IL-17 induces production of IL-6 and IL-8 in rheumatoid arthritis synovial fibroblasts via NF-κB- and PI3-kinase/Akt-dependent pathways (original) (raw)
- Research article
- Published: 21 January 2004
- Ju-Young Kim1,
- Kyoung-Woon Kim1,
- Mi-Kyung Park1,
- Youngmee Moon1,
- Wan-Uk Kim2 &
- …
- Ho-Youn Kim1,2
Arthritis Res Ther volume 6, Article number: R120 (2004)Cite this article
- 17k Accesses
- 315 Citations
- 3 Altmetric
- Metrics details
Abstract
Recent studies of the pathogenesis of rheumatoid arthritis (RA) have revealed that both synovial fibroblasts and T cells participate in the perpetuation of joint inflammation as dynamic partners in a mutual activation feedback, via secretion of cytokines and chemokines that stimulate each other. In this study, we investigated the role of IL-17, a major Th1 cytokine produced by activated T cells, in the activation of RA synovial fibroblasts. Transcripts of IL-17R (IL-17 receptor) and IL-17RB (IL-17 receptor B) were present in fibroblast-like synoviocytes (FLS) of RA patients. IL-17R responded with increased expression upon in vitro stimulation with IL-17, while the level of IL-17RB did not change. IL-17 enhanced the production of IL-6 and IL-8 in FLS, as previously shown, but did not affect the synthesis of IL-15. IL-17 appears to be a stronger inducer of IL-6 and IL-8 than IL-15, and even exerted activation comparable to that of IL-1β in RA FLS. IL-17-mediated induction of IL-6 and IL-8 was transduced via activation of phosphatidylinositol 3-kinase/Akt and NF-κB, while CD40 ligation and p38 MAPK (mitogen-activated protein kinase) are not likely to partake in the process. Together these results suggest that IL-17 is capable of more than accessory roles in the activation of RA FLS and provide grounds for targeting IL-17-associated pathways in therapeutic modulation of arthritis inflammation.
Introduction
Increasing attention is being given to the role of IL-17, a proinflammatory cytokine produced by activated T cells, in the perpetuation of joint inflammation in rheumatoid arthritis (RA) [1–3]. Overproduction of this cytokine has been associated with elevated production of proinflammatory mediators such as IL-6, IL-8, granulocyte/macrophage-colony-stimulating factor, GRO-α, and prostaglandin E2 in various cell types [4, 5]. Of these targets, IL-6 and IL-8 are most likely to act as major instigators of RA joint inflammation, since disruption of their functions either by gene knockout [6] or by systemic IL-4 treatment [7] leads to protection against arthritis in animal models. Early studies have also denominated IL-1β and tumor necrosis factor α (TNF-α) as major inducers of IL-6 and IL-8 in RA synovium, and IL-17 appears to exert an additive and synergistic effect with these two cytokines [5]. However, results from studies using mice and human joint explants suggest that IL-17 is capable of provoking inflammatory responses by itself [8, 9]. Yet by comparison with the vast information about the role of IL-1β and TNF-α in synovial inflammation, relatively little is known about the mode of IL-17-mediated activation.
The cytoplasmic tail of IL-17R (IL-17 receptor) does not contain any known motifs associated with intracellular signaling, and not much is known about the pathway that relays IL-17-mediated stimulation on to the induction of target cytokines. The involvement of JAK/STAT (Janus kinase/signal transducer and activator of transcription) and TRAF6 (TNF-receptor-associated factor 6) has been suggested to transmit IL-17 signaling in human monocyte cell line [10] and embryonic fibroblasts [11], respectively, and yet cytoplasmic players transmitting IL-17-mediated activation in RA synovial fibroblasts remain to be investigated. Moreover, recent searches using the characteristic 'four-cysteine motif' of IL-17 identified a panoply of IL-17 family members, listed as IL-17B to F, as well as novel isoforms of IL-17 receptors, in various cell types [1]. Given the role of IL-17 in the propagation of arthritis inflammation, it would be highly relevant to investigate the potential contribution of other members of the IL-17 family as well.
While not much is known about intracellular targets of IL-17 that are associated with RA pathogenesis, it is generally believed that IL-17 shares downstream transcription factors with IL-1 and TNF-α. The versatile transcription factor NF-κB is markedly increased in the RA synovium [12, 13]. IL-17 has been shown to instigate a rapid degradation of inhibitor of κB in RA synovial fibroblasts [4], indicating that activation of NF-κB is involved in IL-17 signaling. Studies of IL-1β-stimulated synovial fibroblasts showed that NF-κB plays a dominant role in the expression of IL-6 and IL-8 [14]; however, it is not known whether IL-17 also employs NF-κB activation to elevate the production of target cytokines in these cells.
In the present study, we found that two forms of IL-17R, namely IL-17R and IL-17RB (IL-17 receptor B), are expressed in fibroblast-like synoviocytes (FLS) of RA patients. IL-17 stimulated increased production of IL-6 and IL-8 from FLS but not of IL-15. In comparison with the effect of other proinflammatory cytokines, IL-17 generated stronger induction of IL-6 and IL-8 than did IL-15 or IFN-γ. IL-17-mediated induction of IL-6 and IL-8 appears to involve activation of phosphatidylinositol 3-kinase (PI3-kinase), Akt, and NF-κB in FLS, among other signaling pathways. Together, these data provide us with basic knowledge about how this T-cell-derived proinflammatory mediator participates in the activation of synovial fibroblasts in inflamed RA joints.
Materials and methods
Reagents
Recombinant human IL6, IL-8, IL-15, IFN-γ, transforming growth factor β (TGF-β), IL-18, and IL-1β were purchased from R&D Systems Inc (Minneapolis, MN, USA). LY294002, wortmannin, and SB203580 were obtained from Calbiochem (Schwalbach, Germany), and pyrrolidine dithiocarbamate (PDTC) was from Sigma (St Louis, MO, USA). Soluble recombinant CD40L (sCD40L) was provided by R&D Systems.
Isolation and establishment of fibroblast-like synoviocyte cell lines from RA patients
FLS cell lines were prepared from synovectomized tissue of nine RA patients undergoing joint replacement surgery. Informed consent was obtained from each patient enrolled. The mean age of the patients was 46.2 years, and the disease duration was more than 24 months for all patients. All had erosions visible on radiographs of the hand. To set up cell lines, synovial tissues were minced into 2–3-mm pieces and treated for 4 hours with 4 mg/ml type 1 collagenase (Worthington Biochemicals, Freehold, NJ, USA) in Dulbecco's modified Eagle's medium (DMEM) at 37°C in 5% CO2. Dissociated cells were centrifuged at 500 **g**and were resuspended in DMEM supplemented with 10% FCS, 2 mM L-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. Suspended cells were plated in 75-cm2 culture flasks and cultured at 37°C in 5% CO2. Medium was replaced every 3 days, and once the primary culture reached confluence, cells were split weekly. Cells at passages 5 to 8 contained a homogeneous population of FLS (<2.5% CD14+, <1% CD3+, and <1% CD19+ in flow cytometry analysis).
To investigate the effect of cytokines and/or chemical inhibitors, cells were cured for at least 24 hours after the last splitting, washed twice with phosphate-buffered saline (PBS), and incubated in DMEM supplemented with 1 × insulin–transferrin–selenium-A (Invitrogen, Carlsbad, CA, USA) for 24 hours before the addition of cytokines and other reagents.
