Genetic dissection of a behavioral quantitative trait locus shows that Rgs2 modulates anxiety in mice (original) (raw)
Main
Crosses between inbred strains of mice are frequently used to map quantitative trait loci (QTLs) that give rise to the genetic component of quantitative variation in traits of biomedical interest, such as those underlying susceptibility to depression and anxiety1. It has proven difficult to identify genes underlying behavioral QTLs: although 94 such QTLs have been reported to exceed a genome-wide significance threshold, in no case has the responsible gene, or genes, been identified2. One problem is that each QTL individually makes only a modest contribution to the phenotype; on average, a detectable behavioral QTL accounts for ∼5% of the total phenotypic variance2.
Over the past ten years, anxiety-related QTLs in mice have been identified on 13 chromosomes3,4,5. Although the individual effect of each QTL is small, their detection can be replicated6, and one QTL has been mapped to a small interval of ∼1 cM on chromosome 1 (near 145 Mb on the National Center for Biotechnology Information mouse genome build 30) using a genetically heterogeneous stock of mice7,8,9. Despite extensive analysis of the genes and variants at this locus10, however, the molecular nature of QTLs that influence anxiety-like behavior in mice remains obscure.
Positional cloning of small-effect QTLs by purely genetic means is extremely difficult because many recombinants are needed to isolate a single gene. Genetic mapping has the additional problem that it locates a functional variant (or variants) rather than a gene. The positions of genes and sequence variants that affect gene expression do not always coincide. Functionally important elements have been discovered far from their cognate genes11, and regulatory elements for expression of one gene may lie in an intron of another, functionally unrelated, gene12,13.
Alternative strategies to obtain functional evidence that a gene contributes to behavioral variation can also be extremely challenging. In a few cases, the molecular basis of large-effect QTLs (those explaining 40% or more of the phenotypic variance in an intercross) has been identified by the analysis of gene expression differences14,15, but the method has so far not been successful when applied to the much more common small-effect QTLs that are responsible for individual differences in behavior. Moreover, where cellular processes are causally remote from the phenotype, as is the case for behavior, expression differences or altered protein function provide only circumstantial evidence to implicate a gene as a QTL. Variation in gene expression is not necessarily translated into behavioral differences, and a gene's effect may depend on where and when it is expressed in the brain16.
Two approaches might overcome these problems. First, high-resolution mapping in outbred populations, taking advantage of recombination between loci accumulating over many generations, has been successfully applied to mapping small-effect QTLs in fruit flies17,18 and humans19,20,21,22. We reasoned that a similar strategy might work in outbred mice.
Second, a method called quantitative complementation testing has been used to investigate the role of candidate genes in QTL mapping experiments in fruit flies18,23, and a similar method was used in a study of a QTL in yeast (reciprocal hemizygosity analysis24). It has not yet been used in mammals. The method requires no information about the nature of responsible sequence variants, their mode of action or their location with respect to the candidate gene, but it does rely on access to deficiency stocks or recessive mutants. These resources are now becoming available for mouse genetics.
Here we describe the application of both methods to characterize the chromosome 1 QTL, and we show that the gene Rgs2, encoding a regulator of G protein signaling, is a candidate in this region that modulates variation in anxiety-like behavior.
Note: Supplementary information is available on the Nature Genetics website.
Results
Single-marker mapping using MF1 outbred mice
We mapped more precisely the region previously shown to contain a QTL influencing anxiety on chromosome 1 (ref. 9). We measured anxiety in 729 outbred MF1 mice using an open-field arena, a brightly lit white arena that is an unwelcome and potentially threatening environment for the animal. Open-field activity (OFA) and open-field defecation (OFD) are indices of rodent fearfulness or 'emotionality', which has many parallels with human anxiety. We previously showed that an analysis that combines OFA and OFD increases power to detect an effect9. We define a new composite phenotype, 'emotionality' (EMO), constructed by taking the difference between the standardized scores for OFA and OFD and rescaling the scores to a standard normal distribution.
We obtained genotypes for 42 single-nucleotide polymorphisms (SNPs) over the 3.5-Mb region10 of the 729 mice and analyzed the EMO scores using single-marker analysis of variance (Fig. 1). We determined the significance threshold by permutation, and the 5% threshold, expressed as logP, a negative logarithm of the P value, is 2.76 (slightly less than a Bonferroni-corrected 5% threshold of 2.93), equaled by a single marker at 1.3 Mb with a logP value of 2.76. Consequently, single-marker analysis provides only weak evidence of genetic association to this locus.
Figure 1: Single-marker and multipoint HAPPY QTL mapping in MF1 mice.
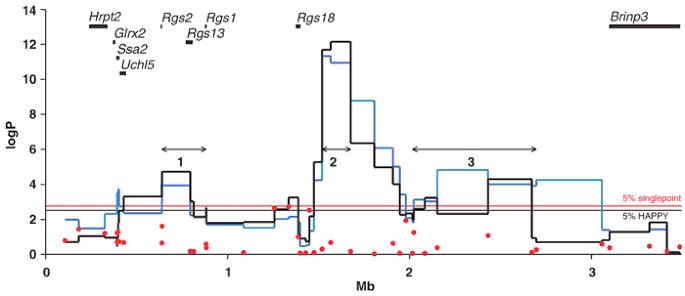
Genetic mapping of two kinds in MF1 mice (single-point analysis of variance results, red dots; HAPPY mapping, black and blue lines) with the physical map of gene positions, shown as horizontal black bars at the top of the figure. HAPPY analysis was carried out in two ways, assuming either four (black line) or eight (blue line) progenitor strains. The horizontal black and red lines indicate 5% significance threshold for HAPPY and single point analyses, respectively. The 95% c.i. for the three QTL peaks are shown as three numbered lines with arrowheads.
