Mechanisms that lessen benefits of β-secretase reduction in a mouse model of Alzheimer’s disease (original) (raw)
Introduction
The β-secretase called BACE1 (β-site amyloid precursor protein-cleaving enzyme 1) is responsible for initiating the generation of amyloid-β (Aβ) peptides.1 Given that cerebral accumulation of Aβ plays a key role in the pathogenesis of Alzheimer’s disease (AD),2, 3 BACE1 represents a prime target for designing therapeutic drugs to prevent or treat this devastating neurodegenerative disorder.4, 5, 6, 7 As a proof of concept for this view, it has been demonstrated that BACE1 gene deletion (BACE1−/−) prevents the development of AD-like pathologies and memory impairments in different lines of amyloid precursor protein (APP) transgenic mice.8, 9, 10, 11 However, as BACE1 has a large catalytic site, it has been challenging to find small-molecule BACE1 inhibitors that can efficiently cross the blood–brain barrier and are large enough to block the substrate-binding site of this enzyme.12, 13, 14 Nevertheless, a recent progress in medicinal chemistry has led to the development of some promising nonpeptidomimetic BACE1 inhibitors that are reported to lower cerebral Aβ levels after systemic administration in various species such as mice, rats, monkeys and humans.15, 16, 17, 18, 19, 20, 21
Our laboratory and others have previously used BACE1 haploinsufficiency (BACE1+/−; that is, 50% reduction) as a therapeutic relevant model to evaluate the efficacy of partial inhibition of this enzyme.11, 22, 23, 24, 25, 26, 27 By crossing BACE1+/− mice with AD transgenic mouse lines, these studies have demonstrated that partial reduction of BACE1 can diminish brain Aβ concentrations and improve AD-like phenotypes including amyloid plaque and tau pathologies, synaptic dysfunction and memory deficits. However, the beneficial outcomes associated with BACE1 haploinsufficiency decline with age or the progression of disease in APP transgenic mice.11, 22, 25, 26 Similarly, although the most recent studies of BACE1 inhibitors successfully show cognitive improvements concomitant with cerebral Aβ reduction following systemic administration in relatively earlier stages of APP mice, the decrease in efficacy is noted with advanced age.28, 29 For preclinical validation of BACE1-inhibiting therapeutic approaches, it is imperative to determine the mechanisms that may lessen benefits from genetic or pharmacologic β-secretase suppression during the course of AD development.
Here we report two distinct mechanisms by which BACE1 haploinsufficiency fails to rescue AD-like memory deficits in 5XFAD mice at 15–18 months of age harboring robust plaque pathology, although it shows benefits at 6 months with moderate Aβ deposition. First, BACE1 haploinsufficiency is not able to block the translational elevation of BACE1 expression through the PERK/eIF2α (PKR-endoplasmic reticulum-related kinase/eukaryotic translation initiation factor-2α) pathway in 15–18-month-old 5XFAD mice. Second, downregulation of the Aβ-degrading enzyme neprilysin in 5XFAD brains becomes no longer responsive to rescue by BACE1 haploinsufficiency during disease progression. Therefore, our results raise a possibility that partial suppression of BACE1 may lose the therapeutic efficacy by encountering both the decreased ability to suppress Aβ overproduction and compromised Aβ clearance during advanced stages of AD.
Materials and methods
Animals
We used 5XFAD mice (Tg6799 line) that co-overexpress familial AD (FAD) mutant forms of human APP (the Swedish mutation: K670N, M671L; the Florida mutation: I716V; the London mutation: V717I) and presenilin 1 (PS1) (M146L; L286V) transgenes under transcriptional control of the neuron-specific mouse Thy-1 promoter.9, 10, 30 Hemizygous 5XFAD transgenic mice (B6/SJL hybrid background) were crossbred to heterozygous BACE1 knockout (BACE1+/−) mice (C57BL/6 background, The Jackson Laboratory, Bar Harbor, ME, USA),11, 31 yielding animals with four different genotypes (wild-type, BACE1+/−, 5XFAD+/− and BACE1+/−·5XFAD+/−). Genotyping was performed by PCR analysis of tail DNA. 5XFAD mice were chosen for rigorously evaluating the therapeutic inhibition of BACE1, as this aggressive amyloid model with multiple FAD mutations can recapitulate some important AD-like phenotypes (for example, robust BACE1 elevation, neuronal loss and so on) lacking in most other APP transgenic mice.10, 25, 30, 32, 33 Experiments were done blind with respect to the genotype of mice at 6 and 15–18 months of age, which demonstrate moderate and massive levels of Aβ deposition in 5XFAD brains, respectively.10, 24, 30 All animal procedures were approved by the Nathan Kline Institute Animal Care and Use Committee and conducted in accordance with National Institutes of Health guidelines.
Contextual fear conditioning
Contextual fear conditioning was tested as described previously.24, 34, 35 The experiments were performed using four standard conditioning chambers, each of which was housed in a soundproof isolation cubicle and equipped with a stainless-steel grid floor connected to a solid-state shock scrambler. Each scrambler was connected to an electronic constant-current shock source that was controlled via an interface connected to a Windows XP computer running FreezeFrame software (Coulbourn Instruments, Allentown, PA, USA). A digital camera was mounted on the steel ceiling of each chamber, and video signals were sent to the same computer for analysis. During training, mice were placed in the conditioning chamber for 3 min and then received two unsignaled footshocks (1.0 mA, 2 s) at 1-min intervals. After the last shock delivery, mice were left in the chamber for another 30 s. Contextual fear memory was evaluated by scoring freezing behavior (the absence of all movement except for that needed for breathing) for 3 min when the mice were placed back into the same conditioning chamber 24 h after training. The automated FreezeFrame system (Coulbourn Instruments), which digitizes the video signal at 4 Hz and compares movement frame by frame, was used to score the amount of freezing. After behavioral testing, some mice were killed for immunoblotting experiments and others were perfused for immunohistochemistry.
Immunoblot analysis
Hemibrain samples were taken from the mice under deep isoflurane anesthesia and were snap-frozen for biochemical assays. For western blot analysis, each sample was homogenized in 5 volumes of modified RIPA buffer containing 150 mM NaCl, 50 mM Tris HCl (pH 8.0), 1 mM EDTA, 1% IGEPAL, 0.5% sodium deoxycholate, 0.1% SDS and protease/phosphatase inhibitor cocktail (Calbiochem, La Jolla, CA, USA), and centrifuged at 10 000 g for 10 min to remove any insoluble material. Protein concentrations were determined by a BCA protein assay kit (Pierce, Rockford, IL, USA), and 10–50 μg of protein was run on NuPAGE 4–12% Bis-Tris gels or 6% Tris-Glycine gels (Invitrogen, Carlsbad, CA, USA) and transferred to nitrocellulose membrane. After blocking, membranes were probed with anti-BACE1 (1:1000, MAB5308, Millipore, Billerica, MA, USA), anti-phospho-eIF2α (Ser51) (1:1000, 3398, Cell Signaling Technology, Danvers, MA, USA), anti-eIF2α (1:2000, 9722, Cell Signaling Technology), anti-phospho-PERK (Ser713) (1:1000, 649401, BioLegend, San Diego, CA), anti-PERK (1:1000, 3192, Cell Signaling Technology), anti-phospho-PKR (Thr451) (1:500, 07–886, Millipore), anti-neprilysin (1:1000, ab951, Abcam, Cambridge, MA, USA) and anti-β-actin (1:15 000, AC-15, Sigma-Aldrich, St Louis, MO, USA). They were then incubated with horseradish peroxidase-conjugated secondary IgG. Immunoblot signals were visualized by an ECL chemiluminescence substrate reagent kit (Pierce), and were quantified by densitometric scanning and image analysis using Quantity One software (Bio-Rad Laboratories, Hercules, CA, USA).
