Loss-of-function mutations in LEMD3 result in osteopoikilosis, Buschke-Ollendorff syndrome and melorheostosis (original) (raw)
- Letter
- Published: 17 October 2004
- Olena Preobrazhenska2,
- Andy Willaert1,
- Philippe Debeer3,
- Peter C M Verdonk4,
- Teresa Costa5,
- Katrien Janssens6,
- Bjorn Menten1,
- Nadine Van Roy1,
- Stefan J T Vermeulen1,
- Ravi Savarirayan7,
- Wim Van Hul6,
- Filip Vanhoenacker8,
- Danny Huylebroeck2,
- Anne De Paepe1,
- Jean-Marie Naeyaert9,
- Jo Vandesompele1,
- Frank Speleman1,
- Kristin Verschueren2,
- Paul J Coucke1 &
- …
- Geert R Mortier1
Nature Genetics volume 36, pages 1213–1218 (2004)Cite this article
- 5013 Accesses
- 321 Citations
- 6 Altmetric
- Metrics details
Abstract
Osteopoikilosis, Buschke-Ollendorff syndrome (BOS) and melorheostosis are disorders characterized by increased bone density1. The occurrence of one or more of these phenotypes in the same individual or family suggests that these entities might be allelic2,3,4. We collected data from three families in which affected individuals had osteopoikilosis with or without manifestations of BOS or melorheostosis. A genome-wide linkage analysis in these families, followed by the identification of a microdeletion in an unrelated individual with these diseases, allowed us to map the gene that is mutated in osteopoikilosis. All the affected individuals that we investigated were heterozygous with respect to a loss-of-function mutation in LEMD3 (also called MAN1), which encodes an inner nuclear membrane protein. A somatic mutation in the second allele of LEMD3 could not be identified in fibroblasts from affected skin of an individual with BOS and an individual with melorheostosis. XMAN1, the Xenopus laevis ortholog, antagonizes BMP signaling during embryogenesis5. In this study, LEMD3 interacted with BMP and activin-TGFβ receptor–activated Smads and antagonized both signaling pathways in human cells.
Similar content being viewed by others
Main
Osteopoikilosis (OMIM 166700) is an autosomal dominant skeletal dysplasia characterized by a symmetric but unequal distribution of multiple hyperostotic areas in different parts of the skeleton (Fig. 1)6. These lesions, usually detected incidentally, represent foci of old remodeled bone with lamellar structure, either connected to adjacent trabeculae of spongy bone or attached to the subchondral cortex7. Osteopoikilosis can occur either as an isolated anomaly or in association with other abnormalities of skin and bone. BOS (OMIM 166700), an autosomal dominant disorder, refers to the association of osteopoikilosis with disseminated connective-tissue nevi. Both elastic-type nevi (juvenile elastoma) and collagen-type nevi (dermatofibrosis lenticularis disseminata) have been described in BOS8. Skin or bony lesions can be absent in some family members, whereas other relatives may have both9. The co-occurrence of osteopoikilosis and melorheostosis in the same family has been reported in a few instances2,3,4. Melorheostosis (OMIM 155950) is characterized by a 'flowing' (rheos) hyperostosis of the cortex of tubular bones. These lesions are usually asymmetric: they may involve only one limb or correspond to a particular sclerotome. They are often accompanied by abnormalities of adjacent soft tissues, such as joint contractures, sclerodermatous skin lesions, muscle atrophy, hemangiomas and lymphoedema10,11. Affected individuals may be asymptomatic or may complain of chronic pain in the affected limb12.
Figure 1: Osteopoikilosis lesions and elastic-type nevus in individual III-3 of family A.
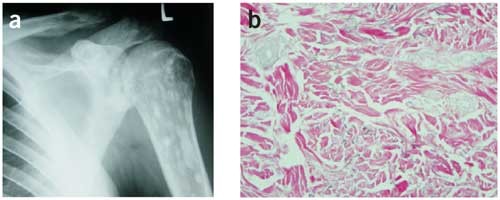
(a) Anteroposterior radiograph of the left shoulder showing multiple osteopoikilosis lesions, best visible in the left humerus. (b) Light micrograph of the elastic-type nevus stained with Van Gieson. Original magnification, × 100. Thick and coarse collagen bundles with numerous broad and irregular elastic fibers are present in the mid-dermis.
To unravel the genetic defect of osteopoikilosis, we started a genome-wide linkage analysis in family A (Fig. 2a). Screening of 400 markers with an average spacing of 10 cM resulted in a significant positive lod score (3.744) for two consecutive markers on chromosome 12q13: D12S368 and D12S83. The results of the linkage analysis in the two other families, families B and C (Fig. 2b,c), were consistent with the linkage data obtained in family A. The centromeric boundary of the genetic interval was determined by a recombination event between D12S1048 and D12S1663 in individual III-1 (of family A). The telomeric boundary was defined by a recombination event between D12S1686 and D12S313 in individual II-5 (of family C), resulting in a candidate region of 23.55 cM on chromosome 12q12–12q14.3. We calculated a combined maximum two-point lod score of 6.691 at θ=0 for markers D12S1661 and D12S1691 (Supplementary Table 1 online).
Figure 2: Pedigree structure and haplotypes of the three families with osteopoikilosis, family A (a), family B (b) and family C (c).
