Effects of currently prescribed LDL-C-lowering drugs on PCSK9 and implications for the next generation of LDL-C-lowering agents (original) (raw)
- Review
- Open access
- Published: 28 February 2011
Lipids in Health and Disease volume 10, Article number: 38 (2011)Cite this article
- 9722 Accesses
- 58 Citations
- 9 Altmetric
- Metrics details
Abstract
Background
During the past decade, proprotein convertase subtilisin kexin type 9 (PCSK9) has been identified as a key regulator of serum LDL-cholesterol (LDL-C) levels. PCSK9 is secreted by the liver into the plasma and binds the hepatic LDL receptor, causing its subsequent degradation. In humans, gain-of-function mutations in PCSK9 cause a form of familial hypercholesterolemia that manifests with dramatically increased serum levels of LDL-C, while loss-of-function mutations in PCSK9 are associated with significantly decreased LDL-C and cardiovascular risk.
Results
Initial studies in animals and cultured cells demonstrated that statins increased PCSK9 mRNA expression, resulting in many research groups exploring the effect of statins on PCSK9 levels in humans. We first reported that statins increased human PCSK9 circulating protein levels. Additional researchers subsequently confirmed these observations, further prompting many laboratories including our own to examine the effect of other lipid lowering medications on PCSK9 levels. Our observation that fenofibrate (200 mg/day) significantly increased PCSK9 levels was confirmed by another laboratory, and an additional group demonstrated that ezetimibe also increased PCSK9 levels.
Conclusions
It has become clear that the major classes of commonly prescribed lipid-lowering medications increase serum PCSK9 levels. These observations almost certainly explain why these agents are not more effective in lowering LDL-C and suggest that efforts should be made toward the development of new LDL-C lowering medications that either do not increase circulating PCSK9 levels or work through decreasing or inhibiting PCSK9.
Discovery of PCSK9, mechanism of action, and genetic findings
The discovery of proprotein convertase subtilisin kexin type 9 (PCSK9) and the subsequent observations that genetic mutations in PCSK9 can drastically affect low density lipoprotein cholesterol (LDL-C) levels and the risk for cardiovascular disease ushered in a new era of understanding about the regulation of circulating LDL-C [1–6]. PCSK9 was first described by Seidah and co-workers in 2003 as a protein important in liver regeneration and neuronal differentiation [7]. These researchers originally named the protein (which was the ninth described member of the proprotein subtilisin kexin family of proteins) neuronal apoptosis regulated convertase-1 (NARC-1).
Later that year, Abifadel and colleagues reported the discovery of 2 mutations in PCSK9 that resulted in familial hypercholesterolemia (FH), a genetic disease characterized by greatly increased levels of LDL-C and early onset of atherosclerosis and cardiovascular events [8]. Prior to that report, FH had been associated with mutations in either the low density lipoprotein receptor (LDLR) or apolipoprotein B (ApoB). Mutations in these 2 genes however, could not account for all cases of FH, and the observation that PCSK9 mutations could result in FH established PCSK9 as third important locus of FH [8].
In 2004, Maxwell and colleagues reported that overexpression of PCSK9 in mice resulted in a phenotype that was very similar to that of an LDLR knockout model, with large increases in circulating non high density lipoprotein cholesterol (HDL-C), and a virtual absence of measureable LDLR protein [9]. In addition, these researchers observed that PCSK9 overexpression had little effect on LDLR mRNA levels suggesting that PCSK9 was acting at the protein level through degradation of the hepatic LDLR [9]. These interesting observations were followed by a number of reports over the next few years further elucidating the mechanism by which PCSK9 decreases LDLR levels. It was gradually recognized that PCSK9 was a protease secreted by the liver into the plasma, which was then able to bind to and cause the degradation of hepatic LDLR [10–19]. The mechanism by which PCSK9 degrades LDLR has turned out to be more complex than originally anticipated. As Figure 1 shows, PCSK9 protein contains a pro-domain that is self-cleaved during secretion and remains tightly associated with the protein, as well as catalytic and C-terminal domains. Recent studies suggest that after self-cleavage and secretion, PCSK9 does not have to be further enzymatically active to cause degradation of the LDLR [20–22]. Instead, PCSK9 has been shown to bind to the LDLR and subsequently target it for lysosomal destruction within the hepatocyte [20–22]. This concept of how PCSK9 acts to decrease hepatic LDLR levels has been further supported by findings that disruption of the binding of PCSK9 to the LDLR using anti-PCSK9 antibody results in preserved LDLR and decreased LDL-C [23–26].
Figure 1
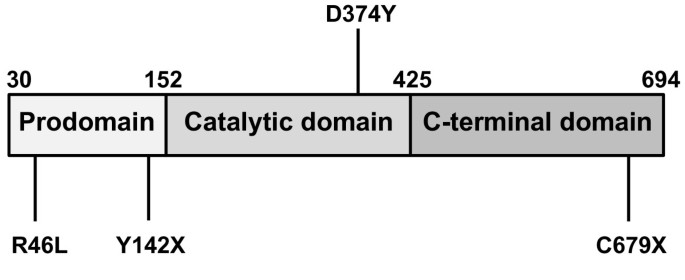
PCSK9 protein structure and selected mutations. PCSK9 protein includes a pro-domain (Pro) that is self-cleaved during secretion and remains tightly associated, a catalytic domain, and a C-terminal domain. Activating mutations such as D374Y cause increased affinity for LDLR. Inactivating mutations such as Y142X and C679X block self-cleavage and secretion of the protein. The R46L mutation results in decreased circulating levels of PCSK9 protein, although the mechanism is not completely understood.
At the same time that the biology of PCSK9 was being increasingly understood, additional different activating PCSK9 mutations causing FH were being reported in humans. It was increasingly recognized that these patients were presenting with gain-of-function mutations of PCSK9, causing LDLR protein levels to be markedly decreased with resulting FH and accompanying increased cardiovascular risk [27–29]. Interest in PCSK9 then dramatically accelerated in 2006 with the report by Hobbs and co-workers that approximately 3% of African-Americans were heterozygous for loss-of-function mutations in PCSK9, including mutations such as Y142X and C679X which prevent the self-cleavage and secretion of the protein, and that these subjects had significantly decreased levels of serum LDL-C and approximately 80-90% reduced cardiovascular risk [30]. Later that same year, Hobbs and co-workers described a compound heterozygote for PCSK9 loss-of-function mutations [31]. Remarkably, the subject who was a healthy 32 year-old female, had a serum LDL-C level of 14 mg/dl [31]. More recently, a second subject, a 49 year-old male, heterozygous for 2 monoallelic dominant negative PCSK9 mutations has also been described and had a LDL-C of 16 mg/dl [32].
Shortly after the loss-of-function findings in African-Americans were described, it was also reported that 3% of Caucasian subjects, while not possessing the complete loss of function mutations observed in African-Americans, did harbor mutations in PCSK9 that resulted in decreased serum cholesterol levels [33]. Among these mutations, one of the most prominent was R46L, which conferred a 10-20% reduction in circulating LDL-C and a 50% reduced risk of cardiovascular disease [33]. It was not intuitively obvious why the R46L mutation should do this. Only when the laboratories of Humphries and co-workers and of Hobbs and co-workers reported that the R46L mutation resulted in significantly decreased levels of circulating PCSK9 did it become apparent that the mutation was somehow lowering the amount of PCSK9 protein in the serum, and that this reduction in serum PCSK9 was likely responsible for the beneficial effects on LDL-C and cardiovascular risk [34, 35].
In light of all of the findings in humans showing that PCSK9 mutations that decrease PCSK9 levels result in decreased serum LDL-C and cardiovascular risk, there has been tremendous increased interest in understanding the normal role of PCSK9. In 2006, Costet and colleagues reported that PCSK9 mRNA expression was decreased 73% in mice after fasting for a 24-hour period and that PCSK9 mRNA expression levels returned to normal following re-feeding with carbohydrate [36]. In the same study, it was also shown that insulin increased PCSK9 mRNA expression levels, suggesting that PCSK9 expression may be regulated by nutritional status via circulating insulin levels [36].