MF1 haplotypes are derived from inbred strains
We next used a more powerful mapping method. We previously showed that a multipoint method, HAPPY, performs substantially better than single-marker analysis in detecting QTLs8, but to apply this technique we need to establish that the MF1 mice can be treated as if they were descended from a small number of known progenitor strains, that is, as a heterogeneous stock8. Each allele in a heterogeneous stock can theoretically be traced back to one of eight progenitor strains. But we do not know the ancestry of the MF1 mice, which were created in the early 1970s by crossing the LACA line, a standard prolific outbred mouse line, with another outbred albino line called CF. Both LACA and CF mice are related to Swiss mice but are not known to share an ancestor with any of the common inbred strains25.
We investigated whether the haplotype structure of the MF1 mice was related to that of the progenitor strains in the heterogeneous stock (C57BL/6J, BALB/cJ, RIII, AKR, DBA/2, I, A/J and C3H) that we had originally used to map the QTLs9. In 12 MF1 mice we sequenced a total of 62 kb surrounding the nine genes in the region and found only four differences with the inbred strain sequences10. All 42 genotyped SNPs were polymorphic and had the same alleles as the heterogeneous stock mice. These data suggested that MF1 haplotypes were very similar to those found in inbred strains.
To test this idea further, we reconstructed the haplotypes of the 729 MF1 mice over the 42 SNPs, using PHASE2 (refs. 26,27) and treating the mice as unrelated. We then devised a dynamic programming algorithm to reconstruct these haplotypes as a mosaic of inbred strains using the least number of chromosomal breakpoints. The mosaics for the 14 most common MF1 haplotypes together account for more than 95% of the chromosome complement (Fig. 2). All haplotypes can be derived from just four inbred strain haplotypes (C3H, AKR, C57BL/6J and I; because there are no sequence differences between C3H and A/J over the region of interest10, these two strains are interchangeable).
Figure 2: Reconstruction of MF1 haplotypes as inbred strain mosaics.
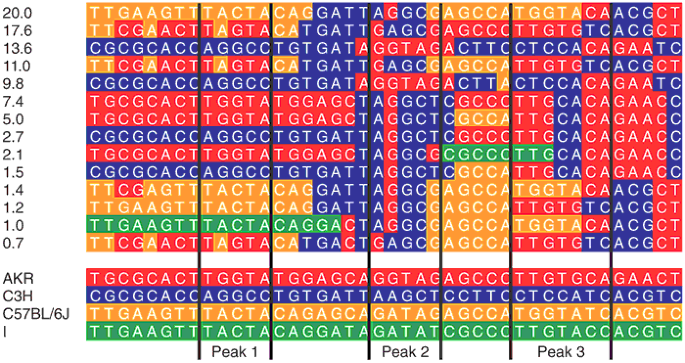
The top part of the figure shows haplotypes that account for 95% of the MF1 chromosomal complement. To the left of each haplotype is its frequency in the population, expressed as a percentage. Each haplotype is represented horizontally as a string of sequence variants at the 42 SNPs used for mapping in the MF1. The bottom part of the figure shows the haplotypes of four inbred strains (C3H, AKR, C57BL/6J and I). The origin of each MF1 haplotype from these inbred strains, as determined by a dynamic programming algorithm, is indicated by color coding of each nucleotide (red for C3H, blue for AKR, yellow for C57BL/6J and green for I). Blocks of contiguous color in the MF1 represent unrecombined haplotypes. The labeled black vertical lines demarcate the 95% c.i. for the three QTL peaks.
If this mosaic is meaningful then we would expect it to have far fewer breakpoints than a mosaic reconstructed from random progenitor strains. We tested whether the number of breakpoints in the mosaic was statistically unlikely by permuting the alleles of the inbred strains at each marker position, reconstructing the optimal mosaic and counting the number of breakpoints. The number of breakpoints using the bona-fide strain haplotypes was less than that observed in each of 10,000 permutations. Consequently, the MF1 haplotypes can be modeled as a mosaic and therefore analyzed like a heterogeneous stock descended from these inbred progenitor strains.
HAPPY analysis of MF1 mice
To avoid assuming a particular mosaic is correct, we searched for QTLs using HAPPY8, which estimates the probability of descent from each inbred strain. HAPPY models the MF1 haplotype mosaics using a hidden Markov model, integrating over all possible mosaic reconstructions weighted according to their relative probabilities8. HAPPY uses unphased genotypes rather than the haplotypes determined by PHASE2 (refs. 26,27) and so does not introduce bias resulting from incorrect specification of the haplotype phase assignment. Hypothesis testing for QTL detection is based on a test for differences between the estimated phenotypic effects attributable to each progenitor strain at the locus of interest.
We carried out the HAPPY analysis using the four strains identified in the mosaic as plausible progenitors of MF1 mice. The method detects genetic effects with much more power than single-marker analysis (Fig. 1). The 5% threshold for region-wide significance for HAPPY logP scores is 2.51; this was exceeded at three places: peak 1 at 0.7 Mb (logP = 5.0, 95% confidence interval (c.i.) = 0.43–0.81); peak 2 at 1.7 Mb (logP = 10.9, 95% c.i. = 1.43–1.67 Mb); and peak 3 at 2.5 Mb (logP = 4.2, 95% c.i. = 2.01–2.69 Mb; confidence intervals are for an additive four-strain QTL model, using a bootstrap procedure8). As expected from previous mapping data, the size of the effect attributable to the locus is small, with each peak contributing less than 5% of the total phenotypic variance. Together the three peaks account for 12% of the variance.