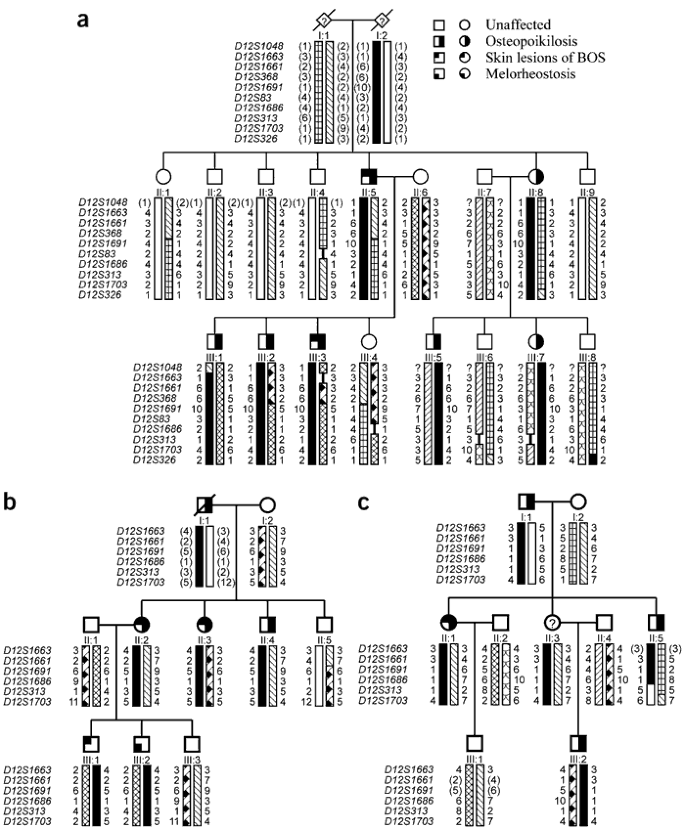
Inferred alleles are shown in brackets. The haplotype cosegregating with the disorder is indicated with a black bar.
The next step in the genetic analysis was the identification of individual G03-1858, who is affected with proportionate short stature, microcephaly, learning disabilities, ectopic kidneys and osteopoikilosis. We hypothesized that this individual might have a microdeletion, resulting in the loss of several contiguous genes, including the gene mutated in osteopoikilosis. We investigated this individual for loss of heterozygosity in the candidate region on 12q12–12q14.3 and found loss of heterozygosity for marker D12S1686, located in the telomeric part of the interval. The testing of additional markers confirmed the presence of a microdeletion with a centromeric boundary at marker D12S329 (Fig. 3). The telomeric boundary of the microdeletion was defined at the single-nucleotide polymorphism tsc0527430 using the GeneChip Mapping 10K Array (results for whole genome in Supplementary Fig. 1 online; results for chromosome 12 in Fig. 4c). We then tested more markers in the region of overlap between the microdeletion and the linkage interval in family C, which allowed us to narrow the linkage interval and define a 3.07-Mb critical region for association with osteopoikilosis between marker D12S329 and μSAT12.10. This region contains 23 known genes (National Center for Biotechnology Information genome viewer; Fig. 3).
Figure 3: Ideogram of chromosome 12 showing the linkage interval, microdeletion and candidate region.
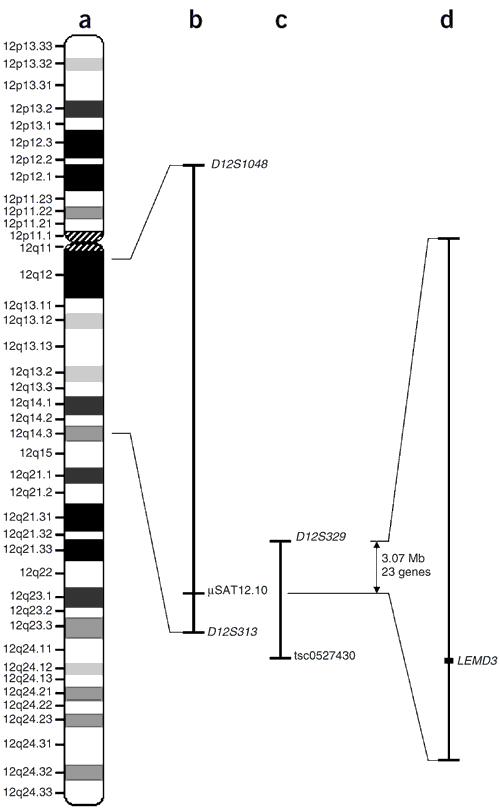
(a) Ideogram of chromosome 12. (b) The 12q12–q14.3 linkage interval with indication of markers at the boundaries. (c) The microdeletion in individual G03-1858 in relation to the linkage interval. The region of interest is shown on the right (d), with LEMD3 as the candidate gene.
Figure 4: Overview of the cytogenetic and molecular defects found in affected individuals.
