LTBP2 and CYP1B1 mutations and associated ocular phenotypes in the Roma/Gypsy founder population (original) (raw)
Introduction
Primary congenital glaucoma (PCG) is the most common type of childhood glaucoma, with an autosomal recessive inheritance1 and incidence ranging from 1:30 000 (MacKinnon et al2) to 1:1250 (Gencik et al3). Signs of the disease include early onset (birth to 3 years) increased intraocular pressure (IOP), increased corneal diameter (megalocornea), enlarged globe (buphthalmos), Haab's striae (breaks in Descemet's membrane), corneal oedema and optic nerve head cupping.4 PCG is a chronic disease, which is a serious cause of blindness worldwide despite available medical and surgical treatments.5
PCG is genetically heterogeneous with three different loci identified to date: GLC3A (2p21),6 GLC3B (1p36)7 and GLC3C (14q24.3).8 Until recently, mutations in CYP1B1 (cytochrome P450, family 1, subfamily B) at the GLC3A locus were the only known aetiology explaining only a proportion of the PCG alleles (reviewed in Sarfarazi et al1) with few exceptions, such as Slovak Gypsies, wherein a single founder mutation (p.E387K) accounts for 100% of disease alleles.9 Digenic inheritance has been proposed in a small number of PCG patients10, 11 carrying variants in both CYP1B1 and MYOC, the gene mutated in some forms of juvenile/primary open angle glaucoma.
In 2008, linkage to a new locus adjacent to GLC3C on 14q24.2-24.3 was reported in consanguineous Pakistani PCG families.12 Analysis of the candidate gene LTBP2, encoding latent transforming growth factor-_β_-binding protein 2, identified truncating mutations in PCG patients from Pakistan and Iran.13, 14 Recently, homozygous mutations in LTBP2 were reported in a syndrome of megalocornea, microspherophakia (small spherical lens), lens dislocation, and secondary glaucoma developing after age 3 years15 and in isolated microspherophakia/lens dislocation.16 In the original studies,13, 14 ectopia lentis was rare, found in some (but not all) affected members in single families. These reports and the LTBP2 expression pattern in the trabecular meshwork, ciliary body and ciliary process13 raise the issue of the range, combinations and evolution of manifestations, and underlying pathophysiology.
In a previous study of 21 patients from 16 Roma/Gypsy families, we identified five CYP1B1 variants, that together accounted for approximately one-third of patients.17 The discovery of LTBP2 allowed the identification of an ancestral Gypsy mutation, p.R299X, which was shared with an affected Pakistani family on an identical haplotype background and explained ∼40% of Gypsy PCG alleles.13 A substantial proportion of alleles remained unknown, an unusual situation in the Gypsy population, characterised by strong founder effect and restricted mutation diversity in Mendelian disorders.18
Here we report a follow-up study, in which we doubled the sample size and performed comprehensive sequence analysis of both LTBP2 and CYP1B1. We describe the mutation spectrum and associated phenotypes, and the identification of double CYP1B1-LTBP2 mutants. We provide a population genetic perspective on the origins and distribution of mutations, in an attempt to explain the unusual genetic heterogeneity.
Subjects and methods
Subjects
Patient recruitment and diagnostic criteria were as described.17 The sample included 34 families with 42 affected subjects. In families with affected first-degree relatives, only the proband was included in mutation frequency estimates; cousins were considered independent, due to observed intra-familial heterogeneity (example in Figure 1). The total number of independent PCG alleles was 74 (Table 1).
Figure 1
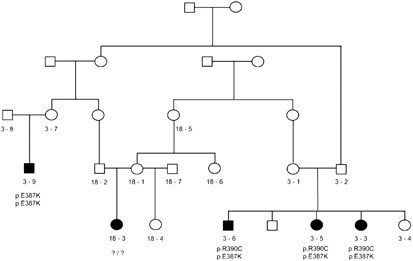
Gypsy PCG kindred, with recently identified relatedness between separately recruited families, PCG 3 and PCG 18. The two branches of family 3 have different CYP1B1 mutations, and none of these is shared by the affected subject in family 18.
Table 1 CYP1B1 and LTBP2 mutant alleles in Gypsy patients with PCG
Carrier testing for the CYP1B1 p.E387K mutation was performed on 827 population controls, of whom 715 have been reported previously17 and 112 were newly added Gypsy samples from Bulgaria and Spain. LTBP2 p.R299X was analysed in 782 samples: 552 control Gypsy individuals from diverse sub-isolates18 and 230 Indians (180 from the northwestern state of Gujarat and 50 members of the Jat Sikh caste from the Northern state of Punjab).
Written informed consent was obtained from all participants. The study complies with the ethics guidelines of the institutions involved.