RT-PCR analysis of IL-17 receptors
FLS lines were cultured for 6 hours in 6-well plates with various stimulants, and mRNAs were extracted using RNAzol B (Tel-Test Inc, Friendswood, TX, USA) in accordance with the manufacturer's protocol. Reverse transcription was performed with 5 μg of total RNA, using Superscript III™ and oligo dT primers (Invitrogen). PCR amplification of IL-17 receptors, as well as glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as a quantitation control, were done by rTaq polymerase (Takara Shuzo, Shiga, Japan) and the following primers: IL-17R, sense 5'-GGGATTACAGGCGTGAGCCA-3', antisense 5'-GCGGTCTGGTTATCGTCTAT-3'; IL-17RB, sense 5'-TCATCTGCACAACTCCGTGG-3', antisense 5'-TCGAATGTTAAGGCTACATT-3'; and GAPDH, sense 5'-CGATGCTGGGCGTGAGTAC-3', antisense 5'-CGT-TCAGCTCAGGGATGACC-3'. The numbers of amplification cycles used were 25 to 30 for GAPDH, and 35 for the receptor molecules.
Immunoassays of IL-6, IL-8, and IL-15
The amounts of secreted cytokines in culture supernatants were measured by sandwich ELISA. Briefly, media containing 4 μg/ml monoclonal antibodies to each cytokine were placed in 96-well culture plates and incubated overnight at 4°C. The next morning, the plates were treated with the blocking solution (1% BSA and 0.05% Tween 20 in PBS) for 2 hours at room temperature, the supernatants to be tested and standard recombinant cytokines were added to each well, and incubation was continued. After 2 hours, 500 ng/ml of biotinylated monoclonal antibodies to each cytokine was added and the reactions were allowed to proceed for another 2 hours at room temperature. Next, streptavidin-conjugated alkaline phosphate (Sigma) was added to make a 1 : 2000 dilution, and cells were incubated again for 2 hours at room temperature. Finally, a color reaction was induced by adding 1 mg/ml of _p_-nitrophenylphosphate (Sigma) dissolved in diethanolamine (Sigma) and was stopped by adding 1 N NaOH. Every time new reagents were added to the well, the plates were washed 4 times with PBS containing 0.1% Tween 20. The optical density of color reactions was measured with a Vmax automated microplate reader (Molecular Devices, Palo Alto, CA, USA) set at 405 nm. Standard curves were drawn by plotting optical density versus the concentration of each recombinant cytokine in a logarithmic scale.
Gel mobility shift assay of NF-κB binding site
FLS nuclear extracts were prepared from about 1 × 106 cells by homogenization in the lysis buffer (20 mM Tris HCl, pH 7.4, 0.5 M NaCl, 0.25% Triton X-100, 1 mM EDTA, 1 mM EGTA, 10 mM β-glycerophosphate, 10 mM NaF, 300 μM Na3VO4, 1 mM benzamidine, 2 M phenylmethylsulfonyl fluoride, 10 μg/ml aprotinin, 1 μg/ml each of leupeptin and pepstatin, and 1 mM dithiothreitol). Cell lysates were centrifuged at 500 **g**for 5 min, and the pellets containing nuclei were retrieved and washed in 1 ml cold PBS. Nuclear extracts were obtained by treatment with 10% NP-40.
Double-stranded oligonucleotide probes encompassing the NF-κB recognition sites in the promoter of IL-6 (5'-TCGACATGTGGGATTTTCCCATGAC-3') and IL-8 (5'-TCGAGCGTGGAATTTCCTCTGG-3'), as well as the AP-1 (activating-protein-1) recognition sites of IL-6 promoter (5'-AAAGTGCTGAGTCACTAATAA-3'), were labeled at the 5' end using [γ-32P]dATP (Amersham Pharmacia Biotech, Uppsala, Sweden) and T4 polynucleotide kinase (Takara) in accordance with the manufacturer's instructions. Unincorporated isotopes were removed by NucTrap purification columns (Stratagene, La Jolla, CA, USA).
For each binding assay, 5-μg nuclear extracts were incubated with 100000 counts per minute of radiolabeled probe containing about 10 ng double-stranded oligonucleotides for 30 min at room temperature in 20 μl of the binding buffer, consisting of 20 mM Tris HCl, pH 7.9, 50 mM KCl, 1 mM dithiothreitol, 0.5 mM EDTA, 5% glycerol, 1 mg/ml BSA, 0.2% NP40, and 50 ng/μl of poly(dIdC). After incubation, the samples were electrophoresed on nondenaturing 5% polyacrylamide gels in 0.5 × Tris-Borate-EDTA buffer (pH 8.0) at 100 V. The gels were dried under vacuum and exposed to Kodak X-OMAT film at -70°C with intensifying screens for 12 to 24 hours.
Western blot analysis of Akt and phosphorylated Akt
Whole-cell lysates of FLS were prepared from about 1 × 106 cells by homogenization in the lysis buffer and centrifuged at 14 000 rpm for 15 min. Protein concentrations in the supernatants were determined using the Bradford method (BioRad, Hercules, CA, USA). Protein samples were separated on 10% SDS–PAGE and transferred to a nitrocellulose membrane (Amersham Pharmacia).
For western hybridization, the membrane was pre-incubated with 0.1% skimmed milk in TTBS (0.1% Tween 20 in Tris-buffered saline) at room temperature for 2 hours; then primary antibodies to either Akt or phosphorylated Akt (Cell Signaling Technology Inc, Beverly, MA, USA), diluted 1 : 200 in PBS, were added and incubated for 1 hour at room temperature. After the preparations had been washed 4 times with TTBS, horseradish-peroxidase-conjugated secondary antibodies (Amersham Pharmacia) were added and allowed to incubate for 30 min at room temperature. After being washed in TTBS, hybridized bands were detected using the ECL detection kit and Hyperfilm-ECL reagents (Amersham Pharmacia).
Results
Expression of IL-17 receptors in RA FLS
It has been shown that the level of IL-17 is elevated in inflamed RA synovium [15, 16]. We examined the expression of IL-17 receptors, e.g. IL-17R and IL-17RB, in FLS cell lines established from three RA patients. Transcripts of both IL-17R and IL-17RB were readily detectable by RT-PCR analyses of RA FLS. While the amount of IL-17R mRNA increased when cells were incubated with recombinant IL-17, the level of IL-17RB transcript remained largely unchanged (Fig. 1). IL-17 appeared to induce the expression of its authentic receptor, IL-17R, most strongly when given at 0.1 ng/ml (Fig. 1a). In a time-course analysis, induction of IL-17 peaked around 3 to 6 hours after adding recombinant IL-17 (Fig. 1b).
