Protozoan encounters with Toll-like receptor signalling pathways: implications for host parasitism (original) (raw)
Key Points
- The cellular compartment of the vertebrate host innate immune system has been shown to have an important role in early resistance and the development of acquired immunity and, in some instances, in pathogenesis during infection with protozoan parasites. Many of these roles are largely mediated by cytokines produced by myeloid cells. However, the means by which protozoans trigger these cells to produce cytokines were mostly unknown. Recent experimental evidence advances our understanding on this important question. This new information includes the characterization of parasite components that activate myeloid cells and identifies receptors and signalling pathways that mediate parasite-induced cytokine synthesis by these cells.
- Glycosylphosphatidylinositol (GPI) anchors and DNA were shown to be the main parasite molecules that trigger the production of cytokines by myeloid cells from vertebrate hosts. They are recognized by Toll-like receptors (TLRs), which are evolutionarily ancient cognate receptors that detect invasive pathogens in mammals. The activation of TLRs results in innate and adaptive immune responses, culminating in host protection.
- Importantly, protozoan parasites are recognized by multiple TLRs, and enhanced susceptibility to infection is clearly observed in Myd88 (myeloid differentiation primary-response gene 88) knockout mice. Also remarkably, mice deficient for both TLR2 (the receptor for GPI anchors) and TLR9 (the receptor for DNA) showed increased susceptibility to Trypanosoma cruzi.
- In contrast to most protozoan infections, _Myd88_-deficient mice effectively clear parasitaemia from mice infected with Plasmodium berghei. Nevertheless, in this rodent malaria model, MyD88 seems to mediate cytokine-induced pathology.
- Interestingly, protozoan parasites prevent excessive TLR activation and pro-inflammatory cytokine responses by various mechanisms, avoiding complete elimination from the host, as well as immunopathology.
- Strategies using chemically defined TLR agonists are currently being used in prophylactic and therapeutic vaccine protocols for infections with protozoan parasites. Conversely, the use of TLR antagonists might be beneficial to treat or prevent sepsis-related symptoms that are observed during acute episodes of malaria.
Abstract
Toll-like receptors (TLRs) have emerged as a major receptor family involved in non-self recognition. They have a vital role in triggering innate immunity and orchestrate the acquired immune response during bacterial and viral infection. However, the role of TLRs during infection with protozoan pathogens is less clear. Nevertheless, our understanding of how these parasitic microorganisms engage the host TLR signalling system has now entered a phase of rapid expansion. This Review describes recent insights into how parasitic protozoans are sensed by TLR molecules, and how the TLR system itself can be targeted by these microbial pathogens for their own survival.
Similar content being viewed by others
Main
Infection with protozoan parasites is a major human health issue, causing vast morbidity and mortality that, particularly in developing countries, contributes to political, social and economic instability. There are currently no vaccines available to prevent these devastating infections, and the development of drug resistance in these pathogens is a growing problem (TDR Diseases web site). Therefore, there is an urgent need for the development of new prophylactic strategies and therapies for patients infected with this class of pathogen. Understanding the complexity of the immunological mechanisms involved in resistance to infection, as well as in pathogenesis caused by protozoan parasites, is essential for the development of effective prophylactic and therapeutic vaccines.
During the early stages of infection, the host innate immune system must rapidly detect and respond to protozoan parasite infection, and this is achieved through innate immune receptors. Furthermore, innate immunity and TLRs orchestrate the development of an acquired immune response, which is necessary for protection against re-infection. Therefore, in the absence of recognition by innate immune receptors, the host will quickly become overwhelmed by the parasitic pathogen, resulting in disease and possibly death. Conversely, if activation of innate immune receptors is excessive, high levels of pro-inflammatory mediators such as interferon-γ (IFNγ), tumour-necrosis factor (TNF) and reactive nitrogen intermediates (RNIs) can be detrimental to the host. Therefore, how the innate immune system detects and responds to protozoan parasites is crucial to understanding how infection is controlled, as well as how excessive immune responses are avoided.
In mammalian cells, the Toll-like receptor (TLR) family is an important group of receptors through which the innate immune system recognizes invasive microbes1,2. TLRs are crucial for many aspects of microbial elimination, including recruitment of phagocytes to infected tissue, and subsequent microbial killing. TLRs expressed by macrophages and dendritic cells (DCs) also have a role in shaping long-term acquired immunity. However, when activated to excess, TLRs mediate pathology, as is the case in septic shock induced by infection with Gram-negative bacteria and by lipopolysaccharide (LPS)3,4.
Although the importance of innate immunity in resistance to parasitic infections, and in the pathogenesis of protozoan infections, is well recognized, the molecular mechanisms underlying recognition of parasitic protozoans by innate immune cells are only now beginning to be understood. Major advances have identified bacterial and viral molecules that act as TLR agonists and have identified how these pathogens can manipulate TLR-induced signalling cascades to prolong their own survival. Now, this area of research is emerging with new and exciting insights into how the TLR signalling system responds to infection by protozoans, including Leishmania spp., Trypanosoma cruzi, Trypanosoma brucei, Plasmodium spp. and Toxoplasma gondii — pathogens that every year account for immense human suffering and death worldwide (Box 1). In this Review, we present recent studies describing protozoan molecules that act as TLR agonists and describe a role for TLRs and Toll/interleukin-1 receptor (TIR)-domain-containing adaptor molecules in resistance to these parasites. Also, emerging examples of the novel mechanisms by which protozoans can manipulate TLR-dependent signal-transduction cascades to establish host parasitism are discussed. Together, these studies argue for the vital role TLR signalling pathways have in innate immune recognition of protozoans, in the emergence of acquired immunity, and conversely in certain cases in the development of immunopathology during protozoan infection.
Protozoan PAMPs
The term pathogen-associated molecular pattern (PAMP) was coined to describe infectious, non-self targets of the innate immune system. Three main features characterize molecules containing PAMPs: they are usually expressed by microbes and not host cells, they are conserved among microorganisms of a given class and their expression is essential for microbial survival. Whereas the first two characteristics allow recognition of microbes and not host cells, the third prevents the development of mutants that escape recognition by the host immune system5. Although the identification of protozoan PAMPs is at an early stage compared with the identification of PAMPs contained within bacterial and viral molecules (Box 2), several TLR agonists derived from protozoans have been identified in recent years6.
Several studies have shown that glycosylphosphatidylinositol (GPI) anchors (or their fragments) from Leishmania major, T. brucei, T. cruzi, Plasmodium falciparum and T. gondii activate cells of both lymphoid and myeloid lineages7,8,9,10,11 (Table 1). GPI moieties function to anchor proteins to the surface of eukaryotic cells, and they are abundantly expressed by many protozoan parasites. GPI anchors are composed of a glycan core and a lipid component. Specificity is conferred through variations in the carbohydrate branches, the lipid inositol portion (glycerol versus ceramide), and the number, length and degree of saturation in the hydrocarbon chains. For T. cruzi trypomastigotes derived from mammalian cells, the pro-inflammatory activity of GPI anchors covalently linked to mucin-like glycoproteins (GPI mucin) expressed on the surface of the parasite depends on the GPI anchor's fine structure. Non-saturated, fatty acyl chains and periodate-sensitive components from the GPI anchor of T. cruzi have been shown to be required to trigger the production of cytokines by macrophages7,12,13.
Table 1 Protozoan pathogen-associated molecular patterns (PAMPs)
Because parasite GPI anchors can initiate host immune responses, this raises the question as to why mammalian GPI anchors do not themselves induce unrestrained autoimmune disease. Mammalian cells typically express 105 copies of GPI anchors per cell, whereas parasitic protozoans express up to 107 copies (and related structures) per cell14. Furthermore, protozoan-derived GPI anchors contain a longer glycan core and a lipid component (which is not present in mammalian GPI anchors)15. Consequently, both the amount and the fine structure of at least some protozoan-derived GPI anchors are important aspects in determining activation of innate defence mechanisms, as well as inflammation in the vertebrate host.
TLR activation by protozoan GPI anchors. The _T. cruzi_-derived GPI anchors trigger the phosphorylation of mitogen-activated protein kinase (MAPK) and inhibitor of nuclear factor-κB (IκB) family members. Activation of both of these pro-inflammatory signalling pathways is triggered by activation of TLRs. By using Chinese hamster ovary (CHO) cells transfected with genes encoding different TLR molecules, _T. cruzi-_derived GPI anchors were shown to trigger nuclear factor-κB (NF-κB) activation through TLR2. In addition, recognition of the GPI anchors required CD14, a host cell-surface molecule involved in the recognition of bacterial LPS by TLR4 (Ref. 16). Furthermore, the induction of pro-inflammatory cytokines by GPI anchors was ablated in macrophages derived from TLR2-deficient mice16. Indeed, live tissue-culture-derived trypomastigotes have been shown to activate CHO cells transfected with human TLR2 (Ref. 16) and promote production of interleukin-1 (IL-1) and cardiomyocyte hypertrophy through TLR2 (Ref. 17). It is well established that TLR2 functions as a heterodimer with either TLR1 or TLR6. Because macrophages that lack TLR6 expression fail to respond to T. cruzi GPI anchors18, the data indicate that a complex of TLR2–TLR6 and CD14 is involved in the recognition of these parasite molecules (Fig. 1).
Figure 1: Activation of Toll-like receptors by protozoan pathogen-associated molecular patterns.