Recently, Horton and colleagues examined the relationship between lathosterol, a marker of hepatic cholesterol biosynthesis and circulating PCSK9 levels in humans [37]. This group observed that lathosterol and PCSK9 levels were highly correlated with each other and declined together during the fasting state. Around the same time, Rudling and colleagues conducted extensive studies on the effect of prolonged fasting as well as the administration of the bile acid binding resin cholestyramine on circulating PCSK9 and lathosterol and observed that PCSK9 levels and lathosterol levels move in virtual lockstep with each other and are both dramatically decreased by fasting, whereas LDL cholesterol was little changed [38]. Together, these results would seem to indicate that PCSK9 may represent a mechanism that has evolved for fine-tuning plasma LDL-C levels, although this idea will have to be further probed. In any event, all of the findings described above certainly established PCSK9 as a major player in the regulation of LDL-C levels.
Statins, PCSK9, and the rule of 6%
Statins represent the most commonly prescribed class of LDL-C lowering medications. Statins work through inhibiting the enzyme 3-Hydroxy-3-methyl-glutaryl-CoA reductase (HMG-CoA reductase) which represents the rate limiting step in cholesterol biosynthesis [39]. By decreasing hepatic intracellular levels of cholesterol, statins increase the activity/nuclear translocation of the transcription factor sterol regulatory element-binding protein-2 (SREBP-2), which activates the LDLR gene [40]. Increased expression of LDLR on the surface of hepatocytes results in increased LDL binding and catabolism which in turn decreases circulating LDL-C levels by 30-40% or even more [40].
In 2003, Maxwell and co-workers first identified PCSK9 as a novel target gene of SREBP and demonstrated that hepatic PCSK9 mRNA levels were significantly increased in SREBP-1a and SREBP-2 transgenic mice [41]. In 2004, Dubuc and colleagues reported in a study using HEPG2 cells and primary hepatocytes that statins upregulate the expression of PCSK9 mRNA [42]. The induction of PCSK9 mRNA was described as being highly dose dependent and was reversed by mevalonate [42]. The authors also observed that human, mouse, and rat PCSK9 promoters contain 2 typical conserved motifs for cholesterol regulation, a sterol regulatory element (SRE) and a Sp1 site. They concluded that PCSK9 regulation is typical of that of the genes implicated in lipoprotein metabolism (such as LDLR) and that PCSK9 was likely a target of SREBP-2 [42]. The following year, Horton and co-workers demonstrated that PCSK9 knockout mice exhibited an exaggerated response to statin administration as demonstrated by increases in hepatic LDLR and increased LDL-C clearance from their plasma [43]. Together with additional results generated in other laboratories [44–46], these data further supported the notion that statins increase the activity/nuclear translocation of SREBP-2, a transcription factor that activates both the LDLR and PCSK9 genes [41–46]. These data gave the first indication in vivo that a PCSK9 inhibitor might be able to reduce plasma cholesterol beyond what was achievable with statins alone [41–46].
These observations led a number of laboratories, including our own, to examine the effects of statins on human PCSK9 levels. In order to do this, there were 2 requirements. The first was a reliable immunoassay for measuring human serum PCSK9 levels. The second was a set of samples to test from patients who had blood drawn at various time points prior to and after the initiation of statin therapy. With regard to the development of a reliable immunoassay, our group had begun working on this goal in early 2006, and by 2007 had established a robust, dual-monoclonal sandwich enzyme linked immunosorbent assay (ELISA) that we were able to use to show that human serum PCSK9 levels were highly correlated with LDL-C levels [47]. Later that same year, we were able to obtain a set of baseline and endpoint samples from patients treated with placebo, 10 mg/day atorvastatin, or 40 mg/day atorvastatin for 12 weeks. Using our sandwich ELISA, we were able to demonstrate that atorvastatin (40 mg/day) increased serum PCSK9 levels by 34% compared to placebo at 12 weeks, while lowering LDL-C by 42% [48]. In contrast, placebo treatment in the same study had no significant effect on either LDL-C levels or serum PCSK9 levels [48].
The fact that atorvastatin significantly increased PCSK9 levels provided evidence that statins increase the activity/nuclear translocation of sterol regulatory element-binding protein-2 (SREBP-2) to such an extent that increased levels of PCSK9 protein were present in the serum. We concluded this was particularly important since the ability to detect any increase in secreted PCSK9 protein could have been negated by the increased hepatic LDLR that would act to bind PCSK9 and remove it from the circulation. Therefore, we hypothesized that atorvastatin treatment must have increased expression and secretion of PCSK9 protein to such an extent that circulating PCSK9 levels exceeded LDLR binding, resulting in increased circulating levels of PCSK9 protein being detected in serum.
Our initial observation that statin treatment in humans caused an increase in serum PCSK9 levels was soon confirmed by other groups, further suggesting the importance of statin-induced increases in serum PCSK9 levels [34, 49, 50]. As interesting as these observations were, they were somewhat limited in that the baseline and endpoint analysis of samples did not allow for the detailed time course of statin-induced PCSK9 increases to be described. It was thus not clear at the time how quickly the effect of statins on PCSK9 levels occurred or whether the effect was sustained over time. As a result, we conducted an additional larger study examining the detailed time course of the effect of 80 mg/day atorvastatin on human serum PCSK9 levels in which we measured serum PCSK9 and lipid levels during a 2-week lead-in baseline period and every 4 weeks thereafter for 16 weeks. We observed in this subsequent study that atorvastatin (80 mg/day) caused a rapid 47% increase in serum PCSK9 after 4 weeks of treatment that was completely sustained throughout 16 weeks of dosing [51]. Interestingly, we also observed that while PCSK9 levels were highly correlated with LDL-C at baseline, atorvastatin (80 mg) completely abolished this correlation [51].
In this study, we also compared baseline PCSK9 levels to atorvastatin-induced changes in LDL-C (from baseline to endpoint) to determine if baseline PCSK9 levels might predict whom would respond most robustly to atorvastatin treatment. We hypothesized that subjects who had the highest PCSK9 levels might also have the largest atorvastatin-induced LDL-C lowering effect. Consistent with this hypothesis, there was a modest relationship between baseline PCSK9 levels and changes in LDL-C, with relatively higher baseline PCSK9 levels tending to be associated with numerically greater decreases in LDL-C [51]. This correlation, however, did not achieve statistical significance [51].
These data, in conjunction with those of Costet and colleagues who recently showed that atorvastatin 10 mg/day increased PCSK9 levels by only 14% in patients after 6 weeks of therapy [52], suggested a clear dose response effect for atorvastatin on PCSK9 levels, with increasing doses of atorvastatin causing larger and larger percentage increases in circulating PCSK9 levels. Available data regarding the effect of atorvastatin on human serum PCSK9 levels in controlled clinical trials where endpoint PCSK9 levels could be compared to baseline levels is summarized in Table 1, which shows that increasing doses of atorvastatin result in dose-dependent increases in circulating PCSK9 protein. Consistent with this pattern, Lakoski and co-workers recently demonstrated in a large trial that at a low dose of simvastatin (10 mg/day), there was no significant increase in median circulating PCSK9 levels [53].
