A Toll-like receptor–independent antiviral response induced by double-stranded B-form DNA (original) (raw)
Main
Although host nucleic acids are normally sequestered from potential signaling receptors, accumulating evidence over the past few decades has suggested that both DNA and RNA can be recognized by and modulate innate immune system1,2. Extensive studies have demonstrated that some Toll-like receptors (TLRs) recognize immunostimulatory nucleic acids during infection or tissue damage, for example. Those studies have shown that the TLRs have distinct specificities for pathogen-derived nucleic acids, with double-stranded RNA (dsRNA) being the ligand for TLR3 and single-stranded RNA (ssRNA) and CpG DNA being the ligands for TLR7 and TLR8 and for TLR9, respectively3. TLR recognition of pathogen-derived nucleic acids is important in both innate and adaptive immune responses to infectious organisms, including bacteria, viruses and parasites. Other less-convincing data have suggested that TLRs may also be important in autoimmune diseases4,5.
Genomic DNA derived from pathogens, such as bacteria or viruses, and certain CpG oligodeoxynucleotides (ODNs) stimulate plasmacytoid dendritic cells (DCs) via TLR9 to produce large amounts of interferon-α (IFN-α)6. TLR9-mediated signaling required for CpG DNA recognition by plasmacytoid DCs requires a specific pathway involving the adaptor molecule MyD88, kinases IRAK1 and IRAK4, adaptor molecule TRAF6 and transcription factor IRF7 (refs. 7–9). Of note, the noncanonical IκB kinase (IKK) family member TBK1 is not required for CpG DNA–induced IFN-α production by plasmacytoid DCs7. In contrast, dsRNA-induced type I interferon production is mediated by TBK1 and partially by IKKi, a second noncanonical IKK family member10,11,12,13.
A growing body of evidence, however, has indicated that DNA, including host DNA, can be recognized independently of TLR9. DNA-immunoglobulin complexes generated during systemic autoimmune diseases, such as systemic lupus erythematosus, activate plasmacytoid DCs to produce type I interferon14. Whereas TLR9 is important for this, Fc γ-receptor IIa is necessary for optimal activation by DNA-immunoglobulin complexes and, notably, TLR9-independent activation is still present15. Mammalian nucleosomes also activate DCs in a MyD88-independent way16.
DNase II, a chief DNase present in the phagosome of macrophages, is important in clearing DNA from phagocytosed apoptotic cells or debris17. Without clearance of the DNA in phagosomes, macrophages begin to produce IFN-β, which can lead to subsequent TLR9-independent inflammatory events18. In addition, dsDNA but not ssDNA derived from either pathogens or the host activates both immune cells (macrophages and DCs) and nonimmune cells (thyroid cells, fibroblasts and muscle cells) when introduced into the cytoplasm by transfection19,20. Also, dsDNA activates a set of genes, including those encoding major histocompatibility complex, costimulatory molecules and immunoproteasome subunits such as TAP1 and LMP2, as well as the transcription factors STAT1 and IRF and protein kinase R19,20. Evidence suggests that induction of interferon-inducible genes and upregulation of costimulatory molecules by dsDNA are mediated in part by a TLR9-independent pathway21,22, indicating a possible DNA-induced, TLR-independent innate immune activation pathway23. The molecular mechanisms involved in such a innate immune pathway, however, have not been elucidated.
Given the findings described above, we have explored here the immunostimulatory element of dsDNA, its intracellular signaling pathway(s), the target genes involved and the possible physiological function of such a pathway in mouse and human stromal cells. We found that double-stranded B-form DNA (B-DNA) but not Z-form DNA (Z-DNA) stimulated mouse and human stromal and DCs, resulting in the production of type I interferon and chemokines. B-DNA activated IRF3 and the Ifnb promoter via both TBK1 and IKKi, whereas it activated transcription factor NF-κB independently of both TBK1 and IKKi. Both of these pathways, moreover, required the adaptor molecule IPS-1 but not TLRs or the helicase RIG-I. In addition, B-DNA activation of fibroblasts conferred resistance to viral infection, suggesting that B-DNA-induced interferon genes might be important in antiviral innate immune responses. Finally, B-DNA activation of the signaling pathways may be involved in the pathogenesis of nucleic acid–dependent autoimmune diseases.
Results
B-DNA induces type I interferon
The dsDNA but not ssDNA derived from either host or pathogen genomes, as well as double-stranded synthetic polynucleotides, stimulate nonimmune cells (such as thyroid cells) and immune cells (such as DCs and macrophages) to express interferon-inducible genes19,20. Those results prompted us to investigate the molecular mechanisms of cell activation triggered by dsDNA. We first examined the effects of dsDNA on mouse embryonic fibroblasts (MEFs) because they do not respond to CpG DNA, a potent TLR9 agonist (data not shown); we confirmed this by adding double-stranded CpG DNA to MEF cell culture media, which did not activate the cells (data not shown). However, when we transfected MEFs with genomic DNA derived from mammals (calf thymus or mouse liver), bacteria (Escherichia coli) or viruses (herpes simplex virus type 1 or 2 or human cytomegalovirus), expression of Ifnb, Cxcl10 (IP-10) and Ccl2 (JE or MCP-1) mRNA was increased considerably in a dose-dependent way within 4 h of stimulation (Fig. 1a,b and data not shown). The comparable ability of the different genomic DNAs to induce the indicated mRNA suggested that specific sequences or nucleotide modifications were not required; we confirmed that conclusion by showing that methylation of calf thymus DNA did not alter the stimulatory effects of the dsDNA (Fig. 1c). These data confirmed published results showing that double-stranded genomic DNA can activate stromal cells19,20 such as MEFs to upregulate type I interferons and chemokines independently of species-specific sequences or DNA modifications.
Figure 1: Activation of MEFs by dsDNA to produce type I interferons and chemokines.
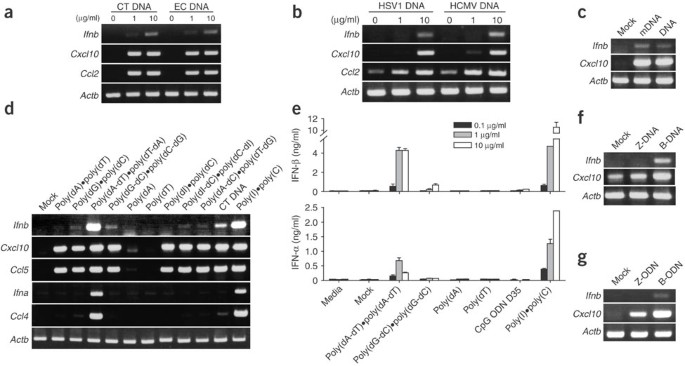
(a–d) Expression of mRNA in MEFs (C57BL/6) stimulated (or transfected) with genomic or synthetic DNA of various origins or with various sequences. Ifnb, Cxcl10, Ccl5, Ccl2, Ifna, Ccl4 and control Actb (β-actin) mRNA expression was assessed by RT-PCR 4 h after stimulation with mammalian DNA (calf thymus (CT DNA)) or bacterial DNA (E. coli (EC DNA); a), herpes simplex virus 1 (HSV DNA) or human cytomegalovirus (HCMV DNA) genomic DNA (b), mock transfection (Mock) or intact (DNA) or methylated (mDNA) calf thymus DNA (c), or synthetic DNA (10 μg/ml) with various sequences and strands, calf thymus DNA and dsRNA (poly(I)•poly(C); d). (e) IFN-α and IFN-β in the culture supernatants of MEFs stimulated with various types of DNA, CpG ODN or poly(I)•poly(C), measured in triplicate by ELISA. (f,g) RT-PCR of expression of Ifnb, Cxcl10 and control Actb mRNA 4 h after stimulation of MEFs with B-form or Z-form dsDNA (poly(dG-dC)•poly(dC-dG); f) or double-stranded ODN ((dG-dC)6; g) with either a B-form or Z-form helical structure. Data are representative of three experiments with similar results.
