c-Abl: activation and nuclear targets (original) (raw)
c-Abl structure and subcellular localization
The proto-oncoprotein c-Abl is a 140 kDa member of the Src family of non-receptor tyrosine kinases. c-Abl was originally identified as the cellular homolog of the v-Abl oncogene of the Abelson murine leukemia virus and has now been cloned from human, mouse, Drosophila, and nematode. In animal cells c-Abl is ubiquitous but with different subcellular localization. In fibroblasts it resides predominantly in the nucleus while in primary haematopoietic cells and neurons c-Abl is more cytoplasmic. In sharp contrast, all the transforming Abl variants are exclusively cytoplasmic. The cellular subcellular localization of c-Abl is controlled by NLSs (nuclear localization signals) and an NES (nuclear export signal). This pattern of cellular distribution of c-Abl hints at its possible involvement in multiple molecular pathways, and indeed various nuclear and cytoplasmic functions have been attributed to c-Abl (reviewed in1). Cellular processes involving the nuclear c-Abl will be discussed below.
Some of the functional domains of c-Abl have been characterized (Figure 1). Common features to this family are the myristoylation site (found at the N terminus of the alternatively spliced human Ib and mouse IV transcripts), the tyrosine kinase domain with substrate specificity,2 the Src homology 2 (SH2) and 3 (SH3), both regulating c-Abl activity by mediating discriminate protein-protein interactions.3,4 The SH3 domain of c-Abl is approximately 50 amino-acids in length, and preferentially interacts with proline rich regions containing the PxxP motif.5,6 This domain determines the interaction of c-Abl with many proteins, such as the Abi family of proteins,7,8 Pag/MSP23,9 ATM,10,11 SHPTP112 and RFX1.13 The SH2 domain, about 100 amino acids long, interacts with tyrosine phosphorylated residues. Although various SH2 domains mediate phosphotyrosine dependent protein-protein interactions, each has distinct binding requirements.14
Figure 1
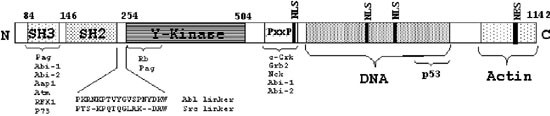
Schematic representation of c-Abl structure. The different functional domains that are discussed in the text are shown. The proteins interacting with the SH3, kinase, PxxP and the c-tail domains are listed. The PxxP region contains several PxxP motifs that may potentially interact with SH3 domains. For more detailed sequence analysis of the DNA-binding region see Miao and Wang.18 The sequence of the linker region in comparison with that of the Src gene is shown. The known NLS (nuclear localization signal) and NES (nuclear export signal) motifs, all found at the c-tail region, are indicated
c-Abl is characterized by its long C-terminal tail. Genetically, this tail is crucial for c-Abl function, and mice homozygous for a C-terminally truncated c-Abl (ablm1 mice) share most of the phenotypic defects of c-Abl null mice.15,16 This region contains three PxxP motifs, conferring interactions with SH3 containing proteins. The C-terminal tail also contains three NLSs and a single NES motif, a putative DNA-binding domain with three high mobility group-like domains17,18 and an actin binding domain.19,20 With all of these structural domains, c-Abl is likely to simultaneously participate in many processes by direct protein-protein interactions.
c-Abl kinase activation
The c-Abl kinase domain is highly regulated and mostly inactive in cells.21,22 The findings that deletions, mutations, or swapping of the SH3 domain often results in c-Abl kinase activation23,24 suggests that this domain inhibits c-Abl kinase activity. The SH3 domain might do so by either interaction with inhibitory proteins or an intramolecular inhibitory mechanism. The former suggests that kinase-inhibitory proteins bind the SH3 domain; however, out of a number of known SH3 binding proteins only a few (Pag/MSP23 and Aap1) inhibit c-Abl kinase activity. Interestingly, Pag/MSP23 also inhibits c-Abl kinase activity in an SH3 domain independent manner, possibly by direct interaction with the kinase domain.9 In addition, Rb (the tumor suppressor retinoblastoma protein) inhibits c-Abl kinase activity by binding the ATP-binding lobe of the Abl kinase and not the SH3 domain.25 Together these findings suggest that inhibiting the kinase activity is not an exclusive function of the SH3 domain. Furthermore, other SH3 binding proteins, such as Abi 1 and 2, are phosphorylated by c-Abl7,8 and are therefore unlikely to inhibit it. Also, the adaptor protein Crk26 and the DNA-binding protein RFX113 support and even potentiate c-Abl kinase activity in vitro. These findings lend support to the possibility that the c-Abl kinase domain is under repression by an intramolecular mechanism. This mechanism is not unique to c-Abl, as it has been demonstrated for both the Src and the Hck tyrosine kinases.27,28 In Src, the SH3 mediated intramolecular inhibition depends on a linker region of 31 amino-acids positioned between the SH2 and kinase domains. It is too early to conclude that c-Abl obeys the same rules, but it is noteworthy that a similar linker sequence is found in c-Abl (Figure 1) and many other tyrosine kinases of the Src family (not shown). Furthermore, c-Abl point mutants at the conserved amino-acids of this linker region are more potent.29 Thus, this mechanism of intramolecular kinase inhibition may be shared by other members of the Src family, including c-Abl.
