Selective modulation of subtype III IP3R by Akt regulates ER Ca2+ release and apoptosis (original) (raw)
Main
Ca2+ efflux from intracellular stores, in particular endoplasmic reticulum (ER), has a fundamental role in modulating different cellular responses. Variations of Ca2+ concentration [Ca2+] works as a trigger, for example, for the secretion of hormones and neurotransmitters, modulation of metabolism and mitotic division in numerous cell types.1 Several extracellular stimuli induce ER depletion, with consequent cytosolic Ca2+ increase, through activation of phospholipase C, which leads to the production of a second messenger, inositol 1,4,5-trisphosphate (IP3). IP3 in turn activates an internal membrane receptor, responsible for ER Ca2+ release, the IP3 receptor (IP3R). In birds and mammals, three different genes encode for three subtypes of IP3R (IP3R I, II and III), which share high similarity (70–80%) in their primary sequences and are expressed to varying degrees in individual cell types. Despite their analogy, the three subtypes possess some peculiarities, which characterize and distinguish them, such as subcellular localization, modulation by binding proteins and kinases, degradation by proteases and regulation by small molecules (i.e., Ca2+ and ATP).2 Phosphorylation/dephosphorylation events regulate IP3Rs Ca2+ release and these modifications assume particular relevance in cell death.3 In fact, it is now generally accepted that Ca2+ flux from the ER and consequent high levels of mitochondrial Ca2+ accumulation – a cationic transfer favored by the physical interconnection between the two organelles4– is linked to the effects of several apoptotic stimuli; ER Ca2+ release is therefore considered an intrinsic mechanism that favors apoptosis.5, 6 Various pro/anti-apoptotic proteins interact with IP3R and modulate ER Ca2+ dynamics to exert their activity: one of these is the anti-apoptotic protein Akt. The proto-oncogene Akt (also known as protein kinase B) is involved in several cellular processes such as glucose metabolism, cell proliferation, apoptosis, transcription and cell migration.7 Akt is a serine/threonine kinase and it is the major downstream target of receptor tyrosine kinases that signal via the phosphoinositide 3-kinase (PI3K). Receptor-activated PI3K leads to membrane attachment and subsequent phosphorylation and activation of Akt.8 Activated Akt exerts its pro-survival activity through phosphorylation of target proteins, inhibiting their pro-apoptotic function.9 All three subtypes of IP3R present, in their C-terminal tail, a robust phosphorylation motif R_X_R_XX_(S/T), highly conserved in different species,10 which is a substrate for Akt kinase activity.10, 11 Both groups cited, using mutants of type I IP3Rs, declared that IP3R phosphorylation by Akt promotes survival, but only the work of Roderick’s group11 connected it to an inhibition of ER Ca2+ release. We demonstrated that Akt activation protects cells from apoptosis by strongly reducing Ca2+ flux from the ER.12
Here, we report a thorough analysis of Akt anti-apoptotic activity through the regulation of IP3R Ca2+ channels. Using cells that display different ratios of IP3Rs, we show that Akt functions specifically on IP3R isoform III, independently of the presence or absence of isoform I. Finally, our data highlight how the Akt-dependent inhibition of Ca2+ transfer from the ER to mitochondria, through IP3R III, preserves the mitochondrial integrity and protects from Ca2+-mediated apoptosis.
Results
We previously reported that Akt protects from Ca2+-dependent apoptotic stimuli through inhibition of ER Ca2+ release in HeLa cells.12 To investigate whether Akt might possess a potential functional role on a specific isoform of IP3R, we used two different cell lines, COS7 and SH-SY 5Y, which lack IP3R type I13 and type III,14 respectively. After confirmation of the effective suitability of our cellular systems (Figure 1a), we measured intracellular calcium homeostasis using aequorin probes, targeted to different subcellular compartments, that is, the cytoplasm and the organelles acting as sources (ER) or targets (mitochondria) of the Ca2+ signal. We compared mock-transfected (control) with myristoylated/palmytolated Akt (m/p-Akt)-overexpressing cells, where m/p-Akt is a plasma membrane-targeted Akt chimera, considered a constitutively active form.15 In COS7 cells, Akt affects neither the total ER Ca2+ content (317.67±15.75 _μ_M control versus 288.33±25.96 _μ_M m/p-Akt) nor the kinetics of Ca2+ accumulation. After stimulation with ATP (which functions on Gq-coupled plasma membrane receptors and causes the production of IP3, thus releasing Ca2+ from the ER through the IP3Rs), Akt-overexpressing cells display a very different behavior in the release phase (Figure 1b), with an 86,7±1.9% decrease in the _V_max of released Ca2+ (_V_max: 14.32±2.11 _μ_M/s control versus 1.9±0.27 _μ_M/s m/p-Akt). Conversely, in SH-SY 5Y cells, the inhibition of ER Ca2+ release mediated by Akt overexpression is negligible (Figure 1c), with no significative differences in the ER plateau values (329.46±11.09 _μ_M control versus 308.25±11.18 _μ_M m/p-Akt), similarly to COS7 cells, and a tiny reduction in the [Ca2+] flow through the IP3-gated channels (_V_max: 19.39±0.97 _μ_M/s control versus 17.49±1.34 _μ_M/s m/p-Akt). We used Carbachol (Cch) as agonist stimulus instead of ATP in SH-SY 5Y cells, because in this cell type, Cch generates higher ER Ca2+ release than ATP (data not shown).