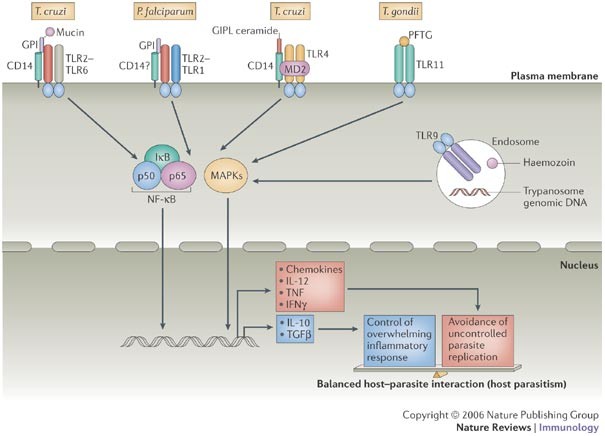
Trypanosoma cruzi glycosylphosphatidylinositol (GPI) covalently linked to mucin-like glycoproteins (GPI mucin) are ligands for the Toll-like receptor 2 (TLR2)–TLR6 heterodimer, and an additional role for CD14 has been implicated. In the case of Plasmodium falciparum GPI anchors, three fatty acyl chains preferentially activate the TLR2–TLR1 heterodimer, but involvement of CD14 has not been determined. The TLR4–MD2 complex recognizes free T. cruzi GPI anchors, known as glycoinositolphospholipids containing ceramide (GIPL ceramide). The TLR11 molecule, which is expressed by mouse but not human cells, recognizes a profilin-like protein (PFTG) from Toxoplasma gondii and possibly other related apicomplexan parasites. TLR9, which is localized in endosomes and the endoplasmic reticulum, recognizes T. cruzi and Trypanosoma brucei genomic DNA, as well as haemozoin derived from red blood cells that are infected with P. falciparum. TLR activation by parasite molecules triggers nuclear factor-κB (NF-κB) and mitogen-activated protein kinase (MAPK) signalling pathways to induce the expression of pro-inflammatory cytokine genes and genes that directly control parasite replication. At the same time, genes that encode anti-inflammatory cytokines are induced. These control an excessive and, therefore, deleterious immune response. An appropriate balance between the pro-inflammatory and anti-inflammatory response facilitates host parasitism and persistent infection. IκB, inhibitor of NF-κB; IL, interleukin; IFNγ, interferon-γ; TGFβ, transforming growth factor-β;TNF, tumour-necrosis factor.
In addition to these T. cruzi TLR2–TLR6 ligands, a subset of free GPI anchors from T. cruzi, known as glycoinositolphospholipids, that contain ceramide activate CHO cells transfected with TLRs. This response is dependent on TLR4 and CD14 but not TLR2 (Ref. 19). In vivo, glycoinositolphospholipids that contain ceramide trigger the production of chemokines, such as CXC-chemokine ligand 2 (CXCL2) in wild-type mice, but not in animals that express a non-functional TLR4 (Ref. 19). Therefore, T. cruzi contains PAMPs that are recognized by both TLR2–TLR6 complexes and TLR4.
Similar to T. cruzi, other kinetoplastids such as Leishmania spp. have GPI-linked molecules that trigger TLR activation. Infectious promastigotes are decorated at their surface with GPI-linked molecules. At this particular stage of development, the main GPI-linked molecules are lipophosphoglycans (LPGs), which contain long carbohydrate branches with repeating phosphoglycan units14,20. LPGs from L. major have been shown to stimulate mouse macrophages and human natural killer (NK) cells through TLR2 (Refs 8, 21). Furthermore, use of RNA interference to knock down the expression of various TLRs revealed that activation of macrophages by Leishmania donovani is also, at least in part, dependent on TLR2 (Ref. 22).
There is evidence that GPI-related molecules from apicomplexan parasites also trigger TLR2 and TLR4 activation. For example, GPI anchors derived from P. falciparum merozoites have been shown to induce TNF synthesis through the interaction of the three fatty acyl chains of the GPI anchor with the TLR2–TLR1 complex, which involves a minor contribution from TLR4 (Refs 23, 24). Native GPI anchors purified from T. gondii tachyzoites, as well as synthetic fragments of the proposed structure of these GPI anchors, were shown to promote NF-κB activation and stimulate TNF synthesis by a mouse macrophage cell line9, and these responses also seem to be mediated through TLR2 and TLR4 (F. Debierre–Grokiego et al., manuscript in preparation). So, parasite GPI anchors and related molecules seem to fit the definition of a PAMP. Collectively, they serve as ligands for the TLR2–TLR6 heterodimer, the TLR2–TLR1 heterodimer and TLR4, and CD14 is involved in at least some of these cases.
Non-GPI-related protozoan TLR ligands. Although most of the limited literature on innate immune activators from parasites has focused on GPI anchors, other protozoan molecules also serve as important orchestrators of pro-inflammatory responses. One example is a _T. cruzi_-derived protein, Tc52, which induces the synthesis of pro-inflammatory cytokines by host cells through TLR2 (Ref. 25). In addition, it is becoming clear that TLR9, well known as a receptor for unmethylated bacterial CpG DNA motifs, is important for the induction of pro-inflammatory cytokines during infection with protozoans. DNA from protozoan parasites such as T. cruzi, T. brucei and Babesia bovis stimulates both macrophage and DC activation26,27 probably through unmethylated CpG motifs28. Recently, the pro-inflammatory activity of T. cruzi and T. brucei genomic DNA has been shown to be mediated by TLR9 (Refs 29, 30) (Fig. 1).
The genomic DNA from Plasmodium spp. has a high AT (70–80%) and low GC (20–30%) content, and its role in TLR9 activation has not been determined. In fact, several studies have shown that non-DNA components of P. falciparum can activate innate immune cells through TLR9. Therefore, a non-DNA heat-labile component and haemozoin, which had previously been shown to stimulate the production of pro-inflammatory cytokines by macrophages32,33, were shown to activate human and mouse DCs through TLR9 (Refs 34, 35). An important question that remains to be solved is whether fragments of parasite genomic DNA that are associated with these P. falciparum preparations are involved in TLR9 activation.
A profilin-like protein from T. gondii (PFTG) has been found to activate TLR11 in mouse cells36. PFTG is present as a relatively conserved molecule in a number of apicomplexans, indicating that these proteins might serve as another broad class of protozoan PAMPs. Although its exact function in the parasite is unknown, PFTG is predicted to bind to actin and, like flagellin, might be involved in parasite motility and invasion of the host cell. The induction of IL-12 in DCs exposed to PFTG is mediated by TLR11, as the response was abolished in DCs from TLR11-deficient mice. In addition, mice lacking TLR11 show increased susceptibility to infection with T. gondii, and this is associated with decreased IL-12 production in vivo36. However, the TLR11 gene in humans has a premature stop codon and therefore encodes a non-functional form of TLR11. Accordingly, it would seem that PFTG does not activate human DCs to produce IL-12. Finally, other T. gondii molecules, such as tachyzoite heat-shock proteins and other partially purified tachyzoite preparations, have been shown to activate TLR4 and TLR2, respectively37,38. From these collective studies, the pattern that emerges is that for any given protozoan, multiple TLR-binding molecules are likely to be expressed (Fig. 1).
Role of TLR signalling in resistance to protozoans
It is now well established that TLRs are important for defence against every known category of human microbial pathogen. Once activated by microbial PAMPs, TLRs interact with adaptor proteins. TLR molecules have a cytoplasmic domain that is homologous to the IL-1 receptor, and is known as the TIR domain39. The best-studied TIR-domain-containing adaptor protein is myeloid differentiation primary-response gene 88 (MyD88), which transduces signals for all TLRs except TLR3, as well as the signals for IL-1 and the IL-18 receptors40,41. Other TIR-domain-containing adaptors are MyD88-adaptor-like protein (MAL; also known as TIRAP), TIR-domain-containing adaptor protein inducing interferon-β (TRIF; also known as TICAM1), and TRIF-related adaptor molecule (TRAM)42,43,44. MAL is required for TLR2 and TLR4 signalling, whereas TRIF is used by TLR3 and TLR4 (Refs 44–46). TRAM has been shown to associate only with TLR4 (Ref. 43).
The most convincing data indicating the importance of the TIR signalling pathway in host resistance and pathogenesis during parasitic diseases are those obtained from infections of MyD88-deficient mice with various protozoan parasites30,47,48,49 (Table 2). MyD88-deficient mice are highly susceptible to infection with T. gondii, such that all animals die within 10 days of infection. Increased susceptibility is associated with impaired production of the T helper 1 (TH1)-associated cytokines IFNγ and IL-12 and marked parasitaemia49. Similar results were obtained from MyD88-deficient mice infected with either T. cruzi47 or T. brucei30. Knockout of Myd88 in otherwise resistant C57BL/6 mice confers susceptibility to infection with L. major. This susceptibility is characterized by large non-healing lesions, marked parasitism and the development of a TH2-type immune response, as opposed to the normal IL-12-dependent TH1-type protective immune response that develops in the wild-type C57BL/6 mice48.
Table 2 TLR signalling pathways in host resistance to and pathogenesis of parasitic infection
A distinct scenario was observed in the case of rodent malaria. Similar to infection with other protozoans, innate immunity and activation of MyD88 in particular, have an important role in the production of pro-inflammatory cytokines, such as IL-12, TNF and IFNγ50. However, infection of MyD88-deficient mice with Plasmodium berghei (the causative agent of rodent malaria) resulted in impaired cytokine production but showed less pathology and an improved outcome51. Therefore, although signalling through MyD88 in innate immune cells has a protective role in most cases of protozoan infection by activating a TH1-associated immune response, in other situations decreased pro-inflammatory responses resulting from a lack of MyD88 signalling might be beneficial to the host.