Double immunofluorescence labeling
Mice were transcardially perfused with 0.1 M phosphate-buffered saline (PBS, pH 7.4), followed by 4% paraformaldehyde in PBS under deep isoflurane anesthesia. Brains were postfixed for 24 h in 4% paraformaldehyde in PBS at 4 °C and transferred to PBS. The brain was sectioned coronally at 30 μm using a vibratome (VT1200, Leica Microsystems, Wetzlar, Germany), and successive sections were stored in PBS containing 0.05% sodium azide at 4 °C. Three sections (separated by ∼90 μm) per mouse were taken at levels between –1.7 and –1.9 mm to bregma according to the mouse brain atlas of Franklin and Paxinos.36 The sections were permeabilized with 0.25% Triton X-100, blocked and then incubated overnight at 4 °C with the goat polyclonal antibody against phospho-eIF2α (Ser52) (1:50, sc-12412, Santa Cruz Biotechnology, Santa Cruz, CA, USA) and rabbit polyclonal antibody against phospho-PERK (Ser713) (1:100, 649401, BioLegend). Immunofluorescence labeling was performed by a 1-h reaction with Alexa Fluor 488- or Alexa Fluor 594-conjugated anti-goat and anti-rabbit IgGs (1:750, Invitrogen) at room temperature. The sections were then washed three times in PBS and mounted with anti-fading medium. Control sections were processed with the omission of the primary antibody in the incubation buffer, and these controls yielded no specific labeling in brain sections. Immunostained sections were imaged with a confocal fluorescence microscope (LSM 510 Meta, Zeiss, Oberkochen, Germany) with a × 40 objective.
Aβ immunohistochemistry
The brain sections were stained by the avidin–biotin peroxidase complex method as described previously.23, 24, 37 Briefly, the sections were incubated overnight at 4 °C with monoclonal anti-Aβ1-16 antibody (1: 200, 6E10; Signet, Dedham, MA, USA). The ABC kit (PK-2200; Vector Laboratories, Burlingame, CA, USA) was utilized with 3,3′-diaminobenzidine tetrahydrochloride as a chromogen to visualize the reaction product. The sections were then mounted on charged slides, dehydrated in a series of alcohol, cleared in xylene and covered with a coverslip. Light microscopy was conducted on an Axioskop 2 microscope equipped with an AxioCaM HRc digital camera (Zeiss) for capturing images. Semiquantitative analysis was performed using AxioVision imaging software with the AutoMeasure module (Zeiss). Identified objects after thresholding were individually inspected to confirm the object as a plaque or not in a blinded manner. Percentage area occupied by Aβ deposits in the hippocampus and cortex was assessed bilaterally to compare plaque burden between 5XFAD control and BACE1+/−·5XFAD mice.
Data analysis
The significance of differences between the groups was determined by a one-way analysis of variance and post hoc Fisher’s protected least significant difference tests were performed when appropriate. Data were presented as mean±s.e.m. and the level of significance was set for _P-_value <0.05.
Results
BACE1 haploinsufficiency and memory impairments in 5XFAD mice
We used 5XFAD APP/PS1 transgenic mice that represent a rapid-onset and aggressive amyloid model based on a combination of five FAD mutations and the consequent acceleration of Aβ42 production.9, 10, 30 5XFAD mice begin to develop visible amyloid deposition as early as 2 months of age, and exhibit memory declines on a battery of hippocampus-dependent tasks between 4 and 6 months concomitant with synaptic dysfunction at the Schaffer collateral-CA1 pathway.9, 23, 30, 35, 38, 39 We first tested the mice with the contextual fear conditioning paradigm, and compared the efficacy of BACE1+/− gene deletion in ameliorating memory deficits in 5XFAD mice at 6 and 15–18 months of age that develop moderate and massive Aβ plaque pathology, respectively (Figure 1).
Figure 1
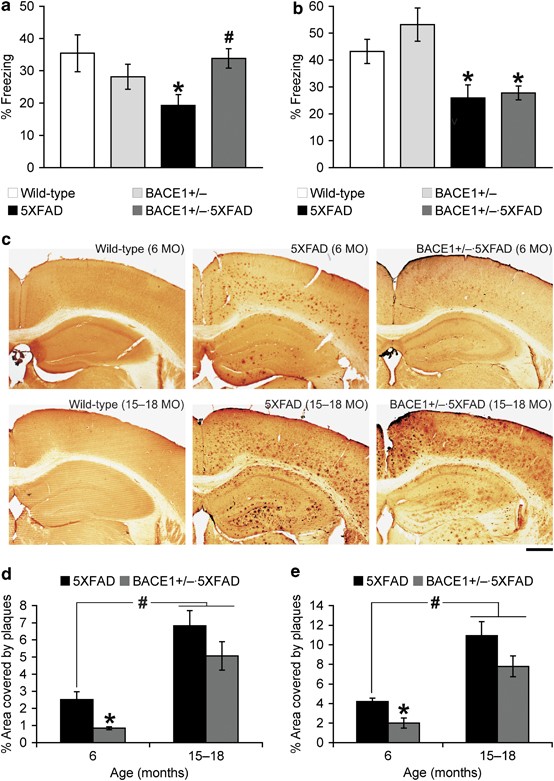
Beneficial effects of BACE1 (β-site amyloid precursor protein-cleaving enzyme 1) haploinsufficiency on memory deficits and β-amyloidosis decline in 5XFAD mice with advanced age. Mice at (a) 6 months (MO) and (b) 15–18 months of age were trained with two conditioned stimulus/unconditioned stimulus pairings for contextual fear conditioning. 5XFAD mice show significantly lower levels of contextual freezing than wild-type mice irrespective of age when tested 24 h after training (*P<0.05). BACE1+/−·5XFAD mice are rescued completely back to wild-type control levels of contextual memory at 6 months but not at 15–18 months of age (#P<0.05 versus age-matched 5XFAD); _n_=10–19 mice per group. (c) Brain sections were immunostained with the 6E10 anti-Aβ antibody. Shown are representative photomicrographs of the hippocampus and cortical area. Scale bar=500 μm. Percentage area occupied by amyloid-β (Aβ) deposits in the hippocampus (d) and cerebral cortex (e) was measured for quantification (_n_=4 mice per group). Note that BACE1 haploinsufficiency is no longer able to significantly reduce profound plaque burden in 5XFAD mice with advanced age (*P<0.05 versus age-matched 5XFAD controls). The Aβ load at 15–18-month-old 5XFAD mice with or without BACE1+/− mutation is significantly greater than that of 6-month-old 5XFAD mice (#P<0.05). All data are presented as mean±s.e.m.