Next we tested synthetic dsDNAs comprising various sequences, including poly(dA)•poly(dT), poly(dG)•poly(dC), poly(dA-dT)•poly(dT-dA), poly(dG-dC)•poly(dC-dG), poly(dI)•poly(dC), poly(dI-dC)•poly(dC-dI) and poly(dA-dC)•poly(dT-dG). All of these synthetic dsDNAs stimulated MEFs to produce Cxcl10 and Ccl5 (encoding RANTES) mRNA, whereas ssDNA poly(dA), poly(dT) and poly(dC) failed to upregulate mRNA expression (Fig. 1d and data not shown). Of the dsDNA tested, poly(dA-dT)•poly(dT-dA) was the most potent inducer of Ifna, Ifnb and Ccl4 (encoding MIP1β), and this induction was comparable to the induction of each mRNA by poly(I)•poly(C) (Fig. 1d). We also confirmed induced protein expression in the culture supernatants 24 h after stimulation (Fig. 1e). We included as a control the potent A/D-type CpG ODN D35, which stimulates TLR9-expressing plasmacytoid DCs to produce considerable IFN-α; it was not active in the MEFs (Fig. 1e).
The synthetic dsDNAs had different abilities to upregulate Ifna, Ifnb and Ccl4 mRNA expression (Fig. 1d,e), whereas they all induced Cxcl10 and Ccl5 mRNA equally (Fig. 1d). Notably, this sequence dependence for type I interferon induction was inversely correlated to the susceptibility of the polynucleotides to form the alternative Z-DNA conformation, an unusual left-handed helical structure of dsDNA24. Although the precise physiological mechanisms remain uncertain, dsDNA tends to form B-DNA, not Z-DNA, when in complex with cationic liposome25. We confirmed by circular dichroism that the transfection reagents used did not induce Z-formation of DNA (data not shown). As an additional control, we induced Z-DNA conformation in one of the synthetic DNAs. Poly(dG-dC)•poly(dC-dG) is more susceptible than poly(dA-dT)•poly(dT-dA) to Z-DNA formation in certain conditions such as bromination or high-salt concentration24. Therefore, we modified poly(dG-dC)•poly(dC-dG) to form Z-DNA by bromination as described26. MEFs transfected with the B-DNA form of poly(dG-dC)•poly(dC-dG) upregulated Ifnb and Cxcl10, whereas the Z-DNA form of poly(dG-dC)•poly(dC-dG) produced only modest Cxcl10 mRNA expression (Fig. 1f). We obtained similar results with B-form and Z-form ODNs. Consistent with that, B-form ODN upregulated Ifnb and Cxcl10 mRNA, whereas Z-form ODN27 was much less stimulatory (Fig. 1g). These data suggested that the right-handed helical B-form but not the left-handed Z-form was responsible for optimal stimulatory activity of dsDNA.
B-DNA induces interferon independently of TLRs and RIG-I
We next determined whether the B-DNA-dependent induction of type I interferon and interferon-inducible genes is mediated by TLR or RIG-I28. To address this issue, we prepared MEFs from mice deficient in both MyD88 and TRIF, which lack all known TLR-mediated activation signaling pathways and are refractory to stimulation of TLR3, TLR7 and TLR9 by nucleic acids29. When transfected with B-DNA, both wild-type MEFs and MEFs deficient in both MyD88 and TRIF upregulated expression of Ifnb, Cxcl10, Ccl5 and Ccl2 mRNA (Fig. 2a). These data demonstrated that B-DNA induction of type I interferon and chemokines was independent of TLR signaling. RIG-I, a DExD/H box RNA helicase that contains two caspase-recruitment domains, recognizes dsRNA in the cytoplasm in a TLR3-independent way28,30. RIG-I-deficient and wild-type MEFs responded similarly to B-DNA by upregulating Ifnb, Cxcl10 and Ccl2 mRNA expression (Fig. 2b). These data suggested that B-DNA-induced upregulation of type I interferon and chemokines was not mediated by TLRs or RIG-I and that previously unknown molecules might be involved in the innate immune recognition of B-DNA.
Figure 2: B-DNA activation requires TBK1 and, in part, IKKi, but not TLRs or RIG-I.
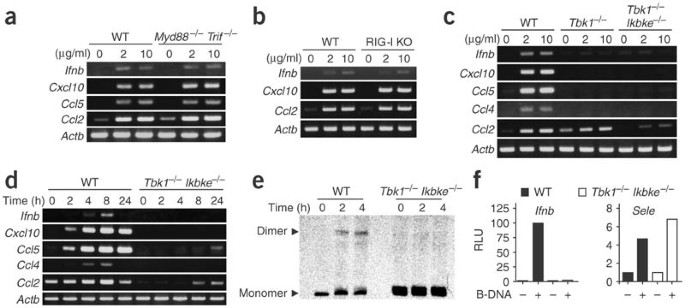
(a–d) RT-PCR of mRNA in MEFs from Myd88 −/− Trif −/− mice (a), RIG-I-deficient (RIG-I KO) mice (b) or Tbk1 −/− or Tbk1 −/− Ikbke −/− mice (c,d) or wild-type litter (WT) littermates. Cells were stimulated with dsDNA (poly(dA-dT)•poly(dT-dA)) at a concentration of 2 or 10 μg/ml for 4 h (a–c) or at a concentration of 10 μg/ml for various times (d), and expression of Ifnb, Cxcl10, Ccl5, Ccl2, Ifna and Ccl4 mRNA was assessed by RT-PCR. Data are representative of three to five independent experiments with similar results. (e) IRF3 activation in Tbk1 −/− Ikbke −/− or wild-type MEFs stimulated with 10 μg/ml of poly(dA-dT)•poly(dT-dA) (times, above lanes), assessed by native PAGE. (f) Promoter activity of Ifnb and Sele in response to poly(dA-dT)•poly(dT-dA) in wild-type or Tbk1 −/− Ikbke −/− MEFs, measured by luciferase assay and presented as relative luciferase units (RLU). Data are representative of three independent experiments with similar results.