Figure 1
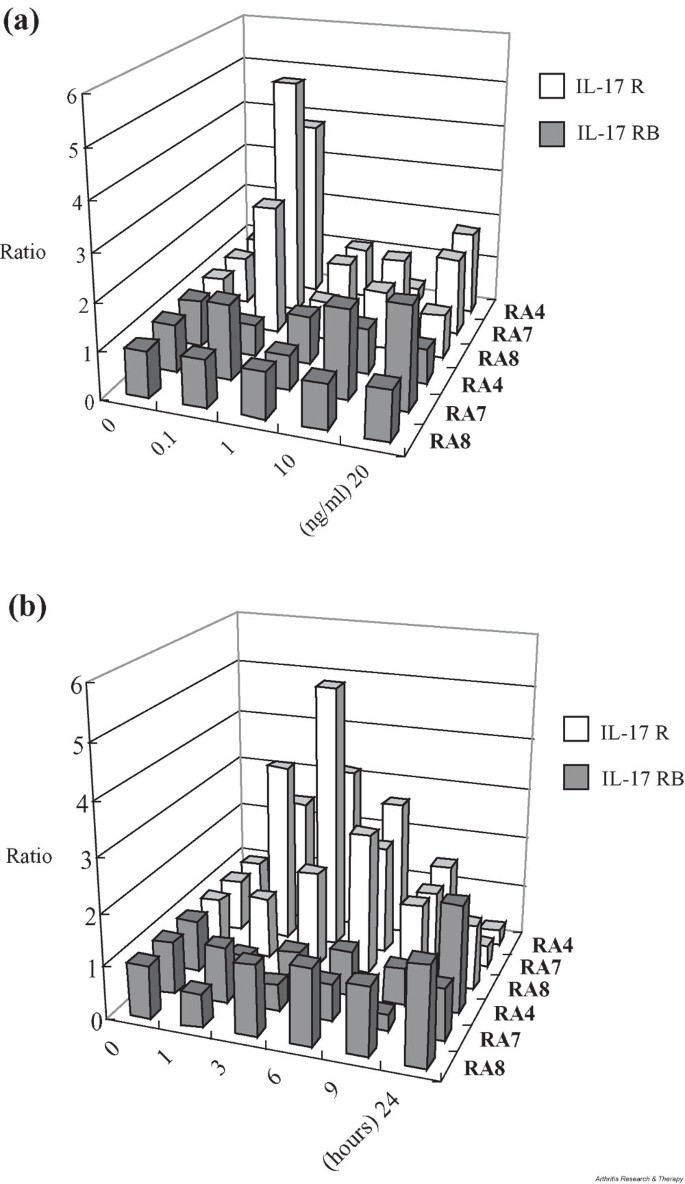
Expression and induction of IL-17R and IL-17RB in IL-17-stimulated FLS from six RA patients. (a) IL-17 dose-dependent changes in the levels of IL-17R and IL-17RB mRNAs. Three independent RA FLS cell lines were stimulated with various amounts of recombinant IL-17 (0 to 20 ng/ml), and subsequent changes in the mRNA levels of IL-17R and IL-17RB were assessed by RT-PCR at 6 hours after the onset of in vitro culture. The relative intensity of each PCR band was normalized against that of GAPDH. Values are the fold increase from the unstimulated cell in each FLS line. (b) Time-dependent changes in the level of IL-17R and IL-17RB mRNAs. Three independent RA FLS cell lines were stimulated with recombinant IL-17, and subsequent changes in the mRNA level of IL-17R and IL-17RB were assessed by RT-PCR 0, 1, 3, 6, 9, and 24 hours after the start of in vitro culture. The relative intensity of each PCR band was normalized against that of GAPDH. Values are the fold increase from the 0 hour measurement in each FLS line. FLS, fibroblast-like synoviocytes; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; IL-17R, IL-17 receptor; IL-17RB, IL-17 receptor B; RA, rheumatoid arthritis.
IL-17 induces production of IL-6 and IL-8 but not IL-15 from fibroblast-like synoviocytes
Previously we have found that coincubation of RA synovial fluid mononuclear cells (SFMCs) with RA patients' FLS induced production of IFN-γ and IL-17 from SFMC T cells [17]. To see whether accumulation of IL-17 in turn exerts any effect on the production of proinflammatory mediators from FLS, we examined changes in the release of IL-15, IL-6, and IL-8 in IL-17-stimulated FLS. We found that in vitro stimulation with 10 ng/ml IL-17 increased production of IL-6 and IL-8 from RA FLS up to six-fold, while production of IL-15 remained unchanged (Fig. 2).
Figure 2
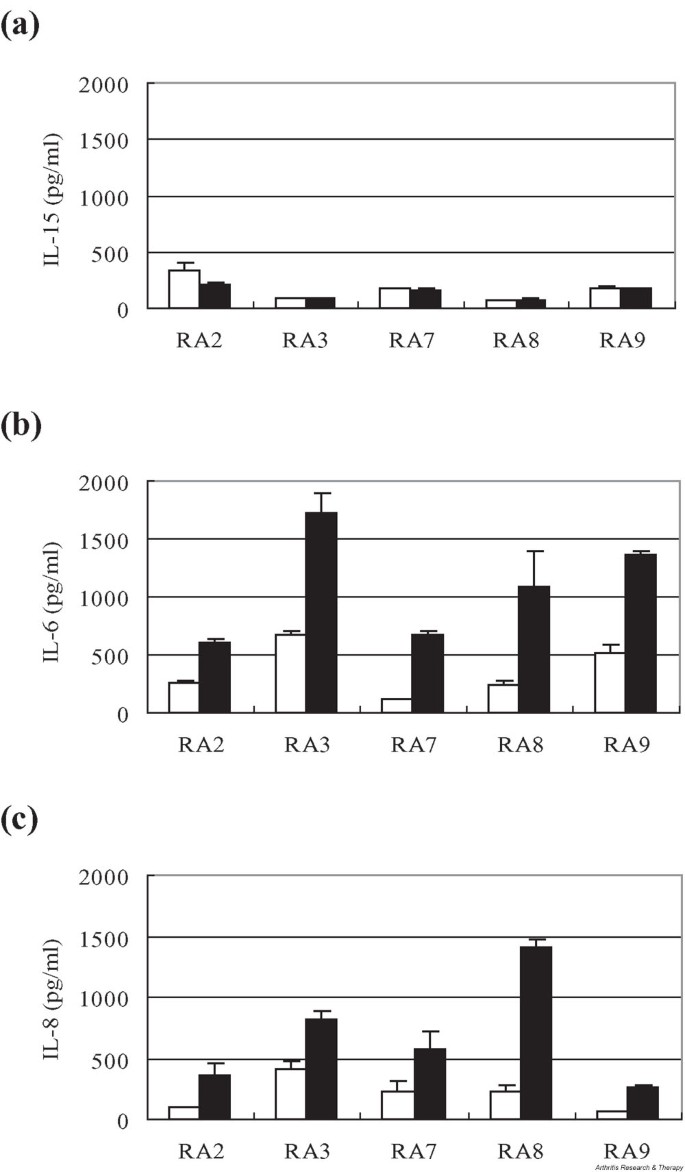
IL-17 induces production of (b) IL-6 and (c) IL-8, but not of (a) IL-15, by synovial fibroblasts from five RA patients. In vitro stimulation with 10 ng/ml IL-17 for 12 hours induced two- to six-fold increases in the levels of IL-6 and IL-8 in the culture supernatant of synovial fibroblasts isolated from RA patients, while the level of IL-15 remained unchanged. Open bar, unstimulated FLS; filled bar, IL-17-stimulated FLS. FLS, fibroblast-like synoviocytes; RA, rheumatoid arthritis.