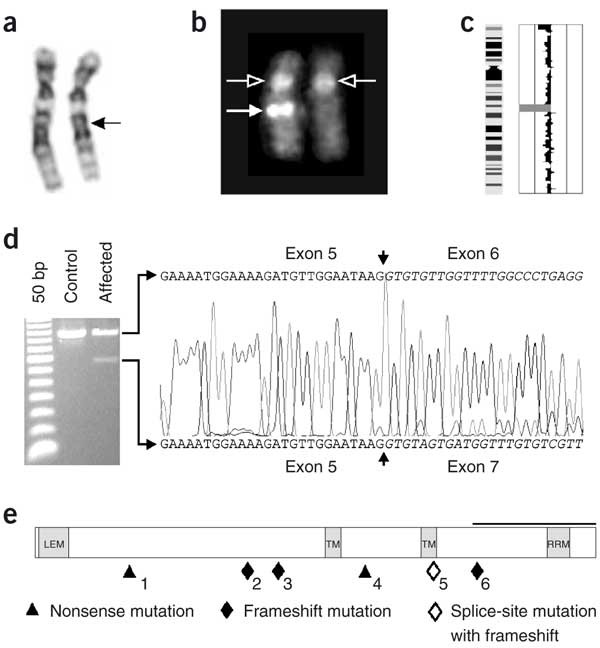
(a) Partial karyotype from individual G03-1858 showing both chromosomes 12. The normal homolog is depicted on the left, and the homolog with the deletion (arrow) is shown on the right. (b) Metaphase FISH analysis with the BAC clone encompassing LEMD3 (RP11-30506; filled arrow) and the centromeric 12 probe (open arrow), showing a microdeletion on the right homolog. (c) Results from the GeneChip Mapping 10K Array analysis for chromosome 12 of individual G03-1858. An ideogram of chromosome 12 is shown on the left; meta-analysis significance of the genetic copy-number variation of each SNP against the reference mean is shown on the right. The region with large negative values (bar to the left) indicates the presence of a microdeletion. (d) Effect of the LEMD3 mutation in individual G03-2881. The electrophoretic analysis of an amplified cDNA fragment containing exons 4–8 shows the presence of a 146-bp shorter fragment as compared to the control. Partial nonsense-mediated decay is probably responsible for the weaker signal of this abnormal fragment. Sequence analysis shows skipping of exon 6 in the mutated allele (causing a frameshift with a premature stop codon in exon 7 at position 2,021; data not shown). (e) The positions of all LEMD3 mutations identified in this study are shown below the structure of LEMD3 (see also Table 1). Functional domains are indicated in gray: the LEM-containing N-terminal domain, the two transmembrane (TM) domains and the C-terminal domain with the RNA-recognition motif (RRM) motif. The black horizontal bar indicates the Smad-interacting part and BMP-antagonizing portion (as shown in XMAN1; ref. 5) of the C-terminal domain.
Two of these genes, WIF1 (Wnt inhibitory factor 1) and LEMD3 (LEM domain–containing 3), are good candidates for involvement in osteopoikilosis. WIF1 is involved in Wnt signaling, and LEMD3 functions in BMP signaling, two pathways important in bone development13,14. Mutation analysis of WIF1 did not identify any abnormalities in the affected individuals. Sequencing of LEMD3 identified loss-of-function mutations in all affected individuals of the three families and in three unrelated individuals with osteopoikilosis (Table 1 and Fig. 4e). The splice-site mutation in individual G03-2881 caused skipping of exon 6, resulting in a frameshift and premature stop codon in exon 7 at position 2,021 (Fig. 4d). The deletion of one of the LEMD3 alleles in individual G03-1858 was corroborated by fluorescence in situ hybridization (FISH) analysis with locus-specific probes (Fig. 4b). Re-evaluation of the karyotype (550-band level) was suggestive of, but not conclusive for, the presence of a deletion in the 12q14–15 region (Fig. 4a).
Table 1 LEMD3 mutations
Some reports have suggested that the asymmetric distribution of skin lesions in BOS and the segmental involvement usually observed in melorheostosis result from a somatic mutation4,15. To investigate this possibility, we took skin biopsy samples from two affected individuals, one from an elastic-type nevus in individual III-3 (of family A) with BOS and a second from a hard sclerodermic-like lesion in individual III-2 (of family B) with melorheostosis. Sequence analysis of LEMD3 on genomic DNA extracted from both skin lesions showed no evidence for an additional somatic mutation in LEMD3 (or 'second hit'). Analysis of intragenic polymorphisms showed no loss of heterozygosity or allelic imbalance and therefore excluded the possible existence of a partial gene deletion as a somatic mutation. In cDNA from normal and affected skin of the individual with BOS, we found only the normal sequence, owing to nonsense-mediated decay of mRNA from the abnormal allele. Because bone specimens were not available, we could not investigate the possibility that a somatic mutation in osteoblasts could explain the spotty occurrence of bone lesions.
LEMD3 is an integral protein of the inner nuclear membrane16. It contains a nucleoplasmic N- and C-terminal domain and two transmembrane segments16. The N-terminal segment shares a conserved globular domain of ∼40 amino acids with other inner nuclear membrane proteins, such as lamina-associated polypeptide 2 (LAP2) and emerin16. The X. laevis ortholog of LEMD3 (XMAN1) antagonizes BMP signaling. This antagonizing activity of XMAN1 resides in the C-terminal region that binds to Smad1, Smad5 and Smad8 (ref. 5).
To investigate whether LEMD3 interacts with BMP receptor–activated and TGFβ receptor–activated Smads, we carried out a yeast two-hybrid analysis using the C-terminal domain of LEMD3 as prey. This analysis identified interactions between the C-terminal domain of LEMD3 and the MH2 domains of Smad1 (BMP-specific) and Smad2 (TGFβ-specific), suggesting that LEMD3 is involved in both BMP and TGFβ signaling (Fig. 5a).
Figure 5: Analysis of normal and mutant LEMD3 in BMP and TGFβ signaling.