Mutation analysis in affected subjects and families
Direct sequencing of the entire coding sequence of CYP1B1 (NM_000104) and of LTBP2 exon 4, harbouring the p.R299X founder mutation, was carried out in all affected individuals. Conditions were as described.13, 17 Where these analyses failed to identify the mutations on both chromosomes, we sequenced all coding regions, parts of the flanking introns and untranslated regions of LTBP2 (NM_000428).13, 14 Novel changes were analysed for potential functional effects using Human Splicing Finder19 and PolyPhen.20 Two LTBP2 variants (Supplementary Figure 1), located ∼1 Kb apart in exons 21 and 23, were analysed by cloning in a pCR4-TOPO cloning vector (Invitrogen, Mulgrave, VIC, Australia) and sequencing with primers T3 and T7. Linkage disequilibrium (LD) was analysed using our genotyping data on 68 controls for 10 SNPs spanning ∼65 kb around LTBP2 (Supplementary Table 1) with HaploView4.2.21
In cases, in whom only a single mutation in either CYP1B1 or LTBP2 was identified, we searched for large rearrangements by family segregation analysis of informative markers, and by copy number state and loss of heterozygosity (Affymetrix SNP 6.0 array data analyses in Genotyping Console 4.0, Affymetrix, Santa Clara, CA, USA).
Population genetic analyses
P.E387K screening was carried out with a PCR-based PsiI RFLP assay,22 and p.R299X was analysed with a custom-designed TaqMan assay (Applied Biosystems, Mulgrave, VIC, Australia) (probes and primers available on request). Heterozygotes were confirmed by sequencing.
For haplotype analysis of the founder mutations, STR markers spanning ∼9 cM around CYP1B1 and LTBP2 (Supplementary Tables 2A and 2B) were genotyped using the Multiplex PCR kit (Qiagen, Doncaster, VIC, Australia). The p.E387K haplotype analysis included 15 independent mutant chromosomes (probands and carrier controls) and 41 normal chromosomes (from unrelated control Gypsy trios and untransmitted parental chromosomes from PCG families). P.R299X was analysed in 37 independent mutant and 61 normal alleles. Most haplotypes were unambiguously assigned using family data; for the remainder, inferences were made using PHASE 2.1.23
Haplotype coalescence time since each mutation was introduced into the Gypsy population of Bulgaria was estimated using DMLE+2.2 (Reeve and Rannala24; 1 000 000 iterations). The demographic growth rate was set at 1.32.25 The allele frequency of LTBP2 p.R299X was 0.013 (Supplementary Table 3) and the proportion sampled was based on 600 000 population size. For p.E387K, whose distribution is limited to a specific geographic region (Figure 3), the allele frequency was set at 0.008 (based on the sub-isolate data in Sivadorai et al17) and the estimates were carried out using a population size of 60K or 100K.
Results
Mutation spectrum
CYP1B1
The increase in sample size, from a total of 38 disease alleles17 to 74 in this study, did not lead to the identification of additional CYP1B1 mutations. Five sequence variants accounted for 25/74 PCG alleles (33.8%; Table 1). Four of these have been reported previously as disease-causing in other populations: p.R390C, p.R368H and p.E229K occur in India,26 and p.E387K is the founder PCG mutation in the Gypsy population of Slovakia.9 The p.F445I mutation, identified in a single homozygous patient,17 can be putatively classified as pathogenic based on its absence in 120 normal chromosomes17 and on the PolyPhen prediction of deleterious functional effects. The larger sample did change the frequency estimates, with the p.E387K contribution increasing to 52% of CYP1B1 mutant alleles and 17.6% of all PCG alleles (Table 1). The frequencies of the other CYP1B1 mutations did not change: p.R390C was found in 9.4% of PCG alleles, and p.F445I, p.R368H and p.E229K remained rare (Table 1).
LTBP2
Pathogenic mutations. The truncating LTBP2 p.R299X mutation13 was present on 25/74 chromosomes in the expanded sample, supporting it as the single most common Gypsy PCG mutation accounting for approximately one-third (33.8%) of disease alleles.
Apart from p.R299X, no obvious pathogenic mutations were detected in LTBP2. Two presumably unrelated patients (PCG 19-3 and PCG 20-3) carried tandem nucleotide substitutions ∼1 kb apart: c.3262G>A in exon 21 and c.3468C>T in exon 23. The first, listed in dbSNP under rs61505039, results in an amino acid substitution, p.G1088S, predicted by PolyPhen to impair protein function. This variant was also observed in Iranian and Indian patients, homozygous for frame-shift LTBP2 mutations (p.Y1793fsX55/family A in Narooie-Nejad et al14 and p.H1816PfsX28/family 1 in Kumar et al16). The second variant is a novel synonymous change (p.G1156G), predicted by Human Splicing Finder to create a donor splicing site that would result in an in-frame deletion of 60 nucleotides (c.3467_3526del). Because family segregation analysis was precluded by lack of parental DNA, haplotype assignment was performed by cloning and sequencing of the DNA fragment containing both mutated sites. The results showed that they occurred on different chromosomes in patient PCG 19-3 and on the same chromosome in PCG 20-3 (Supplementary Figure 1). LD analysis in our population controls showed that these variants reside between two LD blocks, allowing for recombinations (Supplementary Figure 1). Patient PCG 19-3 also carried two pathogenic CYP1B1 mutations – p.E387K and p.R390C – which would be sufficient to cause the disease. No other mutations in LTBP2 or CYP1B1 were identified in PCG 20-3, who was assigned to the group with unknown molecular defects. Although these data allow no definite conclusions on pathogenicity, co-occurrence with known disease-causing mutations in PCG 19-3 and in the Iranian family suggests that the two variants are probably neutral polymorphisms.