Figure 1
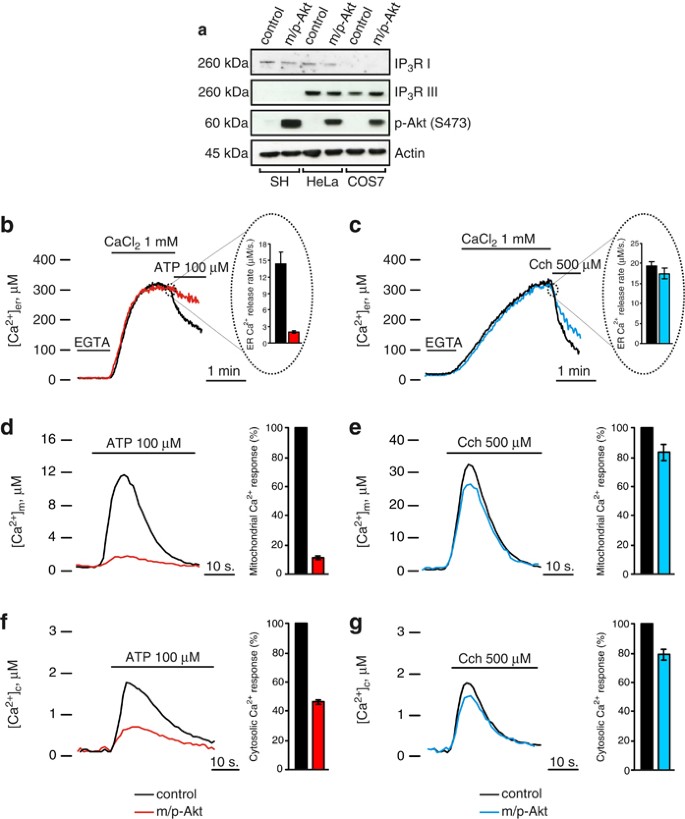
Akt-modulation of the Ca2+ signal in COS7, but not in SH-SY 5Y cells. (a) IP3R levels in COS7, HeLa and SH-SY 5Y cells. HeLa cells were used as reference because they possess both type I and III IP3Rs; the increment of p-Akt S473 levels reflects an effective Akt activation after m/p-Akt overexpression. (b) ER Ca2+ measurements in mock-transfected and m/p-Akt-overexpressing COS7 cells. To induce Ca2+ release from ER, cells were challenged with ATP, an agonist that, through interaction with G-protein-coupled receptors, evokes a rapid discharge from IP3Rs. Akt activation markedly modulates ER Ca2+ kinetics (red trace), diminishing the amount of Ca2+ released, quantified after analysis of maximum rate of calcium release during agonist stimulation (see bars inside the dotted circle). (c) Experiments analogous to b were carried out in SH-SY 5Y cells. In these cells, the agonist used is Cch. (d) Mitochondrial Ca2+ homeostasis modulation after Akt activation in COS7 cells and (e) in SH-SY 5Y cells. (f) Cytosolic Ca2+ homeostasis in control and m/p-Akt-overexpressing COS7 and (g) SH-SY 5Y cells. Where indicated, cells were stimulated with 100 _μ_M ATP or 500 _μ_M Cch. The bars in d–g indicate the levels of Ca2+ uptake, expressed in percentage, control assumed as 100%. All traces are from single representative experiments and [Ca2+] are presented as means of at least 20 experiments (±S.E.)
The close physical association between ER and mitochondria4 explains how mitochondria detect the Ca2+ flux released by ER very efficiently. Using an aequorin form targeted to the mitochondrial matrix, we measured mitochondrial [Ca2+] in COS7 and SH-SY 5Y cells, to have a further read-out system of Akt activity on IP3R Ca2+-releasing properties. As expected, in COS7 cells, Akt overexpression markedly reduces Ca2+ transient in the mitochondrial compartment (11.89±1.31 _μ_M control versus 1.42±0.06 _μ_M m/p-Akt; Figure 1d); this is owing to the IP3R inhibition described above. Figure 1e shows how, in SH-SY 5Y cells, Akt activity does not alter mitochondrial Ca2+ homeostasis in any marked manner, compared with COS7 (peak amplitude: 32.33±4.23 _μ_M control versus 27.34±2.17 _μ_M m/p-Akt), confirming the weak reticular Akt action on Ca2+ release in this cell type. To complete the scenario, we analyzed the cytosolic Ca2+ kinetics: in Akt-transfected COS7 cells, the [Ca2+] increases evoked by stimulation with ATP are significantly smaller than in controls (peak amplitude: 0.8±0.017 _μ_M versus 1.72±0.027 _μ_M; Figure 1f), whereas in SH-SY 5Y this reduction is almost undetectable (peak amplitude: 1.45±0.051 _μ_M versus 1.83±0.064 _μ_M; Figure 1g).
Having shown the very different effect of Akt expression on the modulation of calcium homeostasis in COS7 and SH-SY 5Y cells, we sought to examine the rate of apoptosis after treatment with a Ca2+-mediated apoptotic stimulus. IP3R is an important molecular component regulating apoptosis induced by a number of different stimuli, and IP3R-mediated Ca2+ release can activate apoptotic pathways by inducing the release of a number of pro-apoptotic factors from mitochondria.5 As apoptosis inducer we used arachidonic acid (AA), a very specific stimulus that triggers Ca2+-dependent cell death.16 In neuronal and non-neuronal cell lines, AA leads to apoptosis via a mitochondrial-mediated pathway,17, 18 opening the mitochondrial permeability transition pore, with consequent release of cytochrome c, followed by activation of caspase 3-dependent nuclear condensation and fragmentation. In COS7 cells, Akt overexpression protects from apoptosis induced by AA, with a marked reduction of the amount of cleaved poly-ADP-ribosomal polymerase (PARP) and caspase 3 (Figure 2a, lanes 1 and 4). Moreover, these data were confirmed by direct measurements of caspase activity after application of AA, showing a significant decrease of caspase 3 activity in m/p-Akt-overexpressing cells (Figure 2b). In SH-SY 5Y cells, the Akt anti-apoptotic effect seems to be lost: in fact, no differences were detected both in cleaved PARP levels and in the amount of cleaved caspase 3, comparing control and m/p-Akt-expressing cells after AA treatment (Figure 2c). Similar results are showed in Figure 2d, where AA induces an increase in caspase 3 activity which is mostly comparable between control and m/p-Akt-overexpressing SH-SY 5Y. This assay becomes necessary for a correct visualization of the real caspase 3 activity, because of the difficulty in the detection of cleaved form of the enzyme by immunoblotting in this cellular type.