Table 1 Effect of increasing doses of atorvastatin on serum PCSK9 levels
As Figure 2 demonstrates, these data suggest that the explanation for why increasing doses of statins fail to achieve proportional LDL-C lowering is that statins increase PCSK9 protein levels in a dose-dependent fashion, and that the increased PCSK9 levels largely negate further statin-induced increases in hepatic LDLR levels. For some time, it has been known that statins follow a rule of 6% in that whatever LDL-C reduction is achieved at a starting dose of a given statin is only improved upon an additional approximate 6% with each doubling of the dose. This important observation of non-dose dependent response for commonly prescribed statins has led to much speculation about why statins should affect LDL-C levels in such a manner. Our own data, combined with data that other laboratories have generated, suggest that statin-induced increases in PCSK9 protein levels account for the less than expected, incremental LDL-C lowering observed when the starting dose of a statin is subsequently increased.
Figure 2
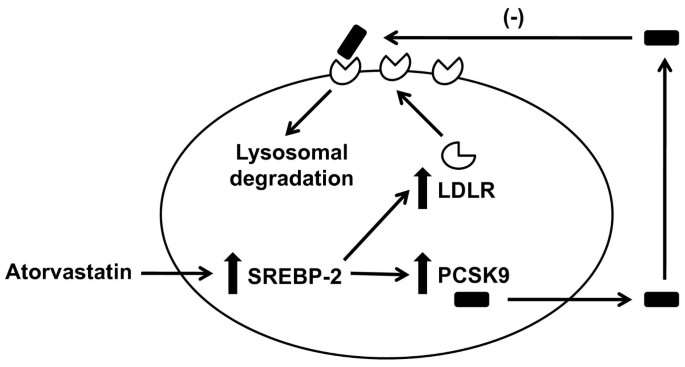
Effect of atorvastatin on hepatocyte LDLR and PCSK9. Atorvastatin increases the activity/nuclear translocation of sterol regulatory element-binding protein-2 (SREBP-2), which is a transcription factor that activates both the LDLR and PCSK9 genes. This results in increased expression and secretion of PCSK9 protein, which binds the LDLR and targets it for lysosomal degradation. This likely prevents atorvastatin from causing as much increased LDLR protein from being present on the hepatocyte plasma membrane as might otherwise be expected.
Besides statins, another commonly prescribed LDL-C lowering medication is ezetimibe, which acts by blocking uptake of dietary cholesterol from the gut by binding the cholesterol transport protein Niemann-Pick C1-Like 1 (NPC1L1) [40, 54–56]. By decreasing the amount of cholesterol supplied to the liver through intestinal uptake, ezetimibe decreases hepatic cholesterol levels, leading to increased hepatic LDLR expression, which results in increased LDL uptake from the plasma and thus decreased circulating LDL-C levels [40, 54–56]. Ezetimibe lowers LDL-C by approximately 15-20%, which is a smaller decrease than that achievable with statins, but its effect appears to be additive to that of statins [40, 54–56].
With regard to the effect of ezetimibe on PCSK9 levels, there have been fewer studies than for statins. A recent reports by Davignon and colleagues indicated that when added onto statin therapy, ezetimibe further increased PCSK9 levels beyond the increases observed with statins alone [57]. In this study, the researchers observed that patients treated with statins alone had an approximately 45% increase in circulating PCSK9 levels, while those treated with a combination of statins and ezetimibe exhibited an approximately 77% increase in PCSK9 protein levels [57]. At the present time, the mechanism by which ezetimibe causes a further increase in PCSK9 levels compared to statin treatment alone is unclear, but is likely associated with reduced intestinal cholesterol absorption and thus reduced hepatic cholesterol to feedback regulate SREBP2 [40]. As a result of these interesting findings, further study in this area will clearly be warranted. Of particular importance will be to determine the effect of ezetimibe alone on serum PCSK9 levels and how it is modulated with the addition of increasing doses of statins.
Fibrates and their effects on PCSK9 levels
Fibrates represent a class of medications that activate the transcription factor peroxisome proliferator-activated receptor α (PPARα) [58, 59]. Activation of this receptor by fibrates alters the transcription of multiple target genes which play a key role in lipid metabolism [58, 59]. In particular fibrates have been shown to reduce plasma levels of triglycerides by approximately 30%-50% and have been demonstrated to increase levels of HDL-C by 5%-15% [58, 59]. The effects on LDL-C are less consistent. In some cases, fibrates may reduce LDL-C by as much as 15%-20% although the effect can be variable, depending on the patient population being studied and the type of hyperlipidemia present [58, 59].
Previous data showing that statins increase PCSK9 levels caused many researchers including our own group to wonder about the effects of fibrates on PCSK9 levels. To address this question, we studied the effect of a commonly prescribed fibrate, fenofibrate, on circulating PCSK9 protein levels in patients treated with either fenofibrate (200 mg/day) or placebo for 12 weeks. We observed that fenofibrate significantly increased circulating PCSK9 levels by 25% compared to baseline, while placebo treatment had no effect on PCSK9 levels [60]. Interestingly, fenofibrate-induced increases in serum PCSK9 levels were highly correlated with fenofibrate-induced changes in LDL-C levels. Similar to what was observed with statins, these results suggested an explanation for why fibrates might not achieve as much LDL-C lowering as might otherwise be expected [60].
The mechanism by which fenofibrate increased PCSK9 levels in this study was unclear. Fenofibrate belongs to a class of drugs called PPAR-α agonists, which affect lipid levels by altering transcription of a number of different genes involved in lipoprotein and fatty acid metabolism [61]. As a PPAR-α agonist, fenofibrate reduces hepatic triglyceride synthesis and increases the breakdown of triglyceride rich lipoproteins, while also enhancing cholesterol efflux from the liver [62]. It is possible that these effects on cholesterol and lipoprotein metabolism may work indirectly within the hepatocyte to decrease intracellular cholesterol levels, thus leading to increased PCSK9 expression and secretion via a mechanism similar to that of statins. In light of the multiple pleiotropic effects of fenofibrate [63], however, it is also possible that fenofibrate may exert a direct effect within hepatocytes to stimulate increased PCSK9 synthesis and secretion independent of SREBP-2.
Making matters somewhat more complex, there have been conflicting observations concerning the effect of fibrates on hepatocyte PCSK9 synthesis and secretion. Previously, Mayne and co-workers reported that fibrates increased human serum PCSK9 levels by 17% [64]. In this study, however, analysis of PCSK9 levels was performed using an immunoprecipitation and western blotting method, which is a semi-quantitative rather than a quantitative technique. In addition, the absolute levels of PCSK9 present in human serum as reported by this method were significantly greater than what our laboratory as well as several others have reported. Also, the group of patients analyzed in this study was relatively small and consisted of a mixture of patients on gemfibrozil and fenofibrate.
In contrast to this report, Kourimate and co-workers demonstrated that fibrate treatment resulted in reduced PCSK9 mRNA levels in hepatocytes [65]. This group also went on to measure levels of PCSK9 protein expression in hepatocytes and concluded that PCSK9 protein expression in hepatocytes was also reduced. Again, similar to the work by Mayne and co-workers, PCSK9 protein levels were measured by western blotting. Kourimate and co-workers concluded from these data that addition of a fibrate to pre-existing statin therapy would result in suppression of circulating PCSK9 levels and thus enhanced LDL-C lowering activity of the statin. On the other hand, Lambert and colleagues used an ELISA method to report that fenofibrate treatment (200 mg per day for 6 weeks) decreased plasma PCSK9 levels by 8% and that this decrease correlated with the fenofibrate-induced decreases in circulating triglycerides [66]. Similar to the observations by Mayne and colleagues, the absolute plasma levels of PCSK9 as reported by this method were significantly greater than what our laboratory and several other groups have reported. The same researchers concluded that fenofibrate-induced decreases in PCSK9 might account for the modest LDL-C reduction observed with fenofibrate [66].