We also compared the IL-17-mediated induction of IL-6 and IL-8 in RA FLS with the effects of other pro- and anti-inflammatory cytokines. As shown in Fig. 3a, IL-17 induced the production of IL-6 as strongly as did IFN-γ and IL-1β, although the relative fold increase tended to vary depending on the cell line. TGF-β, which is known to activate fibroblast-like cells [18], also significantly increased the production of IL-6 from RA FLS. IL-6 production from cells treated with IL-15 was not much different from that of unstimulated controls. IL-17 appeared to be the most potent inducer of IL-8 among the tested cytokines in RA FLS (Fig. 3b). Unlike the pattern seen in IL-6 induction, IFN-γ did not appear to enhance IL-8 synthesis in RA FLS.
Figure 3
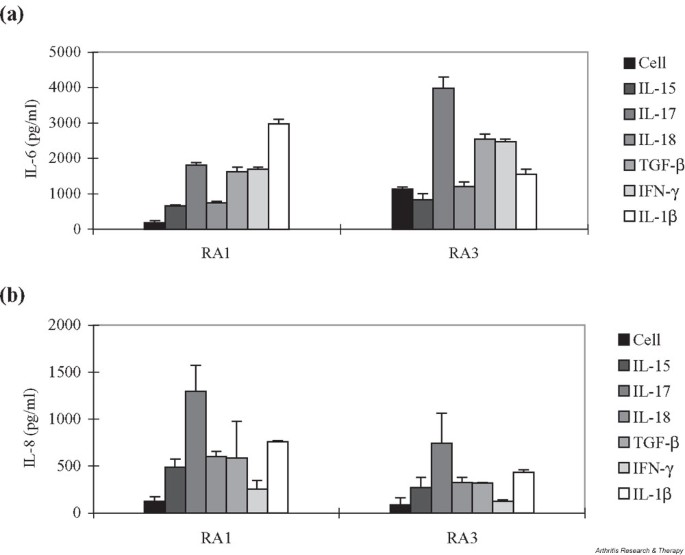
Induction of (a) IL-6 and (b) IL-8 in RA FLS after treatment with various proinflammatory cytokines. Cells were stimulated with 10 ng/ml of IL-15, IL-17, IL-18, TGF-β, or IL-1β, or with 1000 U/ml IFN-γ for 24 hours and assayed for the production of IL-6 and IL-8 in culture supernatants by sandwich ELISA. Values represent average from triplicate cultures. Cell, unstimulated FLS; IFN, interferon; RA, rheumatoid arthritis; FLS, fibroblast-like synoviocyte(s); TGF, transforming growth factor.
NF-κB activation contributes to the increased production of IL-6 and IL-8 from IL-17-stimulated FLS
One previous study reported a rapid degradation of inhibitor of κB in RA FLS stimulated with IL-17, indicating that IL-17 activates NF-κB in these cells [4]. To examine whether signaling pathways that lead to the activation of NF-κB are also employed in the induction of IL-6 and IL-8, we performed gel mobility shift assays of NF-κB recognition sites in the promoters of IL-6 (Fig. 4a) and IL-8 (Fig. 4b). Nuclear extracts from IL-17-stimulated RA FLS showed increased binding of NF-κB to IL-6 and IL-8 promoters, although the degree of activation was lower than that in IL-1β stimulated cells. On the other hand, a significant amount of activating protein-1 was already associated with IL-6 promoter in unstimulated FLS and did not change after IL-17-stimulation (data not shown). To confirm the role of NF-κB activation in the production of IL-6 and IL-8 from RA FLS, we tested the effect of PDTC, a chemical inhibitor of NF-κB activation. Our data show that treatment with 30 μM PDTC reduced the IL-17-mediated induction of IL-6 and IL-8 to their respective levels in unstimulated cells (Fig. 5).
Figure 4
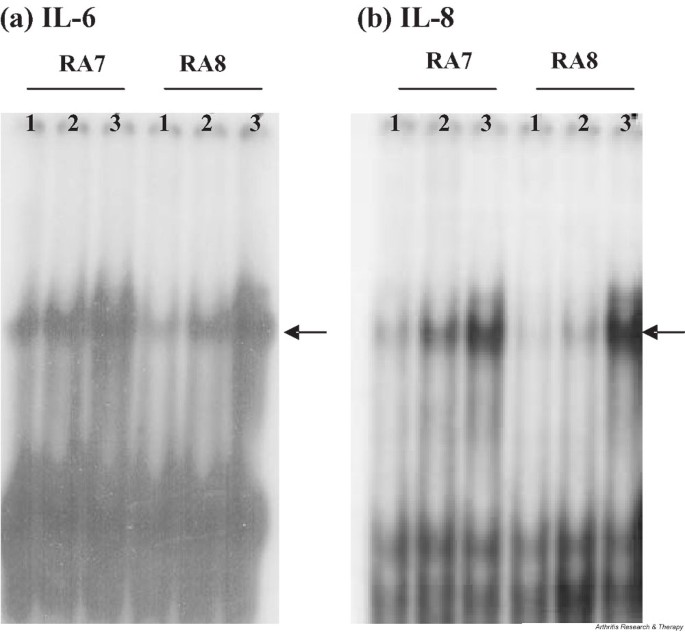
Gel mobility shift analysis of NF-κB recognition sites in the promoters of IL-6 and IL-8, using nuclear extracts from IL-17-stimulated FLS. Changes in the amount of NF-κB in the nuclear extracts isolated from two patients with RA after stimulation of the extracts with 10 ng/ml of IL-17 or IL-1β were tested by gel mobility shift assay of radiolabeled oligonucleotide probes representing the NF-κB sites in the promoter of (a) IL-6 and (b) IL-8. Arrows indicate probe bands shifted by NF-κB binding. Nuclear extracts of IL-1β-stimulated FLS were used as positive controls. Lane 1, unstimulated cells; Lane 2, stimulated with IL-17; lane 3, stimulated with IL-1β. FLS, fibroblast-like synoviocytes; RA, rheumatoid arthritis.
Figure 5
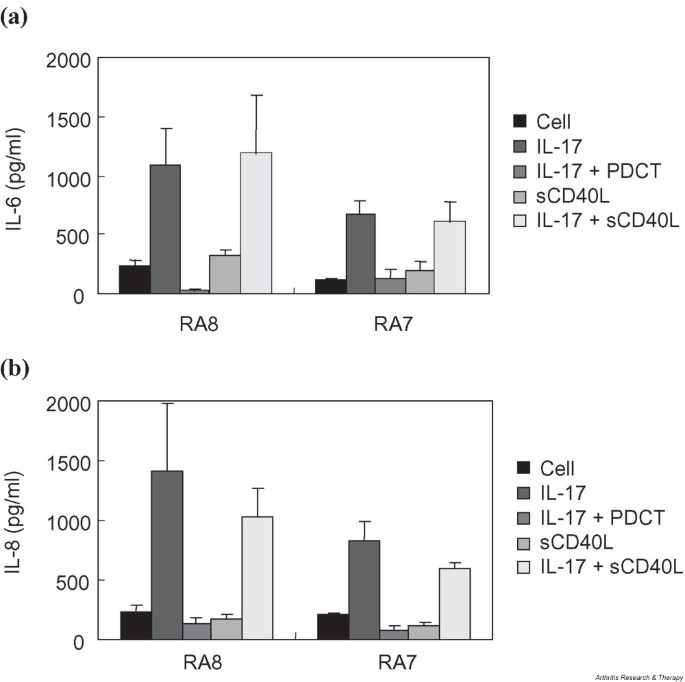
Effect of NF-κB and CD40 blockade on the IL-17-mediated induction of (a) IL-6 and (b) IL-8 in FLS from two patients with RA. FLS were cultured in triplicate with or without 10 ng/ml IL-17 for 24 hours and assayed for the production of IL-6 and IL-8 by sandwich ELISA. Effects of NF-κB blockade and CD40 ligation were investigated by adding 3 μM PDTC and 10 ng/ml sCD40L, respectively, in IL-17-stimulated culture. Cell, unstimulated FLS; FLS, fibroblast-like synoviocytes; PDTC, pyrrolidine dithiocarbamate; RA, rheumatoid arthritis.