Polymorphisms. We observed 47 known LTBP2 polymorphisms and eight novel variants (five intronic and three synonymous coding), seven of which were predicted by Human Splicing Finder to have no impact and were therefore considered new SNPs (Supplementary Table 4). The exception, c.3468C>T, was discussed in the preceding paragraph.
Double mutants
A total of six individuals with mutations in both CYP1B1 and LTBP2 were identified. In family PCG 16, the proband (16-3) and her affected brother (16-4) were homozygous for LTBP2 p.R299X. The proband was also a carrier of the CYP1B1 p.E229K. On the basis of medical histories, there was no difference in disease severity between these two patients who are now in their 30 s. Analysis of the entire family (Figure 2a) identified three double heterozygotes for p.R299X and p.E229K. Ophthalmological examinations failed to detect any pathological changes in the father 16-2 and in sibling 16-6, symptom free at ages 60 and 38 years. The mother, 16-7, was diagnosed at age 59 with sub-acute angle-closure glaucoma with intermittent pupillary block but no evidence of lens dislocation. In family PCG 27 (Figure 2b), both proband and unaffected mother were double p.R299X/p.R368H heterozygotes, with no other mutations in the proband.
Figure 2
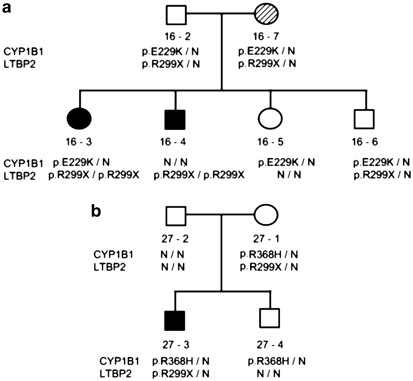
Double CYP1B1 and LTBP2 mutants in (a) family PCG 16 and (b) family PCG 27. Dark shaded symbols indicate PCG affected individuals and dashed symbol (16-7) indicates late-onset angle closure glaucoma.
Genomic rearrangements
No rearrangements were detected in patients 6-3 and 27-3, in whom only one mutation was identified in LTBP2 and/or CYP1B1 (data not shown).
Mutation unknown
The disease-causing mutations remained unidentified or incompletely identified in 14 affected subjects. In 12, we did not detect any changes in either gene; patient 6-3 was only heterozygous for p.R368H in CYP1B1, and 27-3 was a double carrier of LTBP2 p.R299X and CYP1B1 p.R368H. These patients and their families are included in our further studies, aiming at the identification of novel PCG gene(s).
Clinical findings
Affected subjects were grouped according to the mutated gene (CYP1B1; LTBP2; unknown) and the clinical phenotype compared between the groups (Table 2). Because in most cases, the most recent examinations were performed during field trips at the patients' homes, and to avoid the uncertainties of clinical information obtained years ago, the comparison was based on major criteria, for example, number of surgical interventions and outcome. The group with unidentified mutations appeared the most diverse in terms of unilateral/bilateral involvement, corneal clouding and visual acuity. This may be because of further heterogeneity, or to the fact that it was the youngest group (mean age 8.0±5.9 years at most recent examination) and the disease may still be progressing.
Table 2 Summary of clinical findings in primary congenital glaucoma patients grouped by genotype
The CYP1B1 and LTBP2 groups, of comparable size and age, did not differ in terms of bilateral/unilateral involvement, presence of megalocornea and corneal clouding. However, p.R299X homozygotes had undergone more surgical interventions (3.1 vs 0.8 per affected eye) and the outcome in visual acuity was poorer: 11 out of 26 eyes with no light perception as compared with 3 out of 22 among CYP1B1 mutants. At the same time, the PCG diagnosis in p.R299X homozygotes was established at a significantly later age (15.1 months vs 3.6 months in the CYP1B1 group), which could be because of a later onset, similar to the patients described by Désir et al15 One of the Désir et al15 cases was a Gypsy child, p.R299X homozygous, with normal IOP at age 3 years.
We investigated clinical heterogeneity among p.R299X homozygotes in five recently diagnosed children aged 1.5–7 years (Table 3). In patient 32-3 the phenotype was similar to that described by Désir et al,15 except for the earlier onset of glaucoma. The child was referred to the clinic at age 5 months because of buphthalmos and iridodonesis, and diagnosed with bilateral lens dislocation. The IOP at the time was normal but increased to 24/29 mm Hg by age 1.5 years. Patients 33-3 (from the same small sub-isolate as 32-3) and patient 26-3 are examples of unilateral ectopia lentis and bilateral glaucoma. Patient 33-3 was diagnosed at age 1.5 years with megalocornea, increased IOP and sub-luxation of the right lens, whereas the left was normally positioned. Patient 26-3 was diagnosed at 12 months with bilateral megalocornea, bilaterally increased IOP, microspherophakia, Haab's striae, and sub-luxation of the lens of the right eye. At age 2 years, when the child underwent surgery, the left lens was still normally positioned. By age 7 years, when last seen, the patient had undergone five trabeculotomies and presented with megalocornea, corneal clouding, and visual acuity 0.02 in the left and no light perception in the right eye. Finally, affected siblings 17-4 and 17-3 illustrate intra-familial variation. Patient 17-4 had bilateral trabeculotomy at age 38 days for increased IOP. He has undergone four trabeculotomies since, with no ectopia lentis diagnosed during surgery. At follow-up at age 7 years he had bilateral corneal clouding, no perception of light in the right eye and able to count fingers with the left. The brother, 17-3, presented at age 3 months with buphthalmos and normal IOP. Increased IOP and bilateral lens dislocation were diagnosed at age 1.5 years. Follow-up examination at 5 years showed enlarged but clear corneas, visual acuity 0.01 in the left eye, whereas the right eye could follow hand motion.