To test whether our choice of strains was skewing the result, we also analyzed the MF1 mice by using all eight heterogeneous stock progenitors. Both analyses identified the same three peaks (Fig. 1). The much lower significance levels of single-marker association mapping compared with HAPPY reflect the fact that the strain distribution patterns (SDPs) of the SNPs need not coincide with the QTL allele effects, as noted in previous analyses8,28. For example, if the SDP at the functional variant is different from the SDPs of nearby SNP markers (e.g., because it is not diallelic), then no marker is a good surrogate for it. This problem is avoided by multipoint methods such as HAPPY, which consider combinations of markers that induce new SDPs and therefore might coincide with the SDP of the functional variant.
The QTL region has three independent effects
We next asked whether the three peaks were truly independent, as linkage disequilibrium between markers might contribute to interdependence between the peaks. We used our reconstruction of MF1 haplotypes from putative progenitor strains as an index of historical recombination (Fig. 2). On average, 8.4 recombinants separate the MF1 haplotypes from the progenitor haplotypes. The position of the 95% c.i. containing each QTL peak is shown in Figure 2, superimposed on the derivation of the common haplotypes. The haplotypes cannot be reconstructed in such a way that an ancestral haplotype spans all the peaks and no two peaks lie on the same progenitor strain haplotype (Fig. 2), indicating that the peaks are probably independent. We may not have correctly ascertained the founders, however, and so our recombination estimates may be biased. Therefore, we investigated the independence of the three effects by fitting them simultaneously, testing the significance of each QTL peak in the presence of the other two using partial F-tests. All three peaks remained significant (logP = 2.5, 11.9 and 3.3), suggesting that they are independent and real effects, although the significance levels of the first and third peaks were lower and the location estimated for the third peak shifted slightly.
Figure 1 shows the relationship between the QTL peaks and known genes in the region. The second and third QTL peaks are located in a region devoid of known genes, although there are several expressed sequences. Neither the human nor the mouse region was predicted to encode any known microRNA sequences. The 95% c.i. of the second peak, at 0.7 Mb, contains just two genes, Rgs2 and Rgs13 (regulator of G-protein signaling 2 and 13). Only Rgs2 lies completely within a 95% c.i.
Quantitative complementation of Rgs2
On the basis of the MF1 fine-mapping data, Rgs2 is a strong candidate gene. Therefore, we used quantitative complementation to test whether Rgs2 interacts with a functional variant. The test uses four strains: two that bear different QTL alleles (referred to here as high and low lines), a strain bearing a recessive mutation of Rgs2 (m); and a wild-type strain (+) that is ideally coisogenic with the mutant. We determined phenotypes of mice with the four genotypes high/m, low/m, high/+ and low/+ and analyzed them in an experiment with two factors: 'Cross', representing the presence or absence of the mutation, and 'Line', representing natural allelic variation at the QTL. We suppose that the QTL exerts its effect by altering the expression of the gene, as might be the case if it lies in the promoter of the gene or in a more distant enhancer element. In this case, the two effects, one due to the gene and one to the QTL, will not be independent and their joint effect (a failure to complement) will be detected as a significant interaction between Line (high or low) and Cross (m or +) in the analysis of variance. The interaction coefficient between Line and Cross is identical to the contrast (high/m − low/m) − (high/+ − low/+) and measures the failure for the wild type to complement the mutation on different backgrounds (low versus high).
We obtained a recessive mutation of Rgs2 suitable for the quantitative complementation test, but because the Rgs2 mutant was made on a 129/P2 strain and backcrossed to C57BL/6J29, obtaining a wild type on a coisogenic background was difficult. But the genomes of inbred strains of laboratory mice are closely related and can be described as a mosaic structure of alternating segments of sequence similarity and difference30,31,32. We reasoned that the problem of mixed strain background might be overcome if we could show that the genetic effect of any sequence variant in the mutant strain was identical to its effect in C57BL/6J; in other words, even though the sequence might not be identical, the two strains would carry the same QTLs.
We used genotyping data and sequence comparisons to determine whether we could use C57BL/6J as the wild-type control for the complementation test. Analysis of 98 microsatellite markers showed that the genome of the mutant mouse is C57BL/6J, apart from a 37-Mb region on chromosome 1 (between 113 Mb to 150 Mb on the National Center for Biotechnology Information mouse genome build #30). Mapping in the heterogeneous stock indicated that this region contains only the QTLs analyzed here, due to a contrast between two strains (A/J and C3H: low EMO) on one hand and the other six strains on the other (C57BL/6J, DBA/2, I, AKR, RIII and BALB/cJ: high EMO)8,9,28,33.
We investigated the region containing the three QTL peaks, sequencing, in the mutant, amplimers of ∼1.2 kb at an average interval of 8.5 kb across the region. We found no polymorphisms unique to the Rgs2 mutant; the Rgs2 mutant sequence was identical to that of C57BL/6J from 0.5 to 2.95 Mb and identical to that of DBA/2 from 2.95 Mb onward. Mapping experiments identified no QTLs segregating between C57BL/6J and DBA/2 in the region of sequence difference8,28,33,34.