In this behavioral assay, mice learn to associate a distinct context (conditioned stimulus) with aversive footshocks (unconditioned stimulus) through hippocampus-dependent mechanisms.40, 41 Wild-type control mice exhibited a robust conditioned fear response as assessed by freezing (the absence of all but respiratory movements) when placed back into the conditioning chamber 24 h after training with two conditioned stimulus/unconditioned stimulus pairings. At 6 months of age, 5XFAD mice showed significantly reduced levels of freezing compared with wild-type controls, whereas the contextual memory impairment was rescued almost completely back to wild-type levels in 5XFAD mice with BACE1+/− genotype (F(3, 43)=3.16, P<0.05; Figure 1a). In contrast, when mice were tested at 15–18 months of age, levels of contextual freezing were similarly reduced in 5XFAD and BACE1+/−·5XFAD mice as compared with those of age-matched wild-type control mice (F(3, 55)=7.31, P<0.05; Figure 1b), indicating that BACE1+/− reduction was no longer able to improve contextual memory deficits. Meanwhile, contextual freezing levels were indistinguishable between BACE1+/− and wild-type mice irrespective of age, demonstrating that BACE1 haploinsufficiency did not affect baseline memory performances on the wild-type background.
BACE1 haploinsufficiency and amyloid neuropathology in 5XFAD mice
To investigate the relationship between age-dependent cognitive benefits and changes in Aβ accumulation, we next compared the effects of BACE1 haploinsufficiency on plaque burden in 5XFAD mice at 6 and 15–18 months of age (Figure 1c). The amyloid plaque load was significantly reduced in the hippocampus (Figure 1d) and cerebral cortex (Figure 1e) of BACE1+/−·5XFAD mice at 6 months of age as compared with that of age-matched 5XFAD controls (F(1, 6)=13.11, P<0.05 and F(1, 6)=12.77, P<0.05, respectively). Importantly, Aβ deposition was greatly exacerbated in 5XFAD mouse brains from 6 to 15–18 months of age (P<0.05), whereas BACE1 haploinsufficiency became less effective in reducing plaque burden in the hippocampus and cortex of 5XFAD mice with advanced age (F(1, 6)=2.01, _P_=0.20 and F(1, 6)=3.02, _P_=0.13, respectively). Consequently, residual levels of Aβ deposition in 15–18-month-old BACE1+/−·5XFAD mice were significantly higher than those of 6-month-old 5XFAD mice (P<0.05). These observations were consistent with our previous enzyme-linked immunosorbent assay measurements of Aβ peptides in guanidine-solubilized brains showing that total Aβ42 levels were significantly reduced in BACE1+/−·5XFAD mouse brains at 6 months but not at 15–18 months of age as compared with their respective age-matched 5XFAD controls.25 Therefore, the results suggested that BACE1 haploinsufficiency was no longer able to ameliorate memory deficits, most likely because of its limited impact on robust β-amyloidosis in 5XFAD mice with advanced age.
BACE1 haploinsufficiency and BACE1-elevating pathways in 5XFAD mice
To address the mechanisms by which memory impairments as well as Aβ accumulation became less receptive to rescue by BACE1 haploinsufficiency in 5XFAD mice with advanced age, we compared changes in BACE1 expression between two different age groups (Figure 2a). Western blot analysis of hemibrain homogenates from 6-month-old mice demonstrated that BACE1 levels were not different between 5XFAD and wild-type controls, whereas BACE1+/− deletion significantly lowered BACE1 expression (by ∼50%) in concordance with the reduction in gene copy number in 5XFAD and wild-type mice (F(3, 13)=27.51, P<0.05; Figure 2b). In contrast, BACE1 levels in 15–18-month-old 5XFAD mice were significantly increased up to ∼200% of those found in age-matched wild-type control mice (F(3, 13)=22.76, P<0.05). Haploinsufficiency also reduced cerebral BACE1 expression by ∼50% in 5XFAD as well as wild-type mice at this advanced age (P<0.05). However, the residual levels of BACE1 in older BACE1+/−·5XFAD mouse brains were significantly higher than those of BACE1+/− mice (P<0.05) and almost equivalent to wild-type control levels (that is, twice the gene copy number). Taken collectively, these data indicated that the ablation of a single BACE1 allele was not sufficient to prevent BACE1-elevating mechanisms in BACE1+/−·5XFAD mice at 15–18 months of age.
Figure 2
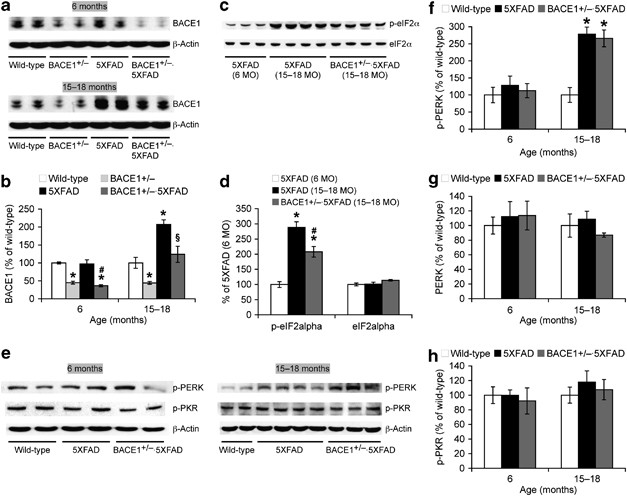
Effects of BACE1 (β-site amyloid precursor protein-cleaving enzyme 1) haploinsufficiency on BACE1 elevations associated with eIF2α (eukaryotic translation initiation factor-2α) phosphorylation pathways in 5XFAD mice. Immunoblot bands are quantified and expressed as the percentage of age-matched wild-type controls except for d, in which 6-month-old 5XFAD is control. (a, b) Haploinsufficiency reduces BACE1 expression by ∼50% in 5XFAD mice irrespective of age, as compared with age-matched 5XFAD mice. However, as BACE1 expression is significantly elevated up to ∼200% in 15–18-month-old 5XFAD mice, levels of BACE1 equivalent to wild-type controls remain in BACE1+/−·5XFAD brains at the advanced age. *P<0.05 versus wild-type, #P<0.05 versus 5XFAD, §P<0.05 versus BACE1+/− and 5XFAD. (c, d) Phospho-eIF2α (p-eIF2α) levels are significantly elevated in 15–18-month-old 5XFAD and BACE1+/−·5XFAD mouse brains compared with those of 6-month-old 5XFAD controls (*P<0.05). BACE1 haploinsufficiency only partially reduces 5XFAD-assocated increase of p-eIF2α at advanced age (#P<0.05 versus 15–18-month-old 5XFAD). (e–h) p-PERK levels, but not total PERK or p-PKR levels, are similarly increased in 15–18-month-old 5XFAD and BACE1+/−·5XFAD mice as compared with those of wild-type controls (*P<0.05). _n_=3–7 mice per group. All data are presented as mean±s.e.m.