Despite studies indicating an important role for TLR signalling in resistance to protozoan infection as is observed in Myd88 −/− mice, mice deficient in a single TLR do not show marked increases in susceptibility to infection in most cases19,30,47,49,51,52,53,54. For example, lack of expression of TLR2, which recognizes protozoan GPI anchors, did not affect the susceptibility of mice to infection with T. cruzi. By contrast, mice that have an inactivating mutation in Tlr4 (C3H/HeJ mice) show a small but significant enhancement in susceptibility to infection with T. cruzi compared with wild-type C3H/HeN mice, which have a functional TLR4 molecule. However, recent data indicate that susceptibility to infection was not significantly altered in _Tlr4_-knockout mice on the C57BL/6 genetic background (R.T.G., unpublished observations), which might point to a role for the genetic background of the mouse in their susceptibility to infection in the absence of TLR4. No major phenotype is observed in terms of parasitism or immune responses when either TLR2-deficient or TLR4-deficient mice are infected with T. brucei30, T. gondii52,54, P. berghei51 or L. major53. By contrast, although not as susceptible as the Myd88 −/− mice, TLR9-deficient mice show increased parasitaemia, as well as accelerated mortality, following infection with either T. cruzi29 or T. brucei30. Furthermore, TLR11-deficient mice are more susceptible to infection with T. gondii, showing an increase in cyst numbers in the central nervous system, and a decrease in IL-12 and IFNγ production, compared with wild-type mice36. However, these mice are not as susceptible as Myd88 −/− mice infected with T. gondii. Therefore, these results indicate that protozoan parasites are recognized by multiple TLRs, and the lack of a specific functional TLR might not be sufficient to result in the dramatic enhancement of host susceptibility to infection that is seen with MyD88 deficiency19.
We propose that host defence against pathogens is orchestrated by multiple TLRs that, in addition to having a redundant role, might lead to distinct responses by different host cells. In support of this hypothesis, it has been shown in vitro that GPI mucin and DNA from T. cruzi can provide synergistic signals to induce cytokine synthesis by macrophages and DCs. In addition, mice that are deficient in both TLR2 and TLR9 are more susceptible to acute infection with T. cruzi than mice that are deficient in one of the receptors29. The inability to control parasite growth was found to correlate with deficient TH1-cell responses in vivo after infection with T. cruzi. However, mice that are deficient in both TLR2 and TLR9 are not as susceptible to T. cruzi as Myd88 −/− mice, indicating that another TLR, possibly TLR4, might contribute to early host resistance to infection with this parasite. Nevertheless, this study clearly indicates a dominant role for TLR9 in the induction of IL-12 and IFNγ expression during infection with T. cruzi. In addition, the B2 receptor for bradykinin was shown to cooperate with TLR2 in inducing TH1-cell responses during infection with T. cruzi55. Furthermore, emerging evidence indicates that other innate immune receptors, which are different from TLRs, might be involved in the initial recognition of protozoan parasites (Box 3). Therefore, future studies with mice that are deficient in multiple TLRs and possibly other host receptors are clearly required to fully understand how TLRs cooperate among themselves and/or with other endogenous host components to trigger pro-inflammatory responses and initiate host defence against protozoan infection.
Protozoan regulation of TLR activation
TLR-dependent pro-inflammatory cascades that are triggered by infections with protozoan parasites and other microbial pathogens must be tightly controlled to avoid immunopathology or possibly death. Also, an over-exuberant immune response is a negative outcome for the parasite, which seeks to keep the host alive long enough to allow its transmission to another host. Accordingly, a common aspect of protozoan infection is the induction of endogenous anti-inflammatory mediators. Not only do these mediators avert an over-exuberant immune response, but, at least in some cases, can directly facilitate parasite persistence. In addition, it is becoming increasingly apparent that protozoan parasites directly interfere with signalling cascades that emanate from cognate recognition receptors such as the TLR family.
Manipulation of TLR signalling pathways by endogenous host mediators. The anti-inflammatory cytokine IL-10, which is induced during infection with protozoan parasites, and is well known for its ability to downregulate pro-inflammatory mediators such as IL-12, TNF and RNIs that are triggered through the TLR signalling pathway56. Infection of IL-10-deficient mice with normally avirulent Toxoplasma strains results in increased mortality, which is associated with hyperproduction of pro-inflammatory cytokines57,58. A similar lethal pro-inflammatory phenotype occurs during infection of IL-10-deficient mice with T. cruzi59, and the severity of both cerebral malaria associated with infection with P. falciparum in humans and malaria in rodents is also associated with a lack of IL-10 production60,61,62. By contrast, lack of IL-10 during infection with L. major is not lethal to the host, but does result in total elimination of the parasite63. This is presumably a consequence of increased microbicidal activity mediated by pro-inflammatory cytokines. So, IL-10 controls pro-inflammatory responses and effector mechanisms during infection, and allows long-term persistence of the parasite within the host.
In addition to IL-10, some parasites induce the production of transforming growth factor-β (TGFβ). This cytokine also has anti-inflammatory properties that promote the survival of protozoans, including Plasmodium chabaudi, Leishmania chagasi, T. cruzi and T. gondii64,65,66,67,68. The activity of TGFβ that favours protozoan infections might be due to suppression of innate cells that produce IFNγ and thereby trigger TH1-cell differentiation69 and might be due to modulation of the effector mechanisms of macrophages70,71. Evidence also indicates that parasitic induction of host lipoxin A4, an anti-inflammatory eicosanoid, might contribute to the downregulation of IL-12 and the avoidance of immunopathology during infection with T. gondii72,73.
Manipulation of TLR signalling pathways by protozoan parasites. In addition to the activation of TLR signalling pathways, there are several examples of interference with pro-inflammatory signal-transduction pathways by protozoans (Fig. 2). Although T. gondii seems to express multiple TLR ligands, macrophages, neutrophils and DCs become unresponsive to LPS-induced activation following infection with this parasite, as measured by the production of IL-12 and TNF, the upregulation of co-stimulatory molecules, and the ability to stimulate T cells74,75,76,77. These effects are mediated, at least in part, through activation of signal transducer and activator of transcription 3 (STAT3; Fig. 2a), a signalling intermediate through which IL-10 exerts its anti-inflammatory effects78. In STAT3-deficient macrophages the ability of T. gondii to block LPS-induced cytokine production is greatly reduced79. Rather than inducing IL-10 directly, T. gondii triggers STAT3 phosphorylation during infection79. Whether this is due to parasite-induced activation of upstream Janus kinase (JAK) molecules, which phosphorylate STATs, or direct STAT3 phosphorylation is not yet clear. Nor is it clear how STAT3 functions during infection with T. gondii or even during IL-10 stimulation. Nevertheless, the implication of these studies is that this parasite can directly hijack the IL-10 signalling pathway to downregulate pro-inflammatory responses.
Figure 2: Manipulation of Toll-like receptor signalling pathways by protozoans.
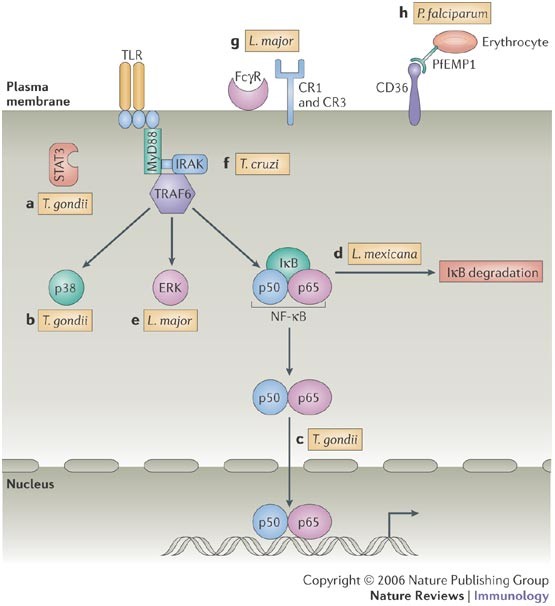
a | Direct activation of signal transducer and activator of transcription 3 (STAT3) by Toxoplasma gondii downregulates interleukin-12 (IL-12) and tumour-necrosis factor (TNF) production. b,c | The parasite also prevents p38 mitogen-activated protein kinase (MAPK) phosphorylation (b) and blocks accumulation of nuclear factor-κB (NF-κB) (c) in the host cell nucleus following the triggering of Toll-like receptor 4 (TLR4). d | A Leishmania mexicana cysteine protease has been identified that is involved in the degradation of inhibitor of NF-κB (IκB) and NF-κB molecules. e | The activation of the extracellular-signal-regulated kinase 1 (ERK1)/ERK2 MAPK pathway has been implicated in the ability of Leishmania major to downregulate IL-12 production by macrophages. f | Trypanosoma cruzi induces tolerance to secondary stimulation, and this is characterized by the induction of type 2A protein phosphatase, which deactivates IL-1-receptor-associated kinase (IRAK), MAPK and NF-κB molecules. g | Antibody- or complement-coated L. major interacts with host receptors for antibodies (such as FcγR), complement receptor 1 (CR1) and CR3, resulting in downregulation of TLR-driven IL-12 production. h | Plasmodium falciparum erythrocyte membrane protein 1 (PfEMP1), which is expressed on the surface of erythrocytes infected with P. falciparum, interacts with CD36 expressed by the host cell, resulting in dendritic-cell deactivation, characterized by unresponsiveness to TLR ligands. MyD88, myeloid differentiation primary-response gene 88; TRAF6, TNF receptor-associated factor 6.