When analyzing the response of c-Abl to ionizing radiation (IR), an additional mechanism for c-Abl activation was revealed. IR activates c-Abl kinase activity by several fold.30 This activity is dependent on the activation of the ataxia-telangiectasia-mutated (atm) gene product.11 Atm is a nuclear member of a family of phosphatidylinositiol-3-kinase like enzymes and binds the c-Abl SH3 domain.10 Atm phosphorylates c-Abl at serine 465 when cells are exposed to IR.11 As Atm and c-Abl constitutively interact, but c-Abl kinase activation is observed only after IR, c-Abl phosphorylation at serine 465, rather than their interaction, is likely to be responsible for c-Abl activation. A similar sequence of events was reported with regard to DNA-PK.31 DNA-PK is a DNA-dependent protein kinase, the product of the severe combined immunodeficiency (scid) gene. It constitutively interacts with c-Abl and, upon IR, phosphorylates and activates c-Abl. A point of interest is the fact that in mismatch repair deficient cells c-Abl is not activated by genotoxic stress,32,33 indicating that there are additional effectors in this process.
c-Abl: DNA-binding and transcription
The large C-terminal segment of c-Abl contains a DNA-binding domain that is a composition of three repeated regions, each displaying sequence similarity with the high mobility group 1-like boxes.18 This region binds cooperatively to the A/T-rich oligonucleotides; however, by using CASTing experiments, others have found a different consensus binding site.34 It has been proposed that the c-Abl-DNA interaction mainly involves contacts within the minor groove of the double helix, reminiscent of those of the high-mobility group. The c-Abl DNA-binding domain interacts with deformed DNA structures such as four-way junctions and bubble DNA containing a large single-stranded loop,34 suggesting a role not only in transcription but also in recombination and DNA repair.
A role for c-Abl in transcription has been proposed, but no direct cellular target genes have been identified. The hepatitis B virus (HBV) enhancer contains the EP element, which generates a nucleo-protein complex containing c-Abl35,36 and RFX1.37 An EP-like box is found in the enhancers of other viruses, such as polyoma and the lentivirus EIAV.38 Interestingly, the EP associated c-Abl is catalytically active in tyrosine phosphorylation.36 c-Abl can generate complexes with other DNA binding proteins. For example, c-Abl binds p53 in vitro and enhances p53-dependent transcription from a promoter containing p53 DNA binding sites. An Abl mutant which no longer binds p53 does not enhance p53 transcriptional activity and fails to suppress growth.39 Also, Abi-1 and 2, the c-Abl binding proteins, have a putative DNA binding domain.7,8 Thus, c-Abl is likely to be associated with different DNA-binding proteins and to target many DNA cis elements.
The role of c-Abl in transcription once targeted to DNA is an open question. One model argues that c-Abl phosphorylates the carboxyl-terminal domain (CTD) of the largest subunit of RNA polymerase II.40,41 Phosphorylation of serine and threonine residues of the CTD occurs during formation of the initiation complex and is correlated with the transition from complex assembly to elongation. It was therefore suggested that tyrosine phosphorylation of the CTD by c-Abl might have a similar role.42,43 However, CTD phosphorylation by c-Abl is not crucial for transcription, since Abl−/− cells are viable.
c-Abl and DNA transactions
DNA replication, recombination and repair, collectively termed DNA transactions, play a fundamental role in balancing between genomic stability and diversity. A large number of effectors, enzymes and auxiliary proteins are involved, and a few studies have suggested a role for c-Abl in this process. Rad51, converting DNA double-strand breaks to recombinational intermediates, interacts with and is phosphorylated by c-Abl.44 It has been suggested that the IR signaling pathway that includes Atm and c-Abl, is required for modifying Rad51 to be assembled in a repair protein complex.45 Also, direct interaction was demonstrated between c-Abl and DNA-PK.31 The latter plays an essential role in DNA recombination. Furthermore, the facts that some genotoxic stresses do not activate c-Abl in DNA mismatch repair deficient cells,32,33 and that IR does not activate c-Abl in Atm deficient cells,10 strongly argue for c-Abl being a downstream effector in a signaling pathway induced by DNA-damaging agents, which targets some of the components of the DNA transaction machineries.
As detailed above, the c-Abl DNA-binding domain shows sequence similarity to HMG proteins. It also recognizes deformed DNA structures such as four-way junctions,34 like other HMG-domain proteins. Interestingly, HMG1 also specifically recognizes the DNA intrastrand crosslinks formed by the DNA-damaging agent cisplatin.46,47 c-Abl, therefore, might have the same activity, and may thus be recruited directly to cisplatin modified DNA regions. This attractive possibility, although not proven, might provide an alternative pathway for c-Abl activation by this genotoxic drug.
c-Abl and G1 arrest
The cell cycle is under surveillance control mechanisms that check to ensure proper completion of early events and genome integrity before progression. These mechanisms are referred to as cell cycle checkpoints and can generate a transient delay, referred to as arrest, which allows DNA transactions to occur before progressing to the next phase of the cycle. The tumor suppressor p53 is a checkpoint protein halting the cell cycle upon DNA damage induced by either ultraviolet radiation, γ-irradiation (IR) or radiomimetic chemicals. In this signaling pathway Atm was suggested to act upstream of p53, since cells defective in the atm gene have a delayed and attenuated p53 response to IR.48 An additional important player in this signaling cascade is p21, whose expression is directly regulated by p53. p21 binds to a number of cyclin and Cdk complexes and inhibits the Cdk kinase activity, and hence cell progression is blocked.