Table 3 Ocular manifestations in pediatric patients homozygous for the p.R299X mutation in LTBP2
In summary, all five patients had megalocornea and early increase in IOP (before age 18 months) regardless of the presence of ectopia lentis. Lens dislocation was bilateral in two patients, in whom the disease evolution suggests that the glaucoma was secondary. The mechanisms appear to be different in the remaining cases, in whom primary dysgenesis of the trabecular meshwork may be responsible for the increased IOP in one or both eyes. Corneal clouding was found in the two patients aged 7 years, but not in the younger ones. The outcome in terms of visual acuity was very poor in all.
Population genetics of PCG in Gypsies
Three founder mutations (p.R299X in LTBP2 and p.E387K and p.R390C in CYP1B1) together account for ∼60% of the 74 unrelated PCG alleles, and three additional rare mutations in CYP1B1 together contribute a further 6.7% (Table 1). Contrary to the expectations based on the individual mutation frequencies, no p.R299X/p.E387K compounds were detected among affected or control subjects, suggesting a differential distribution among endogamous sub-isolates. According to self-declared group identity, most families in this study belonged to the largest Gypsy group in the country, the Yerlia, in which sub-isolates are likely to have existed in the past but are now difficult to tease out. However, the geographical distribution was markedly different, with regional clustering of p.E387K alleles, in contrast to the wide spread of p.R299X (Figure 3). A different history is also suggested by the carrier rates in controls: p.R299X occurs in different Gypsy sub-isolates across Europe at an average rate of ∼2.4% (Supplementary Table 3), whereas p.E387K has so far not been reported in India,26, 27, 28, 29 and its carrier rate among Bulgarian Gypsies is only 0.3% with a limited distribution within Bulgaria and apparently also in Europe (Supplementary Table 5 and Figure 3).
Figure 3
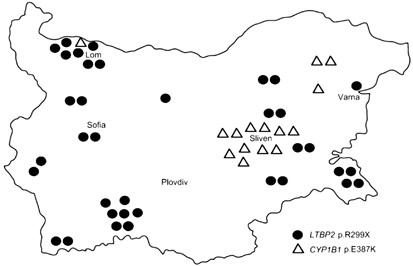
Geographic distribution of the p.R299X and p.E387K alleles in Bulgaria, indicating places of residence of affected subjects and of carriers identified among population controls. The map outline is taken from http://www.worldatlas.com.
Haplotype decay was analysed to estimate the time in generations since the mutations were introduced in the Gypsy population of Bulgaria. A conserved 6.9 cM haplotype (D2S186 to D2S414) was shared by 9/15 p.E387K chromosomes (Figure 4a). The coalescence time was ∼21 generations (95% CI, 15.7–30.9), corresponding to ∼630 years (95% CI, 470–926 years, assuming a generation time of 30 years30). P.E387K may thus have arisen around the fourteenth century in a subset of the Balkan Gypsy population which soon afterwards split to spawn sub-isolates in Slovakia and Eastern Bulgaria.
Figure 4
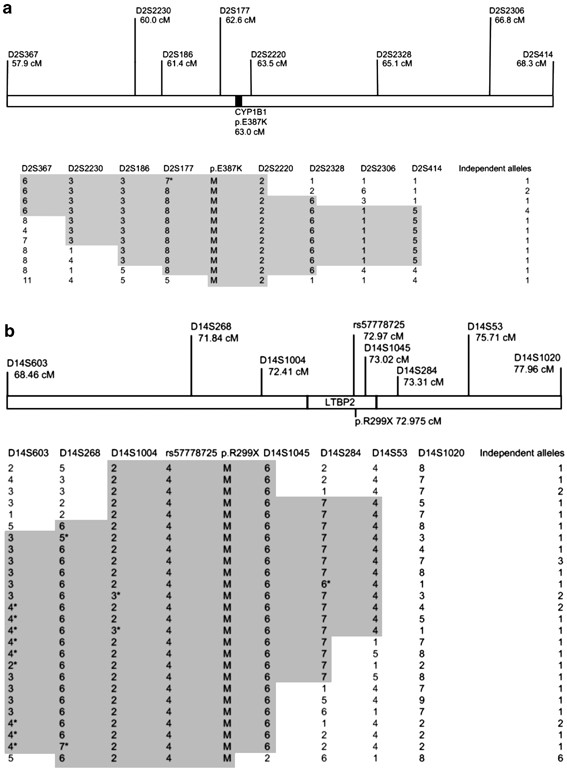
Founder mutation haplotypes on independent chromosomes carrying: (a) CYP1B1 p.E387K and (b) LTBP2 p.R299X. The genetic distances follow the DeCode map. The positions of rs57778725 and of the two mutations are interpolated from the physical distances and positions relative to the DeCode STRs. The conserved (presumably ancestral) haplotypes are highlighted in grey. Asterisks (*) denote potential mutations, rather than recombinations. M denotes mutant allele.