Toxoplasma gondii also regulates LPS-induced MAPK activation80; in particular, the activation of p38 MAPK, which is required for IL-12 production (Fig. 2b). The effect of T. gondii on p38 MAPK signalling, however, seems to be distinct from the phenomenon of LPS tolerance, which is also characterized by MAPK inactivation3,81,82. This is because tolerance induced by LPS targets signalling molecules that are found immediately proximal to the TLR. By contrast, stimulation of _T. gondii-_infected cells with LPS results in the activation of MAPK kinase 3 (MKK3; also known as MAP2K3) and MKK6 (also known as MAP2K6), which are upstream activators of p38 MAPK; degradation of IκB is also observed80. This indicates either that the target for inactivation by T. gondii is another p38-activating kinase (such as MKK4; also known as MAP2K4) or that T. gondii induces a p38 MAPK phosphatase, which prevents the phosphorylation and therefore activation of this kinase.
Activation of NF-κB is also targeted by T. gondii. Although infection induces rapid IκB phosphorylation and degradation, the NF-κB molecule fails to translocate to the nucleus83,84. Similarly, NF-κB fails to translocate when infected cells are stimulated with LPS (Fig. 2c), although this effect is lost within 6 hours of infection74. Recent studies indicate that the apparent lack of translocation might in fact be the result of increased nuclear export rather than decreased nuclear import of NF-κB85. There are also indications that the defect in NF-κB activation might be a characteristic of high-virulence parasite strains, not low-virulence strains86.
Amastigotes of Leishmania mexicana, which are largely devoid of LPGs, specifically suppress IL-12 production by macrophages87. This seems to be due to the expression of amastigote-specific cysteine peptidases88. These molecules, previously identified as virulence factors, modulate the NF-κB signalling pathway by proteolytic breakdown of IκB and NF-κB (Fig. 2d). How these cysteine peptidases exit the vacuole that contains L. mexicana and enter the cytoplasm of the host cell has not yet been determined. Related to these studies, L. major promastigotes also specifically suppress IL-12 production by macrophages during LPS stimulation. In part, this might be due to the stimulation of an extracellular-signal-regulated kinase 1 (ERK1)/ERK2 MAPK pathway by promastigote LPGs, which results in the negative regulation of IL-12 production89 (Fig. 2e). It is of interest that LPG itself is a TLR2 ligand, because other TLR2 ligands, such as Pam3Cys (tripalmitoyl-_S_-glyceryl cysteine), have also been reported to inhibit IL-12 production through the induction of the ERK1/ERK2 MAPK pathway90. Such cross-regulation in TLR pathways is likely to be an important aspect of the innate response to infection.
Nevertheless, it is clear that not all TLR2 ligands act to downregulate IL-12 production directly. GPI mucin from T. cruzi trypomastigotes induces potent IL-12 responses. Similar to LPS, GPI mucin also induces tolerance to secondary stimulation91. Stimulation with either LPS or GPI mucin induces protein phosphatase 2A (PP2A) activity that acts to dismantle the TLR signalling cascade, modulating macrophage pro-inflammatory responses to TLR agonists. This occurs through the deactivation of interleukin-1 receptor-associated kinase 1 (IRAK1), MAPKs and IκB molecules, thereby inducing tolerance92 (Fig. 2f). Control of PP2A occurs through an autoregulatory loop, as induction of its phosphatase activity is dependent on signalling through both p38 MAPK and NF-κB.
IL-12 suppression during infection with Leishmania spp. can also be induced by receptor-mediated uptake of opsonized parasites (Fig. 2g). Signalling through complement receptor 1 (CR1; also known as CD35) and CR3 negatively regulates IL-12 production through impaired tyrosine phosphorylation of STAT1 during activation with LPS and IFNγ93. For infection with L. major, this might be due to the induction of protein tyrosine phosphatases, such as SHP1 (SRC homology 2 (SH2)-domain-containing protein tyrosine phosphatase 1) (Refs 94, 95). Related to these findings, ligation of the macrophage FcγR (a high-affinity receptor for IgG) by opsonized parasites leads to the suppression of IL-12 transcription96. Although this might be a direct inhibitory effect on the IL-12p40 promoter, it was also found that FcγR-mediated uptake of L. major amastigotes stimulated IL-10 release, inhibiting IL-12p40 transcription and IL-12 synthesis. As a consequence, infected macrophages are refractory to the activating effects of IFNγ, enhancing intracellular survival of the parasite97.
DCs that phagocytose red blood cells infected with P. falciparum or Plasmodium yoelii become unresponsive to LPS-induced activation, resulting in defects in T-cell activation98,99,100. In the case of P. falciparum, these effects are mediated by the interaction of the scavenger receptor CD36, which is expressed by DCs, and PfEMP1 (P. falciparum erythrocyte membrane protein 1), which is expressed on infected erythrocytes101 (Fig. 2h), although the link between CD36 and TLR signalling pathways is not clear. On another level, phagocytosis of malaria haemozoin, which is a TLR9 agonist, during P. chabaudi infection results in the non-responsiveness of DCs to LPS and an inability to activate T cells102.
So, protozoans target TLR signalling pathways from within cells (Leishmania spp., T. gondii and T. cruzi) to influence host immune responses and might also exploit host cell-surface receptors (for example, Plasmodium spp. and Leishmania spp.) to suppress these pathways. The diverse mechanisms used by protozoans to downregulate TLR signalling are likely to reflect the need for these organisms to establish long-term residence in their hosts.
TLR-based strategies against protozoan infection
Protozoan infections exert a devastating toll on human health worldwide, and there is an urgent need for new strategies to treat and prevent disease. Exploitation of chemically defined microbial-derived TLR ligands as therapeutic and prophylactic tools holds promise in this regard. As discussed earlier, activation of TLRs leads to expression of co-stimulatory molecules and pro-inflammatory cytokines (for example, IL-12) that promote differentiation of TH1 cells. These cells mediate antigen-specific, cell-mediated immunity and produce IFNγ, which also drive protective antibody production by B cells, and therefore these cells are the basis of protective immunity against microbial infections1. In fact, many of the chemically defined microbial products being used as adjuvants or immunostimulants are TLR agonists (these include polyinosinic–polycytidylic acid (poly I:C) (TLR3 agonist), lipid A (TLR4 agonist), flagellin (TLR5 agonist), imiquimod (TLR7 and TLR8 agonist) and CpG DNA (TLR9 agonist))2. However, a crucial issue is to dissociate the beneficial effects (potentiation of immune responses) from the detrimental effects (oedema and pain) caused by these adjuvants103. In the case of infection with protozoan parasites, monophosphoryl lipid A (MPL) is in advanced stages of development for use in vaccine formulations using recombinant antigens of P. falciparum104 and Leishmania spp.105 to protect against these infections (Table 3). In addition, CpG-containing oligodeoxynucleotides have been successful in vaccine formulations that induce effective protective immunity against challenge from different protozoan parasites (such as Leishmania spp., Plasmodium spp., T. gondii and T. cruzi) in experimental models106,107,108,109,110. Bacteria-derived flagellin has been shown to be highly effective in stimulating mucosal immunity111. This might be of potential value for vaccine development against the subset of protozoan parasites, such as T. gondii, that infect through the intestinal tract.
Table 3 Areas of potential for the use of TLR-based strategies to prevent or treat diseases caused by protozoan parasites
A second area of interest with regard to clinical use of immunostimulatory TLR agonists is in the treatment of persistent chronic infections that are often refractory to conventional chemotherapy. Many chemotherapeutic drugs currently in use, especially those used for leishmaniasis and Chagas disease, are more effective when used in combination with vaccines or immunostimulants such as IL-12 or Mycobacterium bovis bacillus Calmette–Guérin112,113. Therefore, it might be valuable to use therapeutic vaccines in combination with TLR agonists, or simply to use TLR agonists in combination with commercially available anti-parasite drugs. In fact, studies using imiquimod114,115 or CpG-containing oligodeoxynucleotides116 are apparently effective in the treatment of cutaneous leishmaniasis in experimental models.
Another attractive area of immunological intervention in parasite disease is the use of TLR antagonists in cases in which activation of a TLR pathway is involved in pathology. MyD88 seems to be involved in the pathology associated with malaria51. So, use of a TLR antagonist during acute malaria episodes might potentially be of clinical effectiveness, as MyD88 and TLRs might not be crucial for protective immunity and parasite clearance during infection with Plasmodium spp. As P. falciparum GPI anchors are recognized by TLRs23, it is of interest that synthetic GPI anchors mimicking those of the parasite are effective as an anti-toxic vaccine, blocking the pathological effects of malaria117. However, an important issue is whether blocking TLR–TLR ligand interactions might interfere with the development of protective immunity during infection with other protozoans. This might be the case during infection with trypanosomatids or T. gondii that seem to depend largely on activation of MyD88 for resistance30,47,48,49.
The beneficial effect of the TLR-based interventions discussed here are at present largely hypothetical, and their potential as therapeutic agents needs to be further tested in experimental models and in the field to help define their efficacy. Nevertheless, the information accumulated in recent years regarding the cellular and molecular biology of TLRs, and their importance in the immunology to infectious diseases, brings high expectations in terms of the development of new strategies for prophylaxis of infections with protozoan parasites.