Figure 2
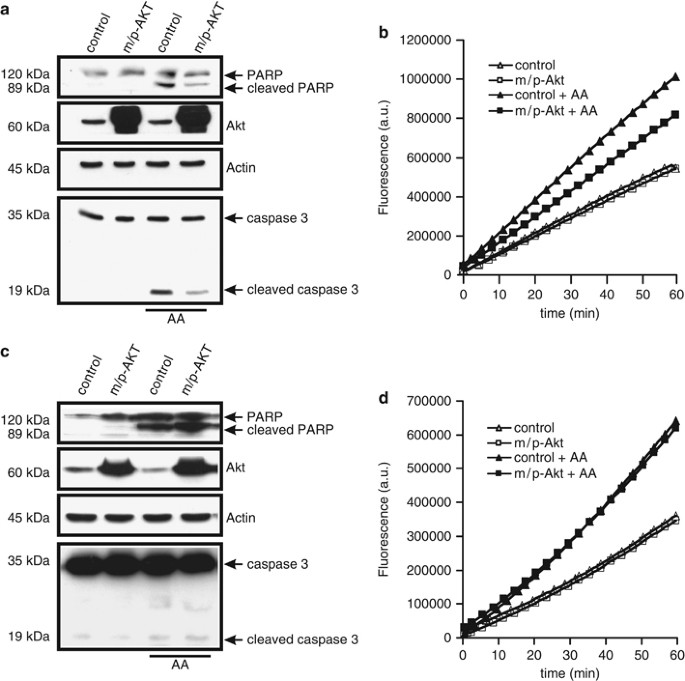
Akt protects from Ca2+-dependent apoptosis in COS7, but not in SH-SY 5Y cells. (a) COS7 cells were transfected with the indicated gene (control: mock-transfected), harvested after 36 h, and the endogenous cleavages of PARP and caspase 3 were revealed through immunoblotting. Apoptosis was induced by treatment with 80 _μ_M AA for 40 min. (b) COS7 cells were transfected and treated in the same manner of a, aliquots of cells were centrifuged, and lysates were assayed for caspase 3 activity as described in Materials and Methods. Traces are from a single representative experiment. (c) Immunoblotting showed different levels of cleaved PARP and caspase 3 in SH-SY 5Y after apoptosis induction and m/p-Akt overexpression. control, mock transfected. (d) Similar experiment to b but performed in SH-SY 5Y cells. The experiments were repeated at least five times
To assess if the protective role of Akt in COS7 cells may be ascribed to a minor amount of reticular Ca2+ released, we analyzed the kinetics of cytosolic Ca2+ generated by AA application. Given that the increase in cytosolic [Ca2+] ([Ca2+]c) evoked by apoptotic stimuli is in the nanomolar range, the [Ca2+]c was measured with the fluorescent indicator Fura-2/AM,19 as aequorin is not accurate enough to reveal small arises of [Ca2+]. We performed a cotransfection (in a 1 : 1 ratio) using green fluorescent protein with a mitochondrial presequence (mtGFP) and m/p-Akt, to identify Akt-positive cells and, therefore, to compare changes in Fura-2/AM 340/380 ratio on the same coverslip. Thus, GFP-positive cells were distinguished from controls by the typical fluorescence emitted upon illumination with blue light. AA addition causes a cytosolic Ca2+ increment that is significantly reduced in Akt-expressing cells (Figure 3a, red trace), with a reduction of 47.8%. Parallel experiments were performed in SH-SY 5Y (Figure 3b), but in these cells m/p-Akt overexpression does not alter the amount of cytosolic Ca2+ evoked by AA application (blue trace). This result is in perfect agreement with the weak Akt activity at the reticular level (Figure 1c) and its lack of anti-apoptotic function (Figures 2c and d).
Figure 3
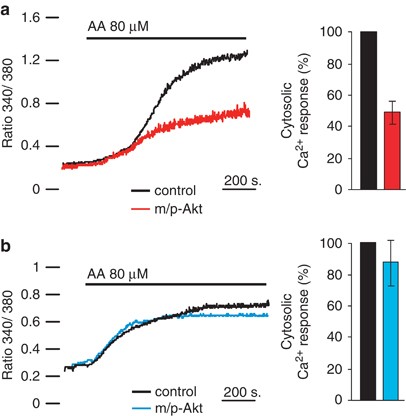
Akt modulates [Ca2+]c elevation generated by AA. (a) Effects of m/p-Akt on [Ca2+]c increase induced by AA in COS7 or (b) in SH-SY 5Y cells. Cells were loaded with the Ca2+ indicator Fura-2/AM and the kinetics of the [Ca2+]c response is presented as the ratio of fluorescence at 340 and 380 nm. The coverslips with the cells were maintained in 1 mM Ca2+/KRB and, where indicated, the cells were challenged with 80 _μ_M AA. To distinguish Akt-positive cells and compare changes in the 340/380 Fura-2/AM ratio on the same coverslip, cells were co-transfected with mtGFP and m/p-Akt in a 1 : 1 ratio. Traces are from a single representative experiment. The data are the mean of different angular coefficients±S.E. of, at least, 15 independent experiments; the bars in the graph show the change in percentage of cytosolic Ca2+ released
Mitochondrial fragmentation is one of early events that occur in apoptotic pathway, it is evolutionarily conserved,20 and it represents a clear hallmark of mitochondrial perturbation, which precedes effector caspase activation and cell death.21 We investigated if the Ca2+ transfer from ER to mitochondria promoted by AA might lead to fragmentation of the organelle and if Akt, modulating ER calcium release, might preserve the mitochondrial integrity, minimizing the apoptotic damage. First of all, we used HeLa cells not only because these cells express mostly type I and III IP3R, with no detected levels of subtype II,13 but mainly because it is a well-characterized cellular system in which it has been shown that Akt carries out its anti-apoptotic role.11, 12 To ensure the overexpression of the protein of interest in all observed mitochondrial networks, we co-transfected cells with the indicated vector (empty vector for control cells) and mtGFP in a 3 : 1 ratio. Figure 4 in toto shows mitochondrial structures of different cells before and 20 min after treatment with AA; this duration of time reflects the maximum peak of cytosolic Ca2+ released by the ER. AA induces a robust mitochondrial fragmentation (Figure 4a), whereas only minor changes in morphology were detected in m/p-Akt-overexpressing HeLa cells (Figure 4b). Analogously, in COS7 cells, AA application leads to fragmentation of the organelle (Figure 4c) and Akt expression preserved the three-dimensional mitochondrial integrity (Figure 4d). On the contrary, on SH-SY 5Y cells, no effect of m/p-Akt was detected, with similar morphological modifications both in control and Akt-expressing cells (Figures 4e and f), once more revealing the Akt inability to exert its ‘usual’ anti-apoptotic activity in this cellular setting. Mathematical description of mitochondrial fragmentation is reported in Figure 4g.