A haplotype of similar size (7.3 cM, D14S603-D14S53, conservatively assuming microsatellite mutations rather than recombinations at some STRs) was present around p.R299X (Figure 4b) in a smaller proportion of chromosomes (13/37). Coalescence time was 33.5 generations (95% CI, 27.8–43.6) or 1005 years (95% CI, 834–1308), that is, around the arrival in Europe, supporting the ancestral origins of the mutation and its presence in the Gypsy population before its fissions occurred.18, 31
Discussion
To our knowledge, the present data are the first comprehensive investigation of the two currently known PCG genes in a relatively large sample of affected families. The spectrum of molecular defects in the Gypsies comprises several ancestral mutations, among which p.R299X in LTBP2 and p.E387K in CYP1B1 stand out as the most common defects, and are thus the primary targets in a diagnostic setting. The possibility of positive selection has been raised in cases of allelic heterogeneity in genetically isolated populations and extensively discussed in the case of lysosomal storage disorders in the Ashkenazim.32 In addition to its hypothesised roles in eye development33 CYP1B1, similar to other members of the P450 family, is involved in the metabolism of environmental chemicals, which can be enzymatically converted to toxic compounds.34 One could, therefore, speculate on the selective advantage of heterozygotes for deleterious CYP1B1 alleles, who would have lower metabolising capacity of environmental chemicals with potentially toxic effect. Given the low global frequency of PCG-causing CYP1B1 alleles, this would imply the unlikely scenario of a specific environment for the traditionally nomadic Gypsies that is not shared with surrounding populations. Genetic drift, as proposed by Risch et al35 to explain the findings in Ashkenazi Jews, seems also the most plausible scenario in the case of the Gypsies.
The population genetics of PCG is peculiar and different from other Mendelian disorders in the Gypsies. Although four of the currently known mutations seem to have been ‘taken out’ of the Indian subcontinent, supporting our previous genetic data on the origin of the Gypsies,31, 36, 37 the allelic diversity in PCG contradicts our previous data suggesting a small number of related founders.31 The existing information does not allow conclusions on the parental population(s) in India/Pakistan and time of separation. Current knowledge about the distribution of p.R299X, the major founder mutation in the Gypsies, is confined to a single Pakistani family, with no carriers identified in our Gujarati and Jat samples. The ancestral Gypsy CYP1B1 mutations occur in India on different haplotype backgrounds, across geographical regions, in Indo-European as well as Dravidian speakers, and in subjects of diverse ethnic, religious and caste affiliations (Supplementary Table 6). This pattern, which points to ancient origins preceding the formation of the current population and social structure of India, is also seen in other ancestral Gypsy mutations – 1267delG in CHRNE36 and W24X in GJB2, the latter with an origin estimated at around 8000 years ago.38 Further population genetic studies will have to await extensive data on multiple genetic system and markers in all populations involved.
Our systematic analysis of CYP1B1 and LTBP2 allowed the identification of double mutants, carrying the pathogenic p.R299X in combination with known changes in CYP1B1: p.E229K or p.R368H. Our current data indicate that double carrier status, in the observed combinations of mutations, is clinically innocuous. The possibility of digenic inheritance was raised in PCG double heterozygotes for mutations in CYP1B1 and MYOC,10, 11 and these two genes have been proposed to interact in a common biochemical pathway.39 Our early observations of lack of clinical manifestations in CYP1B1/LTBP2 double carriers may be attributed to the involvement of the two genes in independent pathways of eye development and glaucoma pathogenesis. However, it is important to note that both CYP1B1 variants – p.E229K and p.R368H – lead to reduced, not abolished, enzyme activity and protein stability,40, 41 and display incomplete penetrance.42, 43 CYP1B1_–_LTBP2 interactions and their clinical implications need to be addressed in larger samples of double carriers of well-supported pathogenic mutations. In the meantime, a conservative approach to diagnosis and counselling may be warranted, under the assumption that some CYP1B1 mutations, for example, p.R368H, may confer susceptibility but are not sufficient to cause PCG.42 Such an approach implies continued molecular investigations in cases with only one identified defect.22, 26, 27, 28, 44
Our current data suggest that the truncating p.R299X mutation in LTBP2 is associated with greater phenotype severity and poorer outcome. Follow-up of this observation should be possible given the increased incidence of PCG in the Gypsies with equal proportions of patients with mutations in the known and yet uncharacterised genes.