Induction of interferon requires TBK1 and, partially, IKKi
Reports have suggested that the IKK-related serine-threonine kinases TBK1 and IKKi are essential for type I interferon induction by dsRNA generated during viral replication10,11,12. Those reports prompted us to investigate the involvement of TBK1 and IKKi in B-DNA-dependent type I interferon induction. B-DNA induced mRNA expression of Ifnb and interferon-inducible chemokines, including Cxcl10, Ccl5 and Ccl4, in wild-type MEFs but not in MEFs deficient in TBK1 (Tbk1 −/−) or deficient in both TBK1 and IKKi (Tbk1 −/− Ikbke −/−; Fig. 2c). In contrast, B-DNA induction of Ccl2 mRNA, although present in Tbk1 −/− MEFs, was much lower in Tbk1 −/− Ikbke −/− MEFs. Stimulation with maximal DNA concentration for 24 h produced similar results, except that Ccl5 and Ccl2 mRNA were slightly induced in Tbk1 −/− Ikbke −/− MEFs (Fig. 2d). These data suggested that B-DNA-stimulated MEF production of Ifnb, Cxcl10 and Ccl4 was totally dependent on TBK1, whereas production of Ccl5 and Ccl2 was only partially dependent on TBK1 and IKKi.
Pathways dependent on and independent of TBK1 and IKKi
To further clarify the signaling pathways involved in B-DNA-dependent induction of type I interferon, we assessed whether B-DNA activated IRF3. There was dimerization of IRF3 2–4 h after B-DNA stimulation in wild-type MEFs but not Tbk1 −/− Ikbke −/− MEFs (Fig. 2e). We also assessed activation of the promoters of Ifnb and the gene encoding endothelial cell–leukocyte adhesion molecule 1 (Sele). We transfected wild-type and Tbk1 −/− Ikbke −/− MEFs with a plasmid expressing luciferase under control of the Ifnb or Sele promoter and then stimulated the cells with B-DNA. B-DNA stimulated the Ifnb promoter 100-fold in wild-type MEFs but had no effect on Tbk1 −/− Ikbke −/− MEFs (Fig. 2f). However, there was a fivefold increase in Sele promoter activity in B-DNA-stimulated EF in both wild-type and Tbk1 −/− Ikbke −/− MEFs (Fig. 2f). These data suggested that B-DNA activates IRF3 and the Ifnb promoter via TBK1 and IKKi, whereas the substantial B-DNA-induced NF-κB activation was independent of TBK1 and IKKi.
Involvement of IPS-1
IPS-1 (also called MAVS, VISA or Cardif), an adaptor molecule of RIG-I, has been identified as the mediator of virus-induced IFN-β and NF-κB activation31,32,33,34. We therefore sought to determine whether IPS-1 is involved in B-DNA-induced innate immune activation. We first evaluated this by reducing IPS-1 expression with small interfering RNA (siRNA) specific to IPS-1. Preliminary experiments assessing the effect of siRNA-mediated 'knockdown' of IPS-1 suggested that HEK 293 cells were better than primary MEFs for this (data not shown). Moreover, B-DNA-induced type I interferon and NF-κB activation were reproducible in HEK293 cells (Fig. 3). When we transfected HEK293 cells with various forms of DNA, dsDNA but not ssDNA triggered the production of endogenous IFN-β and interleukin 8 (IL-8) in a dose-dependent way (Fig. 3a). Of note, poly(dA-dT)•poly(dT-dA) was the most potent inducer of both IFN-β and IL-8, being even better than poly(I)•poly(C) (Fig. 3a). We prepared two double-stranded 21-nucelotide RNA molecules targeting different portions of human IPS-1 mRNA. We transfected HEK293 cells with IPS-1-1, IPS-1-2 or control siRNA and evaluated expression of IPS-1 mRNA. Both IPS-1-1 and IPS-1-2 siRNA reduced IPS-1 mRNA expression but had no effect on the expression of glyceraldehyde phosphodehydrogenase mRNA, as measured by RT-PCR after 48 h (data not shown). We then assessed the effects of these siRNA molecules on B-DNA-stimulated promoter activity of Ifnb and Sele. We first treated HEK293 cells with siRNA and then transfected them with the plasmids expressing luciferase under control of the Ifnb or Sele promoter and then with poly(dA-dT)•poly(dT-dA). 'Knocked-down' expression of IPS-1 resulted in a reduction in both Ifnb and Sele promoter activity after stimulation with B-DNA (P < 0.01; Fig. 3b). These data suggested that B-DNA-induced production of IFN-β and IL-8 as well as activation of the Ifnb and Sele promoters was mediated at least in part by IPS-1.
Figure 3: B-DNA-induced IFN-β and NF-κB requires IPS-1.
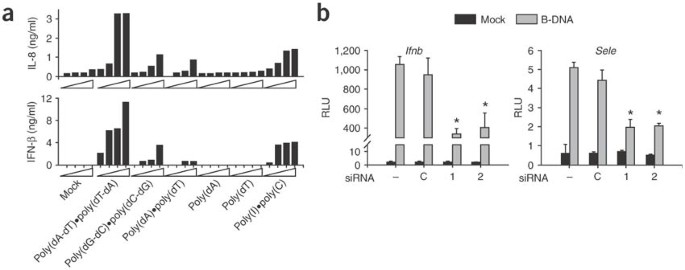
(a) ELISA of IL-8 and IFN-β in culture supernatants of HEK293 cells stimulated for 24 h with various types of DNA or poly(I)•poly(C) at various concentrations (0.01, 0.1, 1, 10 mg/ml; wedges). (b) Ifnb and Sele promoter activity after 'knockdown' of IPS-1 expression in HEK293 cells treated with IPS-1 siRNA (1 and 2) and control cells (C). Cells were transiently transfected with Ifnb promoter plasmid and were stimulated 24 h later with 10 μg/ml of poly(dA-dT)•poly(dT-dA); luciferase activity of cell lysates was measured 18 after stimulation. Data represent mean + s.d. of quadruple samples from one representative of three independent experiments. *, P < 0.01.
Microarray analysis of B-DNA-stimulated cells
To further assess the genes induced or upregulated by B-DNA transfection of MEFs, we did DNA microarray experiments. We obtained MEFs from wild-type, Tbk1 −/−, Ikbke −/− and Tbk1 −/− Ikbke −/− mice and transfected the cells with B-DNA. We assessed mRNA expression profiles 4 h later by DNA microarray. We compared 'fold increases' in mRNA in dsDNA-transfected cells versus mock-transfected cells for wild-type and single- and double-deficient MEFs (Table 1). Genes upregulated by B-DNA were mostly interferon-inducible antiviral genes that are mostly dependent on TBK1, including the gene encoding Vig-1 (Rsad2); Mx1 and Mx2; genes encoding chemokines (Cxcl11 and Cxcl10), GTPases and RNA helicases (D11Lgp1e and Ifih1); Tlr3; genes encoding Z-DNA-binding proteins (Zbp1 and Adar); and Irf7 (Table 1 and data not shown). Some differences in gene expression, such as expression of Il15 and the gene encoding phospholipid scramblase 2 (Plscr2), required either TBK1 or IKKi, as their mRNA was not upregulated in Tbk1 −/− Ikbke −/− MEFs. Among the B-DNA-induced genes, those encoding killer cell lectin-like receptor members 8 and 16 (Klra8 and Klra16, respectively) were dependent only on IKKi. Some interferon-inducible genes were further upregulated by B-DNA in Ikbke −/− MEFs, which suggested a negative regulation mechanism of IKKi for TBK1-mediated gene regulation. These results showed that MEF mRNAs upregulated by B-DNA were differentially and cooperatively regulated by TBK1 and IKKi.