The conclusions reached by these two groups were different from those that we reached with regard to the effect of fibrates on circulating PCSK9 protein levels. One possibility for this difference may have been the use of different analytical techniques than those used by our laboratory and other laboratories. This possibility seems even more likely in light of a very recent, thorough study exploring the effects of fenofibrate on PCSK9 serum levels in patients with diabetes. In their recent report using a robust and specific assay, Costet and colleagues demonstrated that 6 weeks of treatment with fenofibrate (160 mg/day) increased serum PCSK9 by 26% [52]. This increase in circulating PCSK9 levels was very similar to the increase of 25% that we observed after 12 weeks of treatment using our own assay.
Therefore, at the current time, it seems likely that fibrates share with statins the common property of increasing serum PCSK9 protein levels. Unlike the situation for statins though, in which the mechanism for the increase in PCSK9 is likely mediated through SREBP-2, it is much less understood how fibrates actually increase circulating PCSK9 protein levels. Certainly additional studies will be needed to better understand the biology of fibrate-induced PCSK9 increases.
Future directions for LDL-lowering therapies
In light of the genetic studies in humans showing PCSK9 mutations that decrease PCSK9 levels result in decreased serum LDL-C and cardiovascular risk and the findings that the utility of both statins and fibrates may be limited due to the fact that they increase serum PCSK9 levels, there has been tremendous increased interest in developing a PCSK9 inhibitor. One approach that has been tested is to use an antisense oligonucleotide (ASO) to target PCSK9 mRNA. Graham and colleagues demonstrated that this approach reduced total cholesterol by 53% in mice fed a high-fat diet, while also causing a 2-fold increase in hepatic LDLR protein levels [14]. Similarly, Gupta and co-workers reported that an ASO lowered PCSK9 mRNA expression in mice by 60% and increased hepatic LDLR protein expression by almost 3-fold [67]. Comparable results have also been reported using a siRNA approach by Fitzgerald and colleagues, who demonstrated 60% reductions in PCSK9 mRNA and total cholesterol in mice [68]. This same group also demonstrated that such an approach was possible in a non-human primate model as well by showing that a single siRNA injection produced long-lasting reductions in both serum PCSK9 and LDL-C levels [68].
Due to the nature of PCSK9 protein and the way in which it functions by binding the LDLR, several groups have also attempted to generate neutralizing, fully humanized anti-PCSK9 monoclonal antibodies to block the interaction of PCSK9 with the LDLR at the protein level. Preliminary results so far have been encouraging. Chan and colleagues developed a monoclonal antibody that binds a PCSK9 epitope required for recognition by the LDLR. When administered to cynomolgus monkeys, one injection of this antibody decreased serum LDL-C by approximately 80%, and a decrease in LDL-C was observed over the course of 10 days [25]. In a separate, recent report, Ni and co-workers developed a monoclonal PCSK9 antibody that was neutralizing not only to wild type PCSK9 but also to gain of function mutants including D374Y [26]. When this antibody was administered to rhesus monkeys, LDL-C was decreased from 20-50% over a 2-week long period, even though the antibody had a relatively short half-life.
These observations clearly established a proof-of-concept that a neutralizing anti-PCSK9 monoclonal antibody is capable of reducing LDL-C levels in primates. One of the big remaining questions, however, relates to the durability of such a therapeutic approach in actual patients. In order to understand how feasible such an approach will ultimately be in the clinic, it is important to understand 2 key characteristics of PCSK9 - its half-life and what the absolute circulating levels are. With regard to the first issue, the half-life of PCSK9 appears to be relatively short, presumably because PCSK9 in the plasma is rapidly bound by LDLR and/or similar type receptors. The best current estimates for the circulating half-life are somewhere between 5 and 30 minutes [11, 19].
With regard to the absolute levels of circulating PCSK9 protein, there is currently less agreement. Part of the problem has been the relative difficulty of developing robust methods to measure PCSK9 in human serum. When analyzed by western blotting under reducing, denaturing conditions, intact PCSK9 protein co-migrates almost precisely with human albumin, which is present in the serum at a concentration of approximately 4 g/dl. Because of this, it is extremely difficult to accurately gauge human serum levels when performing straight western blotting procedures alone. As Figure 3 demonstrates, when suitable immunoprecipitation is performed, followed by western blotting, it becomes easier to visualize the PCSK9 band, but also apparent that there is at least one additional band which almost precisely co-migrates with human IgG heavy chain under reducing denaturing conditions. This second band has been described as a furin cleavage product of PCSK9 [49], and can be almost as intense as the intact protein itself.
Figure 3
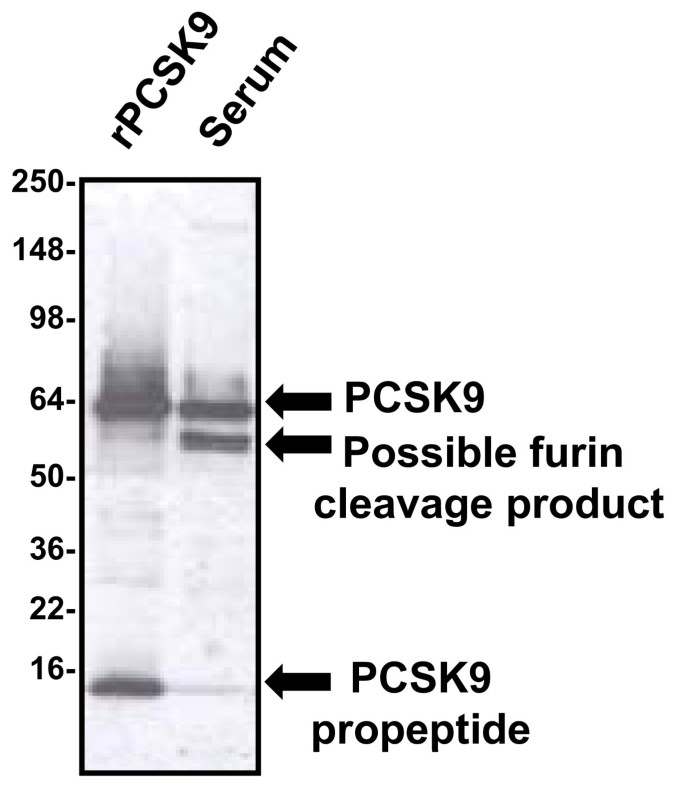
Characterization of PCSK9 protein in human serum. When immunoprecipitated from human serum and analyzed in our laboratory by subsequent western blotting under reducing and denaturing conditions, PCSK9 migrates as an intact band (top arrow) that co-migrates with recombinant PCSK9 protein (rPCSK9) [51, 60]. In addition, a band of approximately 55 kD is also present (middle arrow), and this band had been described as representing a furin cleavage product of the intact protein [49]. The band at the very bottom of the blot represents the propeptide which remains tightly but non-covalently associated during the immunoprecipitation step, but is subsequently separated by denaturation prior to western blotting (lower arrow).
Therefore, when designing a sandwich ELISA to measure PCSK9 protein, it is possible depending on the epitopes recognized by the capture and conjugate antibodies to measure either total circulating PCSK9 or just intact PCSK9. Furthermore, our laboratory has also noticed that some antibodies may have altered affinity for recombinant PCSK9 protein when it is expressed with a His tag versus having no His tag [47, 48, 51, 60]. In addition, it has also been suggested that expressed PCSK9 protein may self-associate to form multimers [69], which might further complicate accurate measurement of PCSK9 levels in serum. To investigate this possibility, we performed experiments pairing each of our monoclonal antibodies with itself in a sandwich ELISA format. In our experience, homomultimeric proteins are able to be detected by such a configuration, but we were not able to measure any signal when using the same anti-PCSK9 monoclonal antibody as both a capture and conjugate antibody in a sandwich ELISA (unpublished data). These results suggested to us that PCSK9 may not form multimers; however, this possibility cannot be excluded. Undoubtedly, these aspects of the molecule may account at least in part for some of the difference in the absolute values of human serum PCSK9 reported using various ELISA methods.
