Clinical Manifestations, Etiology, and Pathogenesis of the Hyper-IgE Syndromes (original) (raw)
Abstract
Autosomal dominant Hyper-IgE syndrome (AD-HIES) is a rare primary immunodeficiency characterized by eczema, recurrent skin and lung infections, elevated serum IgE, and various connective tissue, skeletal, and vascular abnormalities. Mutations in signal transducer and activator of transcription 3 (STAT3) have recently been found to account for most cases; however, the pathogenesis of the varied features remains poorly defined. A distinct syndrome, known as autosomal recessive HIES (AR-HIES) manifests as severe eczema, recurrent bacterial and viral skin infections, and sinopulmonary infections. As opposed to STAT3 deficient HIES, AR-HIES lacks the connective tissue and skeletal manifestations but has an increase in neurologic abnormalities. In this review, we discuss the clinical presentations, genetic etiologies, and immunologic abnormalities of these two syndromes. In addition, we discuss animal models of STAT3 deficiency that provide insight into the pathogenesis of HIES. Further understanding of how STAT3 results in the diverse manifestations of HIES will allow us to develop more specific therapies for HIES as well as for many of the manifestations, such as scoliosis, recurrent staphylococcal infections, and eczema, which are common in the general population.
Main
Job's syndrome was described in 1966 in reference to the Biblical Job who was “smote with sore boils” (1). Buckley et al. (2) refined the syndrome after detection of extremely elevated serum IgE in conjunction with eczema and recurrent boils and pneumonias. Autosomal-dominant hyper-IgE syndrome (AD-HIES) is now recognized as a multisystem disorder with various connective tissue, skeletal, and vascular abnormalities (3,4). Dominant-negative mutations in signal transducer and activator of transcription 3 (STAT3) are responsible for most cases of AD-HIES (5–8). However, the pathogenesis of both the clinical manifestations and the immunologic abnormalities remains poorly understood. Autosomal recessive HIES (AR-HIES) is a distinct clinical entity characterized by elevated IgE, recurrent skin and lung infections, and eczema, susceptibility to viral infections, such as Molluscum contagiosum and an ill-defined CNS involvement but lacks the connective tissue manifestations of AD-HIES (9). The etiology of most cases of AR-HIES remains unknown. We will review the clinical manifestations, genetics, immunologic findings of HIES, and the pathogenesis of these unique diseases.
Clinical Manifestations
Immunologic features.
AD-HIES typically presents in the first few weeks of life with a newborn rash (Table 1). This pustular rash is often most pronounced on the face and scalp, in a pattern most consistent with neonatal acne or eosinophilic dermatitis (10,11). Scrapings of the rash show eosinophilia. The rash may improve or progress to an eczematoid rash often worsened by Staphylococcus aureus infection. Control of eczema is typically most successful with antistaphylococcal therapy, either with antimicrobials or antiseptics, such as dilute bleach. Recurrent S. aureus boils usually start in early childhood. Although frank pus is present upon aspiration of the boils, cardinal features of inflammation, such as heat, erythema, and tenderness, are often absent.
Table 1 Clinical features of AD-HIES
Recurrent pyogenic pneumonias usually start in early childhood. S. aureus is the most common pathogen, with Streptococcus pneumoniae and Haemophilus influenzae also occurring frequently (3,4). Similar to the S. aureus boils, purulent sputum is present, but individuals often lack systemic signs of inflammation, such as fever, and may be quite well appearing. Healing of the pneumonia is often aberrant with pneumatocoeles and bronchiectasis (Fig. 1). These structural abnormalities are then sites of fungal (typically Aspergillus) and Gram-negative (typically Pseudomonas) infection, which are major causes of morbidity and mortality (12). Mold infections can invade blood vessels leading to life-threatening hemoptysis and/or disseminate to extrapulmonary sites of infection.
Figure 1
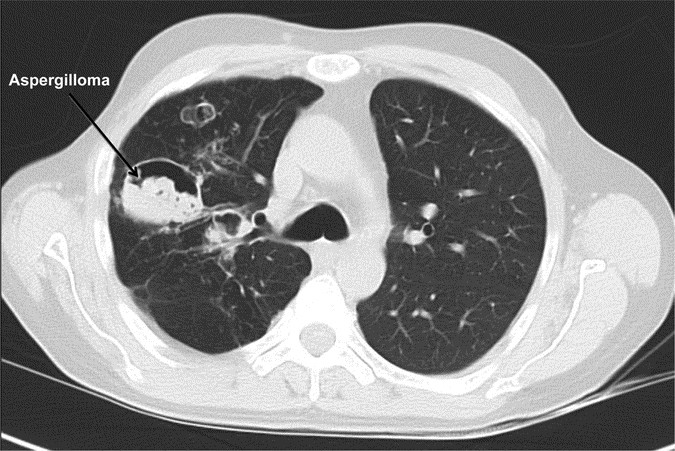
Chest CT of an individual with AD-HIES showing pneumatocoeles complicated by aspergilloma.