References
- Janeway, C. A. Jr. & Medzhitov, R. Innate immune recognition. Annu. Rev. Immunol. 20, 197–216 (2002).
CAS PubMed Google Scholar - Akira, S., Uematsu, S. & Takeuchi, O. Pathogen recognition and innate immunity. Cell 124, 783–801 (2006).
CAS PubMed Google Scholar - Dobrovolskaia, M. A. & Vogel, S. N. Toll receptors, CD14, and macrophage activation and deactivation by LPS. Microbes Infect. 4, 903–914 (2002).
CAS PubMed Google Scholar - Lien, E. & Ingalls, R. R. Toll-like receptors. Crit. Care Med. 30, S1–S11 (2002).
CAS PubMed Google Scholar - Janeway, C. A. Jr. How the immune system works to protect the host from infection: a personal view. Proc. Natl Acad. Sci. USA 98, 7461–7468 (2001).
CAS PubMed PubMed Central Google Scholar - Gazzinelli, R. T., Ropert, C. & Campos, M. A. Role of the Toll/interleukin-1 receptor signaling pathway in host resistance and pathogenesis during infection with protozoan parasites. Immunol. Rev. 201, 9–25 (2004).
CAS PubMed Google Scholar - Camargo, M. M. et al. Glycosylphosphatidylinositol-anchored mucin-like glycoproteins isolated from Trypanosoma cruzi trypomastigotes initiate the synthesis of proinflammatory cytokines by macrophages. J. Immunol. 158, 5890–5901 (1997).
CAS PubMed Google Scholar - de Veer, M. J. et al. MyD88 is essential for clearance of Leishmania major: possible role for lipophosphoglycan and Toll-like receptor 2 signaling. Eur. J. Immunol. 33, 2822–2831 (2003).
CAS PubMed Google Scholar - Debierre-Grockiego, F. et al. Roles of glycosylphosphatidylinositols of Toxoplasma gondii. Induction of tumor necrosis factor-α production in macrophages. J. Biol. Chem. 278, 32987–32993 (2003). An important study revealing that T. gondii GPI anchors also have pro-inflammatory properties. The study uses synthetic GPI anchors and lends support to the idea that protozoan GPI anchors are a major class of PAMPs.
CAS PubMed Google Scholar - Magez, S. et al. The glycosyl-inositol-phosphate and dimyristoylglycerol moieties of the glycosylphosphatidylinositol anchor of the trypanosome variant-specific surface glycoprotein are distinct macrophage-activating factors. J. Immunol. 160, 1949–1956 (1998).
CAS PubMed Google Scholar - Schofield, L. & Hackett, F. Signal transduction in host cells by a glycosylphosphatidylinositol toxin of malaria parasites. J. Exp. Med. 177, 145–153 (1993).
CAS PubMed Google Scholar - Camargo, M. M., Andrade, A. C., Almeida, I. C., Travassos, L. R. & Gazzinelli, R. T. Glycoconjugates isolated from Trypanosoma cruzi but not from Leishmania species membranes trigger nitric oxide synthesis as well as microbicidal activity in IFN-γ-primed macrophages. J. Immunol. 159, 6131–6139 (1997).
CAS PubMed Google Scholar - Talvani, A. et al. Kinetics of cytokine gene expression in experimental chagasic cardiomyopathy: tissue parasitism and endogenous IFN-γ as important determinants of chemokine mRNA expression during infection with Trypanosoma cruzi. Microbes Infect. 2, 851–866 (2000).
CAS PubMed Google Scholar - Ferguson, M. A. J. The structure, biosynthesis and functions of glycosylphosphatidylinositol anchor, and the contributions of trypanosome research. J. Cell Sci. 112, 2799 (1999).
CAS PubMed Google Scholar - Almeida, I. C. & Gazzinelli, R. T. Proinflammatory activity of glycosylphosphatidylinositol anchors derived from Trypanosoma cruzi: structural and functional analyses. J. Leukoc. Biol. 70, 467–477 (2001).
CAS PubMed Google Scholar - Campos, M. A. S. et al. Activation of Toll-like receptor-2 by glycosylphosphatidylinositol anchors from a protozoan parasite. J. Immunol. 167, 416–423 (2001). Establishes that the pro-inflammatory activity of T. cruzi -derived GPI anchors is mediated by TLR2.
CAS PubMed Google Scholar - Petersen, C. A., Krumholz, K. A. & Burleigh, B. A. Toll-like receptor 2 regulates interleukin-1β-dependent cardiomyocyte hypertrophy triggered by Trypanosoma cruzi. Infect. Immun. 73, 6974–6980 (2005).
CAS PubMed PubMed Central Google Scholar - Ropert, C. & Gazzinelli, R. T. Regulatory role of Toll-like receptor 2 during infection with Trypanosoma cruzi. J. Endotoxin Res. 10, 425–430 (2004).
PubMed Google Scholar - Oliveira, A. C. et al. Expression of functional TLR4 confers proinflammatory responsiveness to Trypanosoma cruzi glycoinositolphospholipids and higher resistance to infection with T. cruzi. J. Immunol. 173, 5688–5696 (2004).
CAS PubMed Google Scholar - McConville, M. J., Schnur, L. F., Jaffe, C. & Schneider, P. Structure of Leishmania lipophosphoglycan: inter- and intra-specific polymorphism in Old World species. Biochem. J. 310, 807–818 (1995).
CAS PubMed PubMed Central Google Scholar - Becker, I. et al. Leishmania lipophosphoglycan (LPG) activates NK cells through Toll-like receptor-2. Mol. Biochem. Parasitol. 130, 65–74 (2003).
CAS PubMed Google Scholar - Flandin, J. F., Chano, F. & Descoteaux, A. RNA interference reveals a role for TLR2 and TLR3 in the recognition of Leishmania donovani promastigotes by interferon-γ-primed macrophages. Eur. J. Immunol. 36, 411–420 (2006).
CAS PubMed Google Scholar - Krishnegowda, G. et al. Induction of proinflammatory responses in macrophages by the glycosylphosphatidylinositols of Plasmodium falciparum: cell signaling receptors, glycosylphosphatidylinositol (GPI) structural requirement, and regulation of GPI activity. J. Biol. Chem. 280, 8606–8616 (2005).
CAS PubMed Google Scholar - Naik, R. S. et al. Glycosylphosphatidylinositol anchors of Plasmodium falciparum: molecular characterization and naturally elicited antibody response that may provide immunity to malaria pathogenesis. J. Exp. Med. 192, 1563–1576 (2000).
CAS PubMed PubMed Central Google Scholar - Ouaissi, A. et al. The Trypanosoma cruzi Tc52-released protein induces human dendritic cell maturation, signals via Toll-like receptor 2, and confers protection against lethal infection. J. Immunol. 168, 6366–6374 (2002).
CAS PubMed Google Scholar - Harris, T. H., Cooney, N. M., Mansfield, J. M. & Paulnock, D. M. Signal transduction, gene transcription, and cytokine production triggered in macrophages by exposure to trypanosome DNA. Infect. Immun. 74, 4530–4537 (2006).
CAS PubMed PubMed Central Google Scholar - Shoda, L. K. et al. DNA from protozoan parasites Babesia bovis, Trypanosoma cruzi, and T. brucei is mitogenic for B lymphocytes and stimulates macrophage expression of interleukin-12, tumor necrosis factor α, and nitric oxide. Infect. Immun. 69, 2162–2171 (2001).
CAS PubMed PubMed Central Google Scholar - Brown, W. C. & Corral, R. S. Stimulation of B lymphocytes, macrophages, and dendritic cells by protozoan DNA. Microbes Infect. 4, 969–974 (2002).
CAS PubMed Google Scholar - Bafica, A. et al. TLR9 and TLR2 signaling together account for MyD88-dependent control of parasitemia in Trypanosoma cruzi infection. J. Immunol. 177, 3515–3519 (2006). Shows that activation of innate immune cells, development of T H 1-cell responses and optimal protection against T. cruzi involve the coordinated activity of multiple TLRs, including TLR9 and TLR2.
CAS PubMed Google Scholar - Drennan, M. B. et al. The induction of a type 1 immune response following a Trypanosoma brucei infection is MyD88 dependent. J. Immunol. 175, 2501–2509 (2005).
CAS PubMed Google Scholar - Gardner, M. J. et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature 419, 498–511 (2002).
CAS PubMed Google Scholar - Jaramillo, M., Gowda, D. C., Radzioch, D. & Olivier, M. Hemozoin increases IFN-γ-inducible macrophage nitric oxide generation through extracellular signal-regulated kinase- and NF-κB-dependent pathways. J. Immunol. 171, 4243–4253 (2003).
CAS PubMed Google Scholar - Jaramillo, M. et al. Hemozoin-inducible proinflammatory events in vivo: potential role in malaria infection. J. Immunol. 172, 3101–3110 (2004).
CAS PubMed Google Scholar - Pichyangkul, S., Saengkrai, P. & Webster, H. K. Plasmodium falciparum pigment induces monocytes to release high levels of tumor necrosis factor-α and interleukin-1β. Am. J. Trop. Med. Hyg. 51, 430–435 (1994).
CAS PubMed Google Scholar - Coban, C. et al. Toll-like receptor 9 mediates innate immune activation by the malaria pigment hemozoin. J. Exp. Med. 201, 19–25 (2005). Reports the surprising finding that haemozoin is recognized by TLR9, which is more commonly known as a detector of hypomethylated bacterial DNA containing CpG motifs.