Despite these technical challenges, however, a consensus appears to be forming that normal serum PCSK9 levels are somewhere around 100-1,000 ng/ml [47–53, 57]. If these estimates for half-life and circulating levels are correct, then the liver must be constantly secreting large amounts of PCSK9 protein into the circulation. At one end of the spectrum, assuming a half-life of 30 minutes, a circulating level of 100 ng/ml and a plasma volume of 2.5 liters, the liver would be secreting 6 mg/day of PCSK9 protein into the plasma. At the other end of the spectrum, assuming a half-life of 5 min, a circulating level of 1,000 ng/ml and a plasma volume of 2.5 liters, the liver would be secreting 360 mg/day of PCSK9 protein into the plasma.
The true amount is probably somewhere between these 2 extreme estimates, but going through this exercise demonstrates that there is a significant amount of circulating PCSK9 protein that would have to be neutralized by a monoclonal antibody. As a result, it is far from certain that an anti-PCSK9 monoclonal antibody can be practically developed. The fact that it may be possible, however, combined with the fact that an anti-PCSK9 monoclonal antibody should theoretically be synergistic or at least additive to statins with regard to LDL-C lowering makes the development of such a biotherapeutic a clear goal.
If this goal cannot be attained, however, the data obtained so far showing that statins, fibrates and ezetimibe all increase serum PCSK9 levels strongly suggest that any novel LDL-C-lowering compound that acts to decrease PCSK9 levels should be able to be combined with the currently prescribed medications to result in further LDL-C lowering than has ever been possible to date. Furthermore, it may also be safe to speculate that novel lipid-lowering medications that are PCSK9 neutral (meaning that while they do not decrease, they also do not increase PCSK9 levels) may be combined with currently prescribed medications to enable greater LDL-C lowering than can be achieved at the present time.
Conclusions
Certainly, the past decade has seen an explosion in our knowledge about PCSK9 and the role that it plays in the regulation of LDL-C levels. In less than 10 years, we have witnessed the discovery of the protein, the finding that mutations in PCSK9 can drastically affect LDL-C levels, an understanding of the mechanism by which PCSK9 functions, and the development of immunoassays to measure its circulating levels. These advances have allowed multiple researchers to measure PCSK9 protein levels in human serum and demonstrate that commonly prescribed lipid lowering medications (including statins, fibrates, and ezetimibe) increase circulating PCSK9 levels. Preliminary proof-of-concept studies showing that neutralizing anti-PCSK9 antibodies can significantly lower LDL-C have also occurred, suggesting that addition of PCSK9 inhibitors to current therapies may allow for lowering LDL-C to a greater extent than has previously been achievable. While it is impossible to predict accurately what the next decade will hold, if the past one is any indication, it is safe to bet that the development of novel LDL-C-lowering compounds that decrease or inhibit PCSK9 or that act through PCSK9-independent pathways will be a top priority of multiple research laboratories around the world.
Authors' information
RK, JT, and GC are all employees of Eli Lilly and Company. RK is the Senior Director of the Laboratory for Experimental Medicine. JT is a scientist in the Laboratory for Experimental Medicine. GC is a Senior Research Advisor in the Cardiovascular/Metabolic Disease Drug Hunting Team.
Abbreviations
ASO:
antisense oligonucleotide
HDL:
high density lipoprotein
HDL-C:
high density lipoprotein cholesterol
LDL:
low density lipoprotein
LDLR:
low density lipoprotein receptor
LDL-C:
low density lipoprotein cholesterol
PCSK9:
proprotein convertase subtilisin kexin type 9
TC:
total cholesterol
TG:
triglycerides
VLDL:
very low density lipoprotein
References
- Horton JD, Cohen JC, Hobbs HH: Molecular biology of PCSK9: its role in LDL metabolism. Trends Biochem Sci. 2007, 32: 71-77. 10.1016/j.tibs.2006.12.008
Article PubMed Central CAS PubMed Google Scholar - Cao G, Qian YW, Kowala MC, Konrad RJ: Further LDL cholesterol lowering through targeting PCSK9 for coronary artery disease. Endocr Metab Immune Disord Drug Targets. 2008, 8: 238-243. 10.2174/187153008786848286
Article CAS PubMed Google Scholar - Horton JD, Cohen JC, Hobbs HH: PCSK9: a convertase that coordinates LDL catabolism. J Lipid Res. 2009, 50: 172-177. 10.1194/jlr.R800091-JLR200.
Article Google Scholar - Seidah NG: PCSK9 as a therapeutic target of dyslipidemia. Expert Opin Ther Targets. 2009, 13: 19-28. 10.1517/14728220802600715
Article CAS PubMed Google Scholar - Lambert G, Krempf M, Costet P: PCSK9: a promising therapeutic target for dyslipidemias. Trends Endocrinol Metab. 2006, 17: 79-81. 10.1016/j.tem.2006.02.001
Article CAS PubMed Google Scholar - Mousavi SA, Berge KE, Leren TP: The unique role of proprotein convertase subtilisin/kexin 9 in cholesterol homeostasis. J Intern Med. 2009, 266: 507-519. 10.1111/j.1365-2796.2009.02167.x
Article CAS PubMed Google Scholar - Seidah NG, Benjannet S, Wickham L, Marcinkiewicz J, Jasmin SB, Stifani S, Basak A, Prat A, Chretien M: The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): liver regeneration and neuronal differentiation. Proc Natl Acad Sci USA. 2003, 100: 928-33. 10.1073/pnas.0335507100
Article PubMed Central CAS PubMed Google Scholar - Abifadel M, Varret M, Rabes JP, Allard D, Ouguerram K, Devillers M, Cruaud C, Benjannet S, Wickham L, Erlich D, Derre A, Villeger L, Farnier M, Beucler I, Bruckert E, Chambaz J, Chanu B, Lecerf JM, Luc G, Moulin P, Weissenbach J, Prat A, Krempf M, Junien C, Seidah NG, Boileau C: Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003, 34: 154-156. 10.1038/ng1161
Article CAS PubMed Google Scholar - Maxwell KN, Breslow JL: Adenoviral-mediated expression of Pcsk9 in mice results in a low-density lipoprotein receptor knockout phenotype. Proc Natl Acad Sci USA. 2004, 101: 7100-7105. 10.1073/pnas.0402133101