Although the number of our p.R299X homozygotes is still relatively small, the data point to a spectrum of manifestations in different combinations and with a variable evolution, from strictly defined PCG with dysgenesis of the trabecular meshwork to the ectopia lentis/secondary glaucoma syndrome. LTBP2 is a known interactor of fibrillin 1, the protein mutated in Marfan syndrome (MFS) and other ‘fibrillinopathies’, wherein ectopia lentis occurs in ∼50% of cases and glaucoma can be due to angle closure/pupil block.45 Rarely, glaucoma in MFS can result from abnormalities in the trabecular meshwork,46 and PCG due to primary trabeculodysgenesis may occur in the lethal neonatal MFS.47 So far, both ocular manifestations and the presence/absence of other MFS-like features in LTBP2 defects appear variable and combinatorial.13, 15, 16 We are currently unable to comment on the presence of extra-ocular manifestations: most p.R299X homozygotes in the study have been clinically examined before the identification of the LTBP2 gene with no record of abnormalities found upon physical examination, whereas recently ascertained cases are young children, in whom the diagnosis of MFS-like features is still uncertain. The question whether lens dislocation in p.R229X homozygotes tends to occur in combination with other MFS-like features merits further study, as it may be related to the underlying mechanisms – whether random and unpredictable, or involving sequence variation in the fibrillin 1 gene in addition to the truncating LTBP2 mutation(s). Understanding these mechanisms has important implications for the treatment and management of affected children.
References
- Sarfarazi M, Stoilov I, Schenkman JB : Genetics and biochemistry of primary congenital glaucoma. Ophthalmol Clin North Am 2003; 16: 543–554.
Article Google Scholar - MacKinnon JR, Giubilato A, Elder JE, Craig JE, Mackey DA : Primary infantile glaucoma in an Australian population. Clin Experiment Ophthalmol 2004; 32: 14–18.
Article Google Scholar - Gencik A, Gencikova A, Ferak V : Population genetical aspects of primary congenital glaucoma. I. Incidence, prevalence, gene frequency, and age of onset. Hum Genet 1982; 61: 193–197.
Article CAS Google Scholar - deLuise VP, Anderson DR : Primary infantile glaucoma (congenital glaucoma). Surv Ophthalmol 1983; 28: 1–19.
Article CAS Google Scholar - Ho CL, Walton DS : Primary congenital glaucoma: 2004 update. J Pediatr Ophthalmol Strabismus 2004; 41: 271–288.
Article Google Scholar - Sarfarazi M, Akarsu AN, Hossain A et al: Assignment of a locus (GLC3A) for primary congenital glaucoma (buphthalmos) to 2p21 and evidence for genetic heterogeneity. Genomics 1995; 30: 171–177.
Article CAS Google Scholar - Akarsu AN, Turacli ME, Aktan SG et al.: A second locus (GLC3B) for primary congenital glaucoma (buphthalmos) maps to the 1p36 region. Hum Mol Genet 1996; 5: 1199–1203.
Article CAS Google Scholar - Stoilov I, Sarfarazi M : The third genetic locus (GLC3C) for primary congenital glaucoma (PCG) maps to chromosome 14q24.3. Invest Ophthalmol Vis Sci 2002; 43: 3015.
Google Scholar - Plasilova M, Stoilov I, Sarfarazi M, Kadasi L, Ferakova E, Ferak V : Identification of a single ancestral CYP1B1 mutation in Slovak Gypsies (Roms) affected with primary congenital glaucoma. J Med Genet 1999; 36: 290–294.
CAS PubMed PubMed Central Google Scholar - Kaur K, Reddy AB, Mukhopadhyay A et al: Myocilin gene implicated in primary congenital glaucoma. Clin Genet 2005; 67: 335–340.
Article CAS Google Scholar - Chakrabarti S, Kaur K, Komatireddy S et al: Gln48His is the prevalent myocilin mutation in primary open angle and primary congenital glaucoma phenotypes in India. Mol Vis 2005; 11: 111–113.
CAS PubMed Google Scholar - Firasat S, Riazuddin SA, Hejtmancik JF, Riazuddin S : Primary congenital glaucoma localizes to chromosome 14q24.2-24.3 in two consanguineous Pakistani families. Mol Vis 2008; 14: 1659–1665.
CAS PubMed PubMed Central Google Scholar - Ali M, McKibbin M, Booth A et al: Null mutations in LTBP2 cause primary congenital glaucoma. Am J Hum Genet 2009; 84: 664–671.
Article CAS Google Scholar - Narooie-Nejad M, Paylakhi SH, Shojaee S et al: Loss of function mutations in the gene encoding latent transforming growth factor beta binding protein 2, LTBP2, cause primary congenital glaucoma. Hum Mol Genet 2009; 18: 3969–3977.
Article CAS Google Scholar - Desir J, Sznajer Y, Depasse F et al: LTBP2 null mutations in an autosomal recessive ocular syndrome with megalocornea, spherophakia, and secondary glaucoma. Eur J Hum Genet 2010; 18: 761–767.
Article CAS Google Scholar - Kumar A, Duvvari MR, Prabhakaran VC, Shetty JS, Murthy GJ, Blanton SH : A homozygous mutation in LTBP2 causes isolated microspherophakia. Hum Genet 2010; 128: 365–371.
