DNA methylation requires a DNMT1 ubiquitin interacting motif (UIM) and histone ubiquitination (original) (raw)
Introduction
Epigenetic mechanisms including DNA and histone modifications are crucial for the regulation of gene expression during development. DNA methylation occurs at the C5 position of cytosine residues, mostly within cytosine-guanine dinucleotides (CpG), and is involved in imprinting, X-chromosome inactivation, stable transcriptional repression, genome stability and tumorigenesis1. DNA methylation patterns are established by the de novo methyltransferases DNMT3A and DNMT3B during gametogenesis and early development, and are propagated by the maintenance methyltransferase DNMT1 after DNA replication in somatic cells.
DNMT1 comprises a regulatory N-terminal domain (NTD), which covers two-thirds of the molecule, and a C-terminal catalytic domain (CD), which contains all essential motifs of active C5 DNA methyltransferases. The NTD controls the subcellular distribution of DNMT1 during the cell cycle and its enzymatic activity. A subdomain in the NTD was initially described as a targeting sequence (TS) as it was found to mediate the association of DNMT1 with late replicating pericentromeric heterochromatin2. Subsequent studies defined a distinct proliferating cell nuclear antigen (PCNA) binding domain (PBD) responsible for the interaction with the replication machinery3. The subnuclear localization of DNMT1 undergoes characteristic changes throughout the cell cycle reflecting PBD-mediated PCNA binding during S phase and TS domain-mediated heterochromatin association during late S and G2 phase4,5. The association of DNMT1 with the replication machinery enhances methylation efficiency, but is not strictly required for postreplicative maintenance DNA methylation6,7. In contrast, the TS domain was found to be required for DNMT1 enzymatic activity8,9. However, the molecular mechanism of TS domain function in the regulation of maintenance DNA methylation remains elusive.
Besides its role in replication-independent heterochromatin binding, the TS domain mediates DNMT1 homodimerization9 and autoinhibition10,11. A recent crystal structure shows that the TS domain inserts into the DNA binding pocket of the CD, indicating a role of intramolecular interactions in the regulation of DNMT1 activity10,11. Moreover, the TS domain interacts with the SET- and RING-associated (SRA) domain of ubiquitin like, containing PHD and RING finger domains 1 (UHRF1)12,13,14. In contrast to UHRF2, the interaction of UHRF1 with DNMT1 was found to be S phase-dependent15.
UHRF1, also known as NP95 (mouse) or ICBP90 (human), has been reported as a crucial cofactor for maintenance DNA methylation. Mice lacking UHRF1 show a similar phenotype as Dnmt1 null (Dnmt1 −/−) mice that manifests in genomic DNA hypomethylation and developmental arrest at embryonic day 9.516,17,18. The SRA domain of UHRF1 preferentially binds to hemimethylated DNA resulting from semiconservative DNA replication and is, therefore, thought to play an important role in loading DNMT1 onto newly synthesized DNA substrates16,17,19,20,21,22. The heterochromatin association of UHRF1 is also mediated by the tandem Tudor domain (TTD), which forms an aromatic cage for specific binding of histone H3 tails containing a trimethylated lysine 9 (H3K9me3) residue22,23,24,25. The plant homeodomain (PHD) was reported to act in combination with the TTD to read the H3K9me3 mark26 and to contribute to large-scale reorganization of pericentromeric heterochromatin27. In addition, UHRF1 harbors a really interesting new gene (RING) domain endowed with ubiquitin E3 ligase activity in vitro, which is required for growth regulation of tumor cells24,28. The ubiquitination state and stability of DNMT1 is controlled by UHRF1 and the ubiquitin-specific protease USP729,30. UHRF1 overexpression leads to DNA hypomethylation by the destabilization and delocalization of DNMT131. Besides its role in marking DNMT1 for proteasomal degradation, UHRF1 also exerts its ubiquitin E3 ligase activity on histone substrates24,25.
A recent study describes replication-dependent H3K23 ubiquitination by UHRF1 in Xenopus extracts32. Knockdown and rescue experiments in HeLa cells showed that SRA domain-mediated DNA binding as well as RING domain-dependent E3 ubiquitin ligase activity of UHRF1 are required for H3 ubiquitination. Expression of the SRA and RING domain mutants in Uhrf1 −/− mouse cells could neither restore DNMT1 replication targeting nor DNA methylation levels. A deletion of large parts of the DNMT1 TS domain abolished binding to ubiquitinated H3K23 in vitro, but effects on enzymatic activity were not investigated. In particular, the structure and function of the rather large TS domain with its multiple roles and interactions remain to be clarified.
In this study, we elucidate the complex interplay between UHRF1 and DNMT1. While we could confirm the general role of UHRF1 in recruiting DNMT1 to substrate sites by direct interaction, we found that DNMT1 targeting and activities are essentially controlled by specific binding to histone tails ubiquitinated by UHRF1. We generated defined mutations in different UHRF1 domains that retained SRA domain-mediated binding to hemimethylated DNA substrate sites, TTD-mediated recognition of H3K9me3 and binding of DNMT1, but did not allow maintenance DNA methylation. We could show that binding to unmodified H3R2 via the PHD and ubiquitination of H3K18 via the RING domain are required for UHRF1 to mediate maintenance DNA methylation. In turn, we identified a ubiquitin interacting motif (UIM) in the TS domain of DNMT1 that reads this ubiquitin mark and is strictly required for maintenance DNA methylation in vivo. These results show the manifold regulatory mechanisms controlling DNMT1 activity and illustrate the multifaceted interplay between DNA and histone modifications.
Results
The interaction of DNMT1 with UHRF1 is required for maintenance DNA methylation
To test whether the interaction of DNMT1 with UHRF1 is indeed required for maintenance DNA methylation, we generated stable cell lines based on Dnmt1 −/− ESCs expressing green fluorescent protein (GFP) fusions of either DNMT1 wild-type (GFP-DNMT1 wt) or a truncated TS domain deletion mutant (GFP-DNMT1 Δ458-500) that is defective in binding to UHRF1 (Figure 1A and 1B). The deleted region was determined by a sequence alignment of TS domains from higher eukaryotes and a conserved core region of the domain was chosen for mutational analysis (Supplementary information, Figure S1A). In contrast to GFP-DNMT1 wt, GFP-DNMT1 Δ458-500 did not co-localize with cherry (Ch)-UHRF1 and showed a dispersed distribution in the nucleus (Figure 1C), suggesting that the interaction with UHRF1 is essential for subnuclear localization of DNMT1.
Figure 1
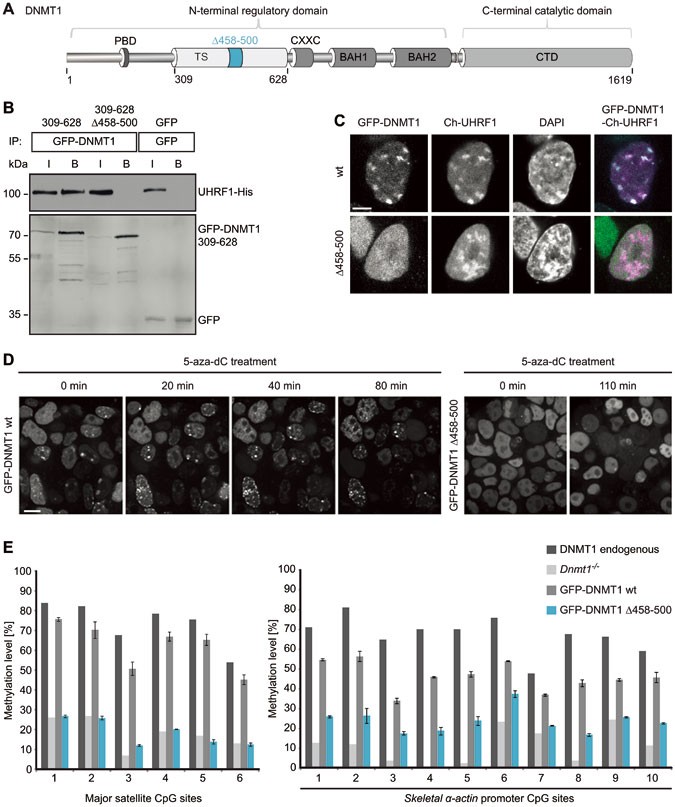
The DNMT1 TS domain is required for UHRF1 interaction, heterochromatin targeting and maintenance DNA methylation. (A) Schematic outline of DNMT1 domains and the TS domain deletion (Δ458-500). DNMT1 comprises a large N-terminal domain (NTD) harboring the PCNA binding domain (PBD), the targeting sequence (TS) domain and two bromo adjacent homology (BAH) domains. The active catalytic center of DNMT1 resides within its C-terminal domain (CD). (B) Co-immunoprecipitation of UHRF1-His and the GFP-DNMT1 TS domain (309-628) wild-type (wt) or Δ458-500 constructs. Both constructs were co-expressed in HEK 293T cells and after immunoprecipitation of GFP fusions, bound proteins were detected by western blot with an anti-UHRF1 and an anti-GFP antibody. GFP was used as negative control. I, input; B, bound. (C) Confocal mid sections of fixed ESCs stably expressing GFP-DNMT1 wt or Δ458-500 mutant constructs. Ch-UHRF1 was transiently co-expressed to illustrate heterochromatic regions, DAPI was used for counterstaining. Scale bar, 5 μm. (D) Covalent complex formation of GFP-DNMT1 wt and GFP-DNMT1 Δ458-500 mutant were analyzed by an in vivo trapping assay. Confocal mid-sections of ESCs stably expressing GFP-DNMT1 wt and deletion mutant constructs before and after treatment with the mechanism-based inhibitor 5-aza-dC are displayed. Scale bar, 10 μm. (E) Local DNA methylation analyses at the major satellite repeats and the skeletal α-actin promoter. CpG methylation levels of mouse Dnmt1 −/− ESCs stably expressing GFP-DNMT1 wt or GFP-DNMT1 Δ458-500 mutant constructs were analyzed by bisulfite treatment of genomic DNA, PCR amplification and direct pyrosequencing. The methylation level of the J1 wt cell line (endogenous DNMT1) and untransfected Dnmt1 −/− cells are shown for comparison. Mean values ± SD from two different clones were calculated, respectively.
Next, we investigated the role of UHRF1 interaction for the catalytic function of DNMT1. Notably, GFP-DNMT1 Δ458-500 that did not interact with UHRF1 was able to fully methylate hemimethylated DNA substrates in vitro (Supplementary information, Figure S1C). To test the DNA methylation activity of this deletion mutant in vivo, we made use of a trapping assay. In this assay, the cytosine analogue 5-aza-2′-deoxycytidine (5-aza-dC) forms an irreversible covalent complex with the methyltransferase at the C6 position of the cytosine residue when incorporated into DNA during replication thereby trapping the enzyme at DNA replication foci. Trapped DNMT1 fractions increase over time and allow monitoring the activity-dependent accumulation of DNMT1 at its target sites33. In ESCs stably expressing GFP-DNMT1 wt, foci of immobilized protein emerged already within 20 min (Figure 1D, left panel). In contrast, GFP-DNMT1 Δ458-500 was not enriched at replication foci even after 110 min, indicating that the deletion mutant is unable to methylate newly replicated DNA in living cells (Figure 1D, right panel). To pursue this idea, we further analyzed site-specific DNA methylation levels of stable GFP-DNMT1 wt and Δ458-500 ESC lines (Supplementary information, Figure S1B). GFP-DNMT1 could restore local DNA methylation at the major satellite repeats in Dnmt1 −/− ESCs leading to an average methylation level of 62% that is comparable to the level of the wt cell line expressing the endogenous protein (74%, Figure 1E, left panel). In contrast, the DNMT1 mutant deficient in UHRF1 binding was unable to reestablish local DNA methylation patterns resulting in decreased levels at the major satellite repeats (average 19%) similar to the Dnmt1 −/− control cell line (average 18%). Consistently, a similar defect of GFP-DNMT1 Δ458-500 in DNA methylation activity was observed at the single-copy sequence of the skeletal α-actin promoter (Figure 1E, right panel). Furthermore, similar results were obtained from DNA methylation analyses at the minor satellite repeats and the Dnmt1o promoter confirming that stable expression of GFP-DNMT1 Δ458-500 could not restore DNA methylation in a Dnmt1 −/− cell line (Supplementary information, Figure S1D).