Figure 4
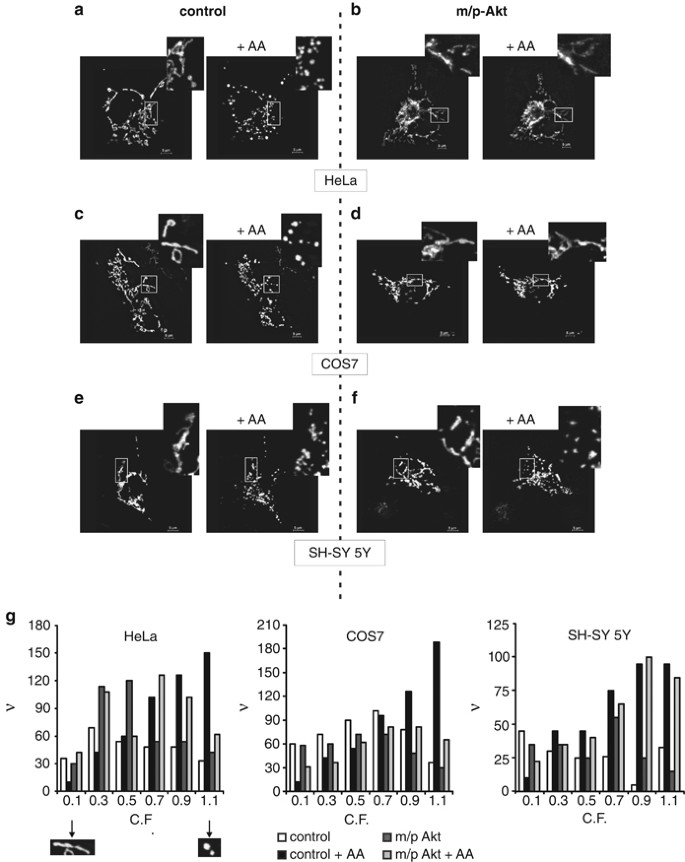
Akt preserves the mitochondrial integrity in an IP3R type III-dependent way. (a–f) The mitochondrial network of HeLa (a and b), COS7 (c and d) and SH-SY 5Y cells (e and f) was analyzed using confocal microscopy, 36 h post-transfection, before and after treatment with 80 _μ_M AA for 20 min. Cells were co-transfected in a 3 : 1 ratio with an empty vector and mtGFP (left panels: control) or m/p-Akt and mtGFP (right panels: m/p-Akt). Greater magnification of the mitochondrial three-dimensional structure is presented in the insets. (g) Frequency distribution of mitochondrial fragmentation: C.F. was analyzed to represent the state of each single mitochondrial object (i.e., C.F.=0.1 ‘elongated’ mitochondrion, considered not fragmented; C.F.=1.1 ‘circular’ mitochondrion, considered fragmented). ν, frequency (number of objects)
We wanted to explore whether the huge discrepancy in Akt behavior between COS7 and SH-SY 5Y might be due to the IP3R isoform III deficiency, which characterizes the latter cellular type. To demonstrate our hypothesis, we used a specific kind of SH-SY 5Y (herein referred as SH 2) that expresses IP3R III (Figure 5a).22 First of all, we analyzed the ER kinetics, with particular attention to the release phase. After agonist induction, m/p-Akt-overexpressing SH 2 cells displayed a significant reduction in the rate of delivered Ca2+ (_V_max: 17.39±1.41 _μ_M/s control versus 9.73±1.62 _μ_M/s m/p-Akt), keeping the ER Ca2+ content unaffected (Figure 5b; release phase magnification in inset). This inhibition of Ca2+ flow through IP3Rs, mediated by Akt, is mirrored at the mitochondrial level, with a marked decrease in mitochondrial Ca2+ uptake (peak amplitude: 45.06±2.94 _μ_M control versus 22.65±2.53 _μ_M m/p-Akt; Figure 5c). We note that this is a very large difference in comparison with SH-SY 5Y IP3R III-deficient cells (Figure 1e). We then asked whether the presence of isoform III of IP3R is the key point of Akt activity on calcium-mediated apoptosis. We measured the intracellular Ca2+ levels after AA treatment in SH 2, using Fura-2/AM as a probe: m/p-Akt overexpression produced minor changes in the cytosolic Ca2+ kinetics generated by AA (Figure 5d, green trace), lowering the ability of arachidonate to increase the intracellular [Ca2+]c. We wanted to directly correlate this evidence with a putative ‘recovered’ Akt ability to protect from apoptosis in SH 2 cells. In fact, Akt hyper-activation in SH 2 cells conferred a significant resistance to AA-apoptosis induction, expressed as caspase 3 activation levels (Figure 5e). Also, immunoblotting revealed a minor amount both in cleaved PARP and caspase 3 (Figure 5f), which further sustained the Akt role in significantly reducing the apoptotic Ca2+ signals promoted by AA. We recorded the Akt capacity to delay/block the transfer of Ca2+-death messages from ER to mitochondria through analysis of the mitochondrial network. As Figure 5g shows, no morphological changes were detected in Akt-expressing SH 2 cells, whereas AA induces mitochondrial fission in control cells, indicating how preservation of the mitochondrial integrity under apoptotic stress is required for correct Akt surviving activity.
Figure 5
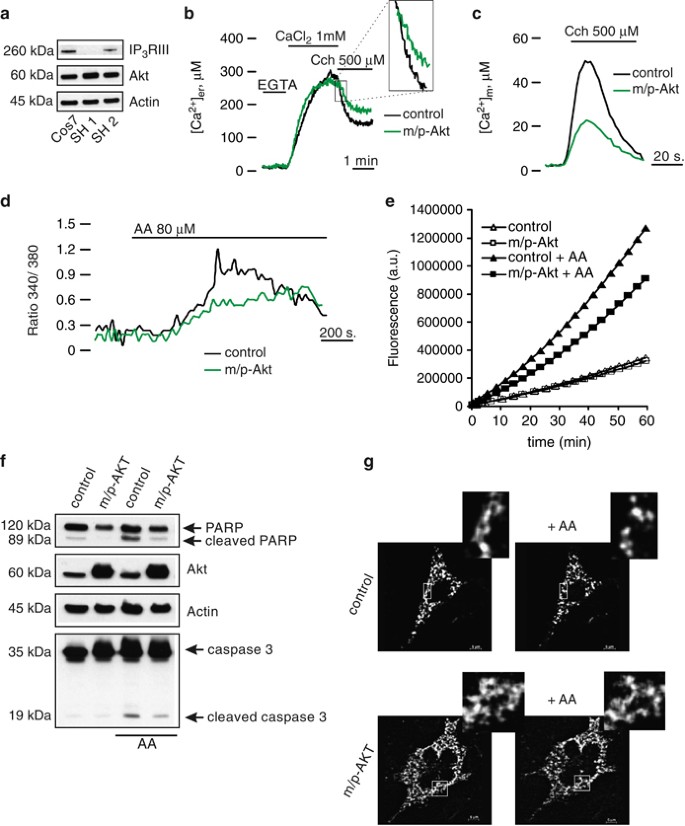
Akt ‘recovers’ its protective function on Ca2+ cell death in SH-SY 5Y-expressing IP3R type III. (a) Immunoblotting showed the effective presence of isoform III of IP3R in a different kind of SH-SY 5Y (termed SH 2). COS7 cells were used as reference for the right molecular weight of IP3R III. SH 1 stands for SH-SY 5Y type III-deficient cells, used in previous experiments. (b) ER Ca2+ measurements in mock-transfected and m/p-Akt-overexpressing SH 2 cells. Magnifications of first part of the release phase are reported (inset). (c) Mitochondrial Ca2+ homeostasis modulation after Akt activation in SH 2 cells. (d) SH 2 cells were loaded with the Ca2+ indicator Fura-2/AM and 340/380 nm ratio changes were measured. The experiment was performed similarly to that reported in Figure 3. (e) SH 2 cells were transfected with the indicated gene (control: mock transfected), harvested after 36 h, and the lysates were assayed for caspase 3 activity as described in Materials and Methods. Apoptosis was induced by treatment with 80 _μ_M AA for 40 min. Traces are from a single representative experiment. (f) SH 2 cells were transfected and treated in the same manner of e, and the endogenous cleavages of PARP and caspase 3 were revealed through immunoblotting. (g) SH 2 cells were co-transfected in a 3 : 1 ratio with empty vector and mtGFP (upper panels) or m/p-Akt and mtGFP (lower panels). Mitochondrial morphology was analyzed using confocal microscopy, 36 h post-transfection, before and after treatment with 80 _μ_M AA, for 20 min. The insets show a greater magnification of the mitochondrial network
Discussion
In this study, we have used different cell lines expressing different subtypes of IP3R ratio, to understand if Akt may have an IP3R isoform-specific action. Our work demonstrates that Akt preferentially exerts its anti-apoptotic role through inhibition of Ca2+ release by IP3R type III (Figure 6), and SH-SY 5Y cells deficient for this receptor isoform appear insensitive to the typical Akt surviving activity (Figures 2c and d), showing no differences in ER calcium release both after physiological (Figure 1c) and apoptotic stimulation (Figure 3b). As apoptosis inducer, we used AA: AA application causes a progressive release of Ca2+ from intracellular stores, thereby directly causing a [Ca2+]c rise and activating capacitative Ca2+ influx, which in turn is responsible for maintaining a long-lasting [Ca2+]c plateau.