We tested for an interaction between Line and Cross at the Rgs2 locus by quantitative complementation, again using the EMO phenotype9 measured in 117 mice. On the basis of our mapping in the heterogeneous stock and from sequence analysis of the QTL region, we knew that C57BL/6J carried the high QTL allele and that either A/J or C3H carried the low QTL allele8,10. We used C57BL/6J and C3H for quantitative complementation, crossing both with the Rgs2 mutant and with the wild type (C57BL/6J). The interaction between Line and Cross was significant (P = 0.009), implicating Rgs2 as a gene involved in the QTL (Table 1).
Table 1 Analysis of variance for quantitative complementation of the Rgs2 mutant
Rgs2 modulates anxiety
If Rgs2 is the quantitative trait gene, then it should have a specific pattern of action35 that affects both OFA and OFD, but in opposite directions as increased anxiety is associated with lower activity and higher defecation. The interaction coefficient should be positive for OFA and negative for OFD. Furthermore, the interaction coefficient for EMO should be larger than those for either OFA or OFD. The gene should also affect other measures of anxiety. In the elevated plus maze, we expected the interaction coefficient to be positive for number of entries and time spent in the open arms of the maze. In another test of novelty, the latency (or amount of time taken) to try a new food, the interaction coefficient should be negative. Last, Rgs2 should not affect activity measured in a nonthreatening environment, such as the distance traveled in 30 min in a home cage (home cage activity). Quantitative complementation of Rgs2 produced the expected pattern of results (Table 1).
Because the control strain used in the complementation test is not identical to the mutant strain, we needed to show that the results were not due to unknown QTL next to Rgs2 that might have been segregating between the DBA/2 and C57BL/6J haplotypes. We directly tested this possibility with another quantitative complementation test using DBA/2, rather than C3H, as the contrasting strain to C57BL/6J. If a QTL segregates between these two strains at the Rgs2 locus, then there should be a failure to complement. We found that the interaction between strain and background was not significant: P = 0.3 for OFA, P = 0.97 for OFD and P = 0.48 for EMO. Furthermore, we did not uncover any functional effect attributable to differences between DBA/2 and C57BL/6J sequence variants in MF1 mice by comparing a model in which a different genetic effect is allowed in each strain with a model in which it is constrained by the strain distribution pattern of the variant, so that strains sharing the same allele must have the same genetic effect. These results indicate that the quantitative complementation result is not compromised by the use of C57BL/6J as a control and that the effect is indeed specific to a small-effect QTL segregating between C3H and C57BL/6J.
Discussion
We report here the identification of a gene, Rgs2, underlying a small-effect QTL that contributes to behavioral variation in the mouse. The variance due to this QTL in the segregating cross is ∼5%, which is typical for behavioral QTLs. Other information about the function of Rgs2 is consistent with this finding. Rgs2 is widely expressed in the brain36, and the Rgs2 mutation has an effect on behavior29. Comparing the behavior of the homozygous Rgs2 mutant with that of C57BL/6J mice indicates that the mutation makes mice more anxious (see Supplementary Table 1 online). Regulators of G-protein signaling are known to have a role in rapid behavioral changes37,38; their involvement in modulating activity levels in the tests used here to measure anxiety in rodents is consistent with these observations. In common with other Rgs genes, Rgs2 affects a wide range of phenotypes including hypertension39, immune response29 and implantation in the womb40.
Although Rgs2 modulates anxiety in the mouse, the genetic data indicate that it is only one component of the QTL. The position of one QTL peak over Rgs2 (Fig. 1) suggests that the functional variant interacting with Rgs2 is close to, or inside, the gene. The positions of the other QTL peaks suggest that Rgs18 and Brinp3 are good candidates for other components (Fig. 1), but confirmation is needed because these peaks lie in intergenic regions, more than 100 kb from the nearest known expressed sequence. We cannot rule out the possibility that these peaks interact with Rgs2 as well. Although several expressed sequence tags align to the genome sequence under the second QTL peak, they probably do not represent protein-coding genes because they have no homology to known protein-coding genes, are not spliced and often contain long and short interspersed element repeats. This observation is important, as it indicates that concentrating solely on known expressed sequences may result in missing important loci.
The complexity of the architecture of the QTL is similar to that reported elsewhere. Studies that isolate genetic effects in congenic and recombinant inbred mouse lines often report that one relatively large effect comprises several loci with much smaller effects41,42,43,44. In Drosophila melanogaster, four different fine-mapping QTL studies reported a similar phenomenon45. Similar complexity will probably be found at other QTLs.
This study establishes two new approaches to genetic mapping in mice. First, we showed that it is possible to use commercially available outbred mice to map small-effect QTLs with a high degree of precision (to within a few hundred kilobases). This success was due to the unexpected finding that MF1 mice can be treated as mosaics of standard inbred strains and analyzed accordingly using probabilistic ancestral reconstruction. It will be of interest to determine whether the genomes of other outbred lines can be treated similarly.
Second, we used a quantitative complementation test to show that Rgs2 modulates anxiety in mice. Whereas genetic fine-mapping locates the functional sequence variants, quantitative complementation identifies the candidate genes. A significant failure to complement implies either allelism (the gene contains the functional variant) or epistasis (the gene interacts with the functional variant, which may be elsewhere in the genome). We cannot exclude the possibility of an interaction between Rgs2 and loci on other chromosomes, but this explanation is unlikely for two reasons. First, we have been unable to detect epistasis between any open-field behavior QTL so far detected in the heterogeneous stock; second, we found no evidence of epistasis acting on open-field or elevated plus maze measures of anxiety in two F2 intercrosses (C75BL/6J × BALB/cJ and DBA/2 × C57BL/6J)28,46.