Based on several observations, it has been proposed that c-Abl, with an intact kinase domain, may be involved in the G1/S checkpoint. Cells expressing a c-Abl kinase mutant and c-Abl nullizygous fibroblasts are impaired in their ability to downregulate Cdk2 or undergo G1 arrest in response to IR.49,50 Also, overexpression of wt c-Abl, which often gives rise to an activated kinase, inhibits cell growth and leads to G1 arrest.39,51,52 In addition it has been shown that cells expressing antisense Abl RNA, hence assumed to have a lower amount of endogenous c-Abl, show shorter G1/S transition.53 However, the suggested role of c-Abl in the G1/S checkpoint is inconsistent with the reports that the Abl−/− fibroblasts do not exhibit defects in the IR-induced cell cycle checkpoint54 and that the G1/S checkpoint is intact in Abl−/−Arg−/− 3T3 cell lines.55 At the moment we do not know the reasons for this discrepancy. It seems that c-Abl is not a crucial effector of the G1/S checkpoint; however, it might be important under certain and limited physiological conditions.
The mechanisms of G1 arrest by c-Abl are not known, and several models have been proposed (Figure 2). A few studies suggest a role for p53 in this process.39,52 This proposal is based on the findings that c-Abl binds p53 both in vitro39 and in co-immunoprecipitation from cell extracts.49 In the latter case low constitutive levels of c-Abl/p53 complexes were identified which were induced upon cell exposure to DNA damaging stress, a behaviour that attributes functional significance to this complex. In accordance with this model, a c-Abl mutant devoid of the p53 binding site failed to induce growth arrest. This model argues that the c-Abl-p53 complex is localized downstream to Atm but upstream to p21 in the DNA damage signaling pathway that leads to G1 arrest. Indeed, c-Abl costimulates the transcription function of p53 to enhance expression of the target gene p21.39 However, detailed analysis has led to the unexpected conclusion that c-Abl's checkpoint role is p21 independent.50 Thus, the downstream effector remains undefined. An alternative and rather indirect role for c-Abl in G1 arrest became evident from the finding that c-Abl can regulate the p53 level by supporting its accumulation, possibly by inhibiting Mdm2-mediated degradation of p53.56 In this case, however, it is expected that the induced G1 arrest would be p21 dependent.
Figure 2
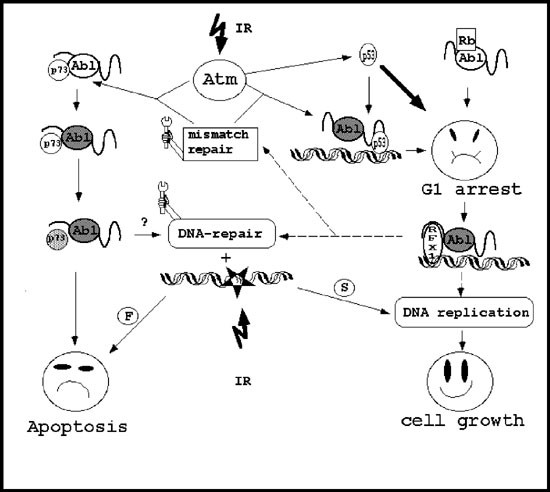
Hypothetical signaling pathways of c-Abl functions under ionizing radiation (IR) stress. Arrows are drawn to indicate the direction of the pathway but not the number of the involved steps. The broken line pathways are at the moment speculative and based on what we have learned from yeast (see the text for more details). The filled circles mark the active form of the protein. According to this model, Atm is activated upon IR, and with the involvement of components of the mismatch repair system, phosphorylates and activates c-Abl (left side of the panel). A fraction of c-Abl is in a complex with p73. The associated p73 is phosphorylated by c-Abl and becomes proapoptotic. Atm can also activate p53 (right side of the panel), which is one of the established pathways to induce G1 arrest (thick arrow). Also, activated c-Abl in complex with p53 and inactive c-Abl in complex with Rb might elicit G1 arrest. Arrested cells activate the DNA-repair machineries. In analogy with yeast, this process might be carried out by RFX1 in association with c-Abl. c-Abl might also directly interact with damaged DNA to recruit the required machineries and effectors. Once the repair is successful (S), DNA replication and cell growth occur. If the repair fails (F), cells undergo apoptosis
The requirement for the Abl kinase activity to induce growth arrest points to the involvement of a c-Abl substrate, yet p53 is not detectably tyrosine phosphorylated. In this regard it is interesting to note that p73, a member of the p53 gene family, directly interacts with c-Abl but, unlike p53, is phosphorylated by c-Abl.57,58 Whether p73 elicits G1-arrest upon c-Abl activation remains to be determined. The fact that p73 can induce p21 expression58,59 is consistent with this possibility.