In summary, we provide strong evidence that the GFP-DNMT1 Δ458-500 mutant deficient in UHRF1 binding, even though able to methylate DNA substrates in vitro, cannot restore DNA methylation patterns in Dnmt1 −/− ESCs. These findings suggest that the interaction of DNMT1 with UHRF1 is required to maintain DNA methylation in vivo.
The PHD and RING domain of UHRF1 are essential for maintenance DNA methylation
Cooperative binding of the UHRF1 TTD to di- and trimethylated histone H3K9 and of the SRA domain to hemimethylated DNA was described as a prerequisite for targeting DNMT1 to its substrate and for subsequent DNA methylation34. Given the regulatory impact of these two domains, we were interested in how the PHD and RING domain of UHRF1 may functionally contribute to maintenance DNA methylation by DNMT1. To this end, we introduced point mutations in the PHD and RING domain (UHRF1-GFP H346G and UHRF1-GFP H730A, respectively) that are expected to prevent coordination of zinc ions by zinc-finger motifs (Figure 2A and Supplementary information, Figure S2A). Consequently, the mutation in the RING domain significantly reduced the E3 ubiquitin ligase activity of UHRF1 in vivo (Supplementary information, Figure S2C and S2D). Notably, the preference of UHRF1-GFP for hemimethylated DNA was not impaired by the PHD and RING domain mutations (Supplementary information, Figure S2B).
Figure 2
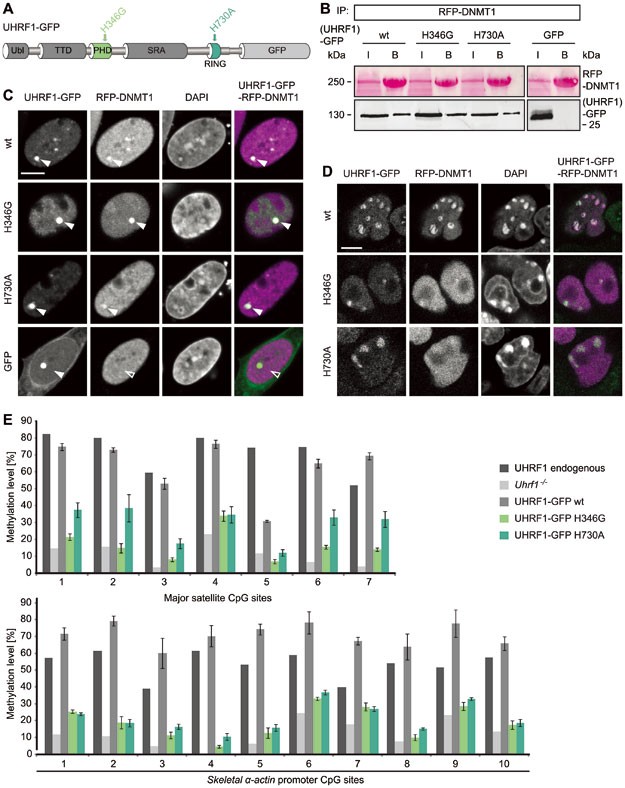
Mutations in the PHD and RING domain of UHRF1 affect DNMT1 targeting and maintenance DNA methylation, but not the interaction with DNMT1. (A) Schematic outline of the multidomain protein UHRF1. UHRF1 harbors a ubiquitin-like (Ubl) domain, a plant homeodomain (PHD) and a tandem Tudor domain (TTD) followed by a SET and RING-associated (SRA) domain and a really interesting new gene (RING) domain. UHRF1-GFP expression constructs carrying point mutations in the PHD (H346G) and RING domain (H730A) are illustrated. (B) Co-immunoprecipitation of UHRF1-GFP wt or PHD and RING domain mutants co-expressed with RFP-DNMT1 in HEK 293T cells. RFP-DNMT1 was immunoprecipitated using the RFP-Trap and bound UHRF1-GFP was detected by western blot with an anti-GFP antibody. GFP was used as negative control. Immunoprecipitated RFP-DNMT1 is shown by Ponceau staining. I, input; B. bound. (C) Fluorescence three-hybrid assay for visualization of the interaction RFP-DNMT1 with UHRF1-GFP wt or PHD and RING domain mutants. Displayed are confocal mid sections of BHK cells carrying a stably integrated Lac-operator array that were triple transfected with LacI fused to the GFP-binder, UHRF1-GFP constructs and RFP-DNMT1. DAPI was used for chromatin counterstaining. Closed arrows indicate the co-localization of both proteins at the _lac_O spot, open arrows indicate no co-localization. GFP was used as negative control. Scale bar, 5 μm. (D) Confocal mid sections of fixed Uhrf1 −/− ESCs stably expressing UHRF1-GFP wt or PHD and RING domain mutant constructs. RFP-DNMT1 was transiently co-expressed and DNA was counterstained by DAPI. Scale bar, 5 μm. (E) Local DNA methylation analyses at major satellite repeats and the skeletalα-actin promoter. CpG site methylation levels of mouse E14 Uhrf1 −/− ESCs stably expressing UHRF1-GFP wt or PHD and RING domain mutant constructs were analyzed by bisulfite treatment of genomic DNA, PCR amplification and direct pyrosequencing. The methylation level of E14 wt ESCs (endogenous UHRF1) and untransfected E14 Uhrf1 −/− cells are shown for comparison. Mean values ± SD from two different clones were calculated, respectively.
First, we tested whether the point mutations in the PHD and RING domain influence the interaction of UHRF1 with DNMT1. UHRF1-GFP wt as well as UHRF1-GFP H346G and UHRF1-GFP H730A still co-precipitated with red fluorescent protein (RFP)-DNMT1, indicating that the mutations do not affect the interaction with DNMT1 directly (Figure 2B). In addition, the unaltered interactions were confirmed by a fluorescent three-hybrid assay35,36. In this assay, UHRF1-GFP fusion constructs were used as baits by tethering them to a lac operator (lacO) array present in baby hamster kidney (BHK) cells that simultaneously express RFP-DNMT1 as a prey. Accumulation of RFP-DNMT1 at the _lac_O spot enriched for UHRF1-GFP wt, UHRF1-GFP H346G or UHRF1-GFP H730A clearly demonstrates that the mutant proteins were still able to interact with DNMT1 in vivo (Figure 2C).
In order to perform functional studies on the PHD and RING domain mutants, we stably expressed GFP-tagged UHRF1 wt, UHRF1 H346G or UHRF1 H730A in Uhrf1 −/− ESCs. Similar to wt, also UHRF1-GFP H346G and UHRF1-GFP H730A showed focal enrichment at heterochromatin (Figure 2D, first panel and Supplementary information, Figure S2E). Thus, the mutations do not affect localization of UHRF1. In contrast to its chromatin association in the UHRF1-GFP wt cell line, transiently co-expressed RFP-DNMT1 did not co-localize with UHRF1-GFP H346G and UHRF1-GFP H730A, but showed a dispersed distribution in the nucleus (Figure 2D, second panel). This observation is consistent with the result of a staining for endogenous DNMT1 (Supplementary information, Figure S2F). Only in the UHRF1-GFP wt cell line, endogenous DNMT1 was enriched at S phase-specific replication sites, whereas it was diffusely distributed in the nucleus of the mutant cell lines pointing towards a defective DNMT1 targeting mechanism. To examine if DNMT1 methylation activity depends on the PHD and RING domain of UHRF1, we performed site-specific methylation analyses at heterochromatic regions. Consistent with defects in targeting DNMT1 to replication sites, DNA methylation levels at the major satellite repeats and the skeletal α-actin promoter revealed that both UHRF1-GFP H346G and UHRF1-GFP H730A were not able to mediate DNA remethylation by DNMT1 in Uhrf1 −/− ESCs in contrast to UHRF1-GFP wt (Figure 2E). Especially at the major satellite repeats, the average DNA methylation in the PHD mutant cell lines remained nearly unchanged (16%) from the Uhrf1 −/− control cell line (11%). Also, the average methylation levels in the RING domain mutant cell lines (29%) did not reach the wt DNA methylation level (62%) at the major satellite repeats. Similar results were obtained for the minor satellite repeats and the Dnmt1o promoter (Supplementary information, Figure S3A). Consistent with this site-specific DNA hypomethylation, the stable UHRF1 mutant cell lines also showed decreased global DNA methylation levels as compared with the wt (Supplementary information, Figure S3B and S3C). Partial rescue of global DNA methylation in the RING domain mutant cell lines could be due to residual E3 ubiquitin ligase activity of UHRF1-GFP H730A (Figure 3B, Supplementary information, Figure S2C and S2D).
Figure 3
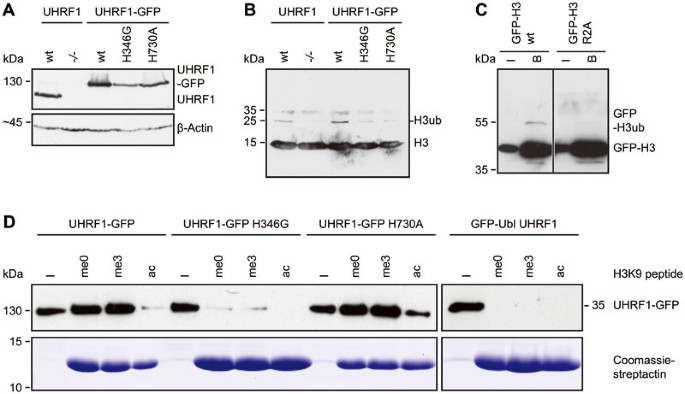
Histone H3 ubiquitination requires the UHRF1 PHD and RING domain. (A) Western blot analyses of endogenous UHRF1 or stably expressed UHRF1-GFP wt or H346G and H730A mutants in E14 Uhrf1 −/− ESCs with an anti-UHRF1 antibody. Equal loading is shown by an anti-β-Actin antibody. (B) Analyses of H3 ubiquitination from acid extracted histones derived from the different cell lines in (A). A specific anti-H3 antibody was used for detection. (C) Ubiquitination of GFP-H3 in dependence on R2. GFP-H3 wt and the arginine to alanine mutant (R2A) were co-expressed with UHRF1-His in HEK 293T cells, respectively, and after immunoprecipitation with the GFP-Trap, the bound fraction was detected by western blotting with a specific anti-H3 antibody. I, input; B, bound. (D) In vitro peptide pull-down assay of UHRF1-GFP wt or the PHD and RING domain mutants from crude cells extracts of HEK 293T cells using H3 peptides (amino acid 1-20) that were either unmodified (me0), K9 trimethylated (me3) or K9 acetylated (ac) and functionalized on streptactin beads. The GFP-Ubl domain of UHRF1 was used as negative control, Coomassie-stained streptactin is shown as loading control. I, input.
To exclude the possibility that DNA hypomethylation might result from lower expression of the PHD and RING domain mutant (Figure 3A), we performed a transient rescue assay in Uhrf1 −/− ESCs. Even though expression levels of the mutant constructs exceeded those of the UHRF1-GFP wt, the PHD and RING domain mutants could not mediate remethylation at the major satellite repeats (Supplementary information, Figure S3D and S3E) arguing for functional rather than expression defects.
In summary, the PHD and RING domain mutants, although not affecting UHRF1 heterochromatin localization or the direct interaction with DNMT1, cannot mediate either targeting of DNMT1 to replication foci nor maintenance DNA methylation. These findings suggest that these UHRF1 domains contribute to the recruitment of DNMT1 by indirect mechanisms.