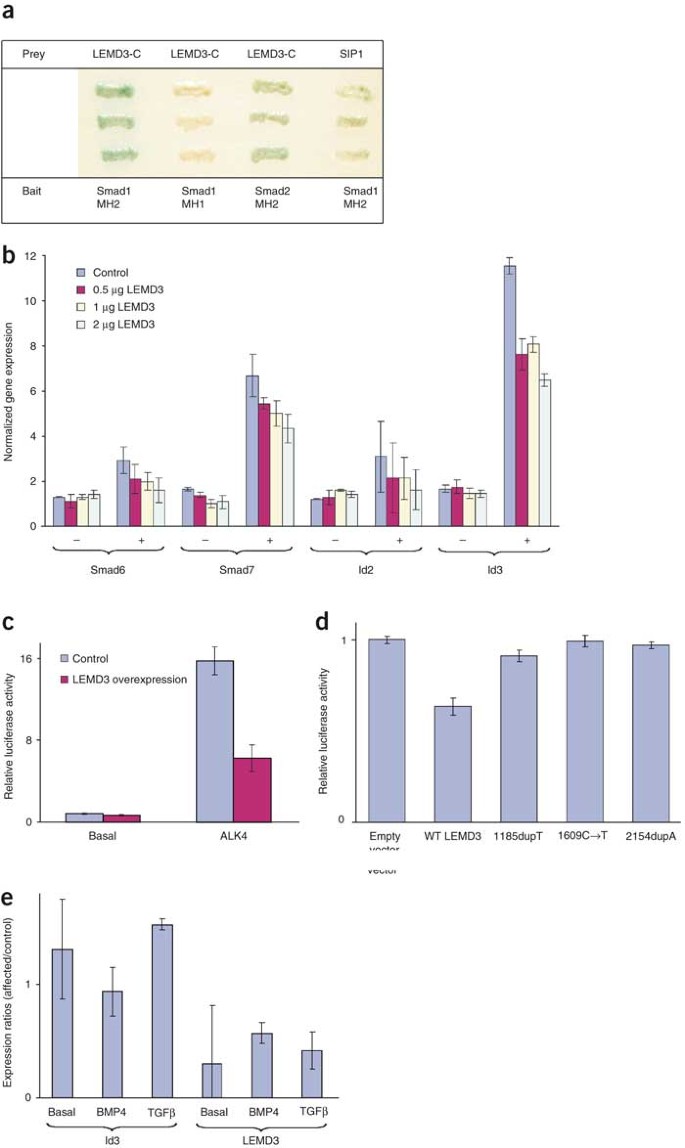
(a) Yeast two-hybrid analysis shows that the C-terminal part of LEMD3 (LEMD3-C) specifically interacts with the MH2 domain of Smad1 and Smad2. SIP1–Smad1 interaction is included as a positive control. Interaction was assessed by activation of the gene MEL1 encoding α-galactosidase, visualized by blue staining of the yeast. (b) Normalized gene expression for four known target genes in basal (−) and BMP4-stimulated (+) HEK293T cells. Overexpression of LEMD3 reduces the capacity of BMP4 to upregulate Smad6, Smad7, Id2 and Id3. (c) Relative luciferase activity from the 3TP-Lux reporter construct in HepG2 cells. Overexpression of LEMD3 reduces activation of the 3TP-Lux reporter gene in the presence of the constitutively active ALK4 receptor. (d) Relative luciferase activity from the (CAGA)12 reporter construct cotransfected with plasmids expressing different LEMD3 mutants in HEK293T cells. Reduction of TGFβ signaling is not observed for the different mutant LEMD3 constructs. (e) Expression ratios between fibroblasts from an affected individual and control fibroblasts for LEMD3 and Id3 as measured by Q-PCR. The ratios for Id3 in basal conditions and after BMP4 or TGFβ stimulation are shown on the left. Significantly (P < 0.05) higher expression of Id3 was observed after TGFβ stimulation. The ratios for LEMD3 are shown on the right and are indicative of haploinsufficiency in fibroblasts from the affected individual (P < 0.05).
We further investigated the role of LEMD3 in both signaling pathways by overexpressing the protein in two different cell lines. In HEK293T cells, we measured the expression of several known target genes in basal conditions and after BMP4 stimulation by quantitative PCR (Q-PCR). Overexpression of LEMD3 reduced the capacity of BMP4 to upregulate Smad6, Smad7, Id2 and Id3 (Fig. 5b). In HepG2 cells, we measured the response of TGFβ using the activin-TGFβ–responsive 3TP-Lux reporter in basal conditions and in the presence of a constitutively active receptor ALK4, which activates Smad2 and Smad3. Overexpression of LEMD3 reduced the ALK4-induced activation of the 3TP-Lux reporter (Fig. 5c). These results indicate that LEMD3 can antagonize both BMP and TGFβ signaling in human cells.
Finally, we investigated the effect of the LEMD3 mutations that we identified in affected individuals. We made three mutant LEMD3 constructs containing the 1185dupT, 1609C → T and 2154dupA mutations, respectively. We carried out a luciferase assay in HEK293T cells transfected with a TGFβ-responsive reporter and found that each mutant construct was unable to reduce TGFβ signaling, unlike the wild-type construct (Fig. 5d). The loss-of-function effect of these mutations most probably resulted from the absence of the Smad-interacting C-terminal domain in the truncated proteins, present on western blots. We therefore believe that the mutations found in this study are hypomorphic, either because of nonsense-mediated decay or because of the production of a truncated protein lacking the C-terminal domain. We measured by Q-PCR the expression of several target genes in skin fibroblasts from individual III-3 of family A with the 2154dupA mutation. This analysis confirmed that fibroblasts from this affected individual were haploinsufficient with respect to LEMD3. In addition, we found that expression of the gene Id3 after TGFβ stimulation was significantly higher in these fibroblasts than in controls (Fig. 5e). This is the first evidence to our knowledge that haploinsufficiency of LEMD3 in human fibroblasts results in enhanced TGFβ signaling with upregulation of target genes downstream in the pathway. We observed no significant differences between fibroblasts from the elastic-type nevus and normal skin of the affected individual in all conditions tested.