c-Abl has also been proposed to play a role in cell cycle progression via interaction with Rb.25 Rb is a nuclear protein regulating cell cycle progression and is a substrate of cyclin-dependent kinases (Cdk4 and 6), the key regulators of G1-S progression. A fraction of c-Abl in G1 phase is complexed with the underphosphorylated Rb. The C-terminal pocket of Rb binds the ATP-binding lobe of the c-Abl kinase domain and inhibits its kinase activity. The Rb/c-Abl complex is disrupted in late-G1/early-S-phase as a result of Rb hyperphosphorylation by the cyclin-D/cdk4/6 kinase. According to this model, the ‘free’ and active c-Abl fraction can promote S1 phase progression, possibly by supporting transcription of S-phase genes.60 However, this model does not explain if and how c-Abl selectively targets the S-phase genes. Also, as the c-Abl SH3 domain is essential for proper c-Abl nuclear activities, its role in context of the c-Abl-Rb interaction has not been clarified. In any case, since c-Abl−/− fibroblasts display no defect in cell cycle progression, this molecular pathway might be relevant under extreme physiological conditions, as exemplified by over production of Rb.61
Recent advances in the study of cell cycle checkpoints in yeast have shed a new light on the possible role of c-Abl in the cell cycle. In S. cerevisiae DNA replication-block and damage induce the expression of over 20 genes.62 The best studied are the RNR genes, encoding ribonucleotide reductase (reviewed in63). The expression of these genes is regulated by Crt1, a DNA-binding protein that binds their promoters and represses transcription by recruiting the general repressors Ssn6 and Tup1.64 In response to DNA damage and replication block the yeast Atm homolog, Mec1 is activated, which sequentially activates the Rad53 and Dun1 protein kinases. The latter phosphorylates Crt1, which dissociates from DNA, resulting in derepression of the RNR gene. Interestingly, CRT1 is the orthologoue (a gene homologus in both structure and function) of the human RFX1 protein and displays the same DNA-binding sequence specificity.64 RFX1 interacts with the c-Abl SH3 domain and stimulates c-Abl kinase activity.13 A limited number of RFX1 target genes have so far been identified, and the products of most of them are involved in DNA replication and repair. These include c-MYC, PCNA and the DNA-repair gene XRCC1. Furthermore, similar to Crt1, RFX1 has been shown to sustain repression activity.65 Thus, it is possible that RFX1 has a Crt1-like function in animal cells to regulate the transcription of replication-block and DNA damage-inducible genes (Figure 2). Since both the upstream effector (Atm) and the downstream DNA-binding protein (RFX1) are conserved from yeast to humans, a pathway analogous to that described in yeast may exist in animal cells. According to this model genotoxic stress activates Atm which in turn activates c-Abl, which targets RFX1 to regulate the expression of the DNA damage inducible genes.
c-Abl and apoptosis
Apoptosis is an essential process in the development of multicellular organisms, in the maintenance of tissue homoestasis and in responding to stress. Molecular pathways have been identified that transmit signals via protein-protein interactions and/or protein covalent modifications such as phosphorylation, leading to apoptosis. Several observations have implicated c-Abl in this process. Dorsch and Goff have shown that B-cell lines from ablm1 mice exhibit increased sensitivity to apoptosis induced either by deprivation of growth factors or by glucocorticoid treatment.66 This finding suggests that c-Abl modulates apoptosis, and that under the employed conditions c-Abl has an antiapoptotic effect. However, the alternative possibility that the c-terminal truncated c-Abl, that is produced by ablm1 mice, is a gain of function mutant with proapoptotic activity was not ruled out. In contrast, others have provided evidence in support of c-Abl proapoptotic activity.57,58,67,68,69,70 Yuan and coworkers have demonstrated that cells null for c-Abl are impaired in the apoptotic response to IR.67 They further showed that cells stably expressing the inactive c-Abl kinase mutant exhibit resistance to IR-induced loss of clonogenic survival and apoptosis. These findings attribute a dominant negative role to the c-Abl kinase mutant in an IR signaling pathway that leads to apoptosis, and therefore suggests that the c-Abl kinase domain plays a pro-apoptotic role.
A known player in DNA damage induced apoptosis is p53, which accumulates following γ-irradiation, cisplatin treatment and UV radiation. Importantly, c-Abl is activated by the two former DNA-damaging agents but not by UV radiation. Therefore, it is unlikely that p53 is involved in the c-Abl induced apoptosis pathway. Also, c-Abl induces apoptosis in both p53-nullizygous cells and p53 positive cells in the presence of E6, a viral protein that facilitates p53 degradation.67 These findings, therefore, do not agree with the possibility that p53 is the downstream c-Abl target in the IR-induced apoptotic pathway. In sharp contrast, over production of p73 together with c-Abl is sufficient to induce apoptosis in fibroblasts.57,58 p73 is a member of the p53 family with similar transcriptional activation, DNA binding, and oligomerization domains.71 Like p53, p73 can induce apoptosis in a variety of cell lines and support transcription from promoters containing a p53-response element.71,72 Unlike p53, p73 is a tyrosine phosphoprotein and its level of phosphorylation is induced by IR.57,58 p73 and c-Abl are in association via a PxxP motif and the SH3 domain, respectively.57 Furthermore, c-Abl phosphorylates p73 both in vitro and in vivo.57,58 Several observations support the possibility that this relationship between the proteins determines their proapoptotic function under genotoxic stress. First, p73 is tyrosine phosphorylated in vivo in response to IR, under conditions whereby c-Abl is activated. Second, the ability of c-Abl to phosphorylate p73 is markedly increased by γ-irradiation.57 Third, disruption of the c-Abl-p73 interaction results in a failure to induce apoptosis by IR.57,58 Finally, a p73 mutant at tyrosine 99 (Y99-F), the site that is phosphorylated by c-Abl, behaves as a dominant negative mutant and blocks the apoptotic response to IR58 (and our unpublished data). Interestingly, collaboration between p73 and c-Abl in inducing apoptosis was also observed by others but with different molecular outcomes. According to this model the half-life of p73 is prolonged by cisplatin treatment, and the accumulated p73 induces apoptosis.69
An interesting question is how activated or accumulated p73 induces apoptosis. The most intriguing model suggests that some of the p73 targets are apoptotic genes. Although the nature of these genes remains elusive, the capacity of the wt p73, but not a mutant that cannot be phosphorylated by c-Abl, to super-induce the transcription of the p21 gene,58 may support this hypothesis. Also, the finding that a p73 mutant in the DNA-binding domain displays no apoptotic activity in Saos-2 cells,72 is in accordance with this model. However, the finding that a p73-PxxP mutant is active in transcription activation but not in apoptosis, argues that transcription activation might be required, but not sufficient, in mediating apoptosis.57 Interestingly, a similar mechanism was proposed for p53. p53 contains a proline rich domain between the activation and the DNA-binding domains which is dispensable for transcriptional activation but important for inhibition of cell growth and apoptosis (reviewed in73). It is, therefore, possible that some of the p53 and p73 target genes, which play a role in cell growth and apoptosis, are activated in a proline rich domain-dependent manner.74 An alternative possibility is that the immediate downstream effector of p73 might be an SH2 containing protein that interacts specifically with the tyrosine residue phosphorylated by c-Abl. A nuclear protein with these attributes has not yet been reported. Given the fact that c-Abl-p73 dependent cell death occurs in multiple cell lines, the p73 downstream target should be a ubiquitous protein. Furthermore, as c-Abl is conserved down to Nematode,75 and a p73 homolog was found in Squid,71 it is possible that this apoptotic pathway is ubiquitous and preserved throughout evolution.