Figure 6
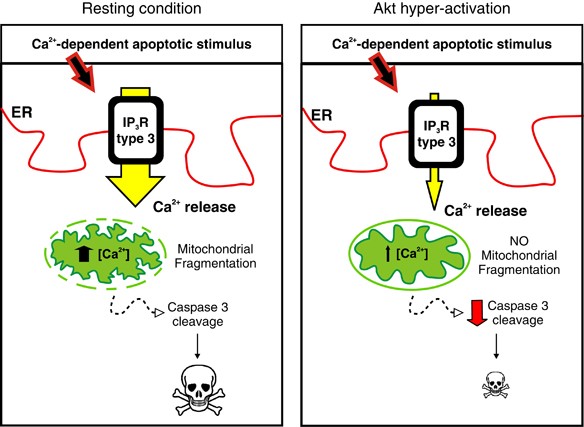
Model depicting Akt anti-apoptotic activity through regulation of IP3R type III-Ca2+ transfer to mitochondria. During basal condition, when Akt activation is physiological, a Ca2+-dependent apoptotic stimulus evokes Ca2+ release from the ER, especially through isoform III, with consequent mitochondrial Ca2+ accumulation, fragmentation of the network and apoptosis. In situations with enhanced Akt activity, a typical condition of many cancers, subtype III is inhibited, therefore decreasing the amount of Ca2+ accumulated by mitochondria. The mitochondrial network remains undamaged and apoptosis is blocked
Interestingly, Akt protection from staurosporine-induced cell death in SH-SY 5Y seems due to the mitochondrial activity of the kinase.23 Nevertheless, the predominant (93%) endogenous IP3R isoform in COS7 cells has been reported to be type III,13 with no detectable levels of type I (Figure 1a). In this cellular type, Akt shows its greater activity, with a marked reduction of Ca2+ released by ER after apoptotic stimulus (Figure 3a) and protection from apoptosis (Figures 2a and b). The inhibition of Ca2+ flux after agonist stimulation is more marked than previously observed in HeLa cells, an aspect which may be ascribed to the amount of IP3R III in COS7 cells. Moreover, in Sf9 insect cells Akt seems to phosphorylate type III IP3R more efficiently than type I.11
The preferential role of Akt on subtype III may be related to the implications of this isoform in a variety of Ca2+-dependent physiological processes. For example, it promotes Ca2+-dependent insulin and somatostatin secretion in pancreatic β and δ cells,24 and it is localized to the apical pole of different type of cells,25, 26 a region responsible for Ca2+ signals generation and thus termed ‘trigger zone’. These observations suggest that type III is more prone to initiate global release of intracellular Ca2+ in intact cells.27 This is further supported by the lack of Ca2+-dependent negative feedback of IP3R III.28 Intriguingly, it has been recently reported that the phosphorylation of the receptor by Akt kinase is enhanced in the presence of Ca2+,29 and, considering the Akt inhibitory effect on ER calcium release, it may represent an intrinsic cellular mechanism to reinstate basal conditions, favoring ER refilling after store depletion due to agonist stimulation.
In a similar way to Akt, cAMP-dependent protein kinase (PKA), another component belonging to the AGC kinase group, is able to bind and phosphorylate all IP3Rs,30 and type III with a certain efficiency.31 PKA is highly homologous to Akt, sharing ∼45% sequence identity with the kinase domain, and rising to ∼80% within the ATP site. Subtype III is phosphorylated by PKA at three different serine residues,31 and, similarly to Akt, a decrease in intracellular Ca2+ release mediated by PKA was observed.32 Conversely, an opposite role of PKA on Ca2+ mobilization by IP3R III has been reported,33 suggesting an alternative mechanism to justify the inhibition.
The most important aspect arising from our data refers to apoptosis. Akt activation does not protect from Ca2+-mediated cell death in cells lacking for IP3R III, a phenomenon which cannot be considered cell-type dependent, because SH-SY 5Y-expressing isoform III showed the typical Akt surviving activity (Figure 5). This result can reasonably be ascribed to a modification in subtype III-Ca2+ release properties, altered by Akt (Figure 5d). As Akt, another negative regulator of apoptosis, the Bcl-2 family member Bcl-xL, seems to be able to interact with all three IP3R isoforms,34 though isoform-specific effects have been described.35 The association between Bcl-xL, amount of ER Ca2+ released and apoptosis is exclusively effective for type III, because although all three IP3R isoforms provided apoptosis resistance when expressed with Bcl-xL, only the type III channel reduced ER [Ca2+].35
The role of IP3R III in apoptosis is, currently, partially controversial, and it is described both as an anti-36 and a pro-apoptotic protein.37 We agree with the latter hypothesis, and we are strongly convinced that it is a key factor in apoptosis, especially in Ca2+-dependent cell death: in fact, siRNA silencing of IP3R III antagonizes apoptosis induced by different Ca2+-mediated stimuli (P Pinton, unpublished data). Further support to this concept is provided by the evidence that, in CHO cells, subtype III co-localizes more closely with mitochondria, and thus preferentially transmits Ca2+ signals to these organelles.38 This observation explains Akt ability to preserve the mitochondrial integrity (Figure 4), delaying Ca2+ transfer from the ER to mitochondria by inhibition of type III channel gating. The Akt isoform-specific activity completes the molecular pathway describing a new role of promyelocytic leukemia (PML) protein as an apoptosis regulator at the ER–mitochondria interface. PML controls Ca2+ transfer to mitochondria through the formation of a complex that, besides PML, includes Akt, IP3R III and PP2A.39
In conclusion, our data collectively suggest the existence of a specific activity of Akt on IP3R type III, leading to channel inhibition, diminished Ca2+ transfer to mitochondria and protection from apoptosis. We suppose that the function of the kinase at the ER–mitochondria level is the early stage of Akt itinerary in promoting cell survival, this concept is also supported by the very recent observation about the reticular localization of mTORC2, the complex responsible for Akt phosphorylation and activation.40 Thus, the modulation of type III-Ca2+ release suggests an additional level of cell death regulation mediated by Akt.