Article PubMed Central CAS PubMed Google Scholar - McNutt MC, Kwon HJ, Chen C, Chen JR, Horton JD, Lagace TA: Antagonism of secreted PCSK9 increases low density lipoprotein receptor expression in HepG2 cells. J Biol Chem. 2009, 284: 10561-10570. 10.1074/jbc.M808802200
Article PubMed Central CAS PubMed Google Scholar - Grefhorst A, McNutt MC, Lagace TA, Horton JD: Plasma PCSK9 preferentially reduces liver LDL receptors in mice. J Lipid Res. 2008, 49: 1303-1311. 10.1194/jlr.M800027-JLR200
Article PubMed Central CAS PubMed Google Scholar - Qian Y, Schmidt RJ, Zhang Y, Chu S, Lin A, Wang H, Wang X, Beyer TP, Bensch WR, Li W, Lu D, Konrad RJ, Eacho PI, Moller DE, Karathanasis SK, Cao G: Secreted proprotein convertase subtilisin/kexin-type 9 reduces low-density lipoprotein receptor through receptor-mediated endocytosis. J Lipid Res. 2007, 48: 1488-1498. 10.1194/jlr.M700071-JLR200
Article CAS PubMed Google Scholar - Kwon HJ, Lagace TA, McNutt MC, Horton JD, Deisenhofer J: Molecular basis for LDL receptor recognition by PCSK9. Proc Natl Acad Sci USA. 2008, 105: 1820-1825. 10.1073/pnas.0712064105
Article PubMed Central CAS PubMed Google Scholar - Graham MJ, Lemonidis KM, Whipple CP, Subramaniam A, Monia BP, Crooke ST, Crooke RM: Antisense inhibition of proprotein convertase subtilisin kexin 9 reduces serum LDL in hyperlipidemic mice. J Lipid Res. 2007, 48: 763-767. 10.1194/jlr.C600025-JLR200
Article CAS PubMed Google Scholar - Lagace TA, Curtis DE, Garuti R, McNutt MC, Park SW, Prather HB, Anderson NN, Ho YK, Hammer RE, Horton JD: Secreted PCSK9 decreases the number of LDL receptors in hepatocytes and in livers of parabiotic mice. J Clin Invest. 2006, 116: 2995-3005. 10.1172/JCI29383
Article PubMed Central CAS PubMed Google Scholar - Maxwell KN, Fisher EA, Breslow JL: Overexpression of PCSK9 accelerates the degradation of the LDLR in a post-endoplasmic reticulum compartment. Proc Natl Acad Sci USA. 2005, 102: 2069-2074. 10.1073/pnas.0409736102
Article PubMed Central CAS PubMed Google Scholar - Park SW, Moon YA, Horton JD: Post-transcriptional regulation of low density lipoprotein receptor protein by proprotein convertase subtilisin/kexin type 9a in mouse liver. J Biol Chem. 2004, 279: 50630-50638. 10.1074/jbc.M410077200
Article CAS PubMed Google Scholar - Benjannet S, Rhainds D, Essalmani R, Mayne J, Wickham L, Jin W, Asselin MC, Hamelin J, Varret M, Allard D, Trillard M, Abifadel M, Tebon A, Attie AD, Rader DJ, Boileau C, Brissette L, Chretien M, Prat A, Seidah NG: NARC-1/PCSK9 and its natural mutants: zymogen cleavage and effects on the low density lipoprotein (LDL) receptor and LDL cholesterol. J Biol Chem. 2004, 279: 48865-48875. 10.1074/jbc.M409699200
Article CAS PubMed Google Scholar - Schmidt RJ, Beyer TP, Bensch WR, Qian YW, Lin A, Kowala M, Alborn WE, Konrad RJ, Cao G: Secreted proprotein convertase subtilisin/kexin type 9 reduces both hepatic and extrahepatic low-density lipoprotein receptors in vivo. Biochem Biophys Res Commun. 2008, 370: 634-640. 10.1016/j.bbrc.2008.04.004
Article CAS PubMed Google Scholar - Li J, Tumanut C, Gavigan JA, Huang WJ, Hampton EN, Tumanut R, Suen KF, Trauger JW, Spraggon G, Lesley SA, Liau G, Yowe D, Harris JL: Secreted PCSK9 promotes LDL receptor degradation independently of proteolytic activity. Biochem J. 2007, 406: 203-207. 10.1042/BJ20070186
Article PubMed Central CAS PubMed Google Scholar - McNutt MC, Lagace TA, Horton JD: Catalytic activity is not required for secreted PCSK9 to reduce low density lipoprotein receptors in HepG2 cells. J Biol Chem. 2007, 282: 20799-20803. 10.1074/jbc.C700095200
Article CAS PubMed Google Scholar - Zhang DW, Lagace TA, Garuti R, Zhao Z, McDonald M, Horton JD, Cohen JC, Hobbs HH: Binding of proprotein convertase subtilisin/kexin type 9 to epidermal growth factor-like repeat A of low density lipoprotein receptor decreases receptor recycling and increases degradation. J Biol Chem. 2007, 282: 18602-18612. 10.1074/jbc.M702027200
Article CAS PubMed Google Scholar - Duff CJ, Scott MJ, Kirby IT, Hutchinson SE, Martin SL, Hooper NM: Antibody-mediated disruption of the interaction between PCSK9 and the low-density lipoprotein receptor. Biochem J. 2009, 419: 577-584. 10.1042/BJ20082407
Article PubMed Central CAS PubMed Google Scholar - Ni YG, Condra J, Orsatti L, Shen X, Di Marco S, Pandit S, Bottomley MJ, Ruggeri L, Cummings RT, Cubbon RM, Santoro JC, Ehrhardt A, Lewis D, Fisher TS, Ha S, Njimoluh L, Wood D, Hammond HA, Wisniewski D, Volpari C, Noto A, Lo Surdo P, Hubbard B, Carfi A, Sitlani A: A proprotein convertase subtilisin-like/kexin type 9 (PCSK9) C-terminal domain antibody antigen binding fragment inhibits PCSK9 internalization and restores LDL-uptake. J Biol Chem. 2010, 285: 12882-12891. 10.1074/jbc.M110.113035
Article PubMed Central CAS PubMed Google Scholar - Chan JC, Piper DE, Cao Q, Liu D, King C, Wang W, Tang J, Liu Q, Higbee J, Xia Z, Di Y, Shetterly S, Arimura Z, Salomonis H, Romanow WG, Thibault ST, Zhang R, Cao P, Yang XP, Yu T, Lu M, Retter MW, Kwon G, Henne K, Pan O, Tsai MM, Fuchslocher B, Yang E, Zhou L, Lee KJ, Daris M, Sheng J, Wang Y, Shen WD, Yeh WC, Emery M, Walker NP, Shan B, Schwarz M, Jackson SM: A proprotein convertase subtilisin/kexin type 9 neutralizing antibody reduces serum cholesterol in mice and nonhuman primates. Proc Natl Acad Sci USA. 2009, 106: 9820-5. 10.1073/pnas.0903849106
Article PubMed Central CAS PubMed Google Scholar - Ni YG, Di Marco S, Condra JH, Peterson LB, Wang W, Wang F, Pandit S, Hammond HA, Rosa R, Cummings RT, Wood DD, Liu X, Bottomley MJ, Shen X, Cubbon RM, Wang SP, Johns DG, Volpari C, Hamuro L, Chin J, Huang L, Zhao JZ, Vitelli S, Haytko P, Wisniewski D, Mitnaul LJ, Sparrow CP, Hubbard B, Carfi A, Sitlani A: A proprotein convertase subtilisin-like/kexin type 9 (PCSK9)-binding antibody that structurally mimics the EGF(A) domain of LDL-receptor reduces free circulating PCSK9 and LDL-cholesterol. J Lipid Res. 2010, 52: 76-86.