Recent findings from our laboratory and others demonstrate that phosphorylation of the translation initiation factor eIF2α plays an important role in the posttranscriptional upregulation of BACE1 in 5XFAD mice and sporadic AD cases.25, 42, 43, 44 We compared phospho-eIF2α levels to test whether this translational mechanism may underlie the persistent BACE1 elevation in BACE1+/−·5XFAD mice at advanced age (Figure 2c). 5XFAD-associated increases in phospho-eIF2α levels were only marginal at 6 months of age but dramatically facilitated at 15–18 months in accordance with the emergence of elevation in BACE1 expression (F(2, 8)=29.10, P<0.05; Figure 2d). Notably, although phospho-eIF2α levels in BACE1+/−·5XFAD mice were significantly reduced as compared with 5XFAD controls at this advanced age (P<0.05), they were much higher than those of 6-month-old 5XFAD mice (P<0.05). Importantly, the changes were specific to the phosphorylated form of eIF2α in the absence of alterations in total eIF2α levels. Therefore, these results indicated that BACE1 haploinsufficiency failed to block phospho-eIF2α-associated BACE1 elevation in 5XFAD mice at 15–18 months of age.
We next explored upstream signaling pathways responsible for the persistent phosphorylation of eIF2α found in BACE1+/−·5XFAD mice with advanced age (Figure 2e). Among the major eIF2α kinases, double-stranded RNA-activated protein kinase (PKR) and PERK are suggested to play a role in AD pathogenesis.43, 44, 45 We found that equivalent levels of PERK activation, as evidenced by dramatic increases in phospho-PERK, were observed specifically in 5XFAD and BACE1+/−·5XFAD mice at 15–18 months of age (F(2, 18)=19.53, P<0.05; Figure 2f) without changes in total PERK levels (Figure 2g). In contrast, phospho-PKR levels were indistinguishable between the groups regardless of age (Figure 2h). Collectively, our results indicated that PERK activation was resistant to rescue by BACE1 haploinsufficiency in advanced stages of 5XFAD mice, resulting in the persistent eIF2α-dependent translational upregulation of BACE1 in brains.
We further conducted double immunofluorescence staining of hippocampal sections to address the direct relationship between PERK activation and eIF2α phosphorylation in 5XFAD mice (Figure 3). In agreement with western blotting data, PERK was activated almost equivalently in the hippocampal CA3 region of 5XFAD (Figure 3d) and BACE1+/−·5XFAD (Figure 3e) mice at 15–18 months of age, whereas no immunoreactivity for phospho-PERK was observed in that of wild-type controls (Figure 3a), 6-month-old 5XFAD mice (Figure 3b) or BACE1+/−·5XFAD mice (Figure 3c). Remarkably, double labeling revealed strong colocalization of phospho-eIF2α and phospho-PERK immunoreactivities in older 5XFAD and BACE1+/−·5XFAD mice. Meanwhile, marginal levels of eIF2α phosphorylation in younger 5XFAD mice, which were abolished by BACE1+/− gene deletion, were not accompanied by PERK activation. Therefore, these findings suggested that PERK-dependent activation of eIF2α occurred specifically in advanced stages of 5XFAD mouse brains suffering from BACE1 elevations and was not affected by BACE1 haploinsufficiency.
Figure 3
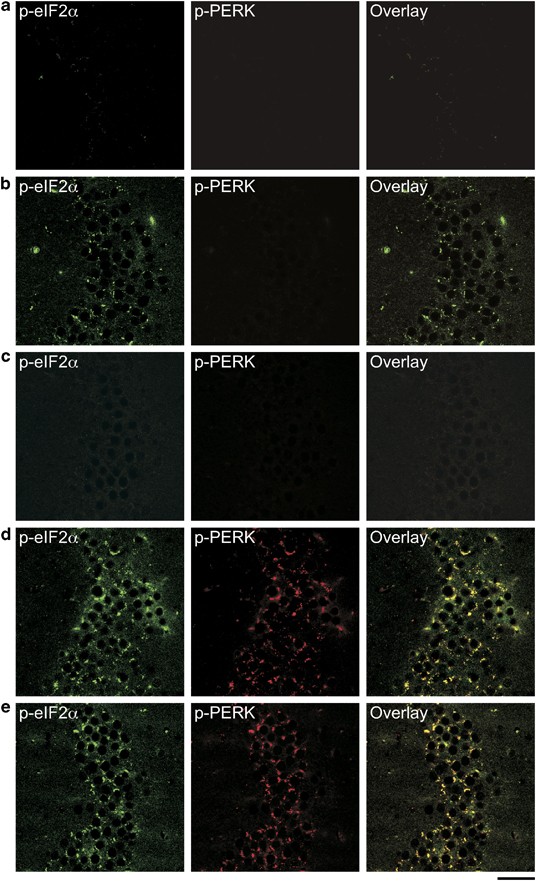
Double immunofluorescence labeling of activated eIF2α (eukaryotic translation initiation factor-2α) and PERK (PKR-endoplasmic reticulum-related kinase) signaling in 5XFAD mice with or without BACE1 (β-site amyloid precursor protein-cleaving enzyme 1) haploinsufficiency. Hippocampal sections were stained with anti-phospho-eIF2α (green) and anti-phospho-PERK (red) antibodies (_n_=4 mice per group). Shown are representative photomicrographs of the CA3 region from wild-type control (a), 6-month-old 5XFAD (b), its BACE1+/−·5XFAD littermate (c), 15–18-month-old 5XFAD (d) and its BACE1+/−·5XFAD littermate mouse (e). Note the high degree of colocalization of p-eIF2α and p-PERK immunoreactivities in neurons of 5XFAD and BACE1+/−·5XFAD mice at 15–18 months of age. Scale bar=50 μm.