In conclusion, we found that loss-of-function mutations in LEMD3 can result in osteopoikilosis, BOS and melorheostosis. The failure to detect a somatic mutation in LEMD3 in affected skin fibroblasts from an individual with BOS and one with melorheostosis may suggest that other genetic factors contribute to the presence and distribution of skin and bone lesions in these disorders. In addition, we found evidence that LEMD3 can antagonize both the BMP and TGFβ signaling pathway. These results are in accordance with previous studies showing that genetic defects in both signaling pathways can result in hyperostotic bone disorders. Loss-of-function mutations in SOST, encoding the extracellular BMP antagonist sclerostin, lead to sclerosteosis17,18 (OMIM 269500), whereas activating missense mutations in TGFB1 can result in Camurati-Engelmann disease19,20 (OMIM 131300). Finally, increased signaling in the TGFβ pathway has been observed in other fibrotic skin disorders, such as scleroderma, and may therefore explain the skin lesions in individuals with BOS and melorheostosis21,22.
Note: Supplementary information is available on the Nature Genetics website.
Methods
Material from affected individuals.
We obtained appropriate informed consent from all subjects involved in the study. Family A is a three-generation family of Belgian origin. All affected individuals had osteopoikilosis. Two affected individuals (II-5 and III-3) also had skin manifestations of BOS. In individual III-3, skin biopsy samples were taken from a connective-tissue nevus on the left thigh and from normal skin on the right thigh. Light microscopy showed more numerous elastin fibers in the middle and deep dermis on the affected side. These fibers had a slightly granular and thickened appearance (Fig. 1b). Ultrastructurally, the elastin fibers were much thicker and more numerous in the affected skin than in the normal skin. They had a well-developed amorphous matrix with delicate peripheral microfibrils. Results of light microscopy and electron microscopy analysis were consistent with the diagnosis of connective-tissue nevus of the elastic-tissue type. Fibroblasts from both biopsies were cultured for molecular analysis.
Family B is another three-generation family of Belgian origin, previously reported2, illustrating the co-occurrence of osteopoikilosis and melorheostosis in one family. Family C is a three-generation British family with autosomal dominant osteopoikilosis and skin lesions reminiscent of BOS. Individual G03-1885 is an Australian with BOS. Individuals G03-2881 and G03-2882 have osteopoikilosis and are both of Belgian origin but are unrelated. Individual G03-1858 had a history of prenatal and postnatal growth retardation and learning disability that was diagnosed as Russell-Silver syndrome in early childhood. Investigations for hypertension in infancy revealed ectopic kidneys and aberrant renal arteries. Evaluation at the age of 16 years showed proportionate short stature with height of 131.5 cm (50th centile for a 9-y-old girl), weight of 31.8 kg (50th centile for a 10-y-old girl) and head circumference of 49 cm (50th centile for a 3-y-old girl). Physical examination revealed a subtly dysmorphic face with synophrys, mild hypertelorism, broad and high nasal bridge, micrognathia and maxillary overbite. In addition, a diffuse hyperpigmentation spot was noted on the left thigh. Osteopoikilosis lesions were found on skeletal survey.
Linkage analysis.
We used all autosomal markers from the Applied Biosystems Linkage mapping set version 2 for a genome-wide linkage analysis. We used an improved protocol from the Centre National de Génotypage for pooling an average of four markers per PCR to carry out all reactions. Additional markers were taken from the Marshfield map or designed based on the simple-tandem-repeat finder in the University of California Santa Cruz genome browser. We carried out genotyping on an Applied Biosystems Prism 3100 Genetic Analyzer running Genemapper v2.0 software. We used the MLINK program of the LINKAGE software package23 to calculate two-point lod scores between the disease phenotype and each of the markers, assuming a dominant mode of inheritance with a penetrance of 0.95 and a disease allele frequency of 0.0001. The frequency of most marker alleles was set to 1/8. For markers with more than eight alleles in the pedigrees, the allele frequency was set to 1/n, where n is the number of alleles.
Cytogenetic and FISH analysis.
We carried out karyotyping in accordance with standard procedures. We analyzed metaphases at the 550-band level. We carried out FISH analysis as described24 using BAC clone RP11-30506 from the 12q14.3 region.
GeneChip Mapping 10K Array analysis.
We analyzed DNA from individual G03-1858 using the GeneChip Mapping 10K Array (Affymetrix) as described25. We analyzed data with both the Affymetrix GCOS and GDAS software and the Affymetrix GeneChip Chromosome Copy Number Tool. We used copy number estimation (meta P value) to identify the microdeletion, the boundaries of which were defined by loss of heterozygosity. Results were visualized with arrayCGHbase (B. Menten et al., unpublished data).
Sequence analysis.
We amplified all exons by PCR using intronic primers and additional exonic primers for larger exons (primer sequences are available on request). We used a touchdown PCR program with an annealing temperature decreasing from 60 °C to 48 °C over 12 cycles, followed by 20 cycles with an annealing temperature of 48 °C. We sequenced PCR products using the BigDye v3.1 ET terminator cycle sequencing kit from Applied Biosystems. Sequencing reactions were loaded onto an Applied Biosystems Prism 3100 Genetic Analyzer and analyzed with Sequencing Analysis v3.7 and SeqScape v1.1 software (Applied Biosystems).
Yeast two-hybrid analysis.
We cloned the C-terminal domain of human LEMD3, encoding amino acids 520–853, as a _Pvu_II-_Xho_I fragment into the _Sma_I and _Xho_I restriction sites of the prey vector pAct2 (Clontech). We cotransformed yeast strain AH109 with this vector or a positive control (SIP1) prey vector with different Smad bait vectors26, using the Yeastmaker yeast transformation system 2 (Clontech). We assayed activation of the gene MEL1 by interacting hybrid proteins in yeast on Trp-Leu drop-out medium containing the chromogenic substrate X-α-Gal (Clontech).
Luciferase assay.