Google Scholar - Maxwell KN, Breslow JL: Proprotein convertase subtilisin kexin 9: the third locus implicated in autosomal dominant hypercholesterolemia. Curr Opin Lipidol. 2005, 16: 167-172. 10.1097/01.mol.0000162321.31925.a3
Article CAS PubMed Google Scholar - Abifadel M, Varret M, Rabes JP, Allard D, Ouguerram K, Devillers M, Cruaud C, Benjannet S, Wickham L, Erlich D, Derre A, Villeger L, Farnier M, Beucler I, Bruckert E, Chambaz J, Chanu B, Lecerf JM, Luc G, Moulin P, Weissenbach J, Prat A, Krempf M, Junien C, Seidah NG, Boileau C: Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003, 34: 154-156. 10.1038/ng1161
Article CAS PubMed Google Scholar - Allard D, Amsellem S, Abifadel M, Trillard M, Devillers M, Luc G, Krempf M, Reznik Y, Girardet JP, Fredenrich A, Junien C, Varret M, Boileau C, Benlian P, Rabes JP: Novel mutations of the PCSK9 gene cause variable phenotype of autosomal dominant hypercholesterolemia. Hum Mutat. 2005, 26: 497- 10.1002/humu.9383
Article PubMed Google Scholar - Cohen JC, Boerwinkle E, Mosley TH, Hobbs HH: Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006, 354: 1264-1272. 10.1056/NEJMoa054013
Article CAS PubMed Google Scholar - Zhao Z, Tuakli-Wosornu Y, Lagace TA, Kinch L, Grishin NV, Horton JD, Cohen JC, Hobbs HH: Molecular characterization of loss-of-function mutations in PCSK9 and identification of a compound heterozygote. Am J Hum Genet. 2006, 79: 514-523. 10.1086/507488
Article PubMed Central CAS PubMed Google Scholar - Cariou B, Ouguerram K, Zaïr Y, Guerois R, Langhi C, Kourimate S, Benoit I, Le May C, Gayet C, Belabbas K, Dufernez F, Chétiveaux M, Tarugi P, Krempf M, Benlian P, Costet P: PCSK9 dominant negative mutant results in increased LDL catabolic rate and familial hypobetalipoproteinemia. Arterioscler Thromb Vasc Biol. 2009, 29: 2191-2197. 10.1161/ATVBAHA.109.194191
Article CAS PubMed Google Scholar - Fasano T, Cefalu AB, Di Leo E, Noto D, Pollaccia D, Bocchi L, Valenti V, Bonardi R, Guardamagna O, Averna M, Tarugi P: A novel loss-of-function mutation of PCSK9 gene in white subjects with low-plasma low-density lipoprotein cholesterol. Arterioscler Thromb Vasc Biol. 2007, 27: 677-681. 10.1161/01.ATV.0000255311.26383.2f
Article CAS PubMed Google Scholar - Lakoski SG, Lagace TA, Cohen JC, Horton JD, Hobbs HH: Genetic and metabolic determinants of plasma PCSK9 levels. J Clin Endocrinol Metab. 2009, 94: 2537-2543. 10.1210/jc.2009-0141
Article PubMed Central CAS PubMed Google Scholar - Humphries SE, Neely RD, Whittall RA, Troutt JS, Konrad RJ, Scartezini M, Li KW, Cooper JA, Acharya J, Neil A: Healthy individuals carrying the PCSK9 p.R46L variant and familial hypercholesterolemia patients carrying PCSK9 D374Y exhibit lower plasma concentrations of PCSK9. Clin Chem. 2009, 55: 2153-2161. 10.1373/clinchem.2009.129759
Article CAS PubMed Google Scholar - Costet P, Cariou B, Lambert G, Lalanne F, Lardeux B, Jarnoux AL, Grefhorst A, Staels B, Krempf M: Hepatic PCSK9 expression is regulated by nutritional status via insulin and sterol regulatory element-binding protein 1c. J Biol Chem. 2006, 281: 6211-8. 10.1074/jbc.M508582200
Article CAS PubMed Google Scholar - Browning JD, Horton JD: Fasting reduces plasma proprotein convertase, subtilisin/kexin type 9 and cholesterol biosynthesis in humans. J Lipid Res. 2010, 51: 3359-63. 10.1194/jlr.P009860
Article PubMed Central CAS PubMed Google Scholar - Persson L, Cao G, Ståhle L, Sjöberg BG, Troutt JS, Konrad RJ, Gälman C, Wallén H, Eriksson M, Hafström I, Lind S, Dahlin M, Amark P, Angelin B, Rudling M: Circulating proprotein convertase subtilisin kexin type 9 has a diurnal rhythm synchronous with cholesterol synthesis and is reduced by fasting in humans. Arterioscler Thromb Vasc Biol. 2010, 30: 2666-72. 10.1161/ATVBAHA.110.214130
Article CAS PubMed Google Scholar - Istvan ES, Deisenhofer J: Structural mechanism for statin inhibition of HMG-CoA reductase. Science. 2001, 292: 1160-4. 10.1126/science.1059344
Article CAS PubMed Google Scholar - Charlton-Menys V, Durrington PN: Human cholesterol metabolism and therapeutic molecules. Exp Physiol. 2008, 93: 27-42.
Article CAS PubMed Google Scholar - Maxwell KN, Soccio RE, Duncan EM, Sehayek E, Breslow JL: Novel putative SREBP and LXR target genes identified by microarray analysis in liver of cholesterol-fed mice. J Lipid Res. 2003, 44: 2109-19. 10.1194/jlr.M300203-JLR200
Article CAS PubMed Google Scholar - Dubuc G, Chamberland A, Wassef H, Davignon J, Seidah NG, Bernier L, Prat A: Statins upregulate PCSK9, the gene encoding the proprotein convertase neural apoptosis-regulated convertase-1 implicated in familial hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2004, 24: 1454-1459. 10.1161/01.ATV.0000134621.14315.43
Article CAS PubMed Google Scholar - Rashid S, Curtis DE, Garuti R, Anderson NN, Bashmakov Y, Ho YK, Hammer RE, Moon YA, Horton JD: Decreased plasma cholesterol and hypersensitivity to statins in mice lacking PCSK9. Proc Natl Acad Sci USA. 2005, 102: 5374-5379. 10.1073/pnas.0501652102
Article PubMed Central CAS PubMed Google Scholar - Berge KE, Ose L, Leren TP: Missense mutations in the PCSK9 gene are associated with hypocholesterolemia and possibly increased response to statin therapy. Arterioscler Thromb Vasc Biol. 2006, 26: 1094-1100. 10.1161/01.ATV.0000204337.81286.1c
Article CAS PubMed Google Scholar - Dong B, Wu M, Li H, Kraemer FB, Adeli K, Seidah NG, Park SW, Liu J: Strong induction of PCSK9 gene expression through HNF1alpha and SREBP2: Mechanism for the resistance to LDL-cholesterol lowering effect of statins in dyslipidemic hamsters. J Lipid Res. 2010, 51: 1486-1495. 10.1194/jlr.M003566
Article PubMed Central CAS PubMed Google Scholar - Schmidt RJ, Ficorilli JV, Zhang Y, Bramlett KS, Beyer TP, Borchert K, Dowless MS, Houck KA, Burris TP, Eacho PI, Liang G, Guo LW, Wilson WK, Michael LF, Cao G: A 15-ketosterol is a liver X receptor ligand that suppresses sterol-responsive element binding protein-2 activity. J Lipid Res. 2006, 47: 1037-44. 10.1194/jlr.M500526-JLR200
Article CAS PubMed Google Scholar - Alborn WE, Cao G, Careskey HE, Qian YW, Subramaniam DR, Davies J, Conner EM, Konrad RJ: Serum Proprotein Convertase Subtilisin Kexin Type 9 Is Correlated Directly with Serum LDL Cholesterol. Clin Chem. 2007, 53: 1814-1819. 10.1373/clinchem.2007.091280
Article CAS PubMed Google Scholar - Careskey HE, Davis RA, Alborn WE, Troutt JS, Cao G, Konrad RJ: Atorvastatin increases human serum levels of proprotein convertase subtilisin/kexin type 9. J Lipid Res. 2008, 49: 394-398. 10.1194/jlr.M700437-JLR200
Article CAS PubMed Google Scholar - Dubuc G, Tremblay M, Paré G, Jacques H, Hamelin J, Benjannet S, Boulet L, Genest J, Bernier L, Seidah NG, Davignon J: A new method for measurement of total plasma PCSK9: clinical applications. J Lipid Res. 2010, 51: 140-149. 10.1194/jlr.M900273-JLR200
Article PubMed Central PubMed Google Scholar - Cariou B, Le Bras M, Langhi C, Le May C, Guyomarc'h-Delasalle B, Krempf M, Costet P: Association between plasma PCSK9 and gamma-glutamyl transferase levels in diabetic patients. Atherosclerosis. 2010, 211: 700-2. 10.1016/j.atherosclerosis.2010.04.015
Article CAS PubMed Google Scholar - Welder G, Zineh I, Pacanowski MA, Troutt JS, Cao G, Konrad RJ: High-dose atorvastatin causes a rapid sustained increase in human serum PCSK9 and disrupts its correlation with LDL cholesterol. J Lipid Res. 2010, 51: 2714-21. 10.1194/jlr.M008144
Article PubMed Central CAS PubMed Google Scholar - Costet P, Hoffmann MM, Cariou B, Guyomarc'h Delasalle B, Konrad T, Winkler K: Plasma PCSK9 is increased by fenofibrate and atorvastatin in a non-additive fashion in diabetic patients. Atherosclerosis. 2010, 212: 246-51. 10.1016/j.atherosclerosis.2010.05.027
Article CAS PubMed Google Scholar - Lakoski SG, Xu F, Vega GL, Grundy SM, Chandalia M, Lam C, Lowe RS, Stepanavage ME, Musliner TA, Cohen JC, Hobbs HH: Indices of cholesterol metabolism and relative responsiveness to ezetimibe and simvastatin. J Clin Endocrinol Metab. 2010, 95: 800-9. 10.1210/jc.2009-1952
Article PubMed Central CAS PubMed Google Scholar - Ge L, Wang J, Qi W, Miao HH, Cao J, Qu YX, Li BL, Song BL: The cholesterol absorption inhibitor ezetimibe acts by blocking the sterol-induced internalization of NPC1L1. Cell Metab. 2008, 7: 508-519. 10.1016/j.cmet.2008.04.001
Article CAS PubMed Google Scholar - Altmann SW, Davis HR, Zhu LJ, Yao X, Hoos LM, Tetzloff G, Iyer SP, Maguire M, Golovko A, Zeng M, Wang L, Murgolo N, Graziano MP: Niemann-Pick C1 like 1 protein is critical for intestinal cholesterol absorption. Science. 2004, 303: 1201-1204. 10.1126/science.1093131
Article CAS PubMed Google Scholar - Weinglass AB, Kohler M, Schulte U, Liu J, Nketiah EO, Thomas A, Schmalhofer W, Williams B, Bildl W, McMasters DR, Dai K, Beers L, McCann ME, Kaczorowski GJ, Garcia ML: Extracellular loop C of NPC1L1 is important for binding to ezetimibe. Proc Natl Acad Sci USA. 2008, 105: 11140-11145. 10.1073/pnas.0800936105
Article PubMed Central CAS PubMed Google Scholar - Davignon J, Dubuc G: Statins and ezetimibe modulate plasma proprotein convertase subtilisin kexin-9 (PCSK9) levels. Trans Am Clin Climatol Assoc. 2009, 120: 163-73.