The combined genetic and functional approaches described here provide a general method for identifying small-effect genes underlying QTLs. Using the analytical techniques we developed, together with information about the sequence structure of inbred strains and available mutants, the entire experiment could be carried out within a year. It should therefore be possible to detect the genes underlying other QTLs in the same way.
Methods
Mice and crosses.
We acquired outbred F2 generation MF1 mice and inbred C3H/HeJ and C57BL/6J mice at 5–6 weeks of age from Harlan UK. We obtained the Rgs2 mutant from J. Penninger (Amgen Institute, University of Toronto, Canada)29. For the quantitative complementation experiment, we made F1 hybrids by crossing C3H/HeJ (low EMO) and C57BL/6J (high EMO) mice to the homozygous Rgs2 mutant (23 and 30 F1 mice, respectively) and crossing C3H/HeJ with C57BL/6J mice (40 mice, low × control). For the remaining component of the cross, we phenotyped 24 C57BL/6J inbred mice (high × control). For the complementation using DBA/2, we used an equivalent number of DBA/C57BL and DBA/Rgs2 mutant F1 hybrids. We phenotyped 729 MF1 mice in 21 families with a mean size of 30 (s.d. = 12). We maintained mice in a vivarium with controlled temperature, light and humidity on a 12-h light-dark cycle. We carried out all behavioral tests during the daylight phase of the cycle. We housed mice in single-sex littermate groups with free access to food and water. All mice were tested when they were between 6 and 8 weeks of age.
Phenotyping.
We measured OFA in a brightly lit, 60-cm-diameter, enclosed white arena with no background noise. We placed mice in the apparatus and monitored them for 5 min by video camera; movements were analyzed using an image analyzer (Videotrack (version NT4.0)) from Viewpoint. At the end of each trial, we recorded the number of fecal boli deposited. We also monitored behavior in the elevated plus maze (apparatus described in ref. 47) using an automated tracking system. We measured the number of entries, time in seconds and the distance traveled in the open and closed arms. We measured food neophobia as the latency to eat a new food (a solution of one-third full-cream sweetened condensed milk and two-thirds water). Mice were restricted to 1 g of food overnight. Mice were given three trials of 2 min each. We stopped the trial when the mouse first tasted the food. The test apparatus has been described47. We measured baseline activity in a home-cage environment using a photo activity system from San Diego Instruments. We measured the number of beam breaks during a 30-min test period.
DNA extraction and genotyping.
We extracted DNA from 0.5-cm tail snips using a phenol-chloroform method48 and separated it into 96-well plates at a concentration of 10 ng μl−1 for genotyping. We designed extension and amplification primers for SNP genotyping using SpectroDESIGNER. Oligonucleotides were synthesized at Metabion. We carried out PCR with Hotstar Taq obtained from Qiagen. Each 5-μl PCR contained 2.5 ng of genomic DNA, 0.2 U of HotStar Taq, 5 pmol of forward and reverse primers, 2 mM of each dNTP, 1 × HotStar Taq PCR buffer as supplied by the enzyme manufacturer (contains 1.5 mM MgCl2, Tris-Cl, KCl and (NH4)2SO4, pH 8.7) and 25 mM MgCl2 (Qiagen). The temperature profile consisted of an initial enzyme activation at 95 °C for 15 min, followed by 45 cycles of 94 °C for 20 s, 56 °C for 30 s and 72 °C for 60 s, and a final incubation at 72 °C for 3 min. We treated PCR products with shrimp alkaline phosphatase (Sequenom) for 20 min at 37 °C first to remove excess dNTPs. We used a thermosequenase (Sequenom) for the base extension reactions. The base extension conditions were 94 °C for 2 min, followed by 55 cycles of 94 °C for 5 s, 52 °C for 5 s, and 72 °C for 5 s. We removed unincorporated nucleotides from extension products using SpectroCLEAN resin. A few nanoliters of each sample were arrayed onto a 384 SpectroCHIP by a SpectroPOINT robot. The chip was read in the Bruker Biflex III Mass Spectrometer system and data analyzed on SpectroTYPER; the resulting genotypes were then automatically uploaded into an Integrated Genotyping System. To determine the relationship between the Rgs2 mutant and C57BL/6J mice, we amplified 98 microsatellite markers, distributed across the genome, that distinguish C57BL/6J from 129/P2 and compared the allele sizes with those present in the Rgs2 mutant. After PCR amplification, we separated products by electrophoresis through 4% agarose gels and scored marker sizes with reference to a size standard.
DNA sequencing.
We used Primer3 to design oligonucleotide primers. We amplified genomic DNA segments in a 50-μl PCR reaction using oligonucleotides synthesized at MWG: 100 ng of DNA, 0.2 U of Gold Taq, 10 pmol of forward and reverse primers, 8 mM of each dNTP, 1 × PCR buffer and 25 mM MgCl2. The PCR conditions were 95 °C for 15 min; 13 cycles of 95 °C for 30 s, 62 °C for 30 s (−0.5 °C per cycle) and 72 °C for 60 s; 29 cycles of 95 °C for 30 s, 58 °C for 30 s and 72 °C for 55 s; and 72 °C for 7 min. We purified PCR products on a 96-well Millipore purification plate and resuspended them in 30 μl of water. We prepared two sequencing reactions for each DNA sample, one with the forward primer and one with the reverse primer. We removed the PCR reagents from solution by an ethanol precipitation in the presence of sodium acetate. All sequencing reactions were carried out on an ABI3700 sequencer.