Opportunistic infections are also seen in HIES. Pneumocystis jiroveci pneumonia can occur in infants as their first pneumonia (13). Disseminated histoplasmosis and Cryptococcus have been described in unusual sites such as focal intestinal disease (14,15). Mucocutaneous candidiasis manifests as fingernail, vaginal, and oral disease (4).
Somatic features.
HIES is distinguished from many of the other primary immunodeficiencies by its many extraimmunologic features. A characteristic facial appearance develops through childhood and adolescence, characterized by facial asymmetry, a broad fleshy nose, and porous skin (3,4). Arnold Chiari I malformations (cerebellar tonsillar protrusion into the foramen magnum) are present in ∼20% of patients and craniosynostosis is seen in some individuals (16–18). Surgical repair of the craniosynostosis and Chiari I malformations is usually not indicated. Failure of primary dentition exfoliation is common and primary teeth often need to be removed for the secondary teeth to emerge (19). Oral mucosal manifestations include central ridges and fissures of the roof of the mouth, central depressions of the tongue, and oral mucosa variations of the cheek (20).
Minimal trauma fractures, osteopenia, hyperextensibility, scoliosis, and degenerative joint disease are common (3,4). Minimal trauma fractures of the ribs and long bones occur in about half of individuals with HIES and may occur independent of osteopenia. Varying degrees of scoliosis are common and may be severe enough to require rod placement. The scoliosis may develop during adolescence in a pattern similar to idiopathic scoliosis, but also occurs secondary to leg length discrepancy, lung disease with volume loss, or lung resections. Degenerative joint disease, especially of the spine, often manifests in the third and fourth decade of life and may require surgical repair (Fig. 2).
Figure 2
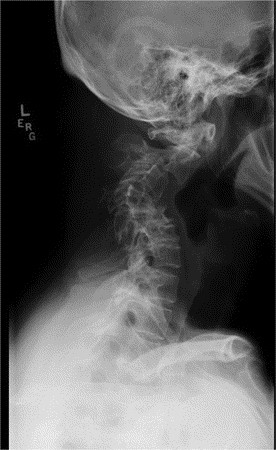
Cervical spine x-ray of a 48-y-old with AD-HIES showing severe degenerative spine disease.
Vascular abnormalities are common in HIES (21–23). One man suffered a myocardial infarction due to a large left anterior descending coronary artery aneurysm (22). On further investigation, coronary artery dilatation, aneurysms, and tortuosity are common in adults with HIES, but are usually asymptomatic. Aneurysms have also been described outside of the coronary arteries, including bilateral berry aneurysms of the carotids in one woman and a mycotic middle cerebral aneurysm in another (12). On brain imaging, lacunar infarctions and T2-weighted hyperintensities, potentially suggestive of small vessel disease, occur at a younger age than expected (Fig. 3) (16).
Figure 3
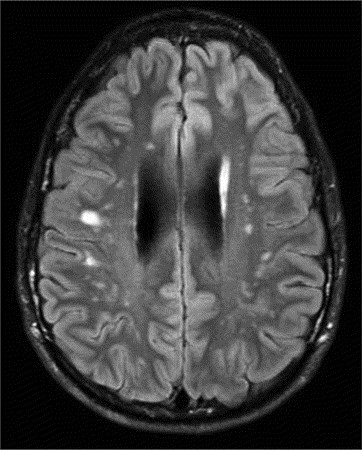
Brain MRI of a 21-y-old with AD-HIES showing multiple focal hyperintensities.
Esophageal dysfunction is poorly understood in adults with HIES (our unpublished observations). Manifestations include choking episodes and inability to tolerate pills or large chunks of food. Esophageal diverticula and strictures occur and esophageal dilation may be required.
Similar to several other primary immunodeficiencies, there is an increased incidence of both Hodgkin's and non-Hodgkin's lymphoma in AD-HIES (24). Other reported malignancies include leukemia and cancers of the vulva, liver, and lung (25,26).
Laboratory abnormalities in HIES are most significant for eosinophilia and elevated serum IgE. The IgE typically peaks over 2000 IU/μL, but may decrease over time and even reach normal values in adulthood (4). Clinical manifestations of HIES may persist despite normalization of IgE. RAST testing for specific antigens may yield positive results; however, this is confounded by the overall high IgE level; clinically apparent allergies do not seem to be increased in HIES. Serum IgG, IgA, and IgM are typically normal.