Table 1 Gene expression profiles of MEFs stimulated with dsDNA
B-DNA protects against viral infection
We further examined the physiological functions of innate immune activation of cells by B-DNA. Modified vaccinia virus Ankara (MVA) is a highly attenuated DNA virus that was developed as a safe vaccine against orthopox virus infections, including human smallpox35. E3l, which encodes a poxvirus regulatory protein with a dsRNA-and-Z-DNA-binding domain, is a critical factor for viral virulence and for inhibiting host type I interferon production36. MVA lacking E3l (MVA ΔE3L) therefore induces type I interferon in infected cells37. We therefore determined whether MVA nucleic acid derived from the wild-type and mutant viruses is involved in virus-induced type I interferon production. Although infection with both viruses stimulated wild-type MEFs to upregulate Cxcl10 mRNA, infection with MVA ΔE3L induced Cxcl10 mRNA much more than did infection with MVA, even at a lower multiplicity of infection (MOI; Fig. 4a). In contrast, Ccl2 mRNA was upregulated only after infection with MVA ΔE3L at a high MOI (Fig. 4a). Notably, genomic DNA isolated and purified from MVA stimulated MEFs more than either wild-type or mutant virus infection (Fig. 4a). Ifnb and Cxcl10 expression induced by either MVA virus and MVA genomic DNA was abrogated in Tbk1 −/− Ikbke −/− MEFs (Fig. 4a). We also noted substantial upregulation of Ccl2 mRNA in MEFs after either high-dose infection with MVA ΔE3L or stimulation with MVA genomic DNA, an effect present in both wild-type and Tbk1 −/− Ikbke −/− MEFs (Fig. 4a).
Figure 4: Effects of B-DNA-induced EF activation on viral infection.
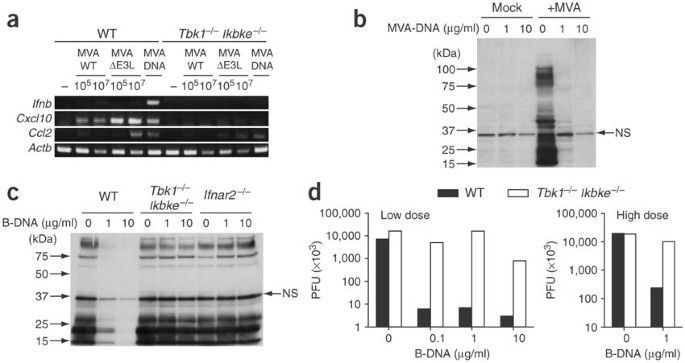
(a) RT-PCR of Ifnb, Cxcl10 and Ccl2 mRNA expression in MEFs from wild-type or Tbk1 −/− Ikbke −/− mice infected with MVA at various plaque-forming units/ml (PFU; above lanes) or transfected with 10 μg/ml of MVA genomic DNA; mRNA was assessed 4 h later. (b,c) Immunoblot of lysates of MEFs from wild-type, Tbk1 −/− Ikbke −/− or Ifnar2 −/− mice. Cells were stimulated for 24 h with 1 or 10 μg/ml of MVA-DNA and were infected for 48 h with 1 × 107 (b) or 1 × 106 (c) plaque-forming units of MVA/ml; lysates were analyzed by immunoblot with antibody to MVA. (d) Virus yield of MEFs from wild-type or Tbk1 −/− Ikbke −/− mice. Cells were stimulated for 4 h with 1 or 10 μg/ml of poly(dA-dT)•poly(dT-dA) and were infected with VSV at low or high dose. Virus yield of culture supernatants was determined by standard plaque assay as described28. NS, nonspecific band.
Next we determined whether the TBK1- and IKKi-dependent immune activation induced by MVA DNA protected MEFs against MVA infection. We stimulated wild-type and Tbk1 −/− Ikbke −/− MEFs with 1 or 10 μg/ml of MVA DNA and then infected the cells with 1 × 107 or 1 × 106 plaque-forming units of wild-type MVA/ml. We analyzed cell lysates of each type of MEF by immunoblot with a vaccinia-specific antibody. Stimulation with MVA DNA did not induce any detectable viral protein production (Fig. 4b). After infection with MVA at a different MOI, however, there were multiple bands of viral proteins on the immunoblot, indicating a permissive molecular life cycle of MVA infection in unstimulated MEFs (Fig. 4b,c). In contrast, cell lysates of wild-type MEFs stimulated with MVA DNA (1 or 10 μg/ml) were almost free of detectable proteins except one nonspecific band at 35 kilodaltons (Fig. 4c). In contrast, there were multiple viral proteins in Tbk1 −/− Ikbke −/− MEFs treated with MVA DNA (Fig. 4c). These data suggested that stimulation with MVA DNA was sufficient to induce protective innate immune activation against MVA infection, which was dependent on TBK1 and IKKi as well as IFN-αβ receptor–mediated signaling. We also determined whether B-DNA-stimulated MEFs were protected against irrelevant RNA virus infection. We infected B-DNA-stimulated wild-type and Tbk1 −/− Ikbke −/− MEFs with vesicular stomatitis virus (VSV) at different multiplicities of infection. We measured virus replication by plaque assay, which showed replication was reduced by 99% after pretreatment with B-DNA, even at a dose of 0.1 μg/ml of B-DNA, a condition in which no type I interferon was detected by enzyme-linked immunosorbent assay (ELISA) or RT-PCR (Fig. 4d and data not shown). Pretreatment with B-DNA suppressed viral replication even at a high MOI in wild-type MEFs, whereas there was no antiviral effect in Tbk1 −/− Ikbke −/− MEFs (Fig. 4d).
B-DNA stimulates DCs via TBK1
Finally, we determined whether B-DNA stimulates immune cells in a TBK-dependent way. To analyze the effect of TBK1 or IKKi on B-DNA-induced innate activation on immune cells such as DCs, we generated mice deficient in tumor necrosis factor (TNF; Tnf −/− mice), Tbk1 −/− mice and Tnf −/− Ikbke −/− mice, because Ikbke −/− mice die in utero, an effect that can be abrogated in the absence of TNF12. We generated bone marrow–derived DCs using granulocyte-monocyte colony-stimulating factor (GM-DCs) or using the ligand for the tyrosine kinase receptor Flt3 (FL-DCs), transfected the cells with B-DNA and monitored the production of IFN-α and IFN-β in culture supernatants by ELISA 24 h after stimulation. B-DNA stimulated Tnf −/− and Tnf −/− Ikbke −/− but not Tnf −/− Tbk1 −/− FL-DCs to produce IFN-α and IFN-β in a dose-dependent way (Fig. 5a). In contrast, poly(I)•poly(C) and A/D-type CpG ODN (D35) stimulated Tnf −/− and Tnf −/− Tbk1 −/− FL-DCs equally to produce IFN-α and IFN-β (Fig. 5a). Similarly, B-DNA stimulated Tnf −/− and Tnf −/− Ikbke −/− but not Tnf −/− Tbk1 −/− GM-DCs to produce IFN-β, although there was still slight upregulation of IFN-β in Tnf −/− Tbk1 −/− GM-DCs (Fig. 5b). In contrast to FL-DCs, GM-DCs did not produce IFN-α in response to B-DNA (Fig. 5b). These data suggested that B-DNA stimulated GM-DCs and FL-DCs to produce type I interferon in a TBK1-dependent way distinct from that of dsRNA and CpG DNA.