Materials and Methods
Cell culture and transfection
HeLa cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum (FCS), L-glutamine and penicillin/streptomycin in 75-cm2 Falcon flasks. Both kinds of SH-SY 5Y cells (with and without IP3R III) were cultured in Ham’s nutrient mixture F12 (HAM’S/F12), supplemented with 10% FCS, 2 mM L-glutamine, penicillin/streptomycin and MEM non-essential amino-acid solution (Sigma-Aldrich, St. Louis, MO, USA), in 75-cm2 Falcon flasks.
For aequorin experiments, cells were seeded onto 13-mm glass coverslips and allowed to grow to 75% confluence; for Fura-2/AM measurements and mitochondrial morphology analysis, cells were seeded on 24-mm glass coverslip in the same conditions of growth.
For aequorin measurements in HeLa and COS7 cells, transfection with 4 _μ_g of total DNA (3 _μ_g of the indicated expression plasmids and 1 _μ_g of aequorin) transfected with a standard calcium–phosphate procedure. In both kinds of SH-SY 5Y cells, we performed transfection with the same 3 : 1 DNA ratio, using Lipofectamine LTX (Invitrogen-Life Technologies, Grand Island, NY, USA). All measurements were performed 36 h after transfection.
Aequorin measurements
Probes used are chimeric aequorins targeted to the ER (erAEQmut), cytosol (cytAEQ) and mitochondria (mtAEQmut). ‘AEQ’ refers to wild-type aequorin, and ‘AEQmut’ refers to a low-affinity D119A mutant of aequorin. For the experiments with cytAEQ and mtAEQmut, cells were incubated with 5 _μ_M coelenterazine for 1–2 h in DMEM supplemented with 1% FCS. A coverslip with transfected cells was placed in a perfused thermostated chamber located in close proximity to a low-noise photomultiplier with a built-in amplifier/discriminator. To reconstitute erAEQmut with high efficiency, the luminal [Ca2+] of the ER first had to be reduced. This was achieved by incubating cells for 1 h at 4 °C in Krebs-Ringer buffer (KRB) supplemented with 5 _μ_M coelenterazine, 5 _μ_M Ca2+ ionophore ionomycin (Sigma-Aldrich) and 600 _μ_M EGTA. After this incubation, cells were extensively washed with KRB supplemented with 2% bovine serum albumin and then transferred to the perfusion chamber. All aequorin measurements were carried out in KRB supplemented with either 1 mM CaCl2 (cytAEQ and mtAEQmut) or 100 _μ_M EGTA (erAEQmut). Agonist was added to the same medium as specified in figure legends. The experiments were terminated by lysing cells with 100 _μ_M digitonin in a hypotonic Ca2+-containing solution (10 mM CaCl2 in H2O), thus discharging the remaining aequorin pool. The output of the discriminator was captured by a Thorn EMI photon-counting board and stored in an IBM-compatible computer for further analyses. The aequorin luminescence data were calibrated offline into [Ca2+] values using a computer algorithm based on the Ca2+ response curve of wild-type and mutant aequorins.
Fura-2/AM measurements
Cytosolic-free [Ca2+]c was evaluated using fluorescent Ca2+ indicator Fura-2/AM (Molecular Probes-Life Technologies, Grand Island, NY, USA). Briefly, cells were incubated in medium supplemented with 2.5 _μ_M Fura-2/AM for 30 min, washed with KRB to remove extracellular probe, supplied with preheated KRB (supplemented with 1 mM CaCl2) and placed in a thermostated incubation chamber at 37 °C on the stage of an inverted fluorescence microscope (Zeiss Axiovert 200, Carl Zeiss, Oberkochen, Germany). Dynamic video imaging was performed using MetaFluor software (Universal Imaging Corporation Ltd, Marlow, Buckinghamshire, UK). Fluorescence was measured every 100 ms with the excitation wavelength alternating between 340 and 380 nm and the emission fluorescence being recorded at 510 nm. At the end of the experiment, a region free of cells was selected, and one averaged background frame was collected at each excitation wavelength for background correction.
Caspase 3 assay
Cells were centrifuged (1000 g for 5 min) and washed once in phosphate-buffered saline. The enzcheck caspase 3 assay kit # 2 by Molecular Probes was used for the determination of caspase 3 activity. Lysate (20 _μ_g), resuspended in a final volume of 100 _μ_l, was assayed using Wallac 1420 Victor3 multitask plate reader (Perkin Elmer, Waltham, MA, USA).
Imaging and analysis of mitochondrial morphology
HeLa, Cos7 and SH-SY5Y cells were seeded and transfected with mtGFP as described previously (see Results). Protein expression was allowed for 36 h and then cells where imaged with Nikon Swept Field Confocal (Nikon Instruments Inc., Melville, NY, USA) equipped with CFI Plan Apo VC60XH objective (n.a. 1.4) and an Andor DU885 EM-CCD camera (Andor, Belfast, Northern Ireland), controlled by the NIS-Elements 3.2. Coverslips were placed in an incubated chamber with controlled temperature, CO2 and humidity and then z-stacks were acquired by 21 planes with 0.6 _μ_m distance, to allow acquisition of the whole cell. After acquisition, images were restored with the Autoquant 3D blind deconvolution module, installed on NIS-Elements (Nikon Instruments Inc.), using a theoretical PSF.
After restoration, images were loaded in Imaris 4.0 (Bitplane AG, Zurich, Switzerland), then subtracted of background and used to generate a threshold based isosurface object group. From each isosurface was calculated the number of objects and the average object volume expressed as voxel number. At the end of each quantitation, representative images were obtained as maximum intensity projection render.
Mathematical analysis of mitochondrial frequency distribution, based on calculation of circularity factor (C.F.), was performed on best-focused plane, using the FIJI software (http://fiji.sc/).