We carried out transfections of HepG2 cells in triplicate in 24-well plates using Fugene (Roche). Each well was transfected with a total amount of 500 ng of plasmid DNA, including 50 ng of 3TP-Lux reporter, 50 ng of RSV-promoter-based lacZ reporter and combinations of 50 ng of the constitutively active ALK4 receptor with 100 ng of LEMD3 or empty expression vector. We maintained a constant amount of DNA by adding pBluescript vector. Forty-eight hours after transfection, we assayed cell extracts for luciferase and β-galactosidase activities in accordance with the manufacturers' protocols (Promega and Clontech, respectively). We normalized the data by calculating the ratio of luciferase activity to β-galactosidase activity.
Q-PCR.
We isolated RNA using RNeasy Mini Kit (Qiagen) and synthesized cDNA using SuperScript II Reverse Transcriptase Kit with random hexamer primers (Invitrogen) in a total volume of 20 μl. We used 5 μl of cDNA (1:10 dilution) in combination with the Q-PCR Core Kit for SYBR Green I (Eurogentec) and 250 nM gene-specific primers to carry out Q-PCR on a GeneAmp 5700 Sequence Detector (Applied Biosystems). The Q-PCR program consists of 40 cycles with 15 s at 95 °C and 1 min at 60 °C, followed by a dissociation run to determine melting curves. We carried out all reactions in duplicate and normalized them to the geometric mean of three stable reference genes (GAPD, HPRT1 and YWHAZ)27.
To investigate the effect of LEMD3 on Smad signaling, we seeded HEK293T cells in 9-cm dishes and transfected them with a total of 2 μg of LEMD3 plasmid DNA (provided by H. Worman; Department of Medicine, College of Physicians and Surgeons, Columbia University, New York, New York, USA) using Fugene (Roche). Twenty-four hours after transfection, cells were serum-starved for 4 h and then stimulated with 5 ng ml−1 of human recombinant BMP4 (R&D Systems) for 1 h followed by RNA extraction.
In the screening for differentially expressed genes, we used fibroblasts from a control individual and from normal and affected skin of affected individual III-3 (of family A). We grew the cells in 13.5-cm dishes to 80% confluency and then serum-starved them for 2 h. We collected cells after 1 h of treatment with 1 ng ml−1 human recombinant TGFβ1 (R&D Systems) or with 5 ng ml−1 of BMP4. Expression levels for all genes were determined in four independent experiments. Differential gene expression was considered significant when the difference was at least 50% and the 95% confidence interval of the mean expression ratio did not include 1 (equivalent with P < 0.05).
We analyzed the following genes (their ID numbers in the RTPrimerDB database28 are given in parentheses): GAPD (3), HPRT1 (5), YWHAZ (9), MMP2 (113), CTGF (596), COL1A1 (1089), COL3A1 (1090), COL5A1 (1091), FN1 (1092), ELN (1093), ID1 (1094), ID2 (1095), ID3 (1096), ID4 (1097), LEMD3 (1098), RUNX2 (1099), SMAD6 (1100), SMAD7 (1101) and SERPINE1 (1102).
Analysis of mutant LEMD3 constructs.
We modified the wild-type LEMD3 construct (provided by H. Worman; Department of Medicine, College of Physicians and Surgeons, Columbia University, New York, New York, USA) to contain the 1185dupT, 1609C → T or 2154dupA mutation using the QuickChange Site-Directed Mutagenesis kit (Stratagene) in accordance with the manufacturer's instructions. We plated HEK293T cells at a density of 4 × 105 cells per well in Dulbecco's modified Eagle medium plus (Invitrogen) and transferred them to Opti-MEM I (Invitrogen) after overnight adherence. We transiently transfected cells with 2 μg of each plasmid and 0.5 μg of the (CAGA)12 TGFβ-responsive reporter construct29 in duplicate using Lipofectamine (Invitrogen). We also transfected cells with 20 ng of pRL-TK (Promega) to correct for transfection efficiency. Cells were serum-starved for 7 h before stimulation with 7 ng ml−1 recombinant human TGFβ (R&D systems). We lysed cells 24 h after transfection. We quantified activities of firefly and Renilla luciferase using the Dual-Luciferase Reporter Assay System (Promega).
URLs.
Improved linkage analysis protocols from the Centre National de Génotypage are available at http://www.cng.fr/. The arrayCGHbase visualization tool and RTPrimerDB database are available at http://medgen.ugent.be/arraycghbase/ and http://medgen.ugent.be/rtprimerdb/, respectively.
References
- Hall, C.M. International nosology and classification of constitutional disorders of bone (2001). Am. J. Med. Genet. 113, 65–77 (2002).
Article PubMed Google Scholar - Debeer, P., Pykels, E., Lammens, J., Devriendt, K. & Fryns, J.P. Melorheostosis in a family with autosomal dominant osteopoikilosis: Report of a third family. Am. J. Med. Genet. 119A, 188–193 (2003).
Article PubMed Google Scholar - Butkus, C.E., Michels, V.V., Lindor, N.M. & Cooney, W.P. 3rd Melorheostosis in a patient with familial osteopoikilosis. Am. J. Med. Genet. 72, 43–46 (1997).
Article CAS PubMed Google Scholar - Nevin, N.C., Thomas, P.S., Davis, R.I. & Cowie, G.H. Melorheostosis in a family with autosomal dominant osteopoikilosis. Am. J. Med. Genet. 82, 409–414 (1999).
Article CAS PubMed Google Scholar - Osada, S., Ohmori, S.Y. & Taira, M. XMAN1, an inner nuclear membrane protein, antagonizes BMP signaling by interacting with Smad1 in Xenopus embryos. Development 130, 1783–1794 (2003).