CAS PubMed PubMed Central Google Scholar - Yarovinsky, F. et al. TLR11 activation of dendritic cells by a protozoan profilin-like protein. Science 308, 1626–1629 (2005). Identifies the first chemical ligand of TLR11, a T. gondii profilin-like protein. Importantly, Tlr11 knockout increases susceptibility to this parasite, although not as dramatically as Myd88 knockout.
CAS PubMed Google Scholar - Aosai, F. et al. _Toxoplasma gondii_-derived heat shock protein HSP70 functions as a B cell mitogen. Cell Stress Chaperones 7, 357–364 (2002).
CAS PubMed PubMed Central Google Scholar - Del Rio, L. et al. Toxoplasma gondii triggers myeloid differentiation factor 88-dependent IL-12 and chemokine ligand 2 (monocyte chemoattractant protein 1) responses using distinct parasite molecules and host receptors. J. Immunol. 172, 6954–6960 (2004).
CAS PubMed Google Scholar - O'Neill, L. A., Fitzgerald, K. A. & Bowie, A. G. The Toll-IL-1 receptor adaptor family grows to five members. Trends Immunol. 24, 286–290 (2003).
PubMed Google Scholar - Adachi, O. et al. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity 9, 143–150 (1998).
CAS PubMed Google Scholar - Takeuchi, O. & Akira, S. MyD88 as a bottle neck in Toll/IL-1 signaling. Curr. Top. Microbiol. Immunol. 270, 155–167 (2002).
CAS PubMed Google Scholar - Fitzgerald, K. A. et al. Mal (MyD88-adapter-like) is required for Toll-like receptor-4 signal transduction. Nature 413, 78–83 (2001).
CAS PubMed Google Scholar - Fitzgerald, K. A. et al. LPS/TLR4 signaling to IRF-3/7 and NF-κB involves the Toll adapters TRAM and TRIF. J. Exp. Med. 198, 1043–1055 (2003).
CAS PubMed PubMed Central Google Scholar - Yamamoto, M. et al. Role of adaptor TRIF in the MyD88-independent Toll-like receptor signaling pathway. Science 301, 640–643 (2003).
CAS PubMed Google Scholar - Horng, T., Barton, G. M., Flavell, R. A. & Medzhitov, R. The adaptor molecule TIRAP provides signalling specificity for Toll-like receptors. Nature 420, 329–333 (2002).
CAS PubMed Google Scholar - Yamamoto, M. et al. Essential role for TIRAP in activation of the signalling cascade shared by TLR2 and TLR4. Nature 420, 324–329 (2002).
CAS PubMed Google Scholar - Campos, M. A. et al. Impaired production of proinflammatory cytokines and host resistance to acute infection with Trypanosoma cruzi in mice lacking functional myeloid differentiation factor 88. J. Immunol. 172, 1711–1718 (2004). Shows the involvement of MyD88-dependent and MyD88-independent pathways in the induction of IL-12, IFNγ and host resistance to the protozoan parasite T. cruzi.
CAS PubMed Google Scholar - Muraille, E. et al. Genetically resistant mice lacking MyD88-adapter protein display a high susceptibility to Leishmania major infection associated with a polarized TH2 response. J. Immunol. 170, 4237–4241 (2003).
CAS PubMed Google Scholar - Scanga, C. A. et al. MyD88 is required for resistance to Toxoplasma gondii infection and regulates parasite-induced IL-12 production by dendritic cells. J. Immunol. 168, 5997–6001 (2002). The first evidence that Myd88 knockout dramatically increases susceptibility to T. gondii . The study directly implicates TLRs in resistance to protozoan infection.
CAS PubMed Google Scholar - Stevenson, M. M. & Riley, E. M. Innate immunity to malaria. Nature Rev. Immunol. 4, 169–180 (2004).
CAS Google Scholar - Adachi, K. et al. Plasmodium berghei infection in mice induces liver injury by an IL-12- and Toll-like receptor/myeloid differentiation factor 88-dependent mechanism. J. Immunol. 167, 5928–5934 (2001). Shows the important principle that signalling through MyD88 can have negative consequences for the host during protozoan infection. This is a significant caveat when considering TLR ligands as immunotherapeutic tools.
CAS PubMed Google Scholar - Chen, M. et al. Involvement of MyD88 in host defense and the down-regulation of anti-heat shock protein 70 autoantibody formation by MyD88 in _Toxoplasma gondii_-infected mice. J. Parasitol. 88, 1017–1019 (2002).
CAS PubMed Google Scholar - Kropf, P. et al. Toll-like receptor 4 contributes to efficient control of infection with the protozoan parasite Leishmania major. Infect. Immun. 72, 1920–1928 (2004).
CAS PubMed PubMed Central Google Scholar - Mun, H. S. et al. TLR2 as an essential molecule for protective immunity against Toxoplasma gondii infection. Int. Immunol. 15, 1081–1087 (2003).
CAS PubMed Google Scholar - Monteiro, A. C. et al. Cooperative activation of TLR2 and bradykinin B2 receptor is required for induction of type-1 immunity in a mouse model of subcutaneous infection by Trypanosoma cruzi. J. Immunol. 177, 6325–6335 (2006).
CAS PubMed Google Scholar - Petska, S. et al. Interleukin-10 and related cytokines and receptors. Annu. Rev. Immunol. 22, 929–979 (2004).
Google Scholar - Gazzinelli, R. T. et al. In the absence of endogenous IL-10, mice acutely infected with Toxoplasma gondii succumb to a lethal immune response dependent upon CD4+ T cells and accompanied by overproduction of IL-12, IFN-γ, and TNF-α. J. Immunol. 157, 798–805 (1996).
CAS PubMed Google Scholar - Suzuki, Y. et al. IL-10 is required for prevention of necrosis in the small intestine and mortality in both genetically resistant BALB/c and susceptible C57BL/6 mice following peroral infection with Toxoplasma gondii. J. Immunol. 164, 5375–5382 (2000).
CAS PubMed Google Scholar - Hunter, C. A. et al. IL-10 is required to prevent immune hyperactivity during infection with Trypanosoma cruzi. J. Immunol. 158, 3311–3316 (1997).
CAS PubMed Google Scholar - Kossodo, S. et al. Interleukin-10 modulates susceptibility in experimental cerebral malaria. Immunology 91, 536–540 (1997).
CAS PubMed PubMed Central Google Scholar - Kurtzhals, J. A. et al. Low plasma concentrations of interleukin 10 in severe malarial anaemia compared with cerebral and uncomplicated malaria. Lancet 351, 1768–1772 (1998).
CAS PubMed Google Scholar - Sanni, L. A., Jarra, W., Li, C. & Langhorne, J. Cerebral edema and cerebral hemorrhages in interleukin-10-deficient mice infected with Plasmodium chabaudi. Infect. Immun. 72, 3054–3058 (2004).
CAS PubMed PubMed Central Google Scholar - Belkaid, Y. et al. The role of interleukin (IL)-10 in the persistence of Leishmania major in the skin after healing and the therapeutic potential of anti-IL-10 receptor antibody for sterile cure. J. Exp. Med. 194, 1497–1506 (2001).
CAS PubMed PubMed Central Google Scholar - Buzoni-Gatel, D. et al. Murine ileitis after intracellular parasite infection is controlled by TGF-β-producing intraepithelial lymphocytes. Gastroenterology 120, 914–924 (2001).
CAS PubMed Google Scholar - Gantt, K. R. et al. Activation of TGF-β by Leishmania chagasi: importance for parasite survival in macrophages. J. Immunol. 170, 2613–2620 (2003).
CAS PubMed Google Scholar - Gazzinelli, R. T., Oswald, I. P., James, S. & Sher, A. IL-10 inhibits parasite killing and nitrogen oxide production by IFN-γ activated macrophages. J. Immunol. 148, 1792–1796 (1992).
CAS PubMed Google Scholar - Li, C., Sanni, L. A., Omer, F., Riley, E. & Langhorne, J. Pathology of Plasmodium chabaudi chabaudi infection and mortality in interleukin-10-deficient mice are ameliorated by anti-tumor necrosis factor α and exacerbated by anti-transforming growth factor β antibodies. Infect. Immun. 71, 4850–4856 (2003).
CAS PubMed PubMed Central Google Scholar - Silva, J. S. et al. Interleukin-10 and interferon-γ regulation of experimental Trypanosoma cruzi infection. J. Exp. Med. 175, 169–174 (1992).
CAS PubMed Google Scholar - Scharton-Kersten, T., Afonso, L. C., Wysocka, M., Trinchieri, G. & Scott, P. IL-12 is required for natural killer cell activation and subsequent T helper 1 cell development in experimental leishmaniasis. J. Immunol. 154, 5320–5330 (1995).
CAS PubMed Google Scholar - Gazzinelli, R. T., Oswald, I. P., Hieny, S., James, S. & Sher, A. The microbicidal activity of interferon-γ-treated macrophages against Trypanosoma cruzi involves an L-arginine-dependent, nitrogen oxide-mediated mechanism inhibitable by interleukin-10 and transforming growth factor-β. Eur. J. Immunol. 22, 2501–2506 (1992).
CAS PubMed Google Scholar - Silva, S. J., Twardzik, D. R. & Reed, S. G. Regulation of Trypanosoma cruzi infections in vitro and in vivo by transforming growth factor-β (TGF-β). J. Exp. Med. 174, 539–545 (1991).
CAS PubMed Google Scholar - Aliberti, J., Serhan, C. & Sher, A. Parasite-induced lipoxin A4 is an endogenous regulator of IL-12 production and immunopathology in Toxoplasma gondii infection. J. Exp. Med. 196, 1253–1262 (2002).