The PHD and RING domain of UHRF1 are required for ubiquitination of histone H3
Histone H3 has been reported as a UHRF1-dependent ubiquitination target in Xenopus egg extracts32, providing a potential mechanism for the recruitment of DNMT1 to chromatin. Thus, we set out to investigate whether H3 ubiquitination required PHD-mediated histone binding and RING domain-mediated ubiquitin E3 ligase activity of UHRF1 in mammalian cells. To this end, we extracted histones from wt or Uhrf1 −/− ESCs and detected modified H3. As expected, histone H3 was less ubiquitinated in the absence of UHRF1 (Figure 3A and 3B), indicating that UHRF1 serves as a ubiquitin E3 ligase for H3 in mammalian cells. We also found that ubiquitination levels of histone H3 in Uhrf1 −/− ESCs stably expressing the RING domain mutant UHRF1-GFP H730A were not rescued to the level of wt cells. Surprisingly, the PHD mutant UHRF1-GFP H346G also could not restore ubiquitination of histone H3 (Figure 3A and 3B).
Since the PHD has been reported to bind to unmodified H3R226,37,38,39, we investigated the role of this histone residue in H3 ubiquitination by mutational analyses. Compared with GFP-H3 wt, ubiquitination of a GFP-H3 R2A mutant expressed in human embryonic kidney (HEK) 293T cells was clearly reduced (Figure 3C) pointing towards an important role of the R2 residue for UHRF1-dependent H3 ubiquitination.
To further test the histone binding properties of the PHD mutant in vitro, we performed a peptide pull-down assay with wt or PHD and RING domain mutant UHRF1-GFP using H3 peptides with an unmodified, trimethylated or acetylated K9 residue. The mutation in the RING domain did not alter the histone binding of UHRF1-GFP showing a preference for unmodified and K9 trimethylated H3 peptides similar to the wt protein (Figure 3D). The mutation in the PHD, however, decreased the binding to both, the unmodified and the K9 trimethylated peptide. We further examined the histone binding preferences of UHRF1-GFP with an in vitro histone tail binding assay. The results revealed the binding of UHRF1-GFP to unmodified but not R2 dimethylated H3 histone tails (Supplementary information, Figure S4), consistent with prior _K_d measurements39. As the PHD of UHRF1 has been shown to bind unmethylated H3R2 residues and to contribute to the K9 methylated H3 histone binding of the TTD26,37,38,39, we propose that PHD-dependent histone binding is required for UHRF1-mediated ubiquitination of histone H3.
UHRF1 ubiquitinates histone H3 on K18 in mammalian cells
Using Xenopus extracts immunodepleted for DNMT1, H3 was shown to be ubiquitinated at the K23 residue32. To map ubiquitination sites on histone H3 tails in mammalian cells, we performed mass spectrometry using human and mouse cells. In contrast to the results from Xenopus extracts, the K18 residue of histone H3 was identified as novel ubiquitination site in mouse ESCs, while the K23 residue was unmodified or acetylated (Figure 4A and 4B). Relative quantification of H3 peptides containing ubiquitinated K18 and an unmodified or acetylated K23 residue showed a reduction of K18 ubiquitination in ESCs lacking UHRF1 (Figure 4C and 4D). Similarly, immunoprecipitation of GFP-UHRF1 from HEK 293T cells and subsequent mass spectrometry also revealed ubiquitination at K18 but not at K23 (Supplementary information, Figure S5A). Comparison of ubiquitination levels of overexpressed GFP-H3 carrying R2A, K18A or K23A mutations suggests that in this constellation K23 could also be modified (Supplementary information, Figure S5B). Interestingly, the GFP-H3 R2A construct showed reduced ubiquitination levels indicating that the R2 residue plays a role in regulating H3 ubiquitination.
Figure 4
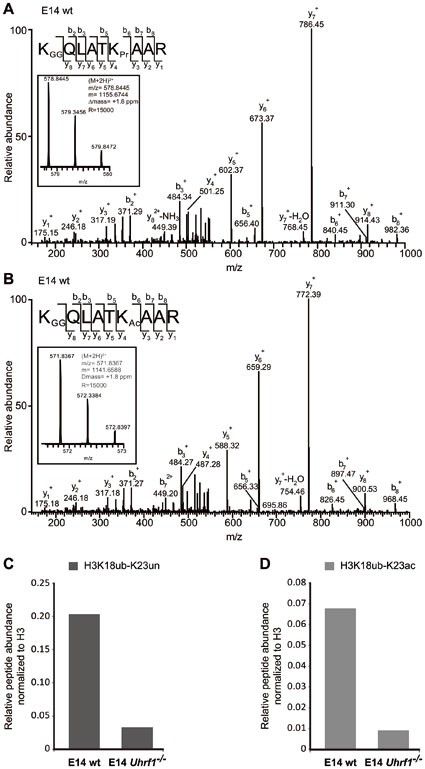
UHRF1 ubiquitinates histone H3 at K18 in mammalian cells. (A) Identification of H3 18-26 peptides carrying ubiquitination (GG) at K18 and no modification (Pr) at K23 by LC-MS/MS. MS2 fragmentation spectrum of the precursor ion is shown in the inset. An almost complete series of b and full y product ions generated by CID fragmentation were detectable providing a high confidence in its correct identification and localization of the ubiquitin modification. Inset: mass, charge and measurement error determination of the H3 18-26 peptides K18GGK23Pr in the E14 wt sample. Displayed is the isotopic distribution of the H3 peptide from which the mass to charge ratio (m/z), the charge (2+) and the monoisotopic mass value (m) were derived. Δ_m_: difference between the expected and the measured masses; R: resolution of the MS measurement. (B) Identification of H3 18-26 peptides carrying ubiquitination (GG) at K18 and acetylation (Ac) at K23 by LC-MS/MS as in (A). (C, D) Quantification of H3 18-26 peptides carrying ubiquitination (ub) at K18 and an unmodified (un) or acetylated (ac) K23 residue from E14 wt and E14 Uhrf1 −/− samples. Extracted ion chromatograms of the ions corresponding to the peptides of interest were used for the quantification. The signals were normalized against the total amount of analyzed H3 proteins.
DNMT1 harbors a UIM that mediates binding to ubiquitinated H3 and is essential for DNA methylation activity in vivo
To unravel how H3 ubiquitination may contribute to maintenance DNA methylation, we screened DNMT1 for potential binding motifs. With bioinformatics analyses, we identified a ubiquitin interacting motif (UIM) in the N-terminal regulatory domain of DNMT1. This motif is located in a region spanning from amino acid 380 to 399 of mouse DNMT1 and shows striking similarity to UIMs of known ubiquitin interacting proteins (Figure 5A). Comparison of the ubiquitin binding properties between GFP-DNMT1 wt and mutants either lacking the UIM (Δ356-404) or containing substitutions of the relevant and conserved amino acids in the motif to alanine (D381A-E382A-S392A, D381A-E382A-M385A-S392A-D395A, Figure 5A and Supplementary information, Figure S6A) showed a defect in the association with ubiquitinated histone H3 and ubiquitinated H2AK119 (Figure 5B, 5C and Supplementary information, Figure S6B-S6D). To further elucidate UIM-dependent ubiquitinated histone binding, we quantified modified H318-26 peptides bound by GFP-DNMT1 wt or the UIM mutants by mass spectrometry. Whereas H3 histone peptides ubiquitinated at K18 and acetylated or unmodified at K23 co-immunoprecipitated with GFP-DNMT1 wt, only little to no ubiquitinated peptide signals were detected for the UIM mutants (Figure 6A, 6B). GFP-DNMT1 Δ458-500 defective in UHRF1 interaction (Figure 1B) showed reduced (Figure 6B) or undetectable (Figure 5B, 5C) binding to ubiquitinated H3 and H2A. This deletion located in a TS domain region C-terminal of the UIM might affect the integrity and functionality of the motif responsible for ubiquitin binding. Therefore, we cannot rule out that apart from disrupted UHRF1 binding also defects in the association with ubiquitinated histones contributed to the observed changes in subnuclear distribution and protein function of GFP-DNMT1 Δ458-500 (Figure 1C-1E).
Figure 5
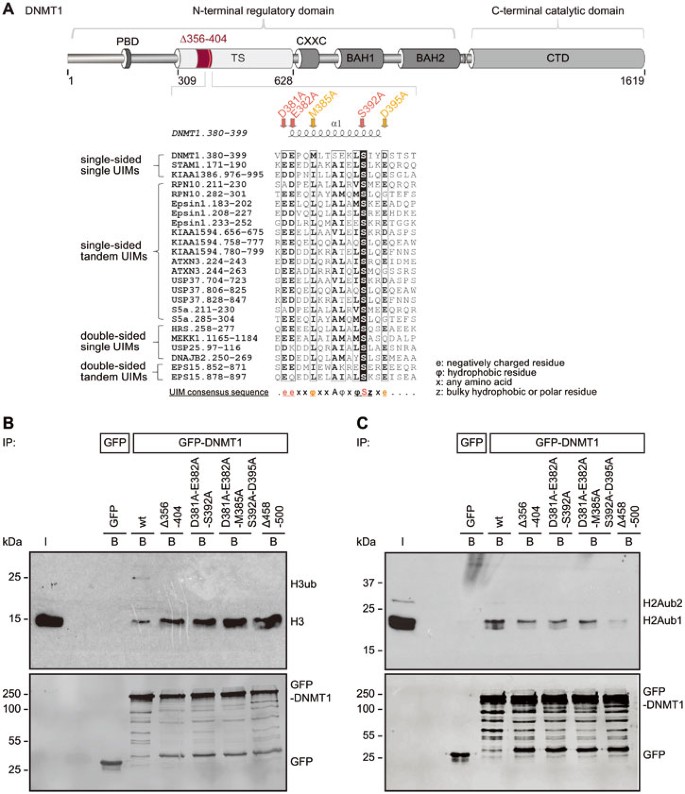
The TS domain of DNMT1 harbors a ubiquitin interacting motif (UIM) that is essential for binding to ubiquitinated H3 and H2A. (A) Schematic outline of the UIM in the TS domain of DNMT1 and indication of the UIM deletion (Δ356-404) and the point mutations (D381A-E382A-S392A and D381A-E382A-M385A-S392A-D395A). A peptide sequence of DNMT1 encompassing amino acid 380-399 was aligned with peptide sequences of proteins previously known to contain UIMs. Identical amino acids are highlighted in black, highly similar amino acids are framed in black. The secondary structure of the DNMT1 region (pdb: 3EPZ10) harboring the UIM is displayed on top of the sequence alignment generated using ESPript78. The consensus sequence for single-sided UIMs58 is shown below. The UIMs were found by scanning the protein primary sequences against a collection of motifs in ExPASy Prosite. Putative subgroups of UIMs are indicated on the left. (B) Ubiquitinated histone H3 binding assay. After extraction of histones from HEK 293T cells, the extracts were incubated with GFP-DNMT1 wt or mutants immobilized on the GFP-Trap and the bound fractions were analyzed by western blotting with specific anti-H3 and anti-GFP antibodies. GFP was used as negative control. I, input; B, bound. (C) Ubiquitinated histone H2A binding assay as in (B). Bound fractions were analyzed by western blotting with specific anti-H2AK119ub and anti-GFP antibodies. Analyses of the anti-H2AK119ub antibody specificity and of peptides isolated from the corresponding band are shown in Supplementary information, Figure S6C and S6D. H2Aub1, monoubiquitinated H2A; H2Aub2, diubiquitinated H2A.