BACE1 haploinsufficiency and Aβ-degrading enzymes in 5XFAD mice
In addition to enhanced β-amyloidogenesis, we investigated a possibility that impairments in Aβ metabolism might contribute to the mechanisms that lessened beneficial effects of BACE1 haploinsufficiency on memory impairments and Aβ accumulation in 5XFAD mice with advanced age (Figure 4). 5XFAD mice showed significant reductions in the major Aβ-degrading enzyme neprilysin (but not insulin-degrading enzyme, data not shown) at 6 months (F(3, 13)=10.49, P<0.05; Figures 4a and b) and 15–18 months of age (F(2, 19)=59.31, P<0.05; Figures 4c and d), as found in human AD brains.46 Remarkably, BACE1 haploinsufficiency restored reduced neprilysin expression almost completely back to wild-type control levels in younger 5XFAD mice (P<0.05). It is important to note that there was no difference in neprilysin levels between BACE1+/− and wild-type control mice (Figures 4a and b), excluding the possibility that BACE1 gene manipulations may directly affect neprilysin expression. In contrast to the observation at 6 months of age, BACE1+/− deletion had no effect on neprilysin deficiency in older 5XFAD mice. Therefore, the results suggested that the beneficial outcomes of partial BACE1 reduction declined in 5XFAD mice with advanced age because of compromised Aβ clearance as well as the persistent BACE1-elevating mechanisms.
Figure 4
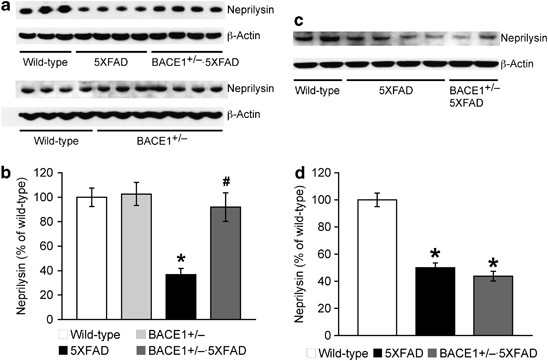
Effects of BACE1 (β-site amyloid precursor protein-cleaving enzyme 1) haploinsufficiency on reduced neprilysin expression in 5XFAD mice. Immunoblot bands are quantified and expressed as the percentage of age-matched wild-type controls. Neprilysin levels in 5XFAD mice at both 6 months (a, b) and 15–18 months of age (c, d) are significantly lower than those of wild-type controls (*P<0.05). Neprilysin reduction is rescued almost back to wild-type control levels in BACE1+/−·5XFAD mice at 6 months but not at 15–18 months of age (#P<0.05 versus age-matched 5XFAD); _n_=3–8 mice per group. All data are presented as mean±s.e.m.
Discussion
A growing body of evidence has demonstrated that BACE1 has a number of substrates besides APP,4, 5, 6, 7 and that endogenous levels of Aβ may be required for physiological functions including learning and memory.47, 48 Given that overinhibition of BACE1 may be associated with potential adverse effects, it is crucial to investigate the efficacy and limitations of partial suppression of the β-secretase enzyme in treating AD and related memory defects. We used BACE1 haploinsufficiency as a therapeutic relevant model, as BACE1+/− reduction does not induce any detrimental phenotypes as observed in BACE1−/− mice,11, 24, 49, 50 and can exert beneficial effects such as cognitive and synaptic improvements concomitant with cerebral Aβ reduction in AD mouse models, at least during relatively earlier pathological stages.11, 22, 23, 24, 25, 26, 27 In this study, we found that the benefits associated with BACE1 haploinsufficiency dramatically decline in 5XFAD mice with advanced age, because of its inability to prevent deleterious mechanisms leading to accelerated Aβ overproduction and compromised Aβ clearance.
Specifically, we first demonstrate that BACE1 haploinsufficiency ameliorates contextual memory deficits as well as cerebral β-amyloidosis in 5XFAD mice at 6 months but not at 15–18 months of age. This is in agreement with the previous findings that BACE1 haploinsufficiency can reduce Aβ concentrations and plaque load during earlier stages of progressive AD-like pathology, whereas it has little or no effects on robust β-amyloidosis at later stages in different APP transgenic mice.11, 22 Our results further reveal that BACE1 expression in 15–18-month-old 5XFAD mouse brains is highly elevated (approximately twofold) compared with wild-type controls, as reported in human AD cases.32, 51, 52, 53 Haploinsufficiency reduces BACE1 levels by ∼50% in 5XFAD mice regardless of age, whereas BACE1 expression remains almost equivalent to that of wild-type control mice (that is, twice the gene copy number) in BACE1+/−·5XFAD mice at advanced age. Therefore, it is apparent that haploinsufficiency is not sufficient to prevent the occurrence of BACE1 elevation during the course of Aβ pathology development.
Mechanistically, phosphorylation of the translation initiation factor eIF2α has been reported to mediate the posttranscriptional upregulation of BACE1 in advanced stages of 5XFAD mice and AD patients.25, 42, 43, 44 In particular, we showed that the increase in phospho-eIF2α induced by Sal 003 (a specific inhibitor of its phosphatase) can elevate BACE1 protein levels in 6-month-old 5XFAD mice that have not yet showed BACE1 upregulation consistent with only marginal changes of eIF2α phosphorylation.25 In this study, 5XFAD mice at 15–18 months of age had elevated levels of BACE1 concomitant with robust activation of eIF2α and its upstream kinase PERK. Immunoblot and double immunostaining experiments clearly demonstrate that levels of not only phospho-eIF2α but also phospho-PERK remain highly increased in 15–18-month-old BACE1+/−·5XFAD mice (relative to 6-month-old 5XFAD levels) in accordance with the high residual level of BACE1. In contrast, phosphorylation of PKR, another eIF2α kinase, is not affected in 5XFAD or BACE1+/−·5XFAD mouse brains. Taken collectively, these findings indicate that BACE1 haploinsufficiency cannot block PERK-mediated activation of the eIF2α phosphorylation pathway responsible for translational elevation of BACE1 in advanced stages of 5XFAD mice.
Net Aβ concentrations in brain are determined by the rate of Aβ production relative to the overall rate of Aβ clearance. It is widely accepted that compromised Aβ clearance in addition to accelerated Aβ generation may also underlie the molecular pathogenesis of AD.54, 55 This view is supported by our results showing that the major Aβ-degrading enzyme neprilysin (but not insulin-degrading enzyme) is significantly decreased in 5XFAD mouse brains consistent with observations in human AD brains.46 Importantly, previous work has shown that genetic deletion and/or reduction of neprilysin in APP transgenic mice further elevate cerebral Aβ levels and dramatically exacerbate AD-like traits such as plaque neuropathology and synaptic/cognitive dysfunctions.56, 57 Therefore, it seems likely that a reduction of neprilysin found in 5XFAD mouse brains contributes to the acceleration of Aβ accumulation and memory impairments. Interestingly, we found that BACE1 haploinsufficiency can rescue reduced neprilysin expression in 5XFAD mice at 6 months of age. Moreover, neprilysin levels are indistinguishable between BACE1+/− and wild-type control mice, indicating no interaction between BACE1 gene manipulations and baseline neprilysin expression. Therefore, it is reasonable to conceive that partial reduction of BACE1 is directly targeted at suppressing Aβ production, whereas it can also consequently prevent Aβ-dependent downregulation of neprilysin expression in 5XFAD mice at this age. Together, our results suggest that cerebral Aβ reductions associated with decreased generation and rescued clearance may underlie therapeutic or cognitive benefits of partial BACE1 inhibition during relatively earlier stages of AD.