Haplotype mosaic generation.
We determined haplotypes of MF1 mice using PHASE2, using the program's default options26,27. All mice were analyzed together, ignoring family information. The derivation of the haplotype mosaic from inbred strains was reconstructed using the following dynamic programming algorithm that finds a mosaic that minimizes the number of breakpoints required. Suppose there are ordered N markers. Let a ij be the allele at marker position j in the _i_th haplotype obtained from PHASE. Let s kj be the allele at marker position j in the _k_th inbred strain. Let x ikj equal 0 if a ij = s kj or equal 1 otherwise. An optimal mosaic is a sequence k(ij) of inbred strains such that the allele a ij = s k(ik) and the number of breakpoints where k(ij) differs from k(i j_−1) is minimal. Let R ijk be the score of an optimal partial mosaic of the i_th haplotype for marker positions 1..j, constrained so that the final j_th position is assigned to strain k. Then R_1_jk = −_ax ik_1 and R ijk = max_n {R i j_−1_n − a x ijn + δ_nk} for j > 1. δ_nk is the delta function, and a is a negative weight parameter chosen such that a breakpoint always occurs in preference to a mismatched allele. Let M ijk be the strain n that maximizes R ijk in this recursion, and M iN the strain k that maximizes R ijk . Then an optimal mosaic is given by the sequence defined as S(iN) = M iN ; S(ij) = M ijS(ij+1); j < N. We carried out permutation analysis by shuffling the alleles at each marker position in the inbred strains and reconstructing the mosaic 10,000 times.
QTL mapping.
We transformed phenotypes into Gaussian deviates by first ranking them and then replacing each rank with its corresponding quantile in the standard normal distribution. We carried out QTL mapping using the HAPPY software package, implemented in C and R 1.9.0. We determined the presence of a QTL at an interval between two adjacent genotyped markers as described8. As progenitors for HAPPY mapping analysis, we used the eight inbred strains that founded the HS strain as well as the four strains identified by the strain reconstruction. We estimated region-wide significance levels by permuting the transformed phenotype values, repeating the single point or HAPPY analysis, recording the maximal logP value and ranking the results of 1,000 analyses to determine significance thresholds. The 5% thresholds for single-point (2.96) and HAPPY (2.51) were close to the Bonferroni approximation assuming independent tests (2.95). We tested the independence of genetic effects by comparing a model with all three QTL peaks fitted simultaneously with three submodels in which each peak was omitted in turn, evaluating significance by a partial F-test. We determined a 95% c.i. for each QTL location by bootstrapping, where the subjects were resampled with replacement 1,000 times and the most significant marker interval was recorded. We estimated the probability that the QTL was in a given marker interval as the frequency with which the interval was most significant in the bootstrapped analyses.
Quantitative complementation testing.
We analyzed quantitative complementation results as a linear model in the R statistical analysis package version 1.9.0 of the form E(y) = μ + C + L + C × L. Here, y is the trait and μ is the intercept, equal to the expected effect for a mouse of genotype high/+, C is the difference between the main effects of Cross (low versus high), L the difference between the main effect of Line (mutant versus wild-type), and C × L the interaction between Cross and Line. Failure to complement is indicated by a significant interaction coefficient in the analysis of variance.
URLs.
An annotated interactive version of the sequence is available at http://bioinformatics.well.ox.ac.uk/project-anxiety/. The database of micro RNA sequences used to search the sequence is available at http://www.sanger.ac.uk/Software/Rfam/mirna/. The HAPPY package is available at http://www.well.ox.ac.uk/happy/, and R software is available from http://www.r-project.org/.
References
- Flint, J. & Mott, R. Finding the molecular basis of quantitative traits: successes and pitfalls. Nat. Rev. Genet. 2, 438–445 (2001).
Article Google Scholar - Flint, J. Analysis of quantitative trait loci that influence animal behavior. J. Neurobiol. 54, 46–77 (2003).
Article CAS Google Scholar - Gershenfeld, H.K. & Paul, S.M. Mapping quantitative trait loci for fear-like behaviors in mice. Genomics 46, 1–8 (1997).
Article CAS Google Scholar - Turri, M.G., De Fries, J.C., Henderson, N.D. & Flint, J. Multivariate analysis of quantitative trait loci influencing variation in anxiety-related behavior in laboratory mice. Mamm. Genome 15, 69–76 (2004).
Article Google Scholar - Henderson, N.D., Turri, M.G., DeFries, J.C. & Flint, J. QTL analysis of multiple behavioral measures of anxiety in mice. Behav. Genet. 34, 267–293 (2004).
Article Google Scholar - Turri, M.G., Henderson, N.D., DeFries, J.C. & Flint, J. Quantitative trait locus mapping in laboratory mice derived from a replicated selection experiment for open-field activity. Genetics 158, 1217–1226 (2001).
CAS PubMed PubMed Central Google Scholar - McClearn, G.E., Wilson, J.R. & Meredith, W. The use of isogenic and heterogenic mouse stocks in behavioral research. in Contributions to Behavior-Genetic Analysis: The Mouse as a Prototype (eds. Lindzey, G. & Thiessen, D.) 3–22 (Appleton Century Crofts, New York, 1970).
Google Scholar - Mott, R., Talbot, C.J., Turri, M.G., Collins, A.C. & Flint, J. A method for fine mapping quantitative trait loci in outbred animal stocks. Proc. Natl. Acad. Sci. USA 97, 12649–12654 (2000).