The nature of the upstream effectors in activating this IR-induced apoptotic pathway is better understood. Radiation-induced c-Abl kinase activity is decreased in cells from AT patients;58 this finding positions c-Abl downstream of Atm (Figure 2). As IR-induced tyrosine phosphorylation of p73 is also decreased in AT cells, it is very likely that p73 is the c-Abl downstream effector in this signaling pathway. However, c-Abl-dependent p73 accumulation under cisplatin is not seen in cells unable to carry out mismatch repair.69 Therefore, mismatch repair must be a component of this signaling pathway, but its upstream-downstream relationship with respect to Atm is yet an open question.
Conclusions and future perspective
The intensive investigation of the many roles of c-Abl has been in part rewarding. We know better how c-Abl is activated, although we badly need the missing 3D structural analysis. We can also predict under which conditions c-Abl is in its active form; however, what exactly the activated c-Abl is programmed to do is still an open question. The identification of the many proteins that interact with c-Abl has helped to implicate c-Abl in the different cellular machineries, but what c-Abl in fact does there is not yet clear. It is becoming more evident that c-Abl plays a role in cell growth and death via interactions with a few key tumor suppressor proteins. Furthermore, c-Abl generates contact with DNA in different contexts. The involvment of c-Abl in DNA transactions is of particular interest, but we need more evidence for its actual role in these processes. A point of concern is that c-Abl null cells do not always fulfill the predictions made from studies that employed overexpression and c-Abl dominant negative. One possible complication emerges from the fact that nuclear c-Abl might have different and even opposite manifestations than the cytoplasmic fraction. Furthermore, the employed c-Abl mutants change their cellular localization and therefore introduce more confusion in the field.
Based on genetic and evolutionary studies the role of c-Abl in development should be further investigated, particularly with respect to the development of the central nervous system. Although in large part this activity is performed by the cytoplasmic c-Abl fraction, and hence not covered in this review, we have a good reason to consider the contribution of nuclear events.
This assumption emerges from the recent findings of physical and functional interaction of c-Abl with p73, the latter being a nuclear protein which, like c-Abl, plays an important role in neuronal development. We can easily address this interesting possibility in the near future by employing the relevant null mice to generate the required crosses.
Abbreviations
ATM:
ataxia-telangiectasia mutant
CTD:
C-terminus domain
EIAV:
equine infectious anaemia virus
HBV:
hepatitis B virus
IR:
ionizing irradiation
NES:
nuclear export signal
NLS:
nuclear localization signal
SH:
Src homology
References
- Van Etten RA . 1999 Cycling, stressed-out and nervous: cellular functions of c-Abl. Trends. Cell Biol. 9: 179–186
Article CAS PubMed Google Scholar - Songyang Z, Shoelson SE, McGlade J et al. 1994 Specific motifs recognized by the SH2 domains of Csk, 3BP2, fps/fes, GRB-2, HCP, SHC, Syk, and Vav. Mol. Cell Biol. 14: 2777–2785
Article CAS PubMed PubMed Central Google Scholar - Cohen GB, Ren R and Baltimore D . 1995 Modular binding domains in signal transduction proteins. Cell 80: 237–248.