In contrast, we found that BACE1 haploinsufficiency is no longer able to rescue reduced levels of neprilysin expression in 5XFAD mice at 15–18 months of age. Therefore, as disease progresses into the more profound stage that suffers from robust amyloid neuropathology, the failure of partial BACE1 reduction to prevent deficient Aβ degradation through neprilysin adds to the problem of persistent BACE1 elevation in 5XFAD mice, further weakening the beneficial impact on Aβ accumulation and memory impairments. In this regard, it should be noted that Aβ accumulation induces BACE1 elevation in neurons surrounding the amyloid core of plaques, which in turn further accelerates Aβ production in 5XFAD mouse and human AD brains.25, 32, 33, 58 Furthermore, previous studies with neprilysin transgenic and knockout mice have clearly shown that neprilysin-dependent degradation of Aβ is essential for suppressing plaque development.56, 57, 59, 60 Therefore, it is interesting to argue that BACE1 haploinsufficiency becomes unable to block a positive feedback pathogenic link between neprilysin reduction, increased levels of Aβ plaque formation and BACE1 elevation during the course of AD progression. Our results suggest that ∼50% of BACE1 suppression does not necessarily represent a reliable level of therapeutic enzyme inhibition for the treatment of AD and related memory deficits. Meanwhile, accumulating evidence demonstrates that BACE1 fulfills important physiological functions,4, 5, 6, 7 and that complete deletion of BACE1 is associated with adverse phenotypes such as some cognitive/noncognitive abnormalities, hypomyelination and retinal pathology.8, 9, 11, 49, 50, 61, 62, 63, 64, 65 Collectively, a combination anti-amyloid strategy aimed at partially inhibiting BACE1 and boosting neprilysin activities or blocking the BACE1-elevating PERK/eIF2α pathway may be useful for increasing the therapeutic benefits while obviating the potential detrimental effects that may be caused by direct overinhibition of BACE1.
References
- Vassar R . The β-secretase, BACE: a prime drug target for Alzheimer’s disease. J Mol Neurosci 2001; 17: 157–170.
Article CAS PubMed Google Scholar - Hardy JA, Higgins GA . Alzheimer’s disease: the amyloid cascade hypothesis. Science 1992; 256: 184–185.
Article CAS Google Scholar - Hardy J, Selkoe DJ . The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 2002; 297: 353–356.
Article CAS PubMed Google Scholar - Ohno M . Genetic and pharmacological basis for therapeutic inhibition of β- and γ-secretases in mouse models of Alzheimer’s memory deficits. Rev Neurosci 2006; 17: 429–454.
Article CAS PubMed Google Scholar - Ohno M . β-Secretase as a prime therapeutic target for Alzheimer’s disease: a perspective from mouse model studies. In Araki W, (ed) Recent Advances in the Biology of Secretases, Key Proteases in Alzheimer’s Disease. Research Signpost: Kerala, India, 2008 pp 1–25.
Google Scholar - Cole SL, Vassar R . The Alzheimer’s disease β-secretase enzyme, BACE1. Mol Neurodegener 2007; 2: 22.
Article PubMed PubMed Central Google Scholar - Vassar R, Kovacs DM, Yan R, Wong PC . The β-secretase enzyme BACE in health and Alzheimer’s disease: regulation, cell biology, function, and therapeutic potential. J Neurosci 2009; 29: 12787–12794.
Article CAS PubMed PubMed Central Google Scholar - Ohno M, Sametsky EA, Younkin LH, Oakley H, Younkin SG, Citron M et al. BACE1 deficiency rescues memory deficits and cholinergic dysfunction in a mouse model of Alzheimer’s disease. Neuron 2004; 41: 27–33.
Article CAS PubMed Google Scholar - Ohno M, Chang L, Tseng W, Oakley H, Citron M, Klein WL et al. Temporal memory deficits in Alzheimer’s mouse models: rescue by genetic deletion of BACE1. Eur J Neurosci 2006; 23: 251–260.
Article PubMed Google Scholar - Ohno M, Cole SL, Yasvoina M, Zhao J, Citron M, Berry R et al. BACE1 gene deletion prevents neuron loss and memory deficits in 5XFAD APP/PS1 transgenic mice. Neurobiol Dis 2007; 26: 134–145.
Article CAS PubMed Google Scholar - Laird FM, Cai H, Savonenko AV, Farah MH, He K, Melnikova T et al. BACE1, a major determinant of selective vulnerability of the brain to amyloid-β amyloidogenesis, is essential for cognitive, emotional, and synaptic functions. J Neurosci 2005; 25: 11693–11709.
Article CAS PubMed PubMed Central Google Scholar - De Strooper B, Vassar R, Golde T . The secretases: enzymes with therapeutic potential in Alzheimer disease. Nat Rev Neurol 2010; 6: 99–107.
Article CAS PubMed PubMed Central Google Scholar - Frisardi V, Solfrizzi V, Imbimbo PB, Capurso C, D'Introno A, Colacicco AM et al. Towards disease-modifying treatment of Alzheimer’s disease: drugs targeting β-amyloid. Curr Alzheimer Res 2010; 7: 40–55.
Article CAS PubMed Google Scholar - Luo X, Yan R . Inhibition of BACE1 for therapeutic use in Alzheimer’s disease. Int J Clin Exp Pathol 2010; 3: 618–628.
CAS PubMed PubMed Central Google Scholar - Chang WP, Koelsch G, Wong S, Downs D, Da H, Weerasena V et al. In vivo inhibition of Aβ production by memapsin 2 (β-secretase) inhibitors. J Neurochem 2004; 89: 1409–1416.
Article CAS PubMed Google Scholar - Hussain I, Hawkins J, Harrison D, Hille C, Wayne G, Cutler L et al. Oral administration of a potent and selective non-peptidic BACE-1 inhibitor decreases β-cleavage of amyloid precursor protein and amyloid-β production in vivo. J Neurochem 2007; 100: 802–809.
Article CAS PubMed Google Scholar - Sankaranarayanan S, Holahan MA, Colussi D, Crouthamel MC, Devanarayan V, Ellis J et al. First demonstration of cerebrospinal fluid and plasma Aβ lowering with oral administration of a β-site amyloid precursor protein-cleaving enzyme 1 inhibitor in nonhuman primates. J Pharmacol Exp Ther 2009; 328: 131–140.
Article CAS PubMed Google Scholar - Lerchner A, Machauer R, Betschart C, Veenstra S, Rueeger H, McCarthy C et al. Macrocyclic BACE-1 inhibitors acutely reduce Aβ in brain after po application. Bioorg Med Chem Lett 2010; 20: 603–607.