Article Google Scholar - Talbot, C.J. et al. High-resolution mapping of quantitative trait loci in outbred mice. Nat. Genet. 21, 305–308 (1999).
Article CAS Google Scholar - Yalcin, B. et al. Unexpected complexity in the haplotypes of commonly used inbred strains of laboratory mice. Proc. Natl. Acad. Sci. USA 101, 9734–9739 (2004).
Article CAS Google Scholar - Nobrega, M.A., Ovcharenko, I., Afzal, V. & Rubin, E.M. Scanning human gene deserts for long-range enhancers. Science 302, 413 (2003).
Article CAS Google Scholar - Higgs, D.R. et al. A major positive regulatory region located far upstream of the human α-globin gene locus. Genes Dev. 4, 1588–1601 (1990).
Article CAS Google Scholar - Lettice, L.A. et al. Disruption of a long-range cis-acting regulator for Shh causes preaxial polydactyly. Proc. Natl. Acad. Sci. USA 99, 7548–7553 (2002).
Article CAS Google Scholar - Aitman, T.J. et al. Quantitative trait loci for cellular defects in glucose and fatty acid metabolism in hypertensive rats. Nat. Genet. 16, 197–201 (1997).
Article CAS Google Scholar - Tafti, M. et al. Deficiency in short-chain fatty acid β-oxidation affects τ oscillations during sleep. Nat. Genet. 34, 320–325 (2003).
Article CAS Google Scholar - Gross, C. et al. Serotonin1A receptor acts during development to establish normal anxiety-like behaviour in the adult. Nature 416, 396–400 (2002).
Article CAS Google Scholar - Lai, C., Lyman, R.F., Long, A.D., Langley, C.H. & Mackay, T.F. Naturally occurring variation in bristle number and DNA polymorphisms at the scabrous locus of Drosophila melanogaster . Science 266, 1697–1702 (1994).
Article CAS Google Scholar - De Luca, M. et al. Dopa decarboxylase (Ddc) affects variation in Drosophila longevity. Nat. Genet. 34, 429–433 (2003).
Article CAS Google Scholar - Zhang, Y. et al. Positional cloning of a quantitative trait locus on chromosome 13q14 that influences immunoglobulin E levels and asthma. Nat. Genet. 34, 181–186 (2003).
Article CAS Google Scholar - Helms, C. et al. A putative RUNX1 binding site variant between SLC9A3R1 and NAT9 is associated with susceptibility to psoriasis. Nat. Genet. 35, 349–356 (2003).
Article CAS Google Scholar - Ueda, H. et al. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature 423, 506–511 (2003).
Article CAS Google Scholar - Tokuhiro, S. et al. An intronic SNP in a RUNX1 binding site of SLC22A4, encoding an organic cation transporter, is associated with rheumatoid arthritis. Nat. Genet. 35, 341–348 (2003).
Article CAS Google Scholar - Long, A.D., Mullaney, S.L., Mackay, T.F.C. & Langley, C.H. Genetic interactions between naturally occurring alleles at quantitative trait loci and mutant alleles at candidate loci affecting bristle number in Drosophila melanogaster . Genetics 144, 1497–1510 (1996).
CAS PubMed PubMed Central Google Scholar - Steinmetz, L.M. et al. Dissecting the architecture of a quantitative trait locus in yeast. Nature 416, 326–330 (2002).
Article CAS Google Scholar - Beck, J.A. et al. Genealogies of mouse inbred strains. Nat. Genet. 24, 23–25 (2000).
Article CAS Google Scholar - Stephens, M. & Donnelly, P. A comparison of Bayesian methods for haplotype reconstruction from population genotype data. Am. J. Hum. Genet. 73, 1162–1169 (2003).
Article CAS Google Scholar - Stephens, M., Smith, N.J. & Donnelly, P. A new statistical method for haplotype reconstruction from population data. Am. J. Hum. Genet. 68, 978–989 (2001).
Article CAS Google Scholar - Talbot, C.J. et al. Fine scale mapping of a genetic locus for conditioned fear. Mamm. Genome 14, 223–230 (2003).
Article Google Scholar - Oliveira-Dos-Santos, A.J. et al. Regulation of T cell activation, anxiety, and male aggression by RGS2. Proc. Natl. Acad. Sci. USA 97, 12272–12277 (2000).
Article CAS Google Scholar - Wade, C.M. et al. The mosaic structure of variation in the laboratory mouse genome. Nature 420, 574–578 (2002).
Article CAS Google Scholar - Lindblad-Toh, K. et al. Large-scale discovery and genotyping of single-nucleotide polymorphisms in the mouse. Nat. Genet. 24, 381–386 (2000).
Article CAS Google Scholar - Wiltshire, T. et al. Genome-wide single-nucleotide polymorphism analysis defines haplotype patterns in mouse. Proc. Natl. Acad. Sci. USA 100, 3380–3385 (2003).
Article CAS Google Scholar - Hitzemann, R. et al. Multiple cross mapping (MCM) markedly improves the localization of a QTL for ethanol-induced activation. Genes Brain Behav. 1, 214–222 (2002).
Article CAS Google Scholar - Plomin, R., McClearn, G.E., Gora-Maslak, G. & Neiderhiser, J.M. Use of recombinant inbred strains to detect quantitative trait loci associated with behavior. Behav. Genet. 21, 99–116 (1991).
Article CAS Google Scholar - Turri, M.G., Datta, S.R., DeFries, J., Henderson, N.D. & Flint, J. QTL analysis identifies multiple behavioral dimensions in ethological tests of anxiety in laboratory mice. Curr. Biol. 11, 725–734 (2001).