Figure 6
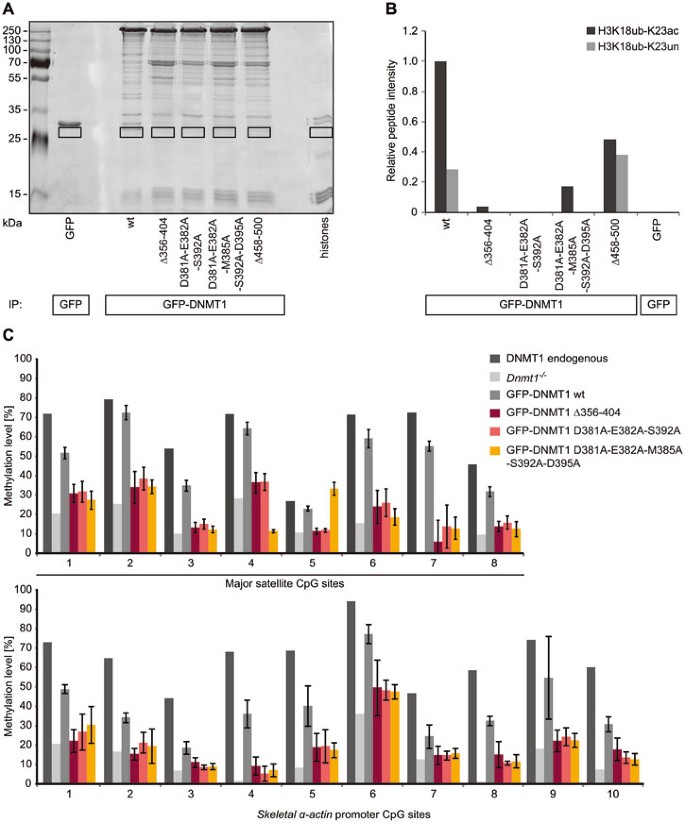
The DNMT1 UIM is required for ubiquitinated H3K18 binding and for DNA methylation. (A) Ubiquitinated histone binding experiments using GFP-DNMT1 wt or UIM mutants as well as the Δ458-500 mutant deficient in binding to UHRF1. Equal amounts of GFP fusions were immobilized on the GFP-Trap and incubated with acid extracted histones. Bound proteins were visualized by Coomassie staining and the fractions highlighted by black rectangles were analyzed by mass spectrometry. GFP was used as negative control. (B) Quantification of H3 18-26 peptides carrying ubiquitination (ub) at K18 and an acetylated (ac) or unmodified (un) K23 residue from histone binding experiment shown in (A). Extracted ion chromatograms of the ions corresponding to the peptides of interest were used for quantification (H3K18ubK23ac: m/z = 571.8353 ± 10 ppm; H3K18ubK23un: m/z = 578.8441 ± 10 ppm). (C) Local DNA methylation analyses of J1 Dnmt1 −/− ESCs expressing GFP-DNMT1 wt or Δ356-404 and UIM point mutants. CpG methylation levels at the major satellite repeats and the skeletal α-actin promoter were analyzed by bisulfite treatment of genomic DNA, PCR amplification and direct pyrosequencing. Methylation levels of untransfected J1 Dnmt1 −/− cells are shown for comparison. Mean values ± SD from three to four biological replicates were calculated, respectively.
Besides a decreased binding to ubiquitinated H3, the TS domain point and deletion mutants exhibited an increased binding to H3 or core histones compared with GFP-DNMT1 wt (Figure 5B). Therefore, specific binding of DNMT1 to ubiquitinated H3 via its UIM might prevent the enzyme from stable chromatin association and thereby facilitate DNA methylation.
To clarify the functional role of the UIM in maintenance DNA methylation in vivo, we performed a functional complementation assay in Dnmt1 −/− ESC lines transiently expressing GFP-DNMT1 wt, GFP-DNMT1 Δ356-404, GFP-DNMT1 D381A-E382A-S392A or GFP-DNMT1 D381A-E382A-M385A-S392A-D395A. Local DNA methylation analyses at the major satellite repeats and the skeletal α-actin promoter showed that the UIM mutants were not able to reestablish DNA methylation patterns (Figure 6C). GFP-DNMT1 wt restored DNA methylation at the major satellite repeats to 48%. By comparison, the UIM deletion and point mutants were not able to rescue resulting in low average methylation levels of 20% to 23% comparable to untransfected Dnmt1 −/− ESCs (15%). Similar results were also observed at the minor satellite repeats and the Dnmt1o promoter (Supplementary information, Figure S7A).
Given that the GFP-DNMT1 TS UIM deletion and point mutants were able to interact with Ch-UHRF1 (Supplementary information, Figure S7B), we were interested in how the UIM in DNMT1 has an influence on the subnuclear localization of the protein. Immunostaining of replicating DNA with a specific anti-PCNA antibody indicated that GFP-DNMT1 wt was enriched at S phase-specific replication foci, while GFP-DNMT1 Δ356-404, GFP-DNMT1 D381A-E382A-S392A and GFP-DNMT1 D381A-E382A-M385A-S392A-D395A showed only weak association with the PCNA-stained replication sites especially in late S phase (Supplementary information, Figure S8). To analyze the UIM-dependent enrichment of DNMT1 at late-replicating heterochromatin, we quantified mean fluorescence intensities at chromocenters compared with the nucleoplasmic region (Figure 7A). In late S phase ES and mouse embryonic fibroblasts (MEF) cells, GFP-DNMT1 wt localized at chromocenters, whereas the UIM mutations abolished heterochromatin enrichment (Figure 7B and 7C). These results clearly demonstrate the key role of the UIM in DNMT1 targeting via ubiquitinated histone H3 binding and for maintenance DNA methylation in mammalian cells.
Figure 7
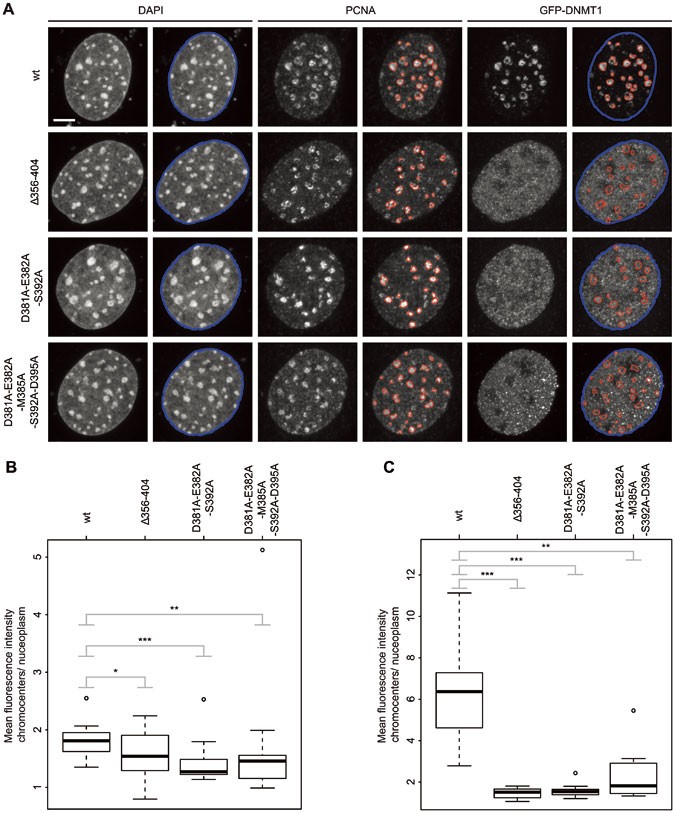
GFP-DNMT1 UIM mutants show a decreased association with PCNA-stained replication sites in late S phase compared with the wt. (A) Maximum intensity projections of MEF cells transiently expressing GFP-DNMT1 wt or UIM mutants. Replicating DNA was stained with a specific anti-PCNA antibody and chromatin was counterstained with DAPI. Replication foci masks (red) match the enrichment of GFP-DNMT1 wt in late S phase, whereas the UIM mutants do not show a focal enrichment. Segmentations were generated in an automated fashion using a machine learning algorithm (WEKA). The nuclear mask outlined in blue was based on the DAPI staining, whereas the replication foci masks outlined in red were based on the PCNA staining. Both masks were superimposed on the GFP channels. The GFP-DNMT1 signal inside the red masks (chromocenters) relative to the remainder of the nucleus (nucleoplasm) was quantified. Scale bar, 5 μm. (B) Quantification of chromocenter association of GFP-DNMT1 wt or UIM mutants in late S phase J1 Dnmt1 −/− ESCs. The ratio of the mean GFP fluorescence intensity at chromocenters over the mean intensity in the nucleoplasm is shown in the box plot from 15 (wt), 16 (Δ356-404), 12 (D381A-E382A-S392A) or 18 (D381A-E382A-M385A-S392A-D395A) cells. The results were further analyzed in R using a Wilcoxon test and considered as statistical significant for P < 0.05 (*) and P < 0.01 (**) or highly significant for P < 0.001 (***). The following P values were calculated: Δ356-404: P = 0.049, D381A-E382A-S392A: P = 0.0016 and D381A-E382A-M385A-S392A-D395A: _P_= 0.0056. (C) Quantification of chromocenter association of GFP-DNMT1 wt or UIM mutants in late S phase MEF cells as in (B). Eleven (wt), 12 (Δ356-404, D381A-E382A-S392A) or 10 (D381A-E382A-M385A-S392A-D395A) cells were analyzed. The following P values were calculated in R using a Wilcoxon test: Δ356-404: P = 0.00000148, D381A-E382A-S392A: P = 0.00000148 and D381A-E382A-M385A-S392A-D395A: P = 0.0012.
Discussion
DNA methylation is an important epigenetic modification regulating gene expression in development and disease. A key question is how methylation marks are set, maintained and removed. According to previous models, DNA methylation marks are set by the de novo methyltransferases DNMT3A and DNMT3B during development and maintained by the maintenance DNA methyltransferase DNMT1 that specifically recognizes and modifies hemimethylated DNA substrates. However, the preference of DNMT1 for hemimethylated DNA measured in vitro40,41,42,43 is not sufficient to explain efficient maintenance of DNA methylation patterns over many cell division cycles in vivo. The interaction of DNMT1 with the replication protein PCNA was shown to enhance maintenance DNA methylation by a factor of two, but not to be essential6,7. In contrast, the interacting factor UHRF1 recruiting and allosterically activating DNMT1 is essential for DNA methylation14,16,17,44. In this study, we have now dissected the distinct role of different UHRF1 and DNMT1 domains in directing DNA methylation.
In line with previous studies, we show that, albeit being weak, the TS domain-mediated interaction of DNMT1 with the SRA domain of UHRF1 is required for targeting and function of DNMT1 in vivo. Accordingly, truncated DNMT1 (Δ458-500) deficient in UHRF1 binding showed weaker association with chromocenters in late S phase mouse fibroblasts4 and failed to maintain DNA methylation in ESC (Figure 1).
Heterochromatin binding of UHRF1 is mediated by the TTD, PHD and SRA domain and defects in any of these three domains lead to decreased DNA methylation by DNMT134,45,46. Accordingly, it was postulated that UHRF1 reads and binds repressive histone marks and hemimethylated DNA and via direct protein-protein interaction recruits DNMT1 for maintenance DNA methylation.
Defects of a RING domain mutant (C713A, C515A and C716A) in restoring ubiquitinated H3 in HeLa cells after knockdown of human DNMT1 and UHRF1 have previously been reported32. We found that the RING domain, though not directly involved in UHRF1 chromatin binding or interaction with DNMT1, is indispensable for DNA methylation by DNMT1. Remarkably, a UHRF1 RING domain mutant (H730A) with diminished ubiquitin E3 ligase activity (Supplementary information, Figure S2C and S2D) that could still bind DNMT1 (Figure 2B), hemimethylated DNA and K9 trimethylated H3 peptides in vitro (Figure 3D and Supplementary information, Figure S2B) and chromocenters in vivo (Supplementary information, Figure S2E), nonetheless failed in recruiting DNMT1 to replication sites (Figure 2D and Supplementary information, Figure S2F). These findings suggest that DNMT1 recruitment to replication forks is not based on direct interaction with UHRF1, but on the catalytic activity of the RING domain. Previously, the RING domain of UHRF1 has been reported to have an autoubiquitination activity28 and, in addition, to ubiquitinate DNMT129,30 and histone substrates24,25. A recent study describes that ubiquitination of H3 by UHRF1 provides docking sites for DNMT1 on chromatin and thus couples maintenance DNA methylation and replication32. While we could confirm the essential role of UHRF1, we obtained new insights into the complex functional interplay of UHRF1 and DNMT1 domains.