In renal epithelial cells, IL-17 has been shown to synergize with CD40 ligation in the induction of IL-6 and IL-8 production [19]. Since the activating signal by CD40L led to the activation of NF-κB in these cells, we tried to find out if similar synergism between IL-17 and CD40 is at work in synovial fibroblasts. Our results showed that stimulating RA FLS with sCD40L did not affect the basal level production of IL-6 and IL-8 (Fig. 5). Also, treating the cells with IL-17 and soluble CD40 did not contribute an additional increase in the production of IL-6 and IL-8 to the effect of IL-17.
Inhibition of MAPK is not likely to affect IL-17-mediated induction of IL-6 and IL-8 in RA FLS
Involvement of p38 mitogen-activated protein kinase (MAPK) in the transduction of IL-17-mediated signaling has been reported from human colonic myofibroblasts [20], where administration of SB203580, a chemical inhibitor of p38, significantly reduced the IL-17-induced secretion of both IL-6 and IL-8. Since IL-17 has also been shown to increase phosphorylation of p38 MAPK in RA FLS [4], we tried to find out if this kinase participates in the induction of IL-6 and IL-8 protein as well. As shown in Fig. 6, occluding MAPK at the time of IL-17 stimulation by SB203580 did not affect the increase in IL-6 production, while a slight reduction was observed in the production of IL-8. These data may reflect the reduced IL-8 mRNA level previously shown in SB203580-treated RA FLS [4], although the level of decline was rather insignificant in both cases.
Figure 6
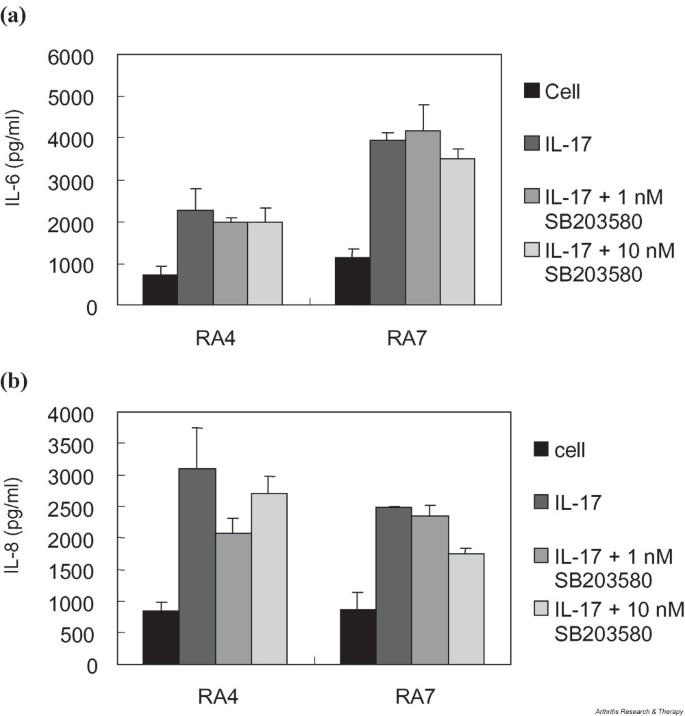
Effect of MAPK blockade on the IL-17-mediated induction of (a) IL-6 and (b) IL-8 in FLS from two patients with RA. FLS were cultured in triplicate with or without 10 ng/ml IL-17 for 24 hours and assayed for the production of IL-6 and IL-8 by sandwich ELISA. Effects of blocking MAPK activation were investigated by adding 1 or 10 nM SB203580 at the time of IL-17 stimulation. Cell, unstimulated FLS; FLS, fibroblast-like synoviocytes; MAPK, mitogen-activated protein kinase; RA, rheumatoid arthritis.
IL-17-mediated induction of IL-6 and IL-8 in FLS involves activation of the PI3-kinase/Akt signaling pathway
It has previously been shown that PI3-kinase and its downstream mediator Akt are involved in the activation of RA FLS by TGF-β [21]. Although TGF-β is widely known for its anti-inflammatory effects on lymphocytes, it provides an opposite signal to fibroblast-like cells, leading to active proliferation and growth. Since we observed that TGF-β induced IL-6 and IL-8 production from FLS (Fig. 3), we were curious to find out if IL-17 also uses the PI3-kinase signaling pathway in FLS. To this end we tested the effect of LY294002, a chemical inhibitor of PI3-kinase, on the production of IL-6 and IL-8 from IL-17-stimulated FLS. We found that LY294002 significantly reduced IL-17-mediated up-regulation of both IL-6 and IL-8 (Fig. 7). IL-17 also activated phosphorylation of Akt in FLS, while the amount of cellular Akt remained unchanged (Fig. 8). As expected, cotreatment with two known chemical inhibitors of PI3-kinase, namely LY294002 and wortmannin, abolished the IL-17-instigated phosphorylation of Akt.
Figure 7
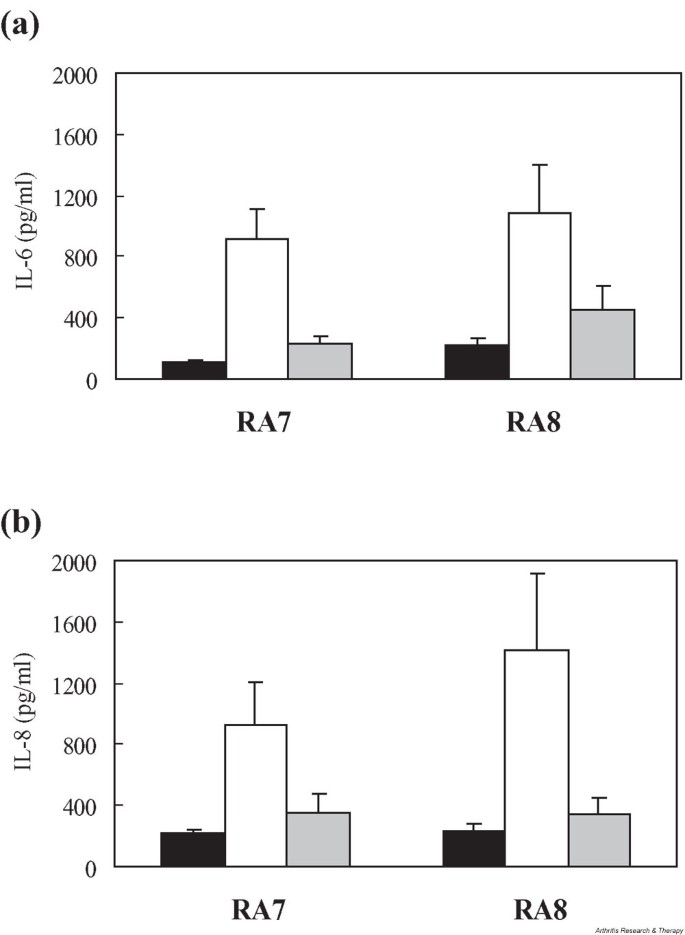
IL-17-mediated induction of (a) IL-6 and (b) IL-8 involves PI3-kinase and Akt signaling in FLS from two patients with RA. Treating the cells with 20 μM LY294002, a chemical inhibitor of PI3-kinase, abolished the IL-17-induced increase in the production of IL-6 and IL-8 from RA FLS. White bars, unstimulated control cells; gray bars, IL-17-stimulated FLS; black bars, cells treated with IL-17 and LY294002. FLS, fibroblast-like synoviocytes; PI3-kinase, phosphatidylinositol 3-kinase; RA, rheumatoid arthritis.