Article CAS Google Scholar - Sivadorai P, Cherninkova S, Bouwer S et al: Genetic heterogeneity and minor CYP1B1 involvement in the molecular basis of primary congenital glaucoma in Gypsies. Clin Genet 2008; 74: 82–87.
Article CAS Google Scholar - Kalaydjieva L, Morar B, Chaix R, Tang H : A newly discovered founder population: the Roma/Gypsies. Bioessays 2005; 27: 1084–1094.
Article CAS Google Scholar - Desmet FO, Hamroun D, Lalande M, Collod-Beroud G, Claustres M, Beroud C : Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res 2009; 37: e67.
Article Google Scholar - Sunyaev S, Ramensky V, Koch I, Lathe III W, Kondrashov AS, Bork P : Prediction of deleterious human alleles. Hum Mol Genet 2001; 10: 591–597.
Article CAS Google Scholar - Barrett JC, Fry B, Maller J, Daly MJ : Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 2005; 21: 263–265.
Article CAS Google Scholar - Dimasi DP, Hewitt AW, Straga T et al: Prevalence of CYP1B1 mutations in Australian patients with primary congenital glaucoma. Clin Genet 2007; 72: 255–260.
Article CAS Google Scholar - Stephens M, Smith NJ, Donnelly P : A new statistical method for haplotype reconstruction from population data. Am J Hum Genet 2001; 68: 978–989.
Article CAS Google Scholar - Reeve JP, Rannala B : DMLE+: Bayesian linkage disequilibrium gene mapping. Bioinformatics 2002; 18: 894–895.
Article CAS Google Scholar - Hunter M, Heyer E, Austerlitz F et al: The P28T mutation in the GALK1 gene accounts for galactokinase deficiency in Roma (Gypsy) patients across Europe. Pediatr Res 2002; 51: 602–606.
Article CAS Google Scholar - Reddy AB, Kaur K, Mandal AK et al: Mutation spectrum of the CYP1B1 gene in Indian primary congenital glaucoma patients. Mol Vis 2004; 10: 696–702.
CAS PubMed Google Scholar - Panicker SG, Mandal AK, Reddy AB, Gothwal VK, Hasnain SE : Correlations of genotype with phenotype in Indian patients with primary congenital glaucoma. Invest Ophthalmol Vis Sci 2004; 45: 1149–1156.
Article Google Scholar - Chakrabarti S, Kaur K, Kaur I et al: Globally, CYP1B1 mutations in primary congenital glaucoma are strongly structured by geographic and haplotype backgrounds. Invest Ophthalmol Vis Sci 2006; 47: 43–47.
Article Google Scholar - Tanwar M, Dada T, Sihota R, Das TK, Yadav U, Dada R : Mutation spectrum of CYP1B1 in North Indian congenital glaucoma patients. Mol Vis 2009; 15: 1200–1209.
CAS PubMed PubMed Central Google Scholar - Tremblay M, Vezina H : New estimates of intergenerational time intervals for the calculation of age and origins of mutations. Am J Hum Genet 2000; 66: 651–658.
Article CAS Google Scholar - Gresham D, Morar B, Underhill PA et al: Origins and divergence of the Roma (gypsies). Am J Hum Genet 2001; 69: 1314–1331.
Article CAS Google Scholar - Zlotogora J, Zeigler M, Bach G : Selection in favor of lysosomal storage disorders? Am J Hum Genet 1988; 42: 271–273.
CAS PubMed PubMed Central Google Scholar - Stoilov I : Cytochrome P450s: coupling development and environment. Trends Genet 2001; 17: 629–632.
Article CAS Google Scholar - Nebert DW, Russell DW : Clinical importance of the cytochromes P450. Lancet 2002; 360: 1155–1162.
Article CAS Google Scholar - Risch N, Tang H, Katzenstein H, Ekstein J : Geographic distribution of disease mutations in the Ashkenazi Jewish population supports genetic drift over selection. Am J Hum Genet 2003; 72: 812–822.
Article CAS Google Scholar - Morar B, Gresham D, Angelicheva D et al: Mutation history of the Roma/Gypsies. Am J Hum Genet 2004; 75: 596–609.
Article CAS Google Scholar - Bouwer S, Angelicheva D, Chandler D, Seeman P, Tournev I, Kalaydjieva L : Carrier rates of the ancestral Indian W24X mutation in GJB2 in the general Gypsy population and individual subisolates. Genet Test 2007; 11: 455–458.
Article CAS Google Scholar - RamShankar M, Girirajan S, Dagan O et al: Contribution of connexin26 (GJB2) mutations and founder effect to non-syndromic hearing loss in India. J Med Genet 2003; 40: e68.
Article CAS Google Scholar - Vincent AL, Billingsley G, Buys Y et al: Digenic inheritance of early-onset glaucoma: CYP1B1, a potential modifier gene. Am J Hum Genet 2002; 70: 448–460.
Article CAS Google Scholar - Pasutto F, Chavarria-Soley G, Mardin CY et al: Heterozygous loss of function variants in CYP1B1 predispose to primary open angle glaucoma. Invest Ophthalmol Vis Sci 2010; 51: 249–254.