Article CAS PubMed Google Scholar - Melnick, J.C. Osteopathia condensans disseminata (osteopoikilosis): study of a family of 4 generations. Am. J. Roentgenol. 82, 229–238 (1959).
CAS Google Scholar - Lagier, R., Mbakop, A. & Bigler, A. Osteopoikilosis: a radiological and pathological study. Skeletal Radiol. 11, 161–168 (1984).
Article CAS PubMed Google Scholar - Ehrig, T. & Cockerell, C.J. Buschke-Ollendorff syndrome: report of a case and interpretation of the clinical phenotype as a type 2 segmental manifestation of an autosomal dominant skin disease. J. Am. Acad. Dermatol. 49, 1163–1166 (2003).
Article PubMed Google Scholar - Berlin, R., Hedensio, B., Lilja, B. & Linder, L. Osteopoikilosis—a clinical and genetic study. Acta. Med. Scand. 181, 305–314 (1967).
Article CAS PubMed Google Scholar - Campbell, C.J., Papademetriou, T. & Bonfiglio, M. Melorheostosis. A report of the clinical, roentgenographic, and pathological findings in fourteen cases. J. Bone Joint Surg. Am. 50, 1281–1304 (1968).
Article CAS PubMed Google Scholar - Rozencwaig, R., Wilson, M.R. & McFarland, G.B. Jr. Melorheostosis. Am. J. Orthop. 26, 83–89 (1997).
CAS PubMed Google Scholar - Freyschmidt, J. Melorheostosis: a review of 23 cases. Eur. Radiol. 11, 474–479 (2001).
Article CAS PubMed Google Scholar - Gong, Y. et al. LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell 107, 513–523 (2001).
Article CAS PubMed Google Scholar - Canalis, E., Economides, A.N. & Gazzerro, E. Bone morphogenetic proteins, their antagonists, and the skeleton. Endocr. Rev. 24, 218–235 (2003).
Article CAS PubMed Google Scholar - Happle, R. Melorheostosis may originate as a type 2 segmental manifestation of osteopoikilosis. Am. J. Med. Genet. 125A, 221–223 (2004).
Article PubMed Google Scholar - Lin, F. et al. MAN1, an inner nuclear membrane protein that shares the LEM domain with lamina-associated polypeptide 2 and emerin. J. Biol. Chem. 275, 4840–4847 (2000).
Article CAS PubMed Google Scholar - Balemans, W. et al. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST). Hum. Mol. Genet. 10, 537–543 (2001).
Article CAS PubMed Google Scholar - Brunkow, M.E. et al. Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am. J. Hum. Genet. 68, 577–589 (2001).
Article CAS PubMed PubMed Central Google Scholar - Janssens, K. et al. Mutations in the gene encoding the latency-associated peptide of TGF-β 1 cause Camurati-Engelmann disease. Nat. Genet. 26, 273–275 (2000).
Article CAS PubMed Google Scholar - Kinoshita, A. et al. Domain-specific mutations in TGF-β1 result in Camurati-Engelmann disease. Nat. Genet. 26, 19–20 (2000).
Article CAS PubMed Google Scholar - Mori, Y., Chen, S.J. & Varga, J. Expression and regulation of intracellular SMAD signaling in scleroderma skin fibroblasts. Arthritis Rheum. 48, 1964–1978 (2003).
Article CAS PubMed Google Scholar - Asano, Y., Ihn, H., Yamane, K., Kubo, M. & Tamaki, K. Impaired Smad7–Smurf-mediated negative regulation of TGF-β signaling in scleroderma fibroblasts. J. Clin. Invest. 113, 253–264 (2004).
Article CAS PubMed PubMed Central Google Scholar - Lathrop, G.M. & Lalouel, J.M. Easy calculations of lod scores and genetic risks on small computers. Am. J. Hum. Genet. 36, 460–465 (1984).
CAS PubMed PubMed Central Google Scholar - Van Roy, N. et al. 1;17 translocations and other chromosome 17 rearrangements in human primary neuroblastoma tumors and cell lines. Genes Chromosomes Cancer 10, 103–114 (1994).
Article CAS PubMed Google Scholar - Matsuzaki, H. et al. Parallel genotyping of over 10,000 SNPs using a one-primer assay on a high-density oligonucleotide array. Genome Res. 14, 414–425 (2004).
Article CAS PubMed PubMed Central Google Scholar - Verschueren, K. et al. SIP1, a novel zinc finger/homeodomain repressor, interacts with Smad proteins and binds to 5′-CACCT sequences in candidate target genes. J. Biol. Chem. 274, 20489–20498 (1999).
Article CAS PubMed Google Scholar - Vandesompele, J. et al. Accurate normalization of real-time quantitative RT–PCR data by geometric averaging of multiple internal control genes. Genome Biol. 3 RESEARCH0034 (2002).
- Pattyn, F., Speleman, F., De Paepe, A. & Vandesompele, J. RTPrimerDB: the real-time PCR primer and probe database. Nucleic Acids Res. 31, 122–123 (2003).
Article CAS PubMed PubMed Central Google Scholar - Dennler, S. et al. Direct binding of Smad3 and Smad4 to critical TGF β-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J. 17, 3091–3100 (1998).