Figure 5: B-DNA stimulates DCs via TBK1.
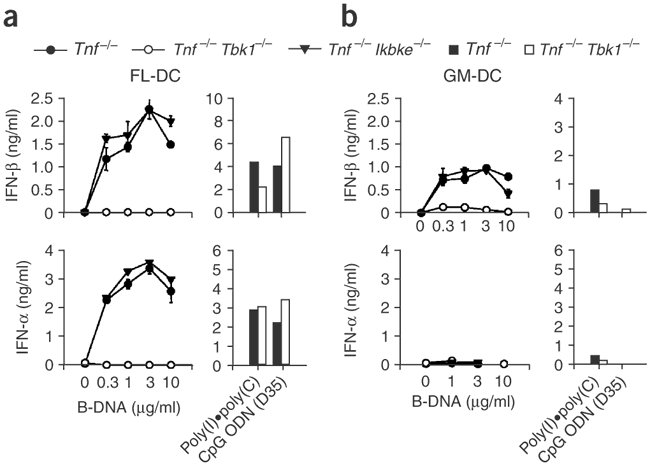
FL-DCs (a) and GM-DCs (b) from Tnf −/−, Tnf −/− Tbk1 −/− or Tbk1 −/− Ikbke −/− mice were transfected for 24 h with various concentrations of B-DNA, 10 μg/ml of poly(I)•poly(C) or 3 μM D-type CpG ODN (D35); IFN-α and IFN-β in culture supernatants were measured by ELISA.
Discussion
We have demonstrated here that double-stranded B-DNA derived from DNA viruses, bacteria, mammals or chemical synthesis can activate mouse and human stromal cells to trigger type I interferon genes through IPS-1 and both TBK1 and IKKi. The results obtained from several mutant strains of mice showed that TLRs and RIG-I, known nucleic acid recognition sensors, are not involved in immune recognition of B-DNA. Furthermore, stimulation of MEFs with viral or synthetic B-DNA conferred protective antiviral responses. The active element of dsDNA, the responding cell types, the signaling pathways involved and the resultant immune responses, including the type I interferon gene program, were distinct from those that have been identified for immunostimulatory nucleic acids such as CpG DNA and dsRNA.
Innate immune activation induced by dsDNA is dependent on a double-stranded structure19. Here we have added the finding that optimal stimulatory activity was dependent on the right-handed B-form helical structure of DNA, as Z-form DNA demonstrated substantially less activity. B-DNA, the right-handed double helical structure of DNA38, is the conformation most commonly found in physiological conditions. Z-DNA, in contrast, forms unusual left-handed helices that can be found near the transcription start site of certain genes in the genome24. There are many structural differences between B-DNA and Z-DNA, suggesting that there may be a unique structure that might explain B-DNA-mediated innate immune recognition. We excluded a third conformational structure of DNA, A-DNA, from this study because it is not clear whether the A-DNA conformation exists in vivo.
Notably, vaccinia virus E3L contains a Z-DNA binding domain that is required for virulence and inhibition of host interferon induction during viral infection37,39,40. Studies have suggested that viral E3L binding of Z-DNA in the host genome near the transcription site might inhibit host antiviral gene inductions36. We have demonstrated that MVA DNA and MVA ΔE3L, but not infection with wild-type MVA, triggered type I interferon and/or chemokine production in MEFs, suggesting that innate recognition of B-DNA and triggering of an antiviral gene program might be targeted by viral E3L as a viral escape mechanism. Moreover, our DNA microarray data showed that B-DNA also upregulated mRNA expression of Z-DNA-binding protein (Zbp1, also called Dlm-1) and double-stranded RNA adenosine deaminase (Adar) in a TBK1-dependent way, indicating that such proteins might also participate in antiviral responses by targeting Z-DNA derived from viruses or by competing with the Z-DNA-binding activity of viral E3L at host antiviral transcription sites. Further studies are needed to clarify the involvement of the Z-DNA-binding activity of viral E3L and host Z-DNA-binding proteins in viral escape from host immune system and in host antiviral immune responses, respectively.
The DExD/H box RNA helicase RIG-I is involved in the innate immune activation induced by RNA viruses and/or dsRNA28,30. RIG-I-deficient MEFs do not respond to RNA viruses, including VSV, Newcastle disease virus and Sendai virus28; in contrast, these MEFs responded to B-DNA by upregulating the mRNA of type I interferons and chemokines. Although our data indicated that RIG-I was not involved in the B-DNA response, IPS-1, the RIG-I-associated adaptor molecule, was required for IFN-β and NF-κB activation, suggesting that the signaling pathways involved in dsRNA- and B-DNA-induced innate immune activation are closely related, though their respective receptor molecules are probably different. Protein kinase R, another antiviral receptor-like molecule that can bind dsRNA and is involved in host cell shutoff of protein synthesis, was not involved in B-DNA-induced innate immune activation (protein kinase R–deficient MEFs responded normally to B-DNA; data not shown). Furthermore, Mda-5, another DExD/H box RNA helicase closely related to RIG-I, associates with IPS-1 (ref. 31). Although RIG-I and Mda-5 have been suggested to be receptor-like molecules for viral RNA41,42,43, preliminary experiments have demonstrated that Mda-5-deficient MEFs responded normally to B-DNA (unpublished data). Moreover, ectopic expression of RIG-I or Mda-5 did not enhance B-DNA-induced Ifnb promoter activity in HEK293 cells (unpublished data). These preliminary data have indicated that Mda-5 is neither necessary nor sufficient for B-DNA induction of type I interferons and chemokines. Further investigation is needed to determine the relevant signaling pathways and the receptor or receptor-like molecules that recognize B-DNA and that are distinct from TLRs, RIG-I or Mda-5.
Our results have also demonstrated that B-DNA has strong antiviral activity. Transfection of MVA genomic DNA conferred considerable resistance to subsequent MVA infection of wild-type MEFs but not Tbk1 −/− Ikbke −/− MEFS. Moreover, B-DNA-induced innate activation of wild-type MEFs was sufficient to protect them from highly virulent VSV infection. Those results strongly suggest that viral DNA, or even host DNA, in infected cells might trigger antiviral responses, such as the production of type I interferons, through the IPS-1–TBK1–IKKi signaling pathway. Further studies will be needed to clarify whether B-DNA-induced antiviral responses are involved in vivo in various DNA virus infections. Investigation of the therapeutic potential of B-DNA for treatment of viral infections, as well as other infectious diseases or cancer in which type I interferons might be beneficial to the host, will also be useful.