Immunoblot
Total cell lysates were prepared in RIPA buffer, likewise separated by SDS-PAGE and the standard immunoblotting procedure was used.
Antibodies used were as follows: rabbit _α_-Akt, rabbit _α_-phospho Akt (S473), mouse _α_-PARP and rabbit _α_-Caspase 3 from Cell Signaling (Danvers, MA, USA); rabbit _α_-actin from Sigma-Aldrich; goat _α_-IP3R I from Santa Cruz Biotechnology (Santa Cruz, CA, USA); and mouse _α_-IP3R III from BD Biosciences (Franklin Lakes, NJ, USA).
Abbreviations
ER:
endoplasmic reticulum
[Ca2+]:
Ca2+ concentration
[Ca2+]c:
cytosolic [Ca2+]
IP3:
inositol 1,4,5-trisphosphate
IP3R:
IP3 receptor
PI3K:
phosphoinositide 3-kinase
m/p Akt:
myristoylated/palmytolated Akt
Cch:
Carbachol
AA:
arachidonic acid
PARP:
poly-ADP-ribosomal polymerase
PKA:
cAMP-dependent protein kinase
PML:
promyelocytic leukemia
C.F.:
circularity factor
References
- Berridge MJ, Bootman MD, Roderick HL . Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev 2003; 4: 517–529.
Article CAS Google Scholar - Thrower EC, Hagar RE, Ehrlich BE . Regulation of Ins(1,4,5)P3 receptor isoforms by endogenous modulators. Trends Pharmacol Sci 2001; 22: 580–586.
Article CAS Google Scholar - Vanderheyden V, Devogelaere B, Missiaen L, De Smedt H, Bultynck G, Parys JB . Regulation of inositol 1,4,5-trisphosphate-induced Ca2+ release by reversible phosphorylation and dephosphorylation. Biochim Biophys Acta 2009; 1793: 959–970.
Article CAS Google Scholar - Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM et al. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science (New York, NY) 1998; 280: 1763–1766.
Article CAS Google Scholar - Joseph SK, Hajnoczky G . IP3 receptors in cell survival and apoptosis: Ca2+ release and beyond. Apoptosis 2007; 12: 951–968.
Article CAS Google Scholar - Pinton P, Giorgi C, Siviero R, Zecchini E, Rizzuto R . Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 2008; 27: 6407–6418.
Article CAS Google Scholar - Manning BD, Cantley LC . AKT/PKB signaling: navigating downstream. Cell 2007; 129: 1261–1274.
Article CAS Google Scholar - Brazil DP, Park J, Hemmings BA . PKB binding proteins. Getting in on the Akt. Cell 2002; 111: 293–303.
Article CAS Google Scholar - Kim D, Chung J . Akt: versatile mediator of cell survival and beyond. J Biochem Mol Biol 2002; 35: 106–115.
CAS PubMed Google Scholar - Khan MT, Wagner L, Yule DI, Bhanumathy C, Joseph SK . Akt kinase phosphorylation of inositol 1,4,5-trisphosphate receptors. J Biol Chem 2006; 281: 3731–3737.
Article CAS Google Scholar - Szado T, Vanderheyden V, Parys JB, De Smedt H, Rietdorf K, Kotelevets L et al. Phosphorylation of inositol 1,4,5-trisphosphate receptors by protein kinase B/Akt inhibits Ca2+ release and apoptosis. Proc Natl Acad Sci USA 2008; 105: 2427–2432.
Article CAS Google Scholar - Marchi S, Rimessi A, Giorgi C, Baldini C, Ferroni L, Rizzuto R et al. Akt kinase reducing endoplasmic reticulum Ca2+ release protects cells from Ca2+-dependent apoptotic stimuli. Biochem Biophys Res Commun 2008; 375: 501–505.
Article CAS Google Scholar - Hattori M, Suzuki AZ, Higo T, Miyauchi H, Michikawa T, Nakamura T et al. Distinct roles of inositol 1,4,5-trisphosphate receptor types 1 and 3 in Ca2+ signaling. J Biol Chem 2004; 279: 11967–11975.
Article CAS Google Scholar - Morita T, Tanimura A, Nezu A, Tojyo Y . Visualization of inositol 1,4,5-trisphosphate receptor type III with green fluorescent protein in living cells. Cell Calcium 2002; 31: 59–64.
Article CAS Google Scholar - Andjelkovic M, Alessi DR, Meier R, Fernandez A, Lamb NJ, Frech M et al. Role of translocation in the activation and function of protein kinase B. J Biol Chem 1997; 272: 31515–31524.
Article CAS Google Scholar - Penzo D, Petronilli V, Angelin A, Cusan C, Colonna R, Scorrano L et al. Arachidonic acid released by phospholipase A(2) activation triggers Ca(2+)-dependent apoptosis through the mitochondrial pathway. J Biol Chem 2004; 279: 25219–25225.
Article CAS Google Scholar - Fang KM, Chang WL, Wang SM, Su MJ, Wu ML . Arachidonic acid induces both Na+ and Ca2+ entry resulting in apoptosis. J Neurochem 2008; 104: 1177–1189.
Article CAS Google Scholar - Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T et al. BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science (New York, NY) 2003; 300: 135–139.
Article CAS Google Scholar - Grynkiewicz G, Poenie M, Tsien RY . A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem 1985; 260: 3440–3450.
CAS PubMed Google Scholar - Jagasia R, Grote P, Westermann B, Conradt B . DRP-1-mediated mitochondrial fragmentation during EGL-1-induced cell death in C. elegans. Nature 2005; 433: 754–760.
Article CAS Google Scholar - Suen DF, Norris KL, Youle RJ . Mitochondrial dynamics and apoptosis. Genes Develop 2008; 22: 1577–1590.
Article CAS Google Scholar - Tovey SC, de Smet P, Lipp P, Thomas D, Young KW, Missiaen L et al. Calcium puffs are generic InsP(3)-activated elementary calcium signals and are downregulated by prolonged hormonal stimulation to inhibit cellular calcium responses. J Cell Sci 2001; 114(Pt 22): 3979–3989.
CAS PubMed Google Scholar - Mookherjee P, Quintanilla R, Roh MS, Zmijewska AA, Jope RS, Johnson GV . Mitochondrial-targeted active Akt protects SH-SY5Y neuroblastoma cells from staurosporine-induced apoptotic cell death. J Cell Biochem 2007; 102: 196–210.
Article CAS Google Scholar - Blondel O, Moody MM, Depaoli AM, Sharp AH, Ross CA, Swift H et al. Localization of inositol trisphosphate receptor subtype 3 to insulin and somatostatin secretory granules and regulation of expression in islets and insulinoma cells. Proc Natl Acad Sci USA 1994; 91: 7777–7781.