Article CAS PubMed Google Scholar - Jeppsson F, Eketjall S, Janson J, Karlstrom S, Gustavsson S, Olsson LL et al. Discovery of AZD3839, a potent and selective BACE1 clinical candidate for the treatment of Alzheimer disease. J Biol Chem 2012; 287: 41245–41257.
Article CAS PubMed PubMed Central Google Scholar - May PC, Dean RA, Lowe SL, Martenyi F, Sheehan SM, Boggs LN et al. Robust central reduction of amyloid-β in humans with an orally available, non-peptidic β-secretase inhibitor. J Neurosci 2011; 31: 16507–16516.
Article CAS PubMed Google Scholar - Takahashi H, Fukumoto H, Maeda R, Terauchi J, Kato K, Miyamoto M . Ameliorative effects of a non-competitive BACE1 inhibitor TAK-070 on Aβ peptide levels and impaired learning behavior in aged rats. Brain Res 2010; 1361: 146–156.
Article CAS PubMed Google Scholar - McConlogue L, Buttini M, Anderson JP, Brigham EF, Chen KS, Freedman SB et al. Partial reduction of BACE1 has dramatic effects on Alzheimer plaque and synaptic pathology in APP transgenic mice. J Biol Chem 2007; 282: 26326–26334.
Article CAS PubMed Google Scholar - Devi L, Ohno M . Genetic reductions of β-site amyloid precursor protein-cleaving enzyme 1 and amyloid-β ameliorate impairment of conditioned taste aversion memory in 5XFAD Alzheimer’s disease model mice. Eur J Neurosci 2010; 31: 110–118.
Article PubMed Google Scholar - Kimura R, Devi L, Ohno M . Partial reduction of BACE1 improves synaptic plasticity, recent and remote memories in Alzheimer’s disease transgenic mice. J Neurochem 2010; 113: 248–261.
Article CAS PubMed PubMed Central Google Scholar - Devi L, Ohno M . Phospho-eIF2α level is important for determining abilities of BACE1 reduction to rescue cholinergic neurodegeneration and memory defects in 5XFAD mice. PLoS One 2010; 5: e12974.
Article PubMed PubMed Central Google Scholar - Devi L, Ohno M . Mitochondrial dysfunction and accumulation of the β-secretase-cleaved C-terminal fragment of APP in Alzheimer’s disease transgenic mice. Neurobiol Dis 2012; 45: 417–424.
Article CAS PubMed Google Scholar - Chabrier MA, Blurton-Jones M, Agazaryan AA, Nerhus JL, Martinez-Coria H, LaFerla FM . Soluble Aβ promotes wild-type tau pathology in vivo. J Neurosci 2012; 32: 17345–17350.
Article CAS PubMed PubMed Central Google Scholar - Fukumoto H, Takahashi H, Tarui N, Matsui J, Tomita T, Hirode M et al. A noncompetitive BACE1 inhibitor TAK-070 ameliorates Aβ pathology and behavioral deficits in a mouse model of Alzheimer’s disease. J Neurosci 2010; 30: 11157–11166.
Article CAS PubMed Google Scholar - Chang WP, Huang X, Downs D, Cirrito JR, Koelsch G, Holtzman DM et al. β-Secretase inhibitor GRL-8234 rescues age-related cognitive decline in APP transgenic mice. FASEB J 2011; 25: 775–784.
Article CAS PubMed PubMed Central Google Scholar - Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J et al. Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci 2006; 26: 10129–10140.
Article CAS PubMed Google Scholar - Cai H, Wang Y, McCarthy D, Wen H, Borchelt DR, Price DL et al. BACE1 is the major β-secretase for generation of Aβ peptides by neurons. Nat Neurosci 2001; 4: 233–234.
Article CAS PubMed Google Scholar - Zhao J, Fu Y, Yasvoina M, Shao P, Hitt B, O'Connor T et al. β-Site amyloid precursor protein cleaving enzyme 1 levels become elevated in neurons around amyloid plaques: implications for Alzheimer’s disease pathogenesis. J Neurosci 2007; 27: 3639–3649.
Article CAS PubMed Google Scholar - Zhang X-M, Cai Y, Xiong K, Cai H, Luo X-G, Feng J-C et al. β-Secretase-1 elevation in transgenic mouse models of Alzheimer’s disease is associated with synaptic/axonal pathology and amyloidogenesis: implications for neuritic plaque development. Eur J Neurosci 2009; 30: 2271–2283.
Article CAS PubMed PubMed Central Google Scholar - Ohno M, Frankland PW, Chen AP, Costa RM, Silva AJ . Inducible, pharmacogenetic approaches to the study of learning and memory. Nat Neurosci 2001; 4: 1238–1243.
Article CAS PubMed Google Scholar - Kimura R, Ohno M . Impairments in remote memory stabilization precede hippocampal synaptic and cognitive failures in 5XFAD Alzheimer mouse model. Neurobiol Dis 2009; 33: 229–235.
Article CAS PubMed Google Scholar - Franklin KBJ, Paxinos G . The Mouse Brain in Stereotaxic Coordinates 3rd edn Academic Press: New York, USA, 2008.
Google Scholar - Devi L, Alldred MJ, Ginsberg SD, Ohno M . Sex- and brain region-specific acceleration of β-amyloidogenesis following behavioral stress in a mouse model of Alzheimer’s disease. Mol Brain 2010; 3: 34.
Article PubMed PubMed Central Google Scholar - Ohno M . Failures to reconsolidate memory in a mouse model of Alzheimer’s disease. Neurobiol Learn Mem 2009; 92: 455–459.
Article PubMed PubMed Central Google Scholar - Jawhar S, Trawicka A, Jenneckens C, Bayer TA, Wirths O . Motor deficits, neuron loss, and reduced anxiety coinciding with axonal degeneration and intraneuronal Aβ aggregation in the 5XFAD mouse model of Alzheimer’s disease. Neurobiol Aging 2012; 33: 196.e129–196.e140.
Article Google Scholar - Fanselow MS . Contextual fear, gestalt memories, and the hippocampus. Behav Brain Res 2000; 110: 73–81.
Article CAS PubMed Google Scholar - Maren S . Neurobiology of Pavlovian fear conditioning. Annu Rev Neurosci 2001; 24: 897–931.
Article CAS PubMed Google Scholar - Chang RC, Wong AK, Ng HK, Hugon J . Phosphorylation of eukaryotic initiation factor-2α (eIF2α) is associated with neuronal degeneration in Alzheimer’s disease. NeuroReport 2002; 13: 2429–2432.
Article CAS PubMed Google Scholar - O'Connor T, Sadleir KR, Maus E, Velliquette RA, Zhao J, Cole SL et al. Phosphorylation of the translation initiation factor eIF2α increases BACE1 levels and promotes amyloidogenesis. Neuron 2008; 60: 988–1009.
Article CAS PubMed PubMed Central Google Scholar - Mouton-Liger F, Paquet C, Dumurgier J, Bouras C, Pradier L, Gray F et al. Oxidative stress increases BACE1 protein levels through activation of the PKR-eIF2α pathway. Biochim Biophys Acta 2012; 1822: 885–896.