First, in contrast to ubiquitination at K23 in Xenopus egg extracts32, our mass spectrometry results identified H3K18 as ubiquitination target of UHRF1 in mammalian cells (Figure 4A, 4B and Supplementary information, Figure S5A). By mutational analysis in HEK 293T cells, we found that in absence of K18, the mutated GFP-tagged H3 might be ubiquitinated at K23 (Supplementary information, Figure S5B). However, by semiquantitative analysis of endogenous ubiquitinated H3 peptides in wt versus Uhrf1 −/− mouse ESCs using mass spectrometry, we clearly show the specificity of K18 ubiquitination by UHRF1 and its reduction by UHRF1 depletion (Figure 4C, 4D). Second, in the previous study, a deletion of 100 amino acids within the DNMT1 TS domain (Δ325-425) caused a loss of histone binding in vitro32. The TS domain is, however, involved in multiple interactions and required for proper folding, stability and activity of DNMT1. The incomplete structural information indicates different TS domain conformations and a role in autoinhibition of the CD, but does not provide any further mechanistic insights10,11,47. With bioinformatics and mutational analyses, we identified a conserved UIM located in the TS domain of DNMT1 (amino acids 381-395) that mediates the recognition of ubiquitinated H3 in vitro (Figure 5, 6A, 6B and Supplementary information, Figure S6B). Localization and activity analyses with specific mutants in vivo clearly indicated that the UIM is required for DNMT1 subnuclear distribution and maintenance DNA methylation (Figures 6C, 7 and Supplementary information, Figure S7A and S8).
Last, we could show that besides hemimethylated DNA binding by the SRA domain32, UHRF1 PHD binding to H3R2 is also required for H3 ubiquitination and subsequent DNA methylation (Figure 2E and 3B). Therefore, we propose that cooperative chromatin binding of the TTD, the PHD and the SRA domain constitutes a prerequisite for H3K18 ubiquitination. These ubiquitinated histone tails are recognized by the UIM and thus mediate DNMT1 chromatin binding. Thereby, UHRF1 acts as a reader and writer of histone marks and via recruitment of DNMT1 dynamically links DNA and histone modification pathways. Based on these results, we propose a ubiquitination-dependent chromatin targeting mechanism for DNMT1 that is essential for maintenance DNA methylation after replication (Figure 8A). The identification and functional characterization of a UIM in DNMT1 not only changes our view of maintenance DNA methylation, but also opens new perspectives for the involvement of DNMT1 in other repressive epigenetic pathways (Figure 8B).
Figure 8
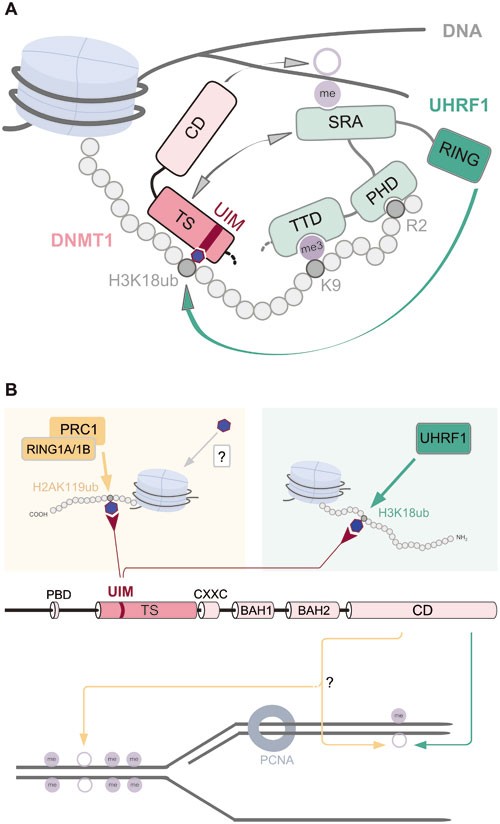
Overview of interactions and modifications controlling DNMT1 activity. (A) UHRF1 is enriched at H3 tails as a result of the PHD-mediated binding to H3R2, the TTD-mediated binding to methylated H3K9 and recognition of hemimethylated CpG sites via the SRA domain. By interaction of the SRA domain with the TS domain, DNMT1 is directly recruited to its target sites. UHRF1 chromatin binding via its TTD, PHD and SRA domain is a prerequisite for subsequent H3 ubiquitination by the RING domain. The UIM of DNMT1 binds to H3 tails ubiquitinated at K18 by UHRF1 and is essential for DNMT1 targeting and DNA methylation in vivo. (B) The previously described direct interaction of DNMT1 with UHRF1 and PCNA is not sufficient for maintenance DNA methylation. Besides the UHRF1-dependent H3K18 ubiquitination recruiting DNMT1 via its UIM for maintenance DNA methylation (right half), we propose an alternative pathway that involves H2AK119 ubiquitination by RING1A/1B of PRC1 (left part). The identification of the DNMT1 UIM now opens the possibility that ubiquitination of histone tail residues by ubiquitin E3 ligases might constitute alternative pathways for DNA methylation by DNMT1 CD beyond classic maintenance. Blue hexagons represent a ubiquitin moiety.
Besides association with ubiquitinated H3, we found that DNMT1 also binds ubiquitinated H2AK119 (Figure 5C and Supplementary information, Figure S6C, S6D). Consistently, DNMT1 was recently detected among proteins binding to H2A ubiquitinated at K118 in Drosophila, corresponding to K119 in mammals48. H2AK119 ubiquitination is catalyzed by RING1A/1B, two components of the Polycomb repressive complex 1 (PRC1), and plays an important role in regulating gene expression49. Similar to UHRF1-dependent H3 ubiquitination, H2A ubiquitination by RING1A/1B might also contribute to DNA methylation. We speculate that UIM-mediated binding of DNMT1 to ubiquitinated H2AK119 might direct DNMT1 to un- or hemimethylated sites dependent on PRC1 ubiquitination activity (Figure 8B, left half).
PRC1-dependent H2A ubiquitination further leads to PRC2 recruitment and subsequent H3K27 methylation50. Enhancer of Zeste homolog 2 (EZH2), a component of PRC2, writes methylated H3K27 and interacts with DNMTs. This interaction was shown to be required for DNA methylation of EZH2 target promoters51. DNMT1 depletion in differentiated cells affects H2A ubiquitination-dependent PRC2 recruitment at pericentromeric heterochromatin52. Thus, UIM binding to ubiquitinated H2A is likely DNA replication independent and DNMT1 might function as adaptor protein mediating PRC2 recruitment and repressive Polycomb domain formation.
Besides recruiting DNMT1 to specific sites on chromatin, the UIM could also play a role in the allosteric activation of the enzyme. The UIM is located within the TS domain of DNMT1 that had been shown to bind the CD and thereby inhibit catalytic activity10,11. It is tempting to speculate that competitive UIM binding to ubiquitinated histone tails displaces the TS domain from the DNA binding pocket and abolishes autoinhibition of DNMT1.
Given the emerging role of ubiquitination in DNA methylation, it is interesting to notice that ubiquitination is a highly dynamic post-translational modification that can be reversed by ubiquitin-specific proteases (USPs). The UHRF1-DNMT1 complex has been reported to contain USP7 that deubiquitinates and stabilizes DNMT129,30. Thus, USP7 might in addition modulate the ubiquitination status of histone H3 and thereby regulate DNMT1 association with chromatin. An alternative pathway controlling DNMT1 chromatin association could involve the recently described chromatin acetylation of H3K18 and K2353,54. Acetylated H3K18 is enriched at the transcriptional start sites of active and poised genes55. Thus, H3K18 acetylation might counteract ubiquitination and thereby prevent binding and silencing of active genes by DNMT1. The dynamic interplay of ubiquitination and acetylation of H3K18 likely controls DNMT1 chromatin binding and thereby directs methylation activity. Studies of UHRF1 and DNMT1 complex composition in different cell cycle phases and cell types should provide further insights into the fine-tuning of DNMT1 activity in vivo.
Given the complex role of the large TS domain on the one hand and the scarce structural and mechanistic data on the other hand, our identification of a well defined UIM provides a concrete basis for functional insights. Ubiquitin binding proteins with defined UIMs have been described in various cellular processes like, e.g., sorting of ubiquitinated membrane proteins for lysosomal degradation. The crystal structure of the signal transducing adaptor molecule 1 (STAM1)56 suggests that three central amino acids in the UIM, L176, A179 and S183 form a hydrophobic interface for ubiquitin binding57. Similar to the UIM in STAM1, the UIM in DNMT1 also harbors a conserved hydrophobic amino acid M385 and S392 flanked by negatively charged amino acids (D381, E382 and D395), which we found to be essential in our analyses (Figures 5, 6 and 7). Different from other UIMs, the central conserved A residue is not present in DNMT1 (Supplementary information, Figure S6A). Based on sequence alignments and structural information, UIMs can be subdivided in single-sided single UIMs, as in STAM1, and in single-sided tandem UIMs, as in the proteasome subunit S5a58 (Figure 5A). The tandem UIMs in S5a provide a model for the recognition of polyubiquitin chains59. In contrast, a double-sided single UIM in the hepatocyte growth factor-regulated tyrosine kinase substrate (HRS) allows for efficient binding of multiple monoubiquitinated receptors in the process of endosomal protein sorting58. Comparison with these known UIMs suggests that the motif in DNMT1 belongs to the group of single-sided single UIMs, which would be compatible with the recognition of single ubiquitinated histone tails.
In summary, the functional analysis of UHRF1 domains and the identification of a UIM in DNMT1 challenge traditional views of maintenance DNA methylation as a simple copying mechanism. Instead, DNA methylation by DNMT1 requires reading of H3R2, H3K9me3 and hemimethylated DNA by UHRF1 and subsequent ubiquitination of H3K18 by its RING domain thereby integrating signals from different epigenetic pathways. These multiple layers controlling DNMT1 activity suggest that overall methylation densities in chromatin domains are maintained rather than specific methylation patterns precisely copied. The functional characterization of the UIM further raises the possibility that other ubiquitin E3 ligases like RING1A/1B of PRC1 might direct DNMT1 activity to repressive chromatin domains beyond simple maintenance.
Materials and Methods
Expression constructs and antibodies
Fusion constructs were generated using enhanced GFP, monomeric RFP or monomeric Ch. The expression constructs for GFP, RFP-DNMT1, GFP-DNMT1 wt, GFP-DNMT1 Δ458-500, GFP-DNMT1 309-628 (GFP-TS) and UHRF1-His have been described previously9,29,33,60,61. GFP-TS Δ458-500, GFP-DNMT1 Δ356-404 and GFP-DNMT1 point mutant (D381A-E382A-S392A and D381A-E382A-M385A-S392A-D395A) expression constructs as well as UHRF1-GFP H346G and H730A were derived from the corresponding wt constructs by overlap extension PCR62. The GFP-UHRF1 single-domain construct for the ubiquitin-like domain (Ubl) was generated by PCR using the corresponding wt full-length construct. Ch-UHRF1 and GFP-UHRF1 expression constructs have been described previously22,63. Expression constructs for GFP-H3 R2A, K18A, K23A as well as K18A-K23A were obtained by overlap extension PCR on the corresponding wt construct. The construct for LacI-GBP has been reported before36,64,65. All constructs were verified by DNA sequencing (MWG Biotech).
For immunofluorescence staining of heterochromatin, a mouse anti-H3K9me3 and an anti-H4K20me3 antibody were used (Active Motif). Endogenous DNMT1 was stained with the rat monoclonal antibody 5A104 and PCNA with the rat monoclonal antibody 16D1066. As secondary antibodies an anti-mouse Alexa Fluor 594 and anti-rat Alexa Fluor 647 antibody were applied, respectively (Invitrogen).
For detection of GFP fusion proteins by western blot, a mouse anti-GFP (Roche) or a rat anti-GFP (Chromotek) antibody was used. RFP or Ch fusion proteins were detected by the rat anti-red antibody 5F867. UHRF1 was visualized by a rabbit anti-UHRF1 antibody24 and HA-ubiquitin by the mouse monoclonal anti-HA antibody 12CA5. Equal loading of cell lysates was assessed by a mouse anti-β-Actin antibody (Sigma-Aldrich). The rabbit anti-H3 antibody was purchased from Abcam and the anti-H2AK119ub from New England Biolabs. Depending on the expected intensity of the signals, secondary antibodies either conjugated to horseradish peroxidase (anti-rabbit (Biorad), anti-rat and anti-mouse (Dianova)) or conjugated to fluorescent dyes (anti-mouse and anti-rat Alexa Fluor 647 as well as anti-rat Alexa Fluor 488 (Invitrogen)) were applied. For detection of HRP-conjugated antibodies, an ECL Plus reagent (GE Healthcare, Thermo Scientific) was used.