Article CAS Google Scholar - Lee MG, Xu X, Zeng W, Diaz J, Wojcikiewicz RJ, Kuo TH et al. Polarized expression of Ca2+ channels in pancreatic and salivary gland cells. Correlation with initiation and propagation of [Ca2+]i waves. J Biol Chem 1997; 272: 15765–15770.
Article CAS Google Scholar - Nathanson MH, Fallon MB, Padfield PJ, Maranto AR . Localization of the type 3 inositol 1,4,5-trisphosphate receptor in the Ca2+ wave trigger zone of pancreatic acinar cells. J Biol Chem 1994; 269: 4693–4696.
CAS PubMed Google Scholar - O'Neill AF, Hagar RE, Zipfel WR, Nathanson MH, Ehrlich BE . Regulation of the type III InsP(3) receptor by InsP(3) and calcium. Biochem Biophys Res Commun 2002; 294: 719–725.
Article CAS Google Scholar - Hagar RE, Burgstahler AD, Nathanson MH, Ehrlich BE . Type III InsP3 receptor channel stays open in the presence of increased calcium. Nature 1998; 396: 81–84.
Article CAS Google Scholar - Anyatonwu G, Khan MT, Schug ZT, da Fonseca PC, Morris EP, Joseph SK . Calcium-dependent conformational changes in inositol trisphosphate receptors. J Biol Chem 2010; 285: 25085–25093.
Article CAS Google Scholar - Wojcikiewicz RJ, Luo SG . Phosphorylation of inositol 1,4,5-trisphosphate receptors by cAMP-dependent protein kinase. Type I, II, and III receptors are differentially susceptible to phosphorylation and are phosphorylated in intact cells. J Biol Chem 1998; 273: 5670–5677.
Article CAS Google Scholar - Soulsby MD, Wojcikiewicz RJ . The type III inositol 1,4,5-trisphosphate receptor is phosphorylated by cAMP-dependent protein kinase at three sites. Biochem J 2005; 392(Pt 3): 493–497.
Article CAS Google Scholar - Giovannucci DR, Groblewski GE, Sneyd J, Yule DI . Targeted phosphorylation of inositol 1,4,5-trisphosphate receptors selectively inhibits localized Ca2+ release and shapes oscillatory Ca2+ signals. J Biol Chem 2000; 275: 33704–33711.
Article CAS Google Scholar - Chaloux B, Caron AZ, Guillemette G . Protein kinase A increases the binding affinity and the Ca2+ release activity of the inositol 1,4,5-trisphosphate receptor type 3 in RINm5F cells. Biol Cell 2007; 99: 379–388.
Article CAS Google Scholar - White C, Li C, Yang J, Petrenko NB, Madesh M, Thompson CB et al. The endoplasmic reticulum gateway to apoptosis by Bcl-X(L) modulation of the InsP3R. Nat Cell Biol 2005; 7: 1021–1028.
Article CAS Google Scholar - Li C, Wang X, Vais H, Thompson CB, Foskett JK, White C . Apoptosis regulation by Bcl-x(L) modulation of mammalian inositol 1,4,5-trisphosphate receptor channel isoform gating. Proc Natl Acad Sci USA 2007; 104: 12565–12570.
Article CAS Google Scholar - Shibao K, Fiedler MJ, Nagata J, Minagawa N, Hirata K, Nakayama Y et al. The type III inositol 1,4,5-trisphosphate receptor is associated with aggressiveness of colorectal carcinoma. Cell Calcium 2010; 48: 315–323.
Article CAS Google Scholar - Khan AA, Soloski MJ, Sharp AH, Schilling G, Sabatini DM, Li SH et al. Lymphocyte apoptosis: mediation by increased type 3 inositol 1,4,5-trisphosphate receptor. Science (New York, NY) 1996; 273: 503–507.
Article CAS Google Scholar - Mendes CC, Gomes DA, Thompson M, Souto NC, Goes TS, Goes AM et al. The type III inositol 1,4,5-trisphosphate receptor preferentially transmits apoptotic Ca2+ signals into mitochondria. J Biol Chem 2005; 280: 40892–40900.
Article CAS Google Scholar - Giorgi C, Ito K, Lin HK, Santangelo C, Wieckowski MR, Lebiedzinska M et al. PML regulates apoptosis at endoplasmic reticulum by modulating calcium release. Science (New York, NY) 2010; 330: 1247–1251.
Article CAS Google Scholar - Boulbes DR, Shaiken T, Sarbassov dos D . Endoplasmic reticulum is a main localization site of mTORC2. Biochem Biophys Res Commun 2011; 413: 46–52.
Article CAS Google Scholar
Acknowledgements
This research was supported by the Italian Association for Cancer Research (AIRC), Telethon (GGP09128 and GGP11139B), local funds from the University of Ferrara, the Italian Ministry of Education, University and Research (COFIN, FIRB and Futuro in Ricerca), the Italian Cystic Fibrosis Research Foundation and Italian Ministry of Health to PP. SM was supported by a FIRC fellowship; AB was supported by a research fellowship FISM – Fondazione Italiana Sclerosi Multipla – Cod. 2010/B/1. We thank all members of the Pinton Lab for stimulating discussion.
SM and PP designed research; SM, MM, AB and MB performed all experiments; SM and PP wrote the paper; SM, AR and CG analyzed data.
Author information
Authors and Affiliations
- Department of Experimental and Diagnostic Medicine, Section of General Pathology, Interdisciplinary Center for the Study of Inflammation (ICSI), Laboratory for Technologies of Advanced Therapies (LTTA), University of Ferrara, Ferrara, Italy
S Marchi, M Marinello, A Bononi, M Bonora, C Giorgi, A Rimessi & P Pinton
Authors
- S Marchi
You can also search for this author inPubMed Google Scholar - M Marinello
You can also search for this author inPubMed Google Scholar - A Bononi
You can also search for this author inPubMed Google Scholar - M Bonora
You can also search for this author inPubMed Google Scholar - C Giorgi
You can also search for this author inPubMed Google Scholar - A Rimessi
You can also search for this author inPubMed Google Scholar - P Pinton
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toP Pinton.
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by D Bano
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Marchi, S., Marinello, M., Bononi, A. et al. Selective modulation of subtype III IP3R by Akt regulates ER Ca2+ release and apoptosis.Cell Death Dis 3, e304 (2012). https://doi.org/10.1038/cddis.2012.45
- Received: 31 January 2012
- Revised: 15 March 2012
- Accepted: 30 March 2012
- Published: 03 May 2012
- Issue Date: May 2012
- DOI: https://doi.org/10.1038/cddis.2012.45