Article CAS PubMed Google Scholar - Devi L, Alldred MJ, Ginsberg SD, Ohno M . Mechanisms underlying insulin deficiency-induced acceleration of β-amyloidosis in a mouse model of Alzheimer’s disease. PLoS One 2012; 7: e32792.
Article CAS PubMed PubMed Central Google Scholar - Wang S, Wang R, Chen L, Bennett DA, Dickson DW, Wang DS . Expression and functional profiling of neprilysin, insulin-degrading enzyme, and endothelin-converting enzyme in prospectively studied elderly and Alzheimer’s brain. J Neurochem 2010; 115: 47–57.
Article CAS PubMed PubMed Central Google Scholar - Puzzo D, Privitera L, Leznik E, Fa M, Staniszewski A, Palmeri A et al. Picomolar amyloid-β positively modulates synaptic plasticity and memory in hippocampus. J Neurosci 2008; 28: 14537–14545.
Article CAS PubMed PubMed Central Google Scholar - Puzzo D, Privitera L, Fa M, Staniszewski A, Hashimoto G, Aziz F et al. Endogenous amyloid-β is necessary for hippocampal synaptic plasticity and memory. Ann Neurol 2011; 69: 819–830.
Article CAS PubMed PubMed Central Google Scholar - Hu X, Hicks CW, He W, Wong P, Macklin WB, Trapp BD et al. Bace1 modulates myelination in the central and peripheral nervous system. Nat Neurosci 2006; 9: 1520–1525.
Article CAS PubMed Google Scholar - Savonenko AV, Melnikova T, Laird FM, Stewart KA, Price DL, Wong PC . Alteration of BACE1-dependent NRG1/ErbB4 signaling and schizophrenia-like phenotypes in BACE1-null mice. Proc Natl Acad Sci USA 2008; 105: 5585–5590.
Article CAS PubMed Google Scholar - Fukumoto H, Cheung BS, Hyman BT, Irizarry MC . β-Secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch Neurol 2002; 59: 1381–1389.
Article PubMed Google Scholar - Holsinger RM, McLean CA, Beyreuther K, Masters CL, Evin G . Increased expression of the amyloid precursor β-secretase in Alzheimer’s disease. Ann Neurol 2002; 51: 783–786.
Article CAS PubMed Google Scholar - Yang LB, Lindholm K, Yan R, Citron M, Xia W, Yang XL et al. Elevated β-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat Med 2003; 9: 3–4.
Article CAS PubMed Google Scholar - Saido TC, Leissring MA . Proteolytic degradation of amyloid-β protein. In Selkoe DJ, Mandelkow E, Holtzman DM, (eds) The Biology of Alzheimer Disease. Cold Spring Harbor Laboratory Press: Cold Spring Harbor, USA, 2012 pp 387–404.
Google Scholar - Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC et al. Decreased clearance of CNS β-amyloid in Alzheimer’s disease. Science 2010; 330: 1774.
Article CAS PubMed PubMed Central Google Scholar - Huang SM, Mouri A, Kokubo H, Nakajima R, Suemoto T, Higuchi M et al. Neprilysin-sensitive synapse-associated amyloid-β peptide oligomers impair neuronal plasticity and cognitive function. J Biol Chem 2006; 281: 17941–17951.
Article CAS PubMed Google Scholar - Farris W, Schutz SG, Cirrito JR, Shankar GM, Sun X, George A et al. Loss of neprilysin function promotes amyloid plaque formation and causes cerebral amyloid angiopathy. Am J Pathol 2007; 171: 241–251.
Article CAS PubMed PubMed Central Google Scholar - Leroy K, Ando K, Laporte V, Dedecker R, Suain V, Authelet M et al. Lack of tau proteins rescues neuronal cell death and decreases amyloidogenic processing of APP in APP/PS1 mice. Am J Pathol 2012; 181: 1928–1940.
Article CAS PubMed Google Scholar - Leissring MA, Farris W, Chang AY, Walsh DM, Wu X, Sun X et al. Enhanced proteolysis of β-amyloid in APP transgenic mice prevents plaque formation, secondary pathology, and premature death. Neuron 2003; 40: 1087–1093.
Article CAS Google Scholar - Meilandt WJ, Cisse M, Ho K, Wu T, Esposito LA, Scearce-Levie K et al. Neprilysin overexpression inhibits plaque formation but fails to reduce pathogenic Aβ oligomers and associated cognitive deficits in human amyloid precursor protein transgenic mice. J Neurosci 2009; 29: 1977–1986.
Article CAS PubMed PubMed Central Google Scholar - Willem M, Garratt AN, Novak B, Citron M, Kaufmann S, Rittger A et al. Control of peripheral nerve myelination by the β-secretase BACE1. Science 2006; 314: 664–666.
Article CAS Google Scholar - Wang H, Song L, Laird F, Wong PC, Lee HK . BACE1 knock-outs display deficits in activity-dependent potentiation of synaptic transmission at mossy fiber to CA3 synapses in the hippocampus. J Neurosci 2008; 28: 8677–8681.
Article CAS PubMed PubMed Central Google Scholar - Cai J, Qi X, Kociok N, Skosyrski S, Emilio A, Ruan Q et al. β-Secretase (BACE1) inhibition causes retinal pathology by vascular dysregulation and accumulation of age pigment. EMBO Mol Med 2012; 4: 980–991.
Article CAS PubMed PubMed Central Google Scholar - Hitt BD, Jaramillo TC, Chetkovich DM, Vassar R . BACE1−/− mice exhibit seizure activity that does not correlate with sodium channel level or axonal localization. Mol Neurodegener 2010; 5: 31.
Article PubMed PubMed Central Google Scholar - Rajapaksha TW, Eimer WA, Bozza TC, Vassar R . The Alzheimer’s β-secretase enzyme BACE1 is required for accurate axon guidance of olfactory sensory neurons and normal glomerulus formation in the olfactory bulb. Mol Neurodegener 2011; 6: 88.
Article CAS PubMed PubMed Central Google Scholar
Acknowledgements
This work was supported by the American Health Assistance Foundation Grant (A2011311 to MO).
Author information
Authors and Affiliations
- Center for Dementia Research, Nathan Kline Institute, Orangeburg, NY, USA
L Devi & M Ohno - Department of Psychiatry, New York University Langone Medical Center, New York, NY, USA
M Ohno
Corresponding author
Correspondence toM Ohno.
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Devi, L., Ohno, M. Mechanisms that lessen benefits of β-secretase reduction in a mouse model of Alzheimer’s disease.Transl Psychiatry 3, e284 (2013). https://doi.org/10.1038/tp.2013.59
- Received: 25 February 2013
- Revised: 22 May 2013
- Accepted: 25 May 2013
- Published: 23 July 2013
- Issue Date: July 2013
- DOI: https://doi.org/10.1038/tp.2013.59