Cell culture, transfection and immunofluorescence staining
HEK 293T and BHK cells were cultured in DMEM supplemented with 10% fetal calf serum and 50 μg/ml gentamycine (PAA). MEF cells were cultured in DMEM supplemented with 15% fetal calf serum, 0.1 mM β-mercaptoethanol (Invitrogen), 2 mM l-glutamine, 1× MEM non-essential amino acids, 100 U/ml penicillin and 100 g/ml streptomycin (PAA). ESCs including J1 wt, Dnmt1 −/−, E14 wt and Uhrf1 −/− were cultured without feeder cells in gelatinized flasks as described33. Culture medium was supplemented with 1 000 U/ml recombinant leukemia inhibitory factor (Millipore). The Dnmt1 −/− ESCs used in this study are homozygous for the c allele68. Mouse E14 wt and Uhrf1 −/− cells have been reported before61. Mouse ESCs and MEF cells were transfected with FuGENE HD (Roche), Lipofectamine® 2 000 or 3 000 reagent (Invitrogen) according to the manufacturer's instructions. HEK 293T cells and BHK cells were transfected using polyethylenimine as transfection reagent (Sigma) according to the manufacturer's instructions. Cell fixation and microscopy were carried out as described35.
Generation of stable ESC lines and DNA methylation analyses
Forty-eight hours after expression of GFP-tagged constructs in Dnmt1 −/− or Uhrf1 −/− ESCs, GFP-positive mouse ESCs were separated using a fluorescence-activated cell sorting (FACS) Aria II instrument (Becton Dikinson). Stably expressing cells were expanded in selection medium containing 10 μg/ml blasticidin (GFP-DNMT1 wt and GFP-DNMT1 Δ458-500) or 500 ng/ml puromycin (UHRF1-GFP wt, H346G and H730A) and GFP-positive cells were FACS sorted a second time. Furthermore, the UHRF1-GFP wt, H346G and H730A cell lines were single-cell sorted. Single clones of GFP-DNMT1 Δ458-500 and corresponding wt29 were picked manually. For all cell lines, clones with low expression levels were chosen for further analyses. The level and the accuracy of the expressed GFP fusion constructs were checked by western blot analyses (Figure 3A and Supplementary information, Figures S1B and S3D). For functional analyses of GFP-DNMT1 wt and GFP-DNMT1 UIM mutants (Δ356-404, D381A-E382A-S392A and D381A-E382A-M385A-S392A-D395A) as well as UHRF1-GFP wt or UHRF1-GFP point mutants (H346G and H730A) by transient rescue assays, 48 h after expression of these proteins in Dnmt1 −/− or Uhrf1 −/− ESCs, respectively, GFP-positive cells were collected with FACS. Genomic DNA isolation, bisulfite conversion and PCR conditions were described before6,60,69. Primer sets used for amplification of minor satellites, major satellites, skeletal α-actin and the Dnmt1o promoter are listed in Supplementary information, Table S1. All PCR products were analyzed by pyrosequencing (Varionostic), which results in a quantitative data set for individual CpG sites70.
Co-immunoprecipitation and western blotting
For co-immunoprecipitation assays, the GFP and RFP, Ch or His fusion constructs were co-expressed in HEK 293T cells and protein extracts were normalized to the same GFP or RFP concentration prior to co-immunoprecipitation with the GFP-Trap or RFP-Trap (Chromotek). Bound fractions were first detected by fluorescence intensity measurements and second by western blot analyses.
Acid extraction and TCA precipitation of histones
Histones were isolated by acid extraction as reported previously71. In brief, 107 mouse ESCs or HEK 293T cells were treated in hypotonic buffer (10 mM Tris-HCl pH 8, 10 mM KCl, 1.5 mM MgCl2, 1 mM DTT and 1× Protease Inhibitor, 2 mM PMSF) for 30 min and centrifuged at 1 000× g at 4 °C to get the intact nuclei. After washing steps, nuclei were resuspended in 0.4 N H2SO4 and incubated on a rotator at 4 °C overnight. After centrifugation, histones in the supernatant were transferred into a fresh reaction tube and precipitated using 33% trichloroacetic acid (TCA). After washing twice with cold acetone, histones were dissolved in H2O. Histone concentrations were measured using the PierceTM 660 nm protein assay kit (Thermo Scientific).
Ubiquitinated histone binding experiment
For ubiquitinated histone binding experiment, acid extracted histones from HEK 293T cells were used. GFP-DNMT1 and its mutants were immobilized on the GFP-Trap (Chromotek) and incubated with equal amounts of acid extracted histones for 30 min at 4 °C. After washing steps, the bound fractions were analyzed by western blot.
Due to unspecific binding of histones to the eppendorf tubes, we used eppendorf tubes with low binding affinity during mass spectrometry sample preparation.
Immunoprecipitation of ubiquitinated GFP-H3
GFP-H3 wt and R2A mutant constructs were co-expressed in HEK 293T cells with UHRF1-His and harvested after treatment with 2 mM N-ethylmaleimide (NEM, AppliChem) for 5 min. Lysates were prepared by firstly isolating nuclei in hypotonic buffer (10 mM HEPES pH 7.9, 10 mM KCl, 0.1 mM MgCl2, 10% glycerol, 0.1 mM EDTA, 0.1 mM DTT, 1× protease inhibitor, 2 mM PMSF, 0.1% NP-40, 0.625 mg/ml NEM) and secondly by lysis of the nuclei in hypertonic buffer (20 mM HEPES pH 7.9, 150 mM KCl, 1.5 mM MgCl2, 10% glycerol, 0.1 mM EDTA, 1 mM DTT, 1× protease inhibitor, 2 mM PMSF, 1 mg/ml DnaseI (AppliChem), 0.625 mg/ml NEM). Prior to immunoprecipitation, the GFP concentration was equalized using lysates from UHRF1-His transfected HEK 293T cells for dilution. After immunoprecipitation of GFP-H3 with the GFP-Trap (Chromotek) and washing (20 mM HEPES pH 7.9, 300 mM KCl, 10% glycerol, 0.1% Triton X-100), the bound fraction was analyzed by western blot.
For semiquantitative analysis of the GFP-H3 wt or K18A, K23A, K18A-K23A and R2A ubiquitination, the GFP fusion constructs were co-expressed with HA-ubiquitin in HEK 293T cells and 2 days after transfection, the cells were harvested as described above and further processed as reported previously29.
F3H assay and trapping assay
The F3H assay was performed as described previously65. In the trapping assay, mouse ESCs stably expressing GFP-DNMT1 wt or Δ458-500 were cultured in Ibidi chambers and incubated with 10 μM of the cytosine analogue 5-aza-2′-deoxycytidine (Sigma). Images were acquired with a UltraVIEW VoX spinning disc microscope (PerkinElmer) assembled to an Axio Observer D1 inverted stand (Zeiss) and using a 63×/1.4 NA Plan-Apochromat oil immersion objective.
In vitro peptide pull-down assay
The peptide pull-down assay from nuclear cell extracts of HEK 293T cells expressing UHRF1-GFP fusion constructs was performed as described72 with the following modifications. C-terminally biotinylated histone peptides were purchased from PSL and are listed in Supplementary information, Table S2. Streptactin beads (Iba) were used for the immobilization of biotinylated peptides in binding buffer (10 mM Tris-HCl, pH 7.5, 300 mM NaCl, 0.5 mM EDTA, 1 mM DTT). After the binding reaction, beads were washed four times with wash buffer (20 mM HEPES pH 7.9, 20% glycerol, 0.2 mM EDTA, 300 mM KCl, 0.1% Triton X-100). Bound fractions were eluted by boiling in 2× Laemmli sample buffer and analyzed by western blot.
Mass spectrometry
The gel was stained with Coomassie and H3 bands were manually excised, propionylated and digested with trypsin as described before73 with minor modifications. For peptide extraction, gel slices were incubated twice with 50 μl of 50% acetonitrile 0.25% TFA and twice more with 50 μl of acetonitrile. The resulting liquid containing the digested peptides was totally evaporated, redissolved with 15 μl of 0.1% formic acid and stored at −20 °C until further processing.
Tryptic peptides were injected (5 μl) in an Ultimate 3 000 HPLC system (LC Packings Dionex). Samples were desalted on-line in a C18 microcolumn (300 m i.d. × 5 mm, packed with C18 PepMap™, 5 μm, 100 Å by LC Packings), and peptides were separated with a gradient from 5% to 60% acetonitrile in 0.1% formic acid over 40 min at 300 nl/min on a C18 analytical column (75 μm i.d. × 15 cm, packed with C18 PepMap™, 3 μm, 100 Å by LC Packings).
The effluent from the HPLC was directly electrosprayed into a linear trap quadrupole-Orbitrap XL mass spectrometer (Thermo Fisher Scientific). The MS instrument was operated in data-dependent mode. Survey full-scan MS spectra (from m/z 300–2 000) were acquired in the Orbitrap with resolution R = 60 000 at m/z 400 (after accumulation to a “target value” of 500 000 in the linear ion trap). The six most intense peptide ions with charge states between two and four were sequentially isolated to a target value of 10 000 and fragmented by collision-induced dissociation and recorded in the linear ion trap. For all measurements with the Orbitrap detector, three lock-mass ions were used for internal calibration74. Typical MS conditions were spray voltage, 1.5 kV; no sheath and auxiliary gas flow; heated capillary temperature, 200 °C; normalized collision-induced dissociation energy 35%; activation q = 0.25; and activation time = 30 ms.
Mascot 2.3.02 was used for protein identification with the following settings: Database: Swissprot 57.7; Taxonomy: Homo sapiens (human); MS tolerance: 10 ppm; MS/MS tolerance: 0.5 Da; peptide FDR: 0.1; protein FDR: 0.01; minimum peptide length: 5; and variable modifications: propionyl (K, N-term), GlyGly (K).
Quantification of modified H3 18-26 peptides was based on the intensities of the MS1 peaks. The spectra depicted in Figure 4A and 4B were used to determine the exact masses (m/z ± 10 ppm) and used as a reference for further quantification.
Quantitative analysis of DNMT1 subnuclear localization
During late S phase, DNMT1 is enriched in replication foci at chromocenters. In order to quantify the subnuclear distribution of GFP-DNMT1 wt and defined UIM mutants the following procedure was used: confocal z-stacks (0.21 μm interval) were acquired with identical scan settings in three color channels to visualize replication foci (anti-PCNA staining, 594 nm excitation), DNMT1 localization (GFP-DNMT1 fusions enhanced with GFP-booster (Chromotek), 488 nm excitation) and DNA counterstaining (DAPI, 405 nm excitation). For each color channel, maximum intensity projections were calculated and only GFP-expressing cells were analyzed. Segmentation of replication foci or whole nuclei was performed with the Weka segmentation plugin75 in Fiji76. Training of the classifier was finalized until the result matched the visual impression (Figure 7A). Due to variations in ESC samples, replication foci were segmented using different classifiers for wt or the different UIM mutants. In contrast, for all somatic cells, one classifier was sufficient to segment replication foci. Whole nuclei were segmented by a classifier based on the DAPI signal. After Weka segmentation, the resulting binary masks were filtered using the particle analyzer of Fiji with a circularity value ≥ 0.25. To select for cells in late S phase, only replication foci ≥ 150 pixel where further analyzed in the ESC samples. In MEF cell samples, only late S phase cells were imaged and analyzed without applying size exclusion for replication foci. Nuclear masks (size ≥ 3 000 pixel) were used to quantify the total amount of GFP fusion protein in a single nucleus. Nuclei were further subsegmented by replication foci masks. For each nucleus, the ratio between the mean GFP signals in replication foci relative to the mean GFP signal outside the foci was calculated. Raw data were corrected for background signals by subtracting the modal grey value. Ratios from all nuclei expressing GFP-DNMT1 wt or UIM mutants were visualized as box plots. Numerical calculations and statistical analysis were performed with R[77](/articles/cr201572#ref-CR77 "R Core Team (2014). R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL http://www.R-project.org/
.").Statistical analysis
Results were expressed as mean values ± SD or as mean values ± SEM from the number of biological replicates indicated in the corresponding figure legend.