Another possible physiological role for B-DNA-induced innate immune activation has been suggested. The inability to degrade DNA derived from erythroid precursors results in IFN-β production18. Our results might well be linked to the observation that such IFN-β production might be mediated by undigested double-stranded B-DNA through an IPS-1–TBK1–dependent signaling pathway. The dsDNA-induced immune activation might thus facilitate the pathogenesis of local or even systemic autoimmune disorders19. In that case, dsDNA derived from pathogens or a damaged host (especially necrotic cells) might act not only on nonimmune cells but also on 'professional' antigen-presenting cells such as DCs as an adjuvant20, thereby influencing the subsequent adaptive immune responses. In gene therapy, alternatively, DNA-induced immune activation of transfected cells might inadvertently, through the induction of type I interferons, limit efficient transcription of the gene of interest or cause local and or systemic inflammatory responses, which could also hamper gene transduction or expression efficacy. Furthermore, DNA-based vaccines in which CpG motifs in a plasmid vector contribute to their immunogenicity as a 'built-in adjuvant' might need to be re-evaluated for their immunogenicity. That possibility stems partly from the fact that Tlr9 −/− mice, which do not respond to CpG-DNA, are nevertheless able to mount both humoral and cellular immune responses after vaccination with DNA44,45. Further studies will be needed to clarify the physiological function of B-DNA-induced, IPS-1–TBK1–IKKi–dependent innate immune activation of host defenses, DNA-associated immune disorders and DNA-based immunotherapy.
Methods
Cells and reagents.
MEFs from C57BL/6, Myd_88−/−_Trif −/−, Tbk −/−, Ikbke −/− and RIG-I knockout mice were prepared as described12,28,29. Ifnar2 −/− mice were purchased from B&K Universal. HEK293 cells were obtained from American Type Culture Collection. Mouse bone marrow DCs were generated by 7 d of culture of bone marrow cells with either granulocyte-monocyte colony-stimulating factor (GM-DCs) or Flt3 ligand (FL-DCs) as described46. These cells were cultured in DMEM supplemented with 10% FCS in an incubator with 5% CO2. Recombinant VSV was gift from T. Abe and Y. Matsuura (Osaka University, Osaka, Japan). Genomic DNA derived from herpes simplex virus types 1 and 2 and human cytomegalovirus was purchased from Advanced Biotechnologies, and DNA from calf thymus and E. coli were purchased from Sigma and Invitrogen, respectively. Synthetic polydeoxynucleotides and poly(I)•poly(C) were purchased from Amersham Biosciences. Synthetic ODNs including CpG ODN D35 (ref. 47) were synthesized by Hokkaido System Bioscience or Invitrogen. All DNA or RNA was mixed for 15 min with Lipofectamine 2000 (Invitrogen) at a ratio of 1:1 (volume/weight) in OptiMEM before use in the stimulation experiments. DNA was methylated as described20. All DNA used was tested and was free of endotoxin (less than 0.001 U/mg DNA).
RT-PCR.
RT-PCR was done as described48. Total RNA was extracted with TRIzol reagent (Invitrogen) according to the manufacturer's protocol, and then 1 μg total RNA was reverse-transcribed; cDNA was generated by standard PCR of 25 cycles with primers as described48,49.
ELISA.
Human IFN-β, mouse IFN-α and IFN-β in culture supernatants was measured by ELISA (PBL) according to the manufacturer's instructions. ELISA for human IL-8 was done as described50.
Native PAGE.
Native PAGE was done as described12. MEFs (1 × 106 cells) were stimulated with 10 μg/ml of DNA for various times and then whole-cell lysates were prepared. Cell lysates in native PAGE buffer (62.5 mM Tris-HCl, pH 6.8, and 15% glycerol) were separated by 7.5% gel electrophoresis followed by immunoblot with anti–IRF-3 (Santa Cruz).
Reporter assay.
HEK293 cells and MEFs were transiently transfected with 100 ng of the luciferase reporter plasmid together with a total of 1 μg expression plasmid or empty control plasmid. Luciferase reporter constructs for Ifnb and Sele have been described7. At 18 h after transfection, luciferase activity in the total cell lysate was measured with the Dual-luciferase reporter assay system (Promega). The renilla-luciferase reporter gene was simultaneously transfected as an internal control.
RNA interference.
Interference of IPS-1 mRNA was done as described31. Dharmacon Research synthesized dsRNA composed of 21–base pair sense and antisense oligonucleotides. The RNA oligonucleotides used for targeting human IPS-1 in this study were as follows: _Ips1_-1 sense, 5′-UAGUUGAUCUCGCGGACGAdTdT-3′, and antisense, 5′-UCGUCCGCGAGAUCAACUd TdT-3′; and _Ips1_-2 sense, 5′-CCGUUUGCUGAAGACAAGAdTdT-3′, and antisense, 5′-UCUUGUCUUCAGCAAACGGdTdT-3′. Control siRNA (siCONTROL Non-targeting siRNA#1) was purchased from Dharmacon Research. HEK293 cells were transfected with 100 nM siRNA using Lipofectamine 2000 (Invitrogen). At 48 h after transfection, cells were used for further experiments. 'Knockdown' of IPS-1 mRNA was verified by RT-PCR (data not shown).
Microarray analysis.
Total RNA was extracted from MEFs stimulated for 4 h with or without poly(dA-dT)•poly(dT-dA), after which cRNA was synthesized. Preparation of cRNA, hybridization and scanning of the microarray were done according to the manufacturer's instructions (Affymetrix). A microarray (MG U74A version 2; Affymetrix) was used with Microarray Suite software (version 5.0; Affymetrix) and GeneSpring software (Silicon Genetics).
MVA infection.
MEFs were transfected with various concentrations of MVA DNA and were infected 24 h later with 1 × 107 or 1 × 106 plaque-forming units of MVA/ml (MOI = 10 or 1, respectively). Then, 48 h after MVA infection, cells were washed with PBS and were lysed with lysate buffer (CellLytic-MT; Sigma) in the presence of a protease inhibitor 'cocktail' (Sigma). Lysates were separated by SDS-PAGE and proteins were then transferred to polyvinyldifluoride membranes. Membranes were blocked and were analyzed by immunoblot with polyclonal rabbit antibody to vaccinia virus diluted 1:1,000 (Biogenesis), followed by horseradish peroxidase–conjugated secondary antibody and LumiGlo reagent (Cell Signaling Technology).
Plaque assay.
Culture supernatants were collected from MEFs infected with VSV. MEFs were pretreated with transfection for 4 h of various concentrations of poly(dA-dT)•poly(dT-dA). Virus yield in the culture supernatants was determined by a standard plaque assay as described28. Prepared baby hamster kidney cells were infected with serial dilutions of the recovered viruses. The cells were overlaid with DMEM containing 1% low-melting agarose and were incubated for 24 h; plaques were then counted.
Statistical analysis.
