Zarate Jr CA, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA et al (2006a). A randomized trial of an _N_-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry 63: 856–864.
Article CAS PubMed Google Scholar - Zarate CA, Singh JB, Carlson PJ, Quiroz J, Jolkovsky L, Luckenbaugh D et al (2007). Efficacy of a protein kinase C Inhibitor (Tamoxifen) in the treatment of acute mania: a pilot study. Bip Disord 9: 561–570.
Article CAS Google Scholar - Zarate Jr CA, Singh JB, Manji HK (2006b). Cellular plasticity cascades: targets for the development of novel therapeutics for bipolar disorder. Biol Psychiatry 59: 1006–1020.
Article CAS PubMed Google Scholar - Zhang F, Phiel CJ, Spece L, Gurvich N, Klein PS (2003). Inhibitory phosphorylation of glycogen synthase kinase-3 (GSK-3) in response to lithium. Evidence for autoregulation of GSK-3. J Biol Chem 278: 33067–33077.
Article CAS PubMed Google Scholar - Zhou FQ, Zhou J, Dedhar S, Wu YH, Snider WD (2004). NGF-induced axon growth is mediated by localized inactivation of GSK-3beta and functions of the microtubule plus end binding protein APC. Neuron 42: 897–912.
Article CAS PubMed Google Scholar - Zhou R, Gray NA, Yuan P, Li X, Chen J, Chen G et al (2005). The anti-apoptotic, glucocorticoid receptor cochaperone protein BAG-1 is a long-term target for the actions of mood stabilizers. J Neurosci 25: 4493–4502.
Article CAS PubMed PubMed Central Google Scholar - Zipursky RB, Seeman MV, Bury A, Langevin R, Wortzman G, Katz R (1997). Deficits in gray matter volume are present in schizophrenia but not bipolar disorder. Schizophr Res 26: 85–92.
Article CAS PubMed Google Scholar
Acknowledgements
We thank Ioline Henter for outstanding editorial assistance. RJS and HKM are supported by the Intramural Program of the NIMH. PSK is supported by grants from the National Institute of Mental Health and the American Federation for Aging Research. JH is supported by a fellowship from the Leukemia and Lymphoma Society.
Author information
Author notes
- Robert J Schloesser and Jian Huang: Dr Schloesser and Dr Huang share first authorship.
Authors and Affiliations
- Laboratory of Molecular Pathophysiology, Mood and Anxiety Disorders Program, National Institute of Mental Health, NIH, Bethesda, MD, USA
Robert J Schloesser & Husseini K Manji - Department of Medicine (Hematology-Oncology), University of Pennsylvania School of Medicine, Philadelphia, PA, USA
Jian Huang & Peter S Klein
Authors
- Robert J Schloesser
You can also search for this author inPubMed Google Scholar - Jian Huang
You can also search for this author inPubMed Google Scholar - Peter S Klein
You can also search for this author inPubMed Google Scholar - Husseini K Manji
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toHusseini K Manji.
Additional information
DISCLOSURE/CONFLICT OF INTEREST
The authors report no conflicts of interest.
PowerPoint slides
Rights and permissions
About this article
Cite this article
Schloesser, R., Huang, J., Klein, P. et al. Cellular Plasticity Cascades in the Pathophysiology and Treatment of Bipolar Disorder.Neuropsychopharmacol 33, 110–133 (2008). https://doi.org/10.1038/sj.npp.1301575
- Received: 16 July 2007
- Revised: 01 August 2007
- Accepted: 14 August 2007
- Published: 03 October 2007
- Issue Date: January 2008
- DOI: https://doi.org/10.1038/sj.npp.1301575