Article CAS Google Scholar - Grafstein-Dunn, E., Young, K.H., Cockett, M.I. & Khawaja, X.Z. Regional distribution of regulators of G-protein signaling (RGS) 1, 2, 13, 14, 16, and GAIP messenger ribonucleic acids by in situ hybridization in rat brain. Brain Res. Mol. Brain Res. 88, 113–123 (2001).
Article CAS Google Scholar - Dong, M.Q., Chase, D., Patikoglou, G.A. & Koelle, M.R. Multiple RGS proteins alter neural G protein signaling to allow C. elegans to rapidly change behavior when fed. Genes Dev. 14, 2003–2014 (2000).
CAS PubMed PubMed Central Google Scholar - Rahman, Z. et al. RGS9 modulates dopamine signaling in the basal ganglia. Neuron 38, 941–952 (2003).
Article CAS Google Scholar - Heximer, S.P. et al. Hypertension and prolonged vasoconstrictor signaling in RGS2-deficient mice. J. Clin. Invest. 111, 445–452 (2003).
Article CAS Google Scholar - Huang, Z.P. et al. Expression of regulator of G-protein signalling protein 2 (RGS2) in the mouse uterus at implantation sites. Reproduction 126, 309–316 (2003).
Article CAS Google Scholar - Legare, M.E., Bartlett, F.S. & Frankel, W.N. A major effect QTL determined by multiple genes in epileptic EL mice. Genome Res. 10, 42–48 (2000).
CAS PubMed PubMed Central Google Scholar - Wanstrat, A. & Wakeland, E. The genetics of complex autoimmune diseases: non-MHC susceptibility genes. Nat. Immunol. 2, 802–809 (2001).
Article Google Scholar - Fijneman, R.J., de Vries, S.S., Jansen, R.C. & Demant, P. Complex interactions of new quantitative trait loci, Sluc1, Sluc2, Sluc3, and Sluc4, that influence the susceptibility to lung cancer in the mouse. Nat. Genet. 14, 465–467 (1996).
Article CAS Google Scholar - van Wezel, T., Ruivenkamp, C.A., Stassen, A.P., Moen, C.J. & Demant, P. Four new colon cancer susceptibility loci, Scc6 to Scc9 in the mouse. Cancer Res. 59, 4216–4218 (1999).
CAS PubMed Google Scholar - Mackay, T.F. The genetic architecture of quantitative traits: lessons from Drosophila . Curr. Opin. Genet. Dev. 14, 253–257 (2004).
Article CAS Google Scholar - Flint, J., De Fries, J.C. & Henderson, N.D. Little epistasis for anxiety-related measures in the DeFries strains of laboratory mice. Mamm. Genome 15, 77–82 (2004).
Article Google Scholar - Contet, C., Rawlins, J.N. & Deacon, R.M. A comparison of 129S2/SvHsd and C57BL/6JOlaHsd mice on a test battery assessing sensorimotor, affective and cognitive behaviours: implications for the study of genetically modified mice. Behav. Brain. Res. 124, 33–46 (2001).
Article CAS Google Scholar - Sambrook, J., Fritsch, E.F. & Maniatis, T. Molecular Cloning: A Laboratory Manual (Cold Spring Harbor Laboratory Press, Cold Spring Harbor, 1989).
Google Scholar
Acknowledgements
We thank J. Penninger for providing the _Rgs2_-mutant mice and S. McCormick for comments on the manuscript. This work was funded by the Wellcome Trust.
Author information
Author notes
- Binnaz Yalcin and Saffron A G Willis-Owen: These authors contributed equally to this work.
Authors and Affiliations
- Wellcome Trust Centre for Human Genetics, University of Oxford, Roosevelt Drive, Oxford, OX3 7BN, UK
Binnaz Yalcin, Saffron A G Willis-Owen, Jan Fullerton, Anjela Meesaq, Richard R Copley, Andrew P Morris, Jonathan Flint & Richard Mott - Department of Experimental Psychology, University of Oxford, Oxford, UK
Robert M Deacon & J Nicholas P Rawlins
Authors
- Binnaz Yalcin
You can also search for this author inPubMed Google Scholar - Saffron A G Willis-Owen
You can also search for this author inPubMed Google Scholar - Jan Fullerton
You can also search for this author inPubMed Google Scholar - Anjela Meesaq
You can also search for this author inPubMed Google Scholar - Robert M Deacon
You can also search for this author inPubMed Google Scholar - J Nicholas P Rawlins
You can also search for this author inPubMed Google Scholar - Richard R Copley
You can also search for this author inPubMed Google Scholar - Andrew P Morris
You can also search for this author inPubMed Google Scholar - Jonathan Flint
You can also search for this author inPubMed Google Scholar - Richard Mott
You can also search for this author inPubMed Google Scholar
Corresponding authors
Correspondence toJonathan Flint or Richard Mott.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Table 1
Comparison between C57BL6/J wild type (wt) and Rgs2 homozygous knockout (Mutant) mice. (PDF 5 kb)
Rights and permissions
About this article
Cite this article
Yalcin, B., Willis-Owen, S., Fullerton, J. et al. Genetic dissection of a behavioral quantitative trait locus shows that Rgs2 modulates anxiety in mice.Nat Genet 36, 1197–1202 (2004). https://doi.org/10.1038/ng1450
- Received: 25 June 2004
- Accepted: 13 September 2004
- Published: 17 October 2004
- Issue Date: 01 November 2004
- DOI: https://doi.org/10.1038/ng1450