Differences were analyzed for statistical significance with Student's _t_-test. A P value of less than 0.05 was considered significant.
Note: Supplementary information is available on the Nature Immunology website.
Change history
10 March 2006
In the version of this article initially published, the GEO database accession number is missing. This should be the final subsection of Methods, as follows: Accession code. GEO: microarray data, GSE4171. The error has been corrected in the PDF version of the article.
References
- Isaacs, A, Cox, R.A. & Rotem, Z. Foreign nucleic acids as the stimulus to make interferon. Lancet 2, 113–116 (1963).
Article CAS Google Scholar - Tokunaga, T. et al. Antitumor activity of deoxyribonucleic acid fraction from Mycobacterium bovis BCG. I. Isolation, physicochemical characterization, and antitumor activity. J. Natl. Cancer Inst. 72, 955–962 (1984).
CAS PubMed Google Scholar - Akira, S. & Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 4, 499–511 (2004).
Article CAS Google Scholar - Iwasaki, A. & Medzhitov, R. Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 5, 987–995 (2004).
Article CAS Google Scholar - Theofilopoulos, A.N., Baccala, R., Beutler, B. & Kono, D.H. Type I interferons (α/β) in immunity and autoimmunity. Annu. Rev. Immunol. 23, 307–336 (2005).
Article CAS Google Scholar - Bauer, M. et al. Bacterial CpG-DNA triggers activation and maturation of human CD11c−, CD123+ dendritic cells. J. Immunol. 166, 5000–5007 (2001).
Article CAS Google Scholar - Kawai, T. et al. Interferon-alpha induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat. Immunol. 5, 1061–1068 (2004).
Article CAS Google Scholar - Uematsu, S. et al. Interleukin-1 receptor-associated kinase-1 plays an essential role for Toll-like receptor (TLR)7− and TLR9-mediated interferon-α induction. J. Exp. Med. 201, 915–923 (2005).
Article CAS Google Scholar - Honda, K. et al. Role of a transductional-transcriptional processor complex involving MyD88 and IRF-7 in Toll-like receptor signaling. Proc. Natl. Acad. Sci. USA 101, 15416–15421 (2004).
Article CAS Google Scholar - Sharma, S. et al. Triggering the interferon antiviral response through an IKK-related pathway. Science 300, 1148–1151 (2003).
Article CAS Google Scholar - Fitzgerald, K.A. et al. IKKε and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 4, 491–496 (2003).
Article CAS Google Scholar - Hemmi, H. et al. The roles of two IκB kinase-related kinases in lipopolysaccharide and double stranded RNA signaling and viral infection. J. Exp. Med. 199, 1641–1650 (2004).
Article CAS Google Scholar - Perry, A.K., Chow, E.K., Goodnough, J.B., Yeh, W.C. & Cheng, G. Differential requirement for TANK-binding kinase-1 in type I interferon responses to toll-like receptor activation and viral infection. J. Exp. Med. 199, 1651–1658 (2004).
Article CAS Google Scholar - Vallin, H., Perers, A., Alm, G.V. & Ronnblom, L. Anti-double-stranded DNA antibodies and immunostimulatory plasmid DNA in combination mimic the endogenous IFN-α inducer in systemic lupus erythematosus. J. Immunol. 163, 6306–6313 (1999).
CAS PubMed Google Scholar - Boule, M.W. et al. Toll-like receptor 9-dependent and -independent dendritic cell activation by chromatin-immunoglobulin G complexes. J. Exp. Med. 199, 1631–1640 (2004).
Article CAS Google Scholar - Decker, P., Singh-Jasuja, H., Haager, S., Kotter, I. & Rammensee, H.G. Nucleosome, the main autoantigen in systemic lupus erythematosus, induces direct dendritic cell activation via a MyD88-independent pathway: consequences on inflammation. J. Immunol. 174, 3326–3334 (2005).
Article CAS Google Scholar - Kawane, K. et al. Impaired thymic development in mouse embryos deficient in apoptotic DNA degradation. Nat. Immunol. 4, 138–144 (2003).
Article CAS Google Scholar - Yoshida, H., Okabe, Y., Kawane, K., Fukuyama, H. & Nagata, S. Lethal anemia caused by interferon-beta produced in mouse embryos carrying undigested DNA. Nat. Immunol. 6, 49–56 (2005).
Article CAS Google Scholar - Suzuki, K. et al. Activation of target-tissue immune-recognition molecules by double- stranded polynucleotides. Proc. Natl. Acad. Sci. USA 96, 2285–2290 (1999).
Article CAS Google Scholar - Ishii, K.J. et al. Genomic DNA released by dying cells induces the maturation of APCs. J. Immunol. 167, 2602–2607 (2001).
Article CAS Google Scholar - Li, S. et al. Induction of IFN-regulated factors and antitumoral surveillance by transfected placebo plasmid DNA. Mol. Ther. 11, 112–119 (2005).
Article Google Scholar - Yasuda, K. et al. Endosomal translocation of vertebrate DNA activates dendritic cells via TLR9-dependent and -independent pathways. J. Immunol. 174, 6129–6136 (2005).
Article CAS Google Scholar - Ishii, K.J. & Akira, S. Innate immune recognition of nucleic acids: Beyond toll-like receptors. Int. J. Cancer 117, 517–523 (2005).
Article CAS Google Scholar - Rich, A. & Zhang, S. Timeline: Z-DNA: the long road to biological function. Nat. Rev. Genet. 4, 566–572 (2003).
Article CAS Google Scholar - Braun, C.S. et al. The structure of DNA within cationic lipid/DNA complexes. Biophys. J. 84, 1114–1123 (2003).
Article CAS Google Scholar - Moller, A., Nordheim, A., Kozlowski, S.A., Patel, D.J. & Rich, A. Bromination stabilizes poly(dG-dC) in the Z-DNA form under low-salt conditions. Biochemistry 23, 54–62 (1984).
Article CAS Google Scholar - Kimura, T., Kawai, K., Tojo, S. & Majima, T. One-electron attachment reaction of B- and Z-DNA modified by 8-bromo-2′-deoxyguanosine. J. Org. Chem. 69, 1169–1173 (2004).
Article CAS Google Scholar - Kato, H. et al. Cell type-specific involvement of RIG-I in antiviral response. Immunity 23, 19–28 (2005).
Article CAS Google Scholar - Yamamoto, M. et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 301, 640–643 (2003).
Article CAS Google Scholar - Yoneyama, M. et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 5, 730–737 (2004).
Article CAS Google Scholar - Kawai, T. et al. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 6, 981–988 (2005).
Article CAS Google Scholar - Seth, R.B., Sun, L., Ea, C.K. & Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF 3. Cell 122, 669–682 (2005).
Article CAS Google Scholar - Xu, L.G. et al. VISA is an adapter protein required for virus-triggered IFN-β signaling. Mol. Cell 19, 727–740 (2005).
Article CAS Google Scholar - Meylan, E. et al. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature (2005).
- Drexler, I. et al. Identification of vaccinia virus epitope-specific HLA-A*0201-restricted T cells and comparative analysis of smallpox vaccines. Proc. Natl. Acad. Sci. USA 100, 217–222 (2003).