Article Google Scholar - Lopez-Garrido MP, Blanco-Marchite C, Sanchez-Sanchez F et al: Functional analysis of CYP1B1 mutations and association of heterozygous hypomorphic alleles with primary open-angle glaucoma. Clin Genet 2010; 77: 70–78.
Article CAS Google Scholar - Suri F, Yazdani S, Narooie-Nejhad M et al: Variable expressivity and high penetrance of CYP1B1 mutations associated with primary congenital glaucoma. Ophthalmology 2009; 116: 2101–2109.
Article Google Scholar - Campos-Mollo E, Lopez-Garrido MP, Blanco-Marchite C et al: CYP1B1 mutations in Spanish patients with primary congenital glaucoma: phenotypic and functional variability. Mol Vis 2009; 15: 417–431.
CAS PubMed PubMed Central Google Scholar - Bagiyeva S, Marfany G, Gonzalez-Angulo O, Gonzalez-Duarte R : Mutational screening of CYP1B1 in Turkish PCG families and functional analyses of newly detected mutations. Mol Vis 2007; 13: 1458–1468.
CAS PubMed Google Scholar - Dureau P : Pathophysiology of zonular diseases. Curr Opin Ophthalmol 2008; 19: 27–30.
Article Google Scholar - Izquierdo NJ, Traboulsi EI, Enger C, Maumenee IH : Glaucoma in the Marfan syndrome. Trans Am Ophthalmol Soc 1992; 90: 111–117;discussion 118-122.
CAS PubMed PubMed Central Google Scholar - Whitelaw CM, Anwar S, Ades LC, Gole GA, Elder JE, Savarirayan R : Primary trabeculodysgenesis in association with neonatal Marfan syndrome. Am J Med Genet A 2004; 128A: 418–421.
Article Google Scholar
Acknowledgements
We thank the affected families for their participation. The study is funded by the Australia–India Strategic Research Fund (grant BF020055). DNA is supported by an NHMRC Training Fellowship (634551).
Author information
Author notes
- Stanislava Dimitrova and Laura Florez: These two authors contributed equally to the work.
Authors and Affiliations
- Laboratory for Molecular Genetics, Centre for Medical Research and Western Australian Institute for Medical Research, QEII Medical Centre, University of Western Australia, Perth, Western Australia, Australia
Dimitar N Azmanov, Laura Florez, Bharti Morar, Rosmawati Saat & Luba Kalaydjieva - Molecular Medicine Centre, Medical University, Sofia, Bulgaria
Stanislava Dimitrova & Radka Kaneva - Department of Neurology, Medical University, Sofia, Bulgaria
Sylvia Cherninkova & Ivailo Tournev - Specialised Eye Hospital, Medical University, Varna, Bulgaria
Dragomir Draganov - Servei d'Immunologia, IDIBAPS-Hospital Clínic, Barcelona, Spain
Manel Juan & Juan I Arostegui - Human Genetics Unit, Indian Statistical Institute, Kolkata, India
Sriparna Ganguly & Partha Majumder - National Health Laboratory Service, University of the Witwatersrand, Johannesburg, South Africa
Himla Soodyall - Brien Holden Eye Research Centre, LV Prasad Eye Institute, Hyderabad, India
Subhabrata Chakrabarti - BV Patel Pharmaceutical Education and Research Development Centre, Thaltej, Ahmedabad, India
Harish Padh - Servicio de Análisis Clínicos, Hospital Universitario Virgen de las Nieves, Universidad de Granada, Granada, Spain
Miguel A López-Nevot - Department of Ophthalmology, Medical University, Sofia, Bulgaria
Violeta Chernodrinska & Botio Anguelov - National Institute of Biomedical Genomics, Kalyani, India
Partha Majumder - Department of Paediatrics and Medical Genetics, Medical University, Varna, Bulgaria
Lyudmila Angelova - Lions Eye Institute, University of Western Australia, Perth, Western Australia, Australia
David A Mackey - Department of Cognitive Science and Psychology, New Bulgarian University, Sofia, Bulgaria
Ivailo Tournev
Authors
- Dimitar N Azmanov
- Stanislava Dimitrova
- Laura Florez
- Sylvia Cherninkova
- Dragomir Draganov
- Bharti Morar
- Rosmawati Saat
- Manel Juan
- Juan I Arostegui
- Sriparna Ganguly
- Himla Soodyall
- Subhabrata Chakrabarti
- Harish Padh
- Miguel A López-Nevot
- Violeta Chernodrinska
- Botio Anguelov
- Partha Majumder
- Lyudmila Angelova
- Radka Kaneva
- David A Mackey
- Ivailo Tournev
- Luba Kalaydjieva
Corresponding author
Correspondence toLuba Kalaydjieva.
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Azmanov, D., Dimitrova, S., Florez, L. et al. LTBP2 and CYP1B1 mutations and associated ocular phenotypes in the Roma/Gypsy founder population.Eur J Hum Genet 19, 326–333 (2011). https://doi.org/10.1038/ejhg.2010.181
- Received: 06 May 2010
- Revised: 28 September 2010
- Accepted: 08 October 2010
- Published: 17 November 2010
- Issue Date: March 2011
- DOI: https://doi.org/10.1038/ejhg.2010.181