Figure 8
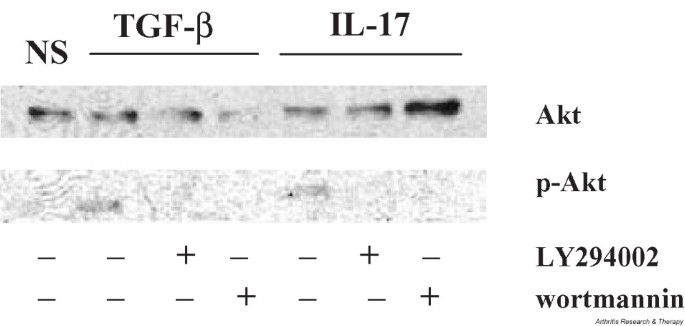
IL-17 stimulation activates phosphorylation of Akt in FLS from patients with RA. The activated form of Akt was detected by western blot analyses using an antibody recognizing phosphorylated Ser473 epitope in RA FLS stimulated with IL-17, while the amount of total Akt remained unchanged. Akt phosphorylation was eliminated in cells treated with two chemical inhibitors of PI3-kinase, LY294002 (20 μM) and wortmannin (200 μM), at the time of IL-17 stimulation. Protein extracts from TGF-β-stimulated FLS were used as positive controls. FLS, fibroblast-like synoviocytes; p-Akt, phosphorylated Akt; PI3-kinase, phosphatidylinositol 3-kinase; RA, rheumatoid arthritis.
Discussion
The current model of RA pathogenesis favors complex interactions among cells in inflamed RA joints, via cytokine secretion and cell-to-cell contact [22, 23], as major instigators of pannus formation and subsequent bone destruction. IL-17 is a proinflammatory cytokine secreted by activated memory T cells and has been shown to be elevated in RA synovium. Studies from OA and skin fibroblasts showed that IL-17 enhanced the effect of IL-1β and TNF-α on the production of IL-6 and IL-8 [24, 5], and the role of IL-17 in arthritis inflammation has usually been addressed in the context of synergism with these Th1 cytokines. However, the fact that exogenous IL-17 can enhance IL-6 production and joint destruction in IL-1-deficient mice [8] demonstrates that IL-17 is capable of launching more than accessory functions in the pathogenic processes of RA. We found that IL-17 stimulated in vitro production of IL-6 and IL-8 better than IL-15, and to a level comparable with that of IL-1β and IFN-γ, but did not affect IL-15 production from RA FLS. Since we previously observed that IL-15 production was elevated when RA FLS are coincubated with antigen-stimulated T cells from RA patients [17], a likely hypothesis is that induction of IL-15 requires the combined influence of other proinflammatory cytokines in addition to IL-17. In view of the fact that IL-1β, TNF-α, and IL-17 are most likely to produce a combined effect on the RA joint, investigation of IL-17-mediated signaling may lead to therapeutic use in addition to the already successful application of IL-1 and TNF-α blockers in RA therapy.
Recently, a systematic homology search throughout the postgenome databases has added a list of genes featuring the characteristic 'four-cysteine residue' of IL-17 [25]. In view of the fact that some of these homologs are also capable of activating NF-κB, it would be highly relevant to investigate their potential contribution to the inflammatory processes in RA synovium. While these proteins are now denominated IL-17B to F, it is not clear which type of membrane receptors recognize these new homologs, except that IL-17B and IL-17E appear to bind IL-17RB [26, 27]. In our experiment, adding recombinant IL-17 induced the level of IL-17R transcript while leaving the amount of IL-17B message largely unchanged, although such data do not rule out the interaction of IL-17 and IL-17RB. By RT-PCR analyses, we detected mRNAs of IL-17C, E, and F, but not IL-17B and D, in SFMC extracts of RA patients (data not shown). Unfortunately, we could not examine the effect of IL-17E on the expression of IL-17RB due to the unavailability of recombinant ligand.
While the induction of IL-6 and IL-8 in fibroblasts is now widely accepted as a functional monitoring system for IL-17 [28], much of the signaling pathway leading to the up-regulation of these proinflammatory mediators in RA FLS still remains to be identified. Considering the rapid activation of NF-κB in IL-17-stimulated cells, together with the fact that inhibition of NF-κB significantly reduced the amount of IL-6 production in pancreatic periacinar myofibroblasts [29], it is most likely that IL-17 also enhances IL-6 production in RA FLS via activation of NF-κB.
In this study we found that binding of NF-κB to its authentic recognition sites in the promoter of IL-6 and IL-8 increased after IL-17 stimulation. Unlike previous experiments done with canonical NF-κB binding oligonucleotides, our result provides a clear demonstration of the involvement of NF-κB in the IL-17-mediated activation of not only IL-6, but also IL-8, production in RA FLS. Our data also suggest that while IL-17-instigated signaling in FLS leads to the activation of NF-κB as in other cell types, it features pathways unique to FLS as well. For example, CD40 ligation did not appear to confer a synergistic effect on the production of IL-6 and IL-8 in our experiment. One possibility is that the monomeric sCD40L we used might not have been efficient, since it has been reported that membrane-bound CD40L [30], and its native soluble variant [31], exist as trimers. The fact that blockade of p38 MAPK did not appear to affect the induction of IL-6 and IL-8 in RA FLS, in contrast with myofibroblasts, may represent another cell-type-dependent characteristic of IL-17 signaling.
PI3-kinase and its downstream kinase Akt, both potent inhibitors of apoptosis in many cell types, have been reported to deliver activating signals from TGF-β [21] and from IL-18 [32] in RA synoviocytes. In this study we examined whether IL-17 also recruits PI3-kinase/Akt-associated signaling molecules to activate synovial fibroblasts. Our data showed that IL-17-induced production of IL-6 and IL-8 in FLS was hampered by a chemical inhibitor of PI3-kinase. The fact that Akt is phosphorylated upon IL-17 stimulation also adds to the possible involvement of PI3-kinase in the propagation of signal through the IL-17R. Interestingly, we observed increased expression of the p85 subunit of PI3-kinase in IL-17-stimulated RA FLS in a differential display analysis (data not shown). Together, these results indicate that PI3-kinase and Akt may serve as the upstream arbitrator of the IL-17-mediated activation in RA FLS. Since signals received by PI3-kinase are often transduced to downstream targets via NF-κB [33], its activation is likely to have contributed to the increased binding of this inflammatory transcription factor to the promoter of IL-6 and IL-8 in IL-17-stimulated FLS.