Article CAS PubMed PubMed Central Google Scholar
Acknowledgements
We thank the affected individuals and families for their interest and cooperation, M. Godfrey for critical review of the manuscript, P. Tylzanowski for suggestions and plasmid stocks and H. Worman for the expression vector encoding human LEMD3. This study was supported, in part, by the Fund for Scientific Research, Flanders, with a mandate fundamental clinical research to P.D. and G.R.M.; a research assistantship to P.C.M.V.; and research projects to F.S., S.J.T.V., W.V.H. and G.R.M. This study was also supported by an Interuniversity Attraction Pole grant to W.V.H. and by the Fifth Framework of the specific research and technological development program “Quality of Life and Management of Living Resources” of the European Commission to J.H., A.D.P. and G.R.M. J.H. is funded by, and J.V. is a postdoctoral researcher with, the Institute for the Promotion of Innovation by Science and Technology in Flanders. N.V.R. and K.J. are postdoctoral researchers of the Fund for Scientific Research, Flanders. The research at Flanders Interuniversity Institute for Biotechnology was supported by the Fund for Scientific Research, Flanders and the University of Leuven. O.P. is holder of a DWTC (Federal Services for Scientific, Technical and Cultural affairs) postdoctoral fellowship and is supported by the Interuniversity Attraction Pole Network. This text presents research results of the Belgian program of Interuniversity Poles of attraction initiated by the Belgian State, Prime Minister's Office, Science Policy Programming. The scientific responsibility is assumed by the authors.
Author information
Authors and Affiliations
- Center for Medical Genetics, Ghent University Hospital, Ghent, Belgium
Jan Hellemans, Andy Willaert, Bjorn Menten, Nadine Van Roy, Stefan J T Vermeulen, Anne De Paepe, Jo Vandesompele, Frank Speleman, Paul J Coucke & Geert R Mortier - Department of Developmental Biology, Flanders Interuniversity Institute for Biotechnology and Laboratory of Molecular Biology, University of Leuven, Leuven, Belgium
Olena Preobrazhenska, Danny Huylebroeck & Kristin Verschueren - Center for Human Genetics, University of Leuven, Leuven, Belgium
Philippe Debeer - Department of Orthopedic Surgery, Ghent University Hospital, Ghent, Belgium
Peter C M Verdonk - Medical Genetics Service, Sainte-Justine Hospital and University of Montréal, Montréal, Canada
Teresa Costa - Department of Medical Genetics, University Hospital and University of Antwerp, Belgium
Katrien Janssens & Wim Van Hul - Genetic Health Services Victoria, Murdoch Childrens Research Institute, and University of Melbourne, Australia
Ravi Savarirayan - Department of Radiology, University Hospital and University of Antwerp, Belgium
Filip Vanhoenacker - Department of Dermatology, Ghent University Hospital, Ghent, Belgium
Jean-Marie Naeyaert
Authors
- Jan Hellemans
You can also search for this author inPubMed Google Scholar - Olena Preobrazhenska
You can also search for this author inPubMed Google Scholar - Andy Willaert
You can also search for this author inPubMed Google Scholar - Philippe Debeer
You can also search for this author inPubMed Google Scholar - Peter C M Verdonk
You can also search for this author inPubMed Google Scholar - Teresa Costa
You can also search for this author inPubMed Google Scholar - Katrien Janssens
You can also search for this author inPubMed Google Scholar - Bjorn Menten
You can also search for this author inPubMed Google Scholar - Nadine Van Roy
You can also search for this author inPubMed Google Scholar - Stefan J T Vermeulen
You can also search for this author inPubMed Google Scholar - Ravi Savarirayan
You can also search for this author inPubMed Google Scholar - Wim Van Hul
You can also search for this author inPubMed Google Scholar - Filip Vanhoenacker
You can also search for this author inPubMed Google Scholar - Danny Huylebroeck
You can also search for this author inPubMed Google Scholar - Anne De Paepe
You can also search for this author inPubMed Google Scholar - Jean-Marie Naeyaert
You can also search for this author inPubMed Google Scholar - Jo Vandesompele
You can also search for this author inPubMed Google Scholar - Frank Speleman
You can also search for this author inPubMed Google Scholar - Kristin Verschueren
You can also search for this author inPubMed Google Scholar - Paul J Coucke
You can also search for this author inPubMed Google Scholar - Geert R Mortier
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toGeert R Mortier.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Fig. 1
Results from the GeneChip Mapping 10K Array analysis for patient G03-1858. (PDF 3 kb)
Supplementary Table 1
Two-point LOD scores for linkage of the osteopoikilosis locus to chromosome 12q12-q14.3. (PDF 82 kb)
Rights and permissions
About this article
Cite this article
Hellemans, J., Preobrazhenska, O., Willaert, A. et al. Loss-of-function mutations in LEMD3 result in osteopoikilosis, Buschke-Ollendorff syndrome and melorheostosis.Nat Genet 36, 1213–1218 (2004). https://doi.org/10.1038/ng1453
- Received: 22 July 2004
- Accepted: 17 September 2004
- Published: 17 October 2004
- Issue Date: 01 November 2004
- DOI: https://doi.org/10.1038/ng1453
This article is cited by
Osteopoikilosis: a case report
- Behzad Mohsenpour
- Amjad Ahmadi
Journal of Medical Case Reports (2023)
A primer on skeletal dysplasias
- Atsuhiko Handa
- Gen Nishimura
- Georges Y. El-Khoury
Japanese Journal of Radiology (2022)
Atypical presentation of melorheostosis with soft tissues involvement: a case report
- Kok King Chia
- Juhara Haron
- Nik Fatimah Salwati Nik Malek
Egyptian Journal of Radiology and Nuclear Medicine (2021)
Deficiency of TMEM53 causes a previously unknown sclerosing bone disorder by dysregulation of BMP-SMAD signaling
- Long Guo
- Aritoshi Iida
- Shiro Ikegawa
Nature Communications (2021)
A 65-year-old Patient with Osteopoikilosis and Bilateral Pleural Effusion. Case Report
- Maria L. Voulgari
- Herbert Kellner
SN Comprehensive Clinical Medicine (2020)