Article CAS PubMed Google Scholar - Pawson T . 1994 SH2 and SH3 domains in signal transduction. Adv. Cancer Res. 64: 87–110
Article CAS PubMed Google Scholar - Cicchetti P, Mayer BJ, Thiel G and Baltimore D . 1992 Identification of a protein that binds to the SH3 region of Abl and is similar to Bcr and GAP-rho. Science 257: 803–806
Article CAS PubMed Google Scholar - Ren R, Mayer BJ, Cicchetti P and Baltimore D . 1993 Identification of a ten-amino acid proline-rich SH3 binding site. Science 259: 1157–1161
Article CAS PubMed Google Scholar - Shi Y, Alin K and Goff SP . 1995 Abl-interactor-1, a novel SH3 protein binding to the carboxy-terminal portion of the Abl protein, suppresses v-abl transforming activity. Genes Dev. 9: 2583–2597
Article CAS PubMed Google Scholar - Dai Z and Pendergast AM . 1995 Abi-2, a novel SH3-containing protein interacts with the c-Abl tyrosine kinase and modulates c-Abl transforming activity. Genes & Dev. 9: 2569–2582
Article CAS Google Scholar - Wen ST and Van Etten RA . 1997 The PAG gene product, a stress-induced protein with antioxidant properties, is an Abl SH3-binding protein and a physiological inhibitor of c-Abl tyrosine kinase activity. Genes Dev. 11: 2456–2467
Article CAS PubMed PubMed Central Google Scholar - Shafman T, Khanna KK, Kedar P et al. 1997 Interaction between ATM protein and c-Abl in response to DNA damage. Nature 387: 520–523
Article CAS PubMed Google Scholar - Baskaran R, Wood LD, Whitaker LL et al. 1997 Ataxia telangiectasia mutant protein activates c-Abl tyrosine kinase in response to ionizing radiation. Nature 387: 516–519
Article CAS PubMed Google Scholar - Kharbanda S, Bharti A, Pei D et al. 1996 The stress response to ionizing radiation involves c-Abl-dependent phosphorylation of SHPTP1. Proc. Natl. Acad. Sci. USA 93: 6898–6901
Article CAS PubMed PubMed Central Google Scholar - Agami R and Shaul Y . 1998 The kinase activity of c-Abl but not v-Abl is potentiated by direct interaction with RFXI, a protein that binds the enhancers of several viruses and cell-cycle regulated genes. Oncogene 16: 1779–1788
Article CAS PubMed Google Scholar - Songyang Z, Shoelson SE, Chaudhuri M et al. 1993 SH2 domains recognize specific phosphopeptide sequences. Cell 72: 767–778
Article CAS PubMed Google Scholar - Tybulewicz VL, Crawford CE, Jackson PK, Bronson RT and Mulligan RC . 1991 Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-abl proto-oncogene. Cell 65: 1153–1163
Article CAS PubMed Google Scholar - Schwatzberg PL, Stall AM, Hardin JD et al. 1991 Mice homozygous for the ablm1 mutation show poor viability and depletion of selected B and T cell populations. Cell 65: 1165–1175
Article Google Scholar - Kipreos ET and Wang JY . 1992 Cell cycle-regulated binding of c-Abl tyrosine kinase to DNA. Science 256: 382–385
Article CAS PubMed Google Scholar - Miao JY and Wang J . 1996 Binding of A/T-rich DNA by three high mobility group-like domains in c-Abl tyrosine kinase. J. Biol. Chem. 271: 22823–22830
Article CAS PubMed Google Scholar - McWhirter JR and Wang JY . 19943 An actin-binding function contributes to transformation by the Bcr-Abl oncoprotein of Philadelphia chromosome-positive human leukemias. EMBO J. 12: 1533–1546
Article Google Scholar - Van Etten R, Jackson PK, Baltimore D, Sanders MC, Matsudaira PT and Janmey PA . 1994 The COOH terminus of the c-Abl tyrosine kinase contains distinct F- and G-actin binding domains with bundling activity. J. Cell Biol. 124: 325–340
Article CAS PubMed Google Scholar - Konopka JB and Witte ON . 1985 Detection of c-abl tyrosine kinase activity in vitro permits direct comparison of normal and altered abl gene products. Mol. Cell Biol. 5: 3116–3123
Article CAS PubMed PubMed Central Google Scholar - Pendergast AM, Muller AJ, Havlik MH, Clark R, McCormick F and Witte ON . 1991 Evidence for regulation of the human ABL tyrosine kinase by a cellular inhibitor. Proc. Natl. Acad. Sci. USA 88: 5927–5931
Article CAS PubMed PubMed Central Google Scholar - Van Etten R, Jackson P and Baltimore D . 1989 The mouse type IV c-abl gene product is a nuclear protein, and activation of transforming ability is associated with cytoplasmic localization. Cell 58: 669–678
Article CAS PubMed Google Scholar - Mayer BJ and Baltimore D . 1994 Mutagenic analysis of the roles of SH2 and SH3 domains in regulation of the Abl tyrosine kinase. Mol. Cell Biol. 14: 2883–2894
Article CAS PubMed PubMed Central Google Scholar - Welch PJ and Wang JY . 1993 A C-terminal protein-binding domain in the retinoblastoma protein regulates nuclear c-Abl tyrosine kinase in the cell cycle. Cell 75: 779–790
Article CAS PubMed Google Scholar - Feller SM, Ren R, Hanafusa H and Baltimore D . 1994 SH2 and SH3 domains as molecular adhesives: the interactions of Crk and Abl. Trends Biochem. Sci. 19: 453–458
Article CAS PubMed Google Scholar - Xu W, Harrision SC and Eck MJ . 1997 Three-dimensional structure of the tyrosine kinase c-Src. Nature 385: 595–602
Article CAS PubMed Google Scholar - Sicheri F, Moarefi I and Kuriyan J . 1997 Crystal structure of the Src family tyrosine kinase Hck. Nature 385: 602–609