Article CAS Google Scholar - Kim, Y.G. et al. A role for Z-DNA binding in vaccinia virus pathogenesis. Proc. Natl. Acad. Sci. USA 100, 6974–6979 (2003).
Article CAS Google Scholar - Hornemann, S. et al. Replication of modified vaccinia virus Ankara in primary chicken embryo fibroblasts requires expression of the interferon resistance gene E3L. J. Virol. 77, 8394–8407 (2003).
Article CAS Google Scholar - Watson, J.D. & Crick, F.H. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature 171, 737–738 (1953).
Article CAS Google Scholar - Smith, E.J., Marie, I., Prakash, A., Garcia-Sastre, A. & Levy, D.E. IRF3 and IRF7 phosphorylation in virus-infected cells does not require double-stranded RNA-dependent protein kinase R or IκB kinase but is blocked by vaccinia virus E3L protein. J. Biol. Chem. 276, 8951–8957 (2001).
Article CAS Google Scholar - Kim, Y.G., Lowenhaupt, K., Oh, D.B., Kim, K.K. & Rich, A. Evidence that vaccinia virulence factor E3L binds to Z-DNA in vivo: Implications for development of a therapy for poxvirus infection. Proc. Natl. Acad. Sci. USA 101, 1514–1518 (2004).
Article CAS Google Scholar - Kovacsovics, M. et al. Overexpression of Helicard, a CARD-containing helicase cleaved during apoptosis, accelerates DNA degradation. Curr. Biol. 12, 838–843 (2002).
Article CAS Google Scholar - Andrejeva, J. et al. The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN-beta promoter. Proc. Natl. Acad. Sci. USA 101, 17264–17269 (2004).
Article CAS Google Scholar - Yoneyama, M. et al. Shared and unique functions of the DExD/H-Box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J. Immunol. 175, 2851–2858 (2005).
Article CAS Google Scholar - Spies, B. et al. Vaccination with plasmid DNA activates dendritic cells via Toll-like receptor 9 (TLR9) but functions in TLR9-deficient mice. J. Immunol. 171, 5908–5912 (2003).
Article CAS Google Scholar - Babiuk, S. et al. TLR9−/− and TLR9+/+ mice display similar immune responses to a DNA vaccine. Immunology 113, 114–120 (2004).
Article CAS Google Scholar - Coban, C. et al. Toll-like receptor 9 mediates innate immune activation by the malaria pigment hemozoin. J. Exp. Med. 201, 19–25 (2005).
Article CAS Google Scholar - Gursel, M., Verthelyi, D., Gursel, I., Ishii, K.J. & Klinman, D.M. Differential and competitive activation of human immune cells by distinct classes of CpG oligodeoxynucleotide. J. Leukoc. Biol. 71, 813–820 (2002).
CAS PubMed Google Scholar - Takeshita, S., Takeshita, F., Haddad, D.E., Ishii, K.J. & Klinman, D.M. CpG oligodeoxynucleotides induce murine macrophages to up-regulate chemokine mRNA expression. Cell. Immunol. 206, 101–106 (2000).
Article CAS Google Scholar - Ogasawara, K. et al. Requirement of the IFN-α/β-induced CXCR3 chemokine signalling for CD8+ T cell activation. Genes Cells 7, 309–320 (2002).
Article CAS Google Scholar - Sugiyama, T. et al. CpG RNA: identification of novel single-stranded RNA that stimulates human CD14+CD11c+ monocytes. J. Immunol. 174, 2273–2279 (2005).
Article CAS Google Scholar
Acknowledgements
We thank Y. Fujita for technical support; K. Matsui, T. Hirotani and H. Kumar for support; K. Sakurai and N. Shimada for circular dichroism measurement of the DNA-lipid complex; T. Abe and Y. Matsuura for providing VSV; H. Shirota for discussions; T. Majima and K. Kawai for providing Z-DNA and for discussions; and other members of Exploratory Research for Advanced Technology, Japan Science and Technology Agency and the Department of Host Defense, Osaka University, for support.
Author information
Authors and Affiliations
- Exploratory Research for Advanced Technology, Japan Science and Technology Agency, Research Institute for Microbial Diseases, Osaka University, Osaka, 565-0871, Japan
Ken J Ishii, Taro Kawai, Osamu Takeuchi & Shizuo Akira - Department of Host Defense, Research Institute for Microbial Diseases, Osaka University, Osaka, 565-0871, Japan
Ken J Ishii, Cevayir Coban, Hiroki Kato, Ken Takahashi, Yuichi Torii, Hiroaki Hemmi, Shintaro Sato, Masahiro Yamamoto, Satoshi Uematsu, Taro Kawai, Osamu Takeuchi & Shizuo Akira - The 21st Century COE, Combined Program on Microbiology and Immunology, Research Institute for Microbial Diseases, Osaka University, Osaka, 565-0871, Japan
Cevayir Coban & Shizuo Akira - Department of Molecular Biodefense Research, Yokohama City University Graduate School of Medicine, Yokohama, 236-0004, Japan
Fumihiko Takeshita - Abteilung fur Virologie, Paul-Ehrlich Institut, Langen, 63225, Germany
Holger Ludwig & Gerd Sutter - Department of Host Defense, Leprosy Research Center, National Institute of Infectious Diseases, Tokyo, 189-0002, Japan
Koichi Suzuki
Authors
- Ken J Ishii
You can also search for this author inPubMed Google Scholar - Cevayir Coban
You can also search for this author inPubMed Google Scholar - Hiroki Kato
You can also search for this author inPubMed Google Scholar - Ken Takahashi
You can also search for this author inPubMed Google Scholar - Yuichi Torii
You can also search for this author inPubMed Google Scholar - Fumihiko Takeshita
You can also search for this author inPubMed Google Scholar - Holger Ludwig
You can also search for this author inPubMed Google Scholar - Gerd Sutter
You can also search for this author inPubMed Google Scholar - Koichi Suzuki
You can also search for this author inPubMed Google Scholar - Hiroaki Hemmi
You can also search for this author inPubMed Google Scholar - Shintaro Sato
You can also search for this author inPubMed Google Scholar - Masahiro Yamamoto
You can also search for this author inPubMed Google Scholar - Satoshi Uematsu
You can also search for this author inPubMed Google Scholar - Taro Kawai
You can also search for this author inPubMed Google Scholar - Osamu Takeuchi
You can also search for this author inPubMed Google Scholar - Shizuo Akira
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toShizuo Akira.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Fig. 1
Mda-5 is not involved in B-DNA-induced type-I IFN and chemokine inductions. (PDF 122 kb)
Rights and permissions
About this article
Cite this article
Ishii, K., Coban, C., Kato, H. et al. A Toll-like receptor–independent antiviral response induced by double-stranded B-form DNA.Nat Immunol 7, 40–48 (2006). https://doi.org/10.1038/ni1282
- Received: 08 August 2005
- Accepted: 01 October 2005
- Published: 13 November 2005
- Issue Date: 01 January 2006
- DOI: https://doi.org/10.1038/ni1282