CAS PubMed PubMed Central Google Scholar - Bannenberg, G. L., Aliberti, J., Hong, S., Sher, A. & Serhan, C. Exogenous pathogen and plant 15-lipoxygenase initiate endogenous lipoxin A4 biosynthesis. J. Exp. Med. 199, 515–523 (2004).
CAS PubMed PubMed Central Google Scholar - Butcher, B. A. & Denkers, E. Y. Mechanism of entry determines ability of Toxoplasma gondii to inhibit macrophage proinflammatory cytokine production. Infect. Immun. 70, 5216–5224 (2002).
CAS PubMed PubMed Central Google Scholar - Denkers, E. Y., Kim, L. & Butcher, B. A. In the belly of the beast: subversion of macrophage proinflammatory signaling cascades during Toxoplasma gondii infection. Cell. Micro. 5, 75–83 (2003).
CAS Google Scholar - McKee, A. S., Dzierszinski, F., Boes, M., Roos, D. S. & Pearce, E. J. Functional inactivation of immature dendritic cells by the intracellular parasite Toxoplasma gondii. J. Immunol. 173, 2632–2640 (2004).
CAS PubMed Google Scholar - Bennouna, S., Sukhumavasi, W. & Denkers, E. Y. Toxoplasma gondii inhibits Toll-like receptor (TLR)4 ligand-induced mobilization of pre-formed TNF-α in mouse peritoneal neutrophils. Infect. Immun. 74, 4274–4281 (2006).
CAS PubMed PubMed Central Google Scholar - Williams, L., Bradley, L., Smith, A. & Foxwell, B. Signal transducer and activator of transcription 3 is the dominant mediator of the anti-inflammatory effects of IL-10 in human macrophages. J. Immunol. 172, 567–576 (2004).
CAS PubMed Google Scholar - Butcher, B. A. et al. IL-10-independent STAT3 activation by Toxoplasma gondii mediates suppression of IL-12 and TNF-α in host macrophages. J. Immunol. 174, 3148–3152 (2005). Shows that T. gondii directly triggers STAT3 activation during infection, resulting in downregulation of pro-inflammatory cytokines and highlights a novel mechanism of interference with TLR signalling.
CAS PubMed Google Scholar - Kim, L., Butcher, B. A. & Denkers, E. Y. Toxoplasma gondii interferes with lipopolysaccharide-induced mitogen-activated protein kinase activation by mechanisms distinct from endotoxin tolerance. J. Immunol. 172, 3003–3010 (2004).
CAS PubMed Google Scholar - Dobrovolskaia, M. A. et al. Induction of in vitro reprogramming by Toll-like receptor (TLR)2 and TLR4 agonists in murine macrophages: effects of TLR 'homotolerance' versus 'heterotolerance' on NF-κB signaling pathway components. J. Immunol. 170, 508–519 (2003).
CAS PubMed Google Scholar - Sato, S. et al. A variety of microbial components induce tolerance to lipopolysaccharide by differentially affecting MyD88-dependent and independent pathways. Int. Immunol. 14, 783–791 (2002).
CAS PubMed Google Scholar - Butcher, B. A., Kim, L., Johnson, P. F. & Denkers, E. Y. Toxoplasma gondii tachyzoites inhibit proinflammatory cytokine induction in infected macrophages by preventing nuclear translocation of the transcription factor NFκB. J. Immunol. 167, 2193–2201 (2001). One of the first studies to show that intracellular T. gondii infection renders macrophages non-responsive to LPS. Simultaneously, the cells have impaired NF-κB nuclear translocation.
CAS PubMed Google Scholar - Shapira, S. S., Speirs, K., Gerstein, A., Caamano, J. & Hunter, C. A. Suppression of NF-κB activation by infection with Toxoplasma gondii. J. Infect. Dis. 185, S66–S72 (2002).
CAS PubMed Google Scholar - Shapira, S. et al. Initiation and termination of NFκB signaling by the intracellular protozoan parasite Toxoplasma gondii. J. Cell Sci. 118, 3501–3508 (2005).
CAS PubMed Google Scholar - Robben, P. M. et al. Production of IL-12 by macrophages infected with Toxoplasma gondii depends on the parasite genotype. J. Immunol. 172, 3686–3694 (2004).
CAS PubMed Google Scholar - Weinheber, N., Wolfram, M., Harbecke, D. & Aebischer, T. Phagocytosis of Leishmania mexicana amastigotes by macrophages leads to a sustained suppression of IL-12 production. Eur. J. Immunol. 28, 2467–2477 (1998).
CAS PubMed Google Scholar - Cameron, P. et al. Inhibition of lipopolysaccharide-induced macrophage IL-12 production by Leishmania mexicana amastigotes: the role of cyteine proteases and the NFκB signaling pathway. J. Immunol. 173, 3297–3304 (2004).
CAS PubMed Google Scholar - Feng, G.-J. et al. Extracellular signal-related kinase (ERK) and p38 mitogen-activated protein (MAP) kinases differentially regulate the lipopolysaccharide-mediated induction of inducible nitric oxide synthase and IL-12 in macrophages: Leishmania phosphoglycans subvert macrophage IL-12 production by targeting ERK MAP kinase. J. Immunol. 163, 6403–6412 (1999).
CAS PubMed Google Scholar - Agrawal, S. et al. Different Toll-like receptor agonists instruct dendritic cells to induce distinct TH responses via differential modulation of extracellular signal-regulated kinase-mitogen-activated protein kinase and c-Fos. J. Immunol. 171, 4984–4989 (2003).
CAS PubMed Google Scholar - Ropert, C. et al. Requirement of mitogen-activated protein kinases and IκB phosphorylation for induction of proinflammatory cytokines synthesis by macrophages indicates functional similarity of receptors triggered by glycosylphosphatidylinositol anchors from parasitic protozoa and bacterial lipopolysaccharide. J. Immunol. 166, 3423–3431 (2001).
CAS PubMed Google Scholar - Ropert, C., Closel, M., Chaves, A. C. & Gazzinelli, R. T. Inhibition of a p38/stress-activated protein kinase-2-dependent phosphatase restores function of IL-1 receptor-associate kinase-1 and reverses Toll-like receptor 2- and 4-dependent tolerance of macrophages. J. Immunol. 171, 1456–1465 (2003).
CAS PubMed Google Scholar - Marth, T. & Kelsall, B. L. Regulation of interleukin-12 by complement receptor 3 signaling. J. Exp. Med. 185, 1987–1995 (1997).
CAS PubMed PubMed Central Google Scholar - Blanchette, J., Racette, N., Faure, R., Siminovitch, K. A. & Olivier, M. _Leishmania_-induced increases in activation of macrophage SHP-1 tyrosine phosphatase are associated with impaired IFN-γ-triggered JAK2 activation. Eur. J. Immunol. 29, 3737–3744 (1999).
CAS PubMed Google Scholar - Forget, G. et al. Role of host phosphotyrosine phosphatase SHP-1 in the development of murine leishmaniasis. Eur. J. Immunol. 31, 3185–3196 (2001).
CAS PubMed Google Scholar - Grazia Cappiello, M., Sutterwala, F. S., Trinchieri, G., Mosser, D. M. & Ma, X. Suppression of Il-12 transcription in macrophages following Fcγ receptor ligation. J. Immunol. 166, 4498–4506 (2001).
CAS PubMed Google Scholar - Kane, M. M. & Mosser, D. M. The role of IL-10 in promoting disease progression in leishmaniasis. J. Immunol. 166, 1141–1147 (2001).
CAS PubMed Google Scholar - Ocana-Morgner, C., Mota, M. M. & Rodriguez, A. Malaria blood stage suppression of liver stage immunity by dendritic cells. J. Exp. Med. 197, 143–151 (2003).
CAS PubMed PubMed Central Google Scholar - Perry, J. A., Olver, C. S., Burnett, R. C. & Avery, A. C. The acquisition of TLR tolerance during malaria infection impacts T cell activation. J. Immunol. 174, 5921–5925 (2005).
CAS PubMed Google Scholar - Urban, B. C. et al. _Plasmodium falciparum_-infected erythrocytes modulate the maturation of dendritic cells. Nature 400, 73–77 (1999). Shows that erythrocytes infected with Plasmodium spp. block LPS-induced DC maturation through adhesive interactions with CD36. Such parasite-induced suppression might help to explain the lack of protective immunity in populations that are continually exposed to the malaria parasite.
CAS PubMed Google Scholar - Urban, B. C., Willcox, N. & Roberts, D. J. A role for CD36 in the regulation of dendritic cell function. Proc. Natl Acad. Sci. USA 98, 8750–8755 (2001).
CAS PubMed PubMed Central Google Scholar - Millington, O. R., Di Lorenzo, C., Phillips, R. S., Garside, P. & Brewer, J. M. Suppression of adaptive immunity to heterologous antigens during Plasmodium infection through hemozoin-induced failure of dendritic cell function. J. Biol. 5, 5 (2006).
PubMed PubMed Central Google Scholar - Pashine, A., Valiante, N. M. & Ulmer, J. B. Targeting the innate immune response with improved vaccine adjuvants. Nature Med. 11, S63–S68 (2005).
CAS PubMed Google Scholar - Richards, R. L. et al. Liposomes containing lipid A serve as an adjuvant for induction of antibody and cytotoxic T-cell responses against RTS,S malaria antigen. Infect. Immun. 66, 2859–2865 (1998).