Article CAS PubMed Google Scholar - Barila D and Superti-Furga G . 1998 An intramolecular SH3-domain interaction regulates c-Abl activity. Nat. Genet. 18: 280–282
Article CAS PubMed Google Scholar - Kharbanda S, Ren R, Pandey P et al. 1995 Activation of the c-Abl tyrosine kinase in the stress response to DNA-damaging agents. Nature 376: 785–788
Article CAS PubMed Google Scholar - Kharbanda S, Pandey P, Jin S et al. 1997 Functional interaction between DNA-PK and c-Abl in response to DNA damage. Nature 386: 732–735
Article CAS PubMed Google Scholar - Nehme A, Baskaran R, Aebi S et al. 1997 Differential induction of c-Jun NH2-terminal kinase and c-Abl kinase in DNA mismatch repair-proficient and -deficient cells exposed to cisplatin. Cancer Res. 57: 3253–3257
CAS PubMed Google Scholar - Nehme A, Baskaran R, Nebel S et al. 1999 Induction of JNK and c-Abl signalling by cisplatin and oxaliplatin in mismatch repair-proficient and -deficient cells. Br. J. Cancer 79: 1104–1110
Article CAS PubMed PubMed Central Google Scholar - David Cordonnier M, Hamdane M, Bailly C and D'Halluin JC . 1998 The DNA binding domain of the human c-Abl tyrosine kinase preferentially binds to DNA sequences containing an AAC motif and to distorted DNA structures. Biochemistry 37: 6065–6076
Article CAS PubMed Google Scholar - Dikstein R, Heffetz D, Ben-Neriah Y and Shaul Y . 1992 c-abl has a sequence-specific enhancer binding activity. Cell 69: 751–757
Article CAS PubMed Google Scholar - Dikstein R, Agami R, Heffetz D and Shaul Y . 1996 p140/c-Abl that binds DNA is preferentially phosphorylated at tyrosine residues. Proc. Natl. Acad. Sci. USA 93: 2387–2391
Article CAS PubMed PubMed Central Google Scholar - Reith W, Ucla C, Barras E et al. 1994 RFX1, a transactivator of hepatitis B virus enhancer I, belongs to a novel family of homodimeric and heterodimeric DNA-binding proteins. Mol. Cell Biol. 14: 1230–1244
Article CAS PubMed PubMed Central Google Scholar - Dikstein R, Faktor O, Ben-Levy R and Shaul Y . 1990 Functional organization of the hepatitis B virus enhancer. Mol. Cell Biol. 10: 3682–3689
Google Scholar - Goga A, Liu X, Hambuch TM et al. 1995 p53 dependent growth suppression by the c-Abl nuclear tyrosine kinase. Oncogene 11: 791–799
CAS PubMed Google Scholar - Baskaran R, Dahmus ME and Wang JY . 1993 Tyrosine phosphorylation of mammalian RNA polymeraseII carboxyl-terminal domain. Proc. Natl. Acad. Sci. USA 90: 11167–11171
Article CAS PubMed PubMed Central Google Scholar - Duyster J, Baskaran R and Wang JY . 1995 Src homology2 domain as a specificity determinant in the c-Abl-mediated tyrosine phosphorylation of the RNA polymeraseII carboxyl-terminal repeated domain. Proc. Natl. Acad. Sci. USA 92: 1555–1559
Article CAS PubMed PubMed Central Google Scholar - Welch PJ and Wang JY . 1995 Disruption of retinoblastoma protein function by coexpression of its C pocket fragment. Genes & Dev. 9: 31–46
Article CAS Google Scholar - Baskaran R, Escobar SR and Wang JY . 1999 Nuclear c-Abl is a COOH-terminal repeated domain (CTD)-tyrosine (CTD)-tyrosine kinase-specific for the mammalian RNA polymeraseII: possible role in transcription elongation. Cell Growth Differ. 10: 387–396
CAS PubMed Google Scholar - Yuan ZM, Huang Y, Ishiko T et al. 1998 Regulation of Rad51 function by c-Abl in response to DNA damage. J. Biol. Chem. 273: 3799–3802
Article CAS PubMed Google Scholar - Chen G, Yuan SS, Liu W et al. 1999 Radiation-induced assembly of rad51 and rad52 recombination complex requires ATM and c-Abl. J. Biol. Chem. 274: 12748–12752
Article CAS PubMed Google Scholar - Chow CS, Barnes CM and Lippard SJ . 1995 A single HMG domain in high mobility group1 protein binds to DNAs as small as 20 base pairs containing the major cisplatin adduct. Biochemistry 34: 2956–2964
Article CAS PubMed Google Scholar - Ohndorf UM, Rould MA, He Q, Pabo CO and Lippard SJ . 1999 Basis for recognition of cisplatin-modified DNA by high-mobility-group proteins. Nature 399: 708–712
Article CAS PubMed Google Scholar - Morgan SE and Kastan MB . 1997 p53 and ATM: cell cycle, cell death, and cancer. Adv. Cancer Res. 71: 1–25
Article CAS PubMed Google Scholar - Yuan ZM, Huang Y, Fan MM, Sawyers C, Kharbanda S and Kufe D . 1996 Genotoxic drugs induce interaction of the c-Abl tyrosine kinase and the tumor suppressor protein p53. J. Biol. Chem. 271: 26457–26460
Article CAS PubMed Google Scholar - Yuan ZM, Huang Y, Whang Y et al. 1996 Role for c-Abl tyrosine kinase in growth arrest response to DNA damage. Nature 382: 272–274
Article CAS PubMed Google Scholar - Sawyers CL, McLauglin J, Goga A, Havlik M and Witte O . 1994 The nuclear tyrosine kinase c-Abl negatively regulates cell growth. Cell 77: 121–131
Article CAS PubMed Google Scholar - Wen ST, Jackson PK and Van Etten RA . 1996 The cytostatic function of c-Abl is controlled by multiple nuclear localization signals and requires the p53 and Rb tumor suppressor gene products. EMBO J. 15: 1583–1595
Article CAS PubMed PubMed Central Google Scholar - Daniel R, Cai Y, Wong PM and Chung SW . 1995 Deregulation of c-abl mediated cell growth after retroviral transfer and expression of antisense sequences. Oncogene 10: 1607–1614