References
- Bird A . DNA methylation patterns and epigenetic memory. Genes Dev 2002; 16:6–21.
Article CAS Google Scholar - Leonhardt H, Page AW, Weier HU, Bestor TH . A targeting sequence directs DNA methyltransferase to sites of DNA replication in mammalian nuclei. Cell 1992; 71:865–873.
Article CAS Google Scholar - Chuang LS, Ian HI, Koh TW, Ng HH, Xu G, Li BF . Human DNA-(cytosine-5) methyltransferase-PCNA complex as a target for p21WAF1. Science 1997; 277:1996–2000.
Article CAS Google Scholar - Schneider K, Fuchs C, Dobay A, et al. Dissection of cell cycle-dependent dynamics of Dnmt1 by FRAP and diffusion-coupled modeling. Nucleic Acids Res 2013; 41:4860–4876.
Article CAS Google Scholar - Easwaran HP, Schermelleh L, Leonhardt H, Cardoso MC . Replication-independent chromatin loading of Dnmt1 during G2 and M phases. EMBO Rep 2004; 5:1181–1186.
Article CAS Google Scholar - Schermelleh L, Haemmer A, Spada F, et al. Dynamics of Dnmt1 interaction with the replication machinery and its role in postreplicative maintenance of DNA methylation. Nucleic Acids Res 2007; 35:4301–4312.
Article CAS Google Scholar - Spada F, Haemmer A, Kuch D, et al. DNMT1 but not its interaction with the replication machinery is required for maintenance of DNA methylation in human cells. J Cell Biol 2007; 176:565–571.
Article CAS Google Scholar - Margot JB, Aguirre-Arteta AM, Di Giacco BV, et al. Structure and function of the mouse DNA methyltransferase gene: Dnmt1 shows a tripartite structure. J Mol Biol 2000; 297:293–300.
Article CAS Google Scholar - Fellinger K, Rothbauer U, Felle M, Langst G, Leonhardt H . Dimerization of DNA methyltransferase 1 is mediated by its regulatory domain. J Cell Biochem 2009; 106:521–528.
Article CAS Google Scholar - Syeda F, Fagan RL, Wean M, et al. The replication focus targeting sequence (RFTS) domain is a DNA-competitive inhibitor of Dnmt1. J Biol Chem 2011; 286:15344–15351.
Article CAS Google Scholar - Takeshita K, Suetake I, Yamashita E, et al. Structural insight into maintenance methylation by mouse DNA methyltransferase 1 (Dnmt1). Proc Natl Acad Sci USA 2011; 108:9055–9059.
Article CAS Google Scholar - Achour M, Jacq X, Ronde P, et al. The interaction of the SRA domain of ICBP90 with a novel domain of DNMT1 is involved in the regulation of VEGF gene expression. Oncogene 2008; 27:2187–2197.
Article CAS Google Scholar - Felle M, Joppien S, Nemeth A, et al. The USP7/Dnmt1 complex stimulates the DNA methylation activity of Dnmt1 and regulates the stability of UHRF1. Nucleic Acids Res 2011; 39:8355–8365.
Article CAS Google Scholar - Berkyurek AC, Suetake I, Arita K, et al. The DNA Methyltransferase Dnmt1 directly interacts with the SET and RING finger associated (SRA) domain of the multifunctional protein Uhrf1 to facilitate accession of the catalytic center to hemi-methylated DNA. J Biol Chem 2013; 289:379–386.
Article Google Scholar - Zhang J, Gao Q, Li P, et al. S phase-dependent interaction with DNMT1 dictates the role of UHRF1 but not UHRF2 in DNA methylation maintenance. Cell Res 2011; 21:1723–1739.
Article CAS Google Scholar - Bostick M, Kim JK, Esteve PO, Clark A, Pradhan S, Jacobsen SE . UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science 2007; 317:1760–1764.
Article CAS Google Scholar - Sharif J, Muto M, Takebayashi S, et al. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature 2007; 450:908–912.
Article CAS Google Scholar - Li E, Bestor TH, Jaenisch R . Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 1992; 69:915–926.
Article CAS Google Scholar - Arita K, Ariyoshi M, Tochio H, Nakamura Y, Shirakawa M . Recognition of hemi-methylated DNA by the SRA protein UHRF1 by a base-flipping mechanism. Nature 2008; 455:818–821.
Article CAS Google Scholar - Avvakumov GV, Walker JR, Xue S, et al. Structural basis for recognition of hemi-methylated DNA by the SRA domain of human UHRF1. Nature 2008; 455:822–825.
Article CAS Google Scholar - Qian C, Li S, Jakoncic J, Zeng L, Walsh MJ, Zhou MM . Structure and hemimethylated CpG binding of the SRA domain from human UHRF1. J Biol Chem 2008; 283:34490–34494.
Article CAS Google Scholar - Rottach A, Frauer C, Pichler G, Bonapace IM, Spada F, Leonhardt H . The multi-domain protein Np95 connects DNA methylation and histone modification. Nucleic Acids Res 2010; 38:1796–1804.
Article CAS Google Scholar - Cheng J, Yang Y, Fang J, et al. Structural insight into coordinated recognition of trimethylated histone H3 lysine 9 (H3K9me3) by the plant homeodomain (PHD) and tandem tudor domain (TTD) of UHRF1 (ubiquitin-like, containing PHD and RING finger domains, 1) protein. J Biol Chem 2013; 288:1329–1339.
Article CAS Google Scholar - Citterio E, Papait R, Nicassio F, et al. Np95 is a histone-binding protein endowed with ubiquitin ligase activity. Mol Cell Biol 2004; 24:2526–2535.
Article CAS Google Scholar - Karagianni P, Amazit L, Qin J, Wong J . ICBP90, a novel methyl K9 H3 binding protein linking protein ubiquitination with heterochromatin formation. Mol Cell Biol 2008; 28:705–717.
Article CAS Google Scholar - Xie S, Jakoncic J, Qian C . UHRF1 double tudor domain and the adjacent PHD finger act together to recognize K9me3-containing histone H3 tail. J Mol Biol 2012; 415:318–328.
Article CAS Google Scholar - Papait R, Pistore C, Grazini U, et al. The PHD domain of Np95 (mUHRF1) is involved in large-scale reorganization of pericentromeric heterochromatin. Mol Biol Cell 2008; 19:3554–3563.
Article CAS Google Scholar - Jenkins Y, Markovtsov V, Lang W, et al. Critical role of the ubiquitin ligase activity of UHRF1, a nuclear RING finger protein, in tumor cell growth. Mol Biol Cell 2005; 16:5621–5629.
Article CAS Google Scholar - Qin W, Leonhardt H, Spada F . Usp7 and Uhrf1 control ubiquitination and stability of the maintenance DNA methyltransferase Dnmt1. J Cell Biochem 2011; 112:439–444.
Article CAS Google Scholar - Du Z, Song J, Wang Y, et al. DNMT1 stability is regulated by proteins coordinating deubiquitination and acetylation-driven ubiquitination. Sci Signal 2010; 3:ra80.
Article Google Scholar - Mudbhary R, Hoshida Y, Chernyavskaya Y, et al. UHRF1 overexpression drives DNA hypomethylation and hepatocellular carcinoma. Cancer Cell 2014; 25:196–209.
Article CAS Google Scholar - Nishiyama A, Yamaguchi L, Sharif J, et al. Uhrf1-dependent H3K23 ubiquitylation couples maintenance DNA methylation and replication. Nature 2013; 502:249–253.
Article CAS Google Scholar - Schermelleh L, Spada F, Easwaran HP, et al. Trapped in action: direct visualization of DNA methyltransferase activity in living cells. Nat Methods 2005; 2:751–756.
Article CAS Google Scholar - Liu X, Gao Q, Li P, et al. UHRF1 targets DNMT1 for DNA methylation through cooperative binding of hemi-methylated DNA and methylated H3K9. Nat Commun 2013; 4:1563.
Article Google Scholar - LDambacher S, Deng W, Hahn M, et al. CENP-C facilitates the recruitment of M18BP1 to centromeric chromatin. Nucleus 2012; 3:101–110.
Article Google Scholar - Zolghadr K, Mortusewicz O, Rothbauer U, et al. A fluorescent two-hybrid assay for direct visualization of protein interactions in living cells. Mol Cell Proteomics 2008; 7:2279–2287.
Article CAS Google Scholar - Hu L, Li Z, Wang P, Lin Y, Xu Y . Crystal structure of PHD domain of UHRF1 and insights into recognition of unmodified histone H3 arginine residue 2. Cell Res 2011; 21:1374–1378.
Article CAS Google Scholar - Rajakumara E, Wang Z, Ma H, et al. PHD finger recognition of unmodified histone H3R2 links UHRF1 to regulation of euchromatic gene expression. Mol Cell 2011; 43:275–284.
Article CAS Google Scholar - Wang C, Shen J, Yang Z, et al. Structural basis for site-specific reading of unmodified R2 of histone H3 tail by UHRF1 PHD finger. Cell Res 2011; 21:1379–1382.
Article CAS Google Scholar - Frauer C, Leonhardt H . A versatile non-radioactive assay for DNA methyltransferase activity and DNA binding. Nucleic Acids Res 2009; 37:e22.
Article Google Scholar - Bestor TH, Ingram VM . Two DNA methyltransferases from murine erythroleukemia cells: purification, sequence specificity, and mode of interaction with DNA. Proc Natl Acad Sci USA 1983; 80:5559–5563.
Article CAS Google Scholar - Yoder JA, Soman NS, Verdine GL, Bestor TH . DNA (cytosine-5)-methyltransferases in mouse cells and tissues. Studies with a mechanism-based probe. J Mol Biol 1997; 270:385–395.
Article CAS Google Scholar - Jeltsch A . On the enzymatic properties of Dnmt1: specificity, processivity, mechanism of linear diffusion and allosteric regulation of the enzyme. Epigenetics 2006; 1:63–66.
Article Google Scholar - Bashtrykov P, Jankevicius G, Jurkowska RZ, Ragozin S, Jeltsch A . The UHRF1 protein stimulates the activity and specificity of the maintenance DNA methyltransferase DNMT1 by an allosteric mechanism. J Biol Chem 2014; 289:4106–4115.
Article CAS Google Scholar - Rothbart SB, Dickson BM, Ong MS, et al. Multivalent histone engagement by the linked tandem Tudor and PHD domains of UHRF1 is required for the epigenetic inheritance of DNA methylation. Genes Dev 2013; 27:1288–1298.
Article CAS Google Scholar - Rothbart SB, Krajewski K, Nady N, et al. Association of UHRF1 with methylated H3K9 directs the maintenance of DNA methylation. Nat Struct Mol Biol 2012; 19:1155–1160.
Article CAS Google Scholar - Song J, Rechkoblit O, Bestor TH, Patel DJ . Structure of DNMT1-DNA complex reveals a role for autoinhibition in maintenance DNA methylation. Science 2011; 331:1036–1040.
Article CAS Google Scholar - Kalb R, Latwiel S, Baymaz HI, et al. Histone H2A monoubiquitination promotes histone H3 methylation in Polycomb repression. Nat Struct Mol Biol 2014; 21:569–571.
Article CAS Google Scholar - Leeb M, Wutz A . RING1B is crucial for the regulation of developmental control genes and PRC1 proteins but not X inactivation in embryonic cells. J Cell Biol 2007; 178:219–229.
Article CAS Google Scholar - Blackledge NP, Farcas AM, Kondo T, et al. Variant PRC1 complex-dependent H2A ubiquitylation drives PRC2 recruitment and polycomb domain formation. Cell 2014; 157:1445–1459.