Conclusion
We have detected two types of receptors for the IL-17 family with known ligand specificity in RA FLS. We also demonstrated that IL-17 alone can induce IL-6 and IL-8 production from RA and FLS to a degree comparable with that for IL-1β. Binding of IL-17 to its membrane receptor on FLS appears to transduce the signal down to IL-6 and IL-8 via activation of PI3-kinase/Akt pathway and NF-κB. Our data provide insights into the cellular mechanisms of how IL-17 participates in the activation of synovial fibroblasts in inflamed RA joints and suggest proinflammatory mediators involved in the process as potential targets of therapeutic modulation of IL-17 function.
Abbreviations
BSA:
bovine serum albumin
DMEM:
Dulbecco's modified Eagle's medium
ELISA:
enzyme-linked immunosorbent assay
FCS:
fetal calf serum
FLS:
fibroblast-like synoviocyte(s)
GAPDH:
glyceraldehyde-3-phosphate dehydrogenase
IFN:
interferon
IL:
interleukin
IL-17R:
IL-17 receptor
IL-17RB:
IL-17 receptor B
MAPK:
mitogen-activated protein kinase
NF-κB:
nuclear factor κB
PBS:
phosphate-buffered saline
PCR:
polymerase chain reaction
PDTC:
pyrrolidine dithiocarbamate
PI3-kinase:
phosphatidylinositol 3-kinase
RA:
rheumatoid arthritis
RT-PCR:
reverse transcriptase-polymerase chain reaction
sCD40L:
soluble recombinant CD40L
SFMC:
synovial fluid mononuclear cells
TGF:
transforming growth factor
Th1:
T helper cell type 1
TNF-α:
tumor necrosis factor α
TTBS:
0.1% Tween 20 in Tris-buffered saline.
References
- Miossec P: Interleukin-17 in rheumatoid arthritis. Arthritis Rheum. 2003, 48: 594-601. 10.1002/art.10816.
Article CAS PubMed Google Scholar - Cai L, Yin J, Starovasnik M, Hogue D, Hillan K, Mort J, Filvarorff E: Pathways by which interleukin 17 induces articular cartilage breakdown in vitro and in vivo. Cytokine. 2001, 16: 10-21. 10.1006/cyto.2001.0939.
Article CAS PubMed Google Scholar - Bush K, Walker J, Lee C, Kirham B: Cytokine expression and synovial pathology in the initiation and spontaneous resolution phases of adjuvant arthritis: Interleukin-17 expression is upregulated in early disease. Clin Exp Immunol. 2001, 123: 487-495. 10.1046/j.1365-2249.2001.01469.x.
Article PubMed Central CAS PubMed Google Scholar - Kehlen A, Thiele K, Riemann D, Langer J: Expression, modulation and signaling of IL-17 receptor in fibroblast-like synoviocytes of patients with rheumatoid arthritis. Clin Exp Immunol. 2002, 127: 539-546. 10.1046/j.1365-2249.2002.01782.x.
Article PubMed Central CAS PubMed Google Scholar - Katz Y, Nadiv O, Beer Y: Interleukin-17 enhances tumor necrosis factor α induced synthesis of interleukins 1, 6, and 8 in skin and synovial fibroblasts. Arthritis Rheum. 2001, 44: 2176-2184. 10.1002/1529-0131(200109)44:9<2176::AID-ART371>3.0.CO;2-4.
Article CAS PubMed Google Scholar - Alonzi T, Fattori E, Lazzaro D, Costa P, Probert L, Kollias G, De Benedetti F, Poli V, Ciliberto G: Interleukin 6 is required for the development of collagen-induced arthritis. J Exp Med. 1998, 187: 461-468. 10.1084/jem.187.4.461.
Article PubMed Central CAS PubMed Google Scholar - Joosten L, Lubberts E, Helsen M, Saxne T, Cornen-de Roo C, Heinegard D, van den Berg WB: Protection against cartilage and bone destruction by systemic interleukin-4 treatment in established murine type II collagen-induced arthritis. Arthritis Res. 1999, 1: 81-91. 10.1186/ar14.
Article PubMed Central CAS PubMed Google Scholar - Lubberts E, Joosten L, Oppers B, van den Bersselaar L, Coenen-de Roo C, Kolls J, Schwarzenberger P, van de Loo F, van den Berg W: IL-1-independent role of IL-17 in synovial inflammation and joint destruction during collagen-induced arthritis. J Immunol. 2001, 167: 1004-1013.
Article CAS PubMed Google Scholar - Chabaud M, Lubberts E, Joosten L, van den Berg W, Miossec P: IL-17 derived from juxta-articular bone and synovium contributes to joint degradation in rheumatoid arthritis. Arthritis Res. 2001, 3: 168-177. 10.1186/ar294.
Article PubMed Central CAS PubMed Google Scholar - Subramaniam S, Cooper R, Adunyah S: Evidence for the involvement of JAK/STAT pathway in the signaling mechanism of interleukin-17. Biochem Biophys Res Commun. 1999, 262: 14-19. 10.1006/bbrc.1999.1156.
Article CAS PubMed Google Scholar - Schwandner R, Yamaguchi K, Cao Z: Requirement of tumor necrosis factor receptor-associated factor (TRAF)6 in interleukin 17 signal transduction. J Exp Med. 2000, 191: 1233-1239. 10.1084/jem.191.7.1233.
Article PubMed Central CAS PubMed Google Scholar - Handel M, McMorrow L, Gravallese E: Nuclear factor-kappa B in rheumatoid synovium. Localization of p50 and p65. Arthritis Rheum. 1995, 38: 1762-1770.
Article CAS PubMed Google Scholar - Marok R, Winyard P, Coumbe A, Kus M, Blades S, Mapp P, Morris C, Blake D, Kaltschmidt C, Baeuerle P: Activation of the transcription factor nuclear factor-kappa B in human inflamed synovial tissue. Arthritis Rheum. 1996, 39: 583-591.
Article CAS PubMed Google Scholar - Georganas C, Liu H, Perlman H, Hoffmana A, Thimmapaya B, Pope R: Regulation of IL-6 and IL-8 expression in rheumatoid arthritis synovial fibroblasts: the dominant role for NF-κB but not C/EBPβ or c-Jun. J Immunol. 2000, 165: 7199-7206.
Article CAS PubMed Google Scholar - Ziolkowska M, Koe A, Luszczykiewicz G, Ksiezopolska-Pietrzak K, Klimczak E, Chwalinska-Sadowska H, Maslinski W: High levels of IL-17 in rheumatoid arthritis patients: IL-15 triggers in vitro IL-17 production via cyclosporine A-sensitive mechanism. J Immunol. 2000, 164: 2832-2838.
Article CAS PubMed Google Scholar - Chabaud M, Durand J, Buchs N, Fossiez F, Page G, Frappart L, Miossec P: Human interleukin 17; a T cell-derived proinflammatory cytokine produced by the rheumatoid arthritis synovium. Arthritis Rheum. 1999, 42: 963-970. 10.1002/1529-0131(199905)42:5<963::AID-ANR15>3.0.CO;2-E.
Article CAS PubMed Google Scholar - Cho ML, Yoon CH, Hwang SY, Park MK, Min SY, Lee SH, Park SH, Kim HY: Effector function of type II collagen-stimulated T cells from rheumatoid arthritis patients. Arthritis Rheum.