PubMed Central PubMed Google Scholar - Goldenberg I, Benderly M, Goldbourt U: Update on the use of fibrates: focus on bezafibrate. Vasc Health Risk Manag. 2008, 4: 131-41. 10.2147/vhrm.2008.04.01.131
Article PubMed Central CAS PubMed Google Scholar - Chapman MJ: Fibrates: therapeutic review. Br J Diabetes Vasc Dis. 2006, 6: 11-18. 10.1177/14746514060060010201.
Article CAS Google Scholar - Troutt JS, Alborn WE, Cao G, Konrad RJ: Fenofibrate treatment increases human serum proprotein convertase subtilisin kexin type 9 levels. J Lipid Res. 2010, 51: 345-351. 10.1194/jlr.M000620
Article PubMed Central CAS PubMed Google Scholar - Farnier M: Update on the clinical utility of fenofibrate in mixed dyslipidemias: mechanisms of action and rational prescribing. Vasc Health Risk Manag. 2008, 4: 991-1000.
PubMed Central CAS PubMed Google Scholar - Keating GM, Croom KF: Fenofibrate. A review of its use in primary dyslipidemia, the metabolic syndrome and type 2 diabetes mellitus. Drugs. 2007, 67: 121-53. 10.2165/00003495-200767010-00013
Article CAS PubMed Google Scholar - Paumelle R, Staels B: Cross-talk between statins and PPARα in cardiovascular diseases: clinical evidence and basic mechanisms. Trends Cardiovasc Med. 2008, 18: 73-8. 10.1016/j.tcm.2008.01.001
Article CAS PubMed Google Scholar - Mayne J, Dewpura T, Raymond A, Cousins M, Chaplin A, Lahey KA, Lahaye SA, Mbikay M, Ooi TC, Chrétien M: Plasma PCSK9 levels are significantly modified by statins and fibrates in humans. Lipids Health Dis. 2008, 7: 1-9. 10.1186/1476-511X-7-22
Article Google Scholar - Kourimate S, Le May C, Langhi C, Jarnoux AL, Ouguerram K, Zaïr Y, Nguyen P, Krempf M, Cariou B, Costet P: Dual mechanisms for the fibrate-mediated repression of proprotein convertase subtilisin/kexin type 9. J Biol Chem. 2008, 283: 9666-73. 10.1074/jbc.M705831200
Article CAS PubMed Google Scholar - Lambert G, Ancellin N, Charlton F, Comas D, Pilot J, Keech A, Patel S, Sullivan DR, Cohn JS, Rye KA, Barter PJ: Plasma PCSK9 concentrations correlate with LDL and total cholesterol in diabetic patients and are decreased by fenofibrate treatment. Clin Chem. 2008, 54: 1038-45. 10.1373/clinchem.2007.099747
Article CAS PubMed Google Scholar - Gupta N, Fisker N, Asselin MC, Lindholm M, Rosenbohm C, Ørum H, Elmén J, Seidah NG, Straarup EM: A locked nucleic acid antisense oligonucleotide (LNA) silences PCSK9 and enhances LDLR expression in vitro and in vivo. PLoS One. 2010, 5: e10682- 10.1371/journal.pone.0010682
Article PubMed Central PubMed Google Scholar - Frank-Kamenetsky M, Grefhorst A, Anderson NN, Racie TS, Bramlage B, Akinc A, Butler D, Charisse K, Dorkin R, Fan Y, Gamba-Vitalo C, Hadwiger P, Jayaraman M, John M, Jayaprakash KN, Maier M, Nechev L, Rajeev KG, Read T, Röhl I, Soutschek J, Tan P, Wong J, Wang G, Zimmermann T, de Fougerolles A, Vornlocher HP, Langer R, Anderson DG, Manoharan M, Koteliansky V, Horton JD, Fitzgerald K: Therapeutic RNAi targeting PCSK9 acutely lowers plasma cholesterol in rodents and LDL cholesterol in nonhuman primates. Proc Natl Acad Sci USA. 2008, 105: 11915-20. 10.1073/pnas.0805434105
Article PubMed Central CAS PubMed Google Scholar - Fan D, Yancey PG, Qiu S, Ding L, Weeber EJ, Linton MF, Fazio S: Self-association of human PCSK9 correlates with its LDLR-degrading activity. Biochemistry. 2008, 47: 1631-9. 10.1021/bi7016359
Article PubMed Central CAS PubMed Google Scholar
Acknowledgements and Funding
All funding for this work was provided by Eli Lilly and Company.
Author information
Authors and Affiliations
- Lilly Research Laboratories, Eli Lilly and Company, Indianapolis, IN, 46285, USA
Robert J Konrad, Jason S Troutt & Guoqing Cao
Authors
- Robert J Konrad
You can also search for this author inPubMed Google Scholar - Jason S Troutt
You can also search for this author inPubMed Google Scholar - Guoqing Cao
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toRobert J Konrad.
Additional information
Competing interests
This work was supported entirely by Eli Lilly and Company.
Authors' contributions
RK conceived of and wrote the manuscript. JT and GC helped to draft and critically edit the manuscript. All authors read and approved the final version of the manuscript.
Authors’ original submitted files for images
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Konrad, R.J., Troutt, J.S. & Cao, G. Effects of currently prescribed LDL-C-lowering drugs on PCSK9 and implications for the next generation of LDL-C-lowering agents.Lipids Health Dis 10, 38 (2011). https://doi.org/10.1186/1476-511X-10-38
- Received: 03 February 2011
- Accepted: 28 February 2011
- Published: 28 February 2011
- DOI: https://doi.org/10.1186/1476-511X-10-38