Article CAS Google Scholar - Vire E, Brenner C, Deplus R, et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 2006; 439:871–874.
Article CAS Google Scholar - Cooper S, Dienstbier M, Hassan R, et al. Targeting polycomb to pericentric heterochromatin in embryonic stem cells reveals a role for H2AK119u1 in PRC2 recruitment. Cell reports 2014; 7:1456–1470.
Article CAS Google Scholar - Kurdistani SK, Tavazoie S, Grunstein M . Mapping global histone acetylation patterns to gene expression. Cell 2004; 117:721–733.
Article CAS Google Scholar - Tsai WW, Wang Z, Yiu TT, et al. TRIM24 links a non-canonical histone signature to breast cancer. Nature 2010; 468:927–932.
Article CAS Google Scholar - Wang Z, Zang C, Rosenfeld JA, et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet 2008; 40:897–903.
Article CAS Google Scholar - Raiborg C, Stenmark H . The ESCRT machinery in endosomal sorting of ubiquitylated membrane proteins. Nature 2009; 458:445–452.
Article CAS Google Scholar - Lim J, Son WS, Park JK, Kim EE, Lee BJ, Ahn HC . Solution structure of UIM and interaction of tandem ubiquitin binding domains in STAM1 with ubiquitin. Biochem Biophys Res Commun 2011; 405:24–30.
Article CAS Google Scholar - Hirano S, Kawasaki M, Ura H, et al. Double-sided ubiquitin binding of Hrs-UIM in endosomal protein sorting. Nat Struct Mol Biol 2006; 13:272–277.
Article CAS Google Scholar - Wang Q, Young P, Walters KJ . Structure of S5a bound to monoubiquitin provides a model for polyubiquitin recognition. J Mol Biol 2005; 348:727–739.
Article CAS Google Scholar - Frauer C, Rottach A, Meilinger D, et al. Different binding properties and function of CXXC zinc finger domains in Dnmt1 and Tet1. PLoS One 2011; 6:e16627.
Article CAS Google Scholar - Meilinger D, Fellinger K, Bultmann S, et al. Np95 interacts with de novo DNA methyltransferases, Dnmt3a and Dnmt3b, and mediates epigenetic silencing of the viral CMV promoter in embryonic stem cells. EMBO Rep 2009; 10:1259–1264.
Article CAS Google Scholar - Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR . Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 1989; 77:51–59.
Article CAS Google Scholar - Pichler G, Wolf P, Schmidt CS, et al. Cooperative DNA and histone binding by Uhrf2 links the two major repressive epigenetic pathways. J Cell Biochem 2011; 112:2585–2593.
Article CAS Google Scholar - Rothbauer U, Zolghadr K, Tillib S, et al. Targeting and tracing antigens in live cells with fluorescent nanobodies. Nat Methods 2006; 3:887–889.
Article CAS Google Scholar - Herce HD, Deng W, Helma J, Leonhardt H, Cardoso MC . Visualization and targeted disruption of protein interactions in living cells. Nat Commun 2013; 4:2660.
Article Google Scholar - Rottach A, Kremmer E, Nowak D, et al. Generation and characterization of a rat monoclonal antibody specific for PCNA. Hybridoma (Larchmt) 2008; 27:91–98.
Article CAS Google Scholar - Rottach A, Kremmer E, Nowak D, Leonhardt H, Cardoso MC . Generation and characterization of a rat monoclonal antibody specific for multiple red fluorescent proteins. Hybridoma (Larchmt) 2008; 27:337–343.
Article CAS Google Scholar - Lei H, Oh SP, Okano M, et al. De novo DNA cytosine methyltransferase activities in mouse embryonic stem cells. Development 1996; 122:3195–3205.
CAS PubMed Google Scholar - Tucker KL, Beard C, Dausmann J, et al. Germ-line passage is required for establishment of methylation and expression patterns of imprinted but not of nonimprinted genes. Genes Dev 1996; 10:1008–1020.
Article CAS Google Scholar - Tost J, Gut IG . DNA methylation analysis by pyrosequencing. Nat Protoc 2007; 2:2265–2275.
Article CAS Google Scholar - Shechter D, Dormann HL, Allis CD, Hake SB . Extraction, purification and analysis of histones. Nat Protoc 2007; 2:1445–1457.
Article CAS Google Scholar - Wysocka J . Identifying novel proteins recognizing histone modifications using peptide pull-down assay. Methods 2006; 40:339–343.
Article CAS Google Scholar - Villar-Garea A, Israel L, Imhof A . Analysis of histone modifications by mass spectrometry. Curr Protoc Protein Sci 2008; Chapter 14:Unit 14.10.
- Olsen JV, de Godoy LM, Li G, et al. Parts per million mass accuracy on an Orbitrap mass spectrometer via lock mass injection into a C-trap. Mol Cell Proteomics 2005; 4:2010–2021.
Article CAS Google Scholar - Mark Hall EF, Geoffrey Holmes, Bernhard Pfahringer, Peter Reutemann, Ian H. Witten . The WEKA data mining software: an update. ACM SIGKDD Explorations Newsletter 2009; 11:10–18.
Article Google Scholar - Schindelin J, Arganda-Carreras I, Frise E, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods 2012; 9:676–682.
Article CAS Google Scholar - R Core Team (2014). R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL http://www.R-project.org/.
- Robert X, Gouet P . Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res 2014; 42:W320–W324.
Article CAS Google Scholar
Acknowledgements
We thank Stefan Jentsch (Max Planck Institute of Biochemistry, Germany) for providing the HA-ubiquitin construct and Peter Becker (Adolf Butenandt Institute, Germany) for the 601 DNA construct. We are grateful to the following colleagues for providing ESCs and somatic cells: Masahiro Muto and Haruhiko Koseki for mouse E14 wt and Uhrf1 −/− ESCs; En Li and T. Chen for mouse J1 wt and Dnmt1 −/−; Thomas Jenuwein for MEF cells; and L. David Spector for providing BHK cells containing a lac operator repeat array. We thank E.M. Baur (Ludwig Maximilians University, Germany) for technical help with the GFP-TS UIM point mutant plasmid constructs. This work was supported by grants from the Deutsche Forschungsgemeinschaft (DFG, SFB1064 A17 to HL and Z03 to AI), the Nanosystem Initiative Munich (NIM to HL) and the Epigenomics Flagship Project (EPIGEN-CNR -IT to IMB). KF and GP were supported by the International Max Planck Research School for Molecular and Cellular Life Sciences (IMPRS-LS). PW, NL and MS are fellows of the Graduate School Life Science Munich (LSM). MS is a fellow of the Integrated Research Training Group (IRTG) of the SFB1064. NL and WQ were also supported by the China Scholarship Council (CSC).
Author information
Author notes
- Garwin Pichler
Present address: Current address: Department of Proteomics and Signal Transduction, Max Planck Institute of Biochemistry, Martinsried, Germany, - Karin Fellinger
Present address: Current address: Intervet International GmbH, Unterschleissheim, Germany, - Fabio Spada
Present address: Current address: Department of Chemistry, Ludwig Maximilians University Munich, Germany, - Weihua Qin and Patricia Wolf: These two authors contributed equally to this work.
Authors and Affiliations
- Department of Biology II, Ludwig Maximilians University Munich, Großhaderner Str. 2, Planegg-Martinsried, 82152, Germany
Weihua Qin, Patricia Wolf, Nan Liu, Stephanie Link, Martha Smets, Federica La Mastra, Garwin Pichler, David Hörl, Karin Fellinger, Fabio Spada, Hartmann Harz & Heinrich Leonhardt - Adolf-Butenandt Institute, Ludwig Maximilians University Munich, Schillerstr. 44, Munich, 80336, Germany
Ignasi Forné & Axel Imhof - Center for Integrated Protein Science Munich (CIPSM), Via Manara 7, Busto Arsizio (VA), 21052, Italy
Weihua Qin, Patricia Wolf, Nan Liu, Stephanie Link, Martha Smets, Federica La Mastra, Ignasi Forné, Garwin Pichler, David Hörl, Karin Fellinger, Fabio Spada, Axel Imhof, Hartmann Harz & Heinrich Leonhardt - Nanosystems Initiative Munich (NIM), Via Manara 7, Busto Arsizio (VA), 21052, Italy
Weihua Qin, Patricia Wolf, Nan Liu, Stephanie Link, Martha Smets, David Hörl, Hartmann Harz & Heinrich Leonhardt - Department of Theoretical and Applied Sciences, University of Insubria, Via Manara 7, Busto Arsizio (VA), 21052, Italy
Federica La Mastra & Ian Marc Bonapace
Authors
- Weihua Qin
You can also search for this author inPubMed Google Scholar - Patricia Wolf
You can also search for this author inPubMed Google Scholar - Nan Liu
You can also search for this author inPubMed Google Scholar - Stephanie Link
You can also search for this author inPubMed Google Scholar - Martha Smets
You can also search for this author inPubMed Google Scholar - Federica La Mastra
You can also search for this author inPubMed Google Scholar - Ignasi Forné
You can also search for this author inPubMed Google Scholar - Garwin Pichler
You can also search for this author inPubMed Google Scholar - David Hörl
You can also search for this author inPubMed Google Scholar - Karin Fellinger
You can also search for this author inPubMed Google Scholar - Fabio Spada
You can also search for this author inPubMed Google Scholar - Ian Marc Bonapace
You can also search for this author inPubMed Google Scholar - Axel Imhof
You can also search for this author inPubMed Google Scholar - Hartmann Harz
You can also search for this author inPubMed Google Scholar - Heinrich Leonhardt
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toHeinrich Leonhardt.
Additional information
( Supplementary information is linked to the online version of the paper on the Cell Research website.)
Supplementary information
Supplementary information, Figure S1
GFP-DNMT1 carrying a deletion of amino acids 458-500 in the TS domain is still active in vitro, but inactive in vivo. (PDF 473 kb)
Supplementary information, Figure S2
The UHRF1-GFP PHD and RING domain mutants preserve their preference for hemimethylated DNA and their heterochromatin association, but cannot recruit endogenous DNMT1 to heterochromatin. (PDF 295 kb)
Supplementary information, Figure S3
The UHRF1-GFP PHD and RING domain mutants cannot mediate DNA remethylation in mouse E14 Uhrf1 –/– ESCs. (PDF 164 kb)
Supplementary information, Figure S4
Histone H3 binding by UHRF1-GFP is sensitive to R2 methylation. (PDF 104 kb)
Supplementary information, Figure S5
H3 histone tail ubiquitination and dependence on R2. (PDF 174 kb)
Supplementary information, Figure S6
DNMT1 UIM deletion and point mutants are defective in ubiquitinated H3 binding. (PDF 318 kb)
Supplementary information, Figure S7
DNMT1 UIM deletion and point mutants still interacting with UHRF1, affect maintenance DNA methylation. (PDF 212 kb)
Supplementary information, Figure S8
DNMT1 UIM mutants show only weak association with PCNA stained replication sites especially in late S phase. (PDF 213 kb)
Supplementary information, Table S1
PCR primers for bisulfite PCR and pyrosequencing. (PDF 93 kb)
Supplementary information, Table S2
Sequence of histone tail peptides used for the histone peptide pull-down and histone peptide binding assays. (PDF 82 kb)
Supplementary information, Table S3
DNA oligonucleotides used for the preparation of double stranded probes for in vitro DNA binding assays with the primer extension reaction. (PDF 82 kb)
Supplementary information, Data S1
Materials and Methods and Supplementary References (PDF 206 kb)
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0
About this article
Cite this article
Qin, W., Wolf, P., Liu, N. et al. DNA methylation requires a DNMT1 ubiquitin interacting motif (UIM) and histone ubiquitination.Cell Res 25, 911–929 (2015). https://doi.org/10.1038/cr.2015.72
- Received: 21 November 2014
- Revised: 27 March 2015
- Accepted: 07 May 2015
- Published: 12 June 2015
- Issue Date: August 2015
- DOI: https://doi.org/10.1038/cr.2015.72