- Scuderi E, Convertino R, Molino N, Provenzano C, Marino M, Zoli A, Bartoccioni E: Effect of pro-inflammatory/anti-inflammatory agents on cytokine secretion by peripheral blood mononuclear cells in rheumatoid arthritis and systemic lupus erythematosus. Autoimmunity. 2003, 36: 71-77. 10.1080/0891693031000079275.
Article CAS PubMed Google Scholar - Woltman A, de Haji S, Boonstra J, Gobin S, Daha M, vab Kooten C: Interleukin-17 and CD40-ligand synergistically enhance cytokine and chemokine production by renal epithelial cells. J Am Soc Nephrol. 2000, 11: 2044-2055.
CAS PubMed Google Scholar - Hata K, Andoh A, Shimada M, Fujino S, Bamba S, Araki Y, Okuno T, Fujiyama Y, Bamba T: IL-17 stimulates inflammatory responses via NF-κB and MAP kinase pathway in human colonic myofibroblasts. Am J Physiol Gastrointest Liver Physiol. 2002, 282: G1035-G1044.
Article CAS PubMed Google Scholar - Kim G, Jun J, Elkon K: Necessary role of phosphatidylinositol 3-kinase in transforming growth factor β-mediated activation of Akt in normal and rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 2002, 46: 1504-1511. 10.1002/art.10314.
Article CAS PubMed Google Scholar - Aidinis V, Plows D, Haralambous S, Armaka M, Papadopoulos P, Kanaki M, Koczan D, Thiesen J, Kollias G: Functional analysis of an arthritogenic synovial fibroblast. Arthritis Res Ther. 2003, 5: R140-R157. 10.1186/ar749.
Article PubMed Central CAS PubMed Google Scholar - Yamamura Y, Gupta R, Morita Y, He X, Pai R, Endres J, Freiberg A, Chung K, Fox D: Effector function of resting T cells: activation of synovial fibroblasts. J Immunol. 2001, 166: 2270-2275.
Article CAS PubMed Google Scholar - Chabaud M, Fossiez F, Taupin J-L, Miossec P: Enhancing effect of IL-17 on IL-1-induced IL-6 and leukemia inhibitory factor production by rheumatoid arthritis synoviocytes and its regulation by Th2 cytokines. J Immunol. 1998, 161: 409-414.
CAS PubMed Google Scholar - Aggarwal S, Gurney A: IL-17: prototype member of an emerging cytokine family. J Leukoc Biol. 2002, 71: 1-8.
CAS PubMed Google Scholar - Shi Y, Ullrich S, Zhang J, Connolly K, Grzegorzewski K, Barber M, Wang W, Wathen K, Hodge V, Fisher C, Olsen H, Ruben S, Knyazev I, Cho Y-H, Kao V, Wilkinson K, Carrell J, Ebner R: A novel cytokine receptor-ligand pair. J Biol Chem. 2000, 275: 19167-19176. 10.1074/jbc.M910228199.
Article CAS PubMed Google Scholar - Lee J, Ho W-H, Maruoka M, Corpuz R, Baldwin D, Foster J, Goddard A, Yansura D, Vandlen R, Wood W, Gurney A: IL-17E, a novel proinflammatory ligand for the IL-17 receptor homolog IL-17Rh1. J Biol Chem. 2001, 276: 1660-1664. 10.1074/jbc.M008289200.
Article CAS PubMed Google Scholar - Kehlen A, Pachnio A, Thiele K, Langner J: Gene expression induced by interleukin-17 in fibroblast-like synoviocytes of patients with rheumatoid arthritis: upregulation of hyaluronan-binding protein TSG-6. Arthritis Res Ther. 2003, 5: R186-R192. 10.1186/ar762.
Article PubMed Central CAS PubMed Google Scholar - Shimada M, Andoh A, Hata K, Tasaki K, Araki Y, Fusiyama Y, Bamba T: IL-6 secretion by human pancreatic periacinar myofibroblasts in response to inflammatory mediators. J Immunol. 2002, 168: 861-868.
Article CAS PubMed Google Scholar - Hsu YM, Lucci J, Su L, Ehrenfels B, Garber E, Thomas D: Heteromultimeric complexes of CD40 ligand are present on the cell surface of human T lymphocytes. J Biol Chem. 1997, 272: 911-915. 10.1074/jbc.272.2.911.
Article CAS PubMed Google Scholar - Pietravalle F, Lecoanet-Henchoz S, Blasey H, Aubry JP, Elson G, Edgerton MD, Bonnefoy JY, Gauchat JF: Human native soluble CD40L is a biologically active trimer, processed inside microsomes. J Biol Chem. 1996, 271: 5965-5967. 10.1074/jbc.271.11.5965.
Article CAS PubMed Google Scholar - Morel J, Park C, Woods J, Koch A: A novel role for interleukin-18 in adhesion molecule induction through NFκB and phosphatidylinositol (PI) 3-kinase-dependent signal transduction pathways. J Biol Chem. 2001, 276: 37069-37075. 10.1074/jbc.M103574200.
Article CAS PubMed Google Scholar - Ozes O, Mayo L, Gustin J, Pfeffer S, Pfeffer L, Donner D: NF-κB activation by tumor necrosis factor requires the Akt serine-threonine kinase. Nature. 1999, 401: 82-85. 10.1038/43466.
Article CAS PubMed Google Scholar
Acknowledgements
This study was supported by a grant from the Korean Health 21 R&D Project, Ministry of Health and Welfare, Republic of Korea (grant no. 02-PJ1-PG3-20905-0011) to H S-Y, and by the Specialized Research Center fund (no. R11-2002-098-01001-0) from the Korea Science and Engineering Foundation (KOSEF) to the Rheumatism Research Center at The Catholic University, Seoul.
Author information
Authors and Affiliations
- Rheumatism Research Center, Catholic Institutes of Medical Science, The Catholic University of Korea, Seoul, Korea
Sue-Yun Hwang, Ju-Young Kim, Kyoung-Woon Kim, Mi-Kyung Park, Youngmee Moon & Ho-Youn Kim - Center for Rheumatic Diseases, Kangnam St Mary's Hospital, The Catholic University of Korea Medical School, Seoul, Korea
Wan-Uk Kim & Ho-Youn Kim
Authors
- Sue-Yun Hwang
You can also search for this author inPubMed Google Scholar - Ju-Young Kim
You can also search for this author inPubMed Google Scholar - Kyoung-Woon Kim
You can also search for this author inPubMed Google Scholar - Mi-Kyung Park
You can also search for this author inPubMed Google Scholar - Youngmee Moon
You can also search for this author inPubMed Google Scholar - Wan-Uk Kim
You can also search for this author inPubMed Google Scholar - Ho-Youn Kim
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toSue-Yun Hwang.
Additional information
Competing interests
None declared.
Authors’ original submitted files for images
Rights and permissions
About this article
Cite this article
Hwang, SY., Kim, JY., Kim, KW. et al. IL-17 induces production of IL-6 and IL-8 in rheumatoid arthritis synovial fibroblasts via NF-κB- and PI3-kinase/Akt-dependent pathways.Arthritis Res Ther 6, R120 (2004). https://doi.org/10.1186/ar1038
- Received: 22 September 2003
- Revised: 02 November 2003
- Accepted: 04 December 2003
- Published: 21 January 2004
- DOI: https://doi.org/10.1186/ar1038