CAS PubMed Google Scholar - Liu ZG, Baskaran R, Lea-Chou ET et al. 1996 Three distinct signalling responses by murine fibroblasts to genotoxic stress. Nature 384: 273–276
Article CAS PubMed Google Scholar - Koleske AJ, Gifford AM, Scott ML et al. 1998 Essential roles for the Abl and Arg tyrosine kinases in neurulation. Neuron. 21: 1259–1272
Article CAS PubMed Google Scholar - Sionov RV, Moallem E, Berger M et al. 1999 c-Abl neutralizes the inhibitory effect of Mdm2 on p53. J. Biol. Chem. 274: 8371–8374
Article CAS PubMed Google Scholar - Agami R, Blandino G, Oren M and Shaul Y . 1999 Interaction of c-Abl and p73alpha and their collaboration to induce apoptosis. Nature 399: 809–813
Article CAS PubMed Google Scholar - Yuan ZM, Shioya H, Ishiko T et al. 1999 p73 is regulated by tyrosine-kinase c-Abl in the apoptotic response to DNA damage [In Process Citation]. Nature 399: 814–817
Article CAS PubMed Google Scholar - Zhu J, Jiang J, Zhou W and Chen X . 1998 The potential tumor suppressor p73 differentially regulates cellular p53 target genes. Cancer Res. 58: 5061–5065
CAS PubMed Google Scholar - Knudsen ES and Wang JY . 1996 Differential regulation of retinoblastoma protein function by specific Cdk phosphorylation sites. J. Biol. Chem. 271: 8313–8320
Article CAS PubMed Google Scholar - Welch PJ and Wang JY . 1995 Abrogation of retinoblastoma protein function by c-Abl through tyrosine kinase-dependent and -independent mechanisms. Mol. Cell Biol. 15: 5542–5551
Article CAS PubMed PubMed Central Google Scholar - Friedberg EC, Bardwell AJ, Bardwell L et al. 1995 Nucleotide excision repair in the yeast Saccharomyces cerevisiae: its relationship to specialized mitotic recombination and RNA polymeraseII basel transcription. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 347: 63–68
Article CAS PubMed Google Scholar - Elledge SJ, Zhou Z, Allen JB and Navas TA . 1993 DNA damage and cell cycle regulation of ribonucleotide reductase. Bioessays 15: 333–339
Article CAS PubMed Google Scholar - Huang M, Zhou Z and Elledge SJ . 1998 The DNA replication and damage checkpoint pathways induce transcription by inhibition of the Crt1 repressor. Cell 94: 595–605
Article CAS PubMed Google Scholar - Katan Y, Agami R and Shaul Y . 1997 The transcriptional activation and repression domains of RFX1, a context-dependent regulator, can mutually neutralize their activities. Nucleic. Acids Res. 25: 3621–3628
Article CAS PubMed PubMed Central Google Scholar - Dorsch M and Goff SP . 1996 Increased sensitivity to apoptotic stimuli in c-abl-deficient progenitor B-cell lines. Proc. Natl. Acad. Sci. USA 23: 13131–13136
Article Google Scholar - Yuan ZM, Huang Y, Ishiko T, Kharbanda S, Weichselbaum R and Kufe D . 1997 Regulation of DNA damage-induced apoptosis by the c-Abl tyrosine kinase. Proc. Natl. Acad. Sci. USA 94: 1437–1440
Article CAS PubMed PubMed Central Google Scholar - Huang Y, Yuan ZM, Ishiko T et al. 1997 Pro-apoptotic effect of the c-Abl tyrosine kinase in the cellular response to 1-beta-D-arabinofuranosylcytosine. Oncogene 15: 1947–1952
Article CAS PubMed Google Scholar - Gong JG, Costanzo A, Yang HQ et al. 1999 The tyrosine-kinase c-Abl regulates p73 in apoptotic response to cisplatin-induced DNA damage. Nature 399: 806–809
Article CAS PubMed Google Scholar - Dan S, Naito M, Seimiya H, Kizaki A, Mashima T and Tsuruo T . 1999 Activation of c-Abl tyrosine kinase requires caspase activation and is not involved in JNK/SAPK activation during apoptosis of human monocytic leukemia U937 cells. Oncogene 18: 1277–1283
Article CAS PubMed Google Scholar - Kaghad M, Bonnet H, Yang A et al. 1997 Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell 90: 809–819
Article CAS PubMed Google Scholar - Jost CA, Marin MC and Kaelin WJ . 1997 p73 is a human p53-related protein that can induce apoptosis. Nature 389: 191–194
Article CAS PubMed Google Scholar - Levine AJ . 1997 p53, the cellular gatekeeper for growth and division. Cell 88: 323–331
Article CAS PubMed Google Scholar - Venot C, Maratrat M, Dureuil C, Conseiller E, Bracco L and Debussche L . 1998 The requirement for the p53 proline-rich functional domain for mediation of apoptosis is correlated with specific PIG3 gene transactivation and with transcriptional repression. EMBO J. 17: 4668–4679
Article CAS PubMed PubMed Central Google Scholar - Schonthal A, Alberts AS, Frost JA and Feramisco JR . 1991 Differential regulation of jun family gene expression by the tumor promoter okadaic acid. New Biol. 3: 977–986
CAS PubMed Google Scholar
Acknowledgements
I would like to thank Y Katan-Khaykovich for critical reading and helpful comments.
Author information
Authors and Affiliations
- Department of Molecular Genetics, Weizmann Institute of Science, Rehovot, 76100, Israel
Y Shaul
Authors
- Y Shaul
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toY Shaul.
Additional information
Edited by R Knight
Rights and permissions
About this article
Cite this article
Shaul, Y. c-Abl: activation and nuclear targets.Cell Death Differ 7, 10–16 (2000). https://doi.org/10.1038/sj.cdd.4400626
- Received: 30 September 1999
- Accepted: 26 October 1999
- Published: 06 March 2000
- Issue Date: 01 January 2000
- DOI: https://doi.org/10.1038/sj.cdd.4400626