CAS PubMed PubMed Central Google Scholar - Skeiky, Y. A. et al. Protective efficacy of a tandemly linked, multi-subunit recombinant leishmanial vaccine (Leish-111f) formulated in MPL adjuvant. Vaccine 20, 3292–3303 (2002). Advanced study using the TLR4 agonist MPL as an adjuvant for a recombinant vaccine against leishmaniasis.
CAS PubMed Google Scholar - Araujo, A. F. et al. CD8+-T-cell-dependent control of Trypanosoma cruzi infection in a highly susceptible mouse strain after immunization with recombinant proteins based on amastigote surface protein 2. Infect. Immun. 73, 6017–6025 (2005). Shows that a vaccine using CpG oligonucleotides and recombinant parasite antigens can induce a strong protective response mediated by CD8+ T cells.
CAS PubMed PubMed Central Google Scholar - Coban, C. et al. Effect of CpG oligodeoxynucleotides on the immunogenicity of Pfs25, a Plasmodium falciparum transmission-blocking vaccine antigen. Infect. Immun. 72, 584–588 (2004).
CAS PubMed PubMed Central Google Scholar - Cunha, M. G., Rodrigues, M. M. & Soares, I. S. Comparison of the immunogenic properties of recombinant proteins representing the Plasmodium vivax vaccine candidate MSP1(19) expressed in distinct bacterial vectors. Vaccine 20, 385–396 (2001).
CAS PubMed Google Scholar - Kumar, S. et al. CpG oligodeoxynucleotide and Montanide ISA 51 adjuvant combination enhanced the protective efficacy of a subunit malaria vaccine. Infect. Immun. 72, 949–957 (2004).
CAS PubMed PubMed Central Google Scholar - Stacey, K. J. & Blackwell, J. M. Immunostimulatory DNA as an adjuvant in vaccination against Leishmania major. Infect. Immun. 67, 3719–3726 (1999).
CAS PubMed PubMed Central Google Scholar - McSorley, S. J., Ehst, B. D., Yu, Y. & Gewirtz, A. T. Bacterial flagellin is an effective adjuvant for CD4+ T cells in vivo. J. Immunol. 169, 3914–3919 (2002).
CAS PubMed Google Scholar - Fernandes, A. P. et al. Combined interleukin-12 and topical chemotherapy for established leishmaniasis drastically reduces tissue parasitism and relapses in susceptible mice. J. Infect. Dis. 183, 1646–1652 (2001).
CAS PubMed Google Scholar - Michailowsky, V. et al. Interleukin-12 enhances in vivo parasiticidal effect of benznidazole during acute experimental infection with a naturally drug-resistant strain of Trypanosoma cruzi. Antimicrob. Agents Chemother. 42, 2549–2556 (1998).
CAS PubMed PubMed Central Google Scholar - Arevalo, I. et al. Successful treatment of drug-resistant cutaneous leishmaniasis in humans by use of imiquimod, an immunomodulator. Clin. Infect. Dis. 33, 1847–1851 (2001). Study showing the effectiveness of adjunct therapy with the TLR8 agonist imiquimod in treating patients with cutaneous leishmaniasis.
CAS PubMed Google Scholar - Buates, S. & Matlashewski, G. Treatment of experimental leishmaniasis with the immunomodulators imiquimod and S-28463: efficacy and mode of action. J. Infect. Dis. 179, 1485–1494 (1999).
CAS PubMed Google Scholar - Flynn, B., Wang, V., Sacks, D. L., Seder, R. A. & Verthelyi, D. Prevention and treatment of cutaneous leishmaniasis in primates by using synthetic type D/A oligodeoxynucleotides expressing CpG motifs. Infect. Immun. 73, 4948–4954 (2005). Study showing the prophylactic and therapeutic effectiveness of therapy with immunostimulatory oligodeoxynucleotides that contain CpG motifs in treating primates infected with L. major.
CAS PubMed PubMed Central Google Scholar - Schofield, L., Hewitt, M. C., Evans, K., Siomos, M. A. & Seeberger, P. H. Synthetic GPI as a candidate anti-toxic vaccine in a model of malaria. Nature 418, 785–789 (2002). Vaccination with the synthetic carbohydrate moiety of P. falciparum GPI anchors protects mice from cytokine-mediate pathology observed during acute malaria.
CAS PubMed Google Scholar - Kim, L. et al. Toxoplasma gondii genotype determines MyD88-dependent signaling in infected macrophages. J. Immunol. 177, 2584–2591 (2006). Shows that the T. gondii strain type has a role in triggering MyD88 signalling pathways, and that virulent strains activate IL-12 production independently of this TLR adaptor molecule.
CAS PubMed Google Scholar - Dziarski, R. & Gupta, D. Peptidoglycan recognition in innate immunity. J. Endotoxin Res. 11, 304–310 (2005).
CAS PubMed Google Scholar - Kato, H. et al. Cell type-specific involvement of RIG-I in antiviral response. Immunity 23, 19–28 (2005).
CAS PubMed Google Scholar - Balachandran, S., Thomas, E. & Barber, G. N. A FADD-dependent innate immune mechanism in mammalian cells. Nature 432, 401–405 (2004).
CAS PubMed Google Scholar
Acknowledgements
We thank M. M. Martins and B. Butcher for enlightening discussion and crucial review of this manuscript. We are also grateful to all the members and collaborators of the R.T.G. and E.Y.D. laboratories for their invaluable contributions to the work reviewed here. R.T.G. is a recipient of fellowships from Conselho Nacional de Pesquisa e Desenvolvimento Tecnológico and the John Simon Guggenheim Memorial Foundation. The R.T.G. laboratory is also funded by the Fundação de Amparo à Pesquisa do Estado de Minas Gerais, the US National Institutes of Health (NIH), the World Health Organization and the Millennium Institute for Vaccine Technology and Development. E.Y.D. is funded by the NIH.
Author information
Authors and Affiliations
- Department of Biochemistry and Immunology, Federal University of Minas Gerais, 31270-901 Belo Horizonte, Brazil
Ricardo T. Gazzinelli - Laboratory of Immunopathology, Rene Rachou Research Center, Oswaldo Cruz Foundation, 30190-002 Belo Horizonte, Brazil
Ricardo T. Gazzinelli - Division of Infectious Diseases and Immunology, University of Massachusetts Medical School, Worcester, 01655, Massachusetts, USA
Ricardo T. Gazzinelli - Department of Microbiology and Immunology, College of Veterinary Medicine, Cornell University, Ithaca, 14853, New York, USA
Eric Y. Denkers
Authors
- Ricardo T. Gazzinelli
You can also search for this author inPubMed Google Scholar - Eric Y. Denkers
You can also search for this author inPubMed Google Scholar
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Related links
Related links
FURTHER INFORMATION
Glossary
Trypomastigote
The extracellular non-replicative stage of Trypanosoma cruzi or the replicative stage of Trypanosoma brucei.
Kinetoplastids
A group of flagellate protozoans that are distinguished by the presence of a kinetoplast, a DNA-containing organelle that is closely associated with the flagellum base. Members of this group include Trypanosoma spp. and Leishmania spp.
Promastigote
The form of the Leishmania parasite that is inoculated into the vertebrate host by the bite of an insect vector. The promastigote enters cells such as macrophages through receptor-mediated uptake and then differentiates into an amastigote.
RNA interference
The use of double-stranded RNAs with sequences that precisely match a given gene, to 'knock down' the expression of that gene by directing RNA-degrading enzymes to destroy the encoded mRNA transcript.
Apicomplexan
A member of the phylum Apicomplexa. These intracellular protozoans are distinguished by a complex of apical organelles that discharge during the process of invasion. Toxoplasma gondii and Plasmodium spp. are prominent members of this phylum.
Merozoite
The intra-erythrocytic replicative stage of the malaria parasite that is associated with pathogenesis.
Tachyzoite
The immunostimulatory replicative form of Toxoplasma gondii found inside vertebrate host cells that is associated with pathogenesis. This contrasts with the quiescent bradyzoite form that is contained within cysts located in skeletal muscle and central nervous tissue.
Haemozoin
The crystalline product resulting from digestion of haemoglobin by intra-erythrocytic Plasmodium merozoites.
Profilin
The actin-binding protein from eukaryotes that has a role in regulating actin polymerization.
Eicosanoids
Biologically active compounds that are primarily derived from arachidonic acid, in part through cyclooxygenases and lipoxygenases, including prostaglandins, prostacyclins, thromboxanes, leukotrienes and lipoxins.
LPS tolerance
A transient, non-responsive cell state induced by Toll-like receptor (TLR) ligands such as liposaccharide (LPS). Cells such as macrophages and dendritic cells that have been made tolerant fail to respond to secondary stimulation with LPS or other TLR ligands. This could be a mechanism to avoid pathology that is associated with overproduction of pro-inflammatory cytokine.
Amastigote
The intracellular replicative stage of Leishmania spp. that is found in the acidified endosomes of macrophages or the replicative stage of Trypanosoma cruzi that is found in the cytoplasm of any nucleated host cells.
Adjuvant
An agent that is mixed with an antigen and increases the immune response to that antigen following immunization.
Rights and permissions
About this article
Cite this article
Gazzinelli, R., Denkers, E. Protozoan encounters with Toll-like receptor signalling pathways: implications for host parasitism.Nat Rev Immunol 6, 895–906 (2006). https://doi.org/10.1038/nri1978
- Published: 17 November 2006
- Issue Date: 01 December 2006
- DOI: https://doi.org/10.1038/nri1978