White blood cell counts in HIES are usually normal, although relative neutropenia has been seen. Leukocytosis and acute phase reactants, such as C-reactive protein during infection, may not be as high as expected. Lymphocyte phenotyping shows decreased memory T cells, and antibody responses are variable (3,23,27).
Genetics
Dominant-negative mutations in STAT3 account for most cases of AD-HIES (5–8,28,29). Many cases are sporadic, but when familial, all individuals carrying the mutation have the HIES phenotype. Mutations have been found in all races and many ethnicities. Most of the reported mutations are missense mutations accounting for one amino acid change, but in-frame deletions of one amino acid, or exon skipping have been reported as well. Mutations have been largely concentrated in the SH2 domain (mediating protein–protein interactions) and DNA binding domain (mediating protein–DNA interactions) of STAT3, with a few reported mutations in the trans-activation domain. There are several mutational hot spots, but no clear phenotypic distinctions have emerged between these two regions. One man has somatic mosaicism for STAT3 mutation and a milder HIES phenotype (5).
Null alleles have not been reported in HIES and heterozygous deletions in mice are phenotypically normal, indicating that the mutations exert a dominant-negative effect on the normal allele. Laboratory constructs of AD-HIES mutations show dominant-negative effect with STAT3 target binding activity ∼25% of normal (7).
STAT3 Function
STAT3 is one of six STAT proteins that are major signal transducers involved in many diverse pathways (30). Many cytokines transmit signals through Janus kinase (Jak)-STAT pathways (31). The intracellular components of cytokine receptors often bind to one of four Jak proteins (Jak1, Jak2, Jak3, Tyk2) leading to STAT recruitment and phosphorylation. Phosphorylated STAT then dimerizes by the Src homology 2 (SH2) domain, translocates to the nucleus leading to activation of STAT-regulated genes. STAT3 is involved in the signal transduction of many cytokines, including but not limited to IL-6, IL-10, IL-21, IL-22, and IL-23, and plays an integral role in wound healing, angiogenesis, cancer, and immunity (Fig. 4) (30).
Figure 4
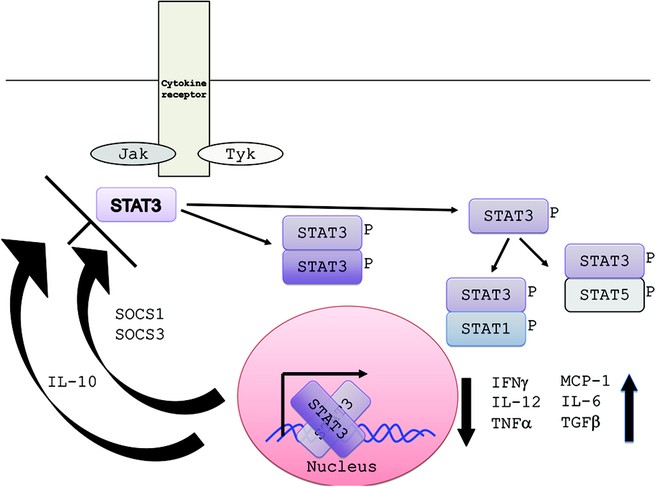
STAT3 signaling pathway. Multiple cytokines, including IL-6, IL-10, IL-21, IL-22, and IL-23 activate STAT3 phosphorylation. Dimerized STAT3 enters the nucleus to affect transcription of various genes. STAT3 can also dimerize with other STATs. Normal STAT3 signaling leads to the up-regulation and down-regulation of the cytokines shown in the right lower corner. IL-10 and SOCS (suppressors of cytokine signaling) can down-regulate STAT3 activity.
The clinical phenotype of HIES is one of both excessive and inadequate inflammation. The prolonged local inflammation in the lung leading to pneumatocoeles is an example of excessive inflammation, with the susceptibility to infections including opportunists and the characteristic “cold” boils being examples of inadequate inflammation. This dichotomy is expected as both pro- (e.g., IL-6) and anti-inflammatory (e.g., IL-10) cytokines signal through STAT3.
Evidence of proinflammatory cytokines is present in the cytokine and microarry data of individuals with HIES as well as through myeloid specific Stat3 knock out mice (5,32,33). Increased expression of TNF-alpha and IFN-gamma, likely due in part to impaired IL-6 signaling, is evident in myeloid-specific Stat3 knockout mice (33). These findings reflect the immunologic data of individuals with STAT3 mutations in whom leukocyte microarrays show increased proinflammatory activity, such as increased TNF-alpha at rest and after stimulation (5). In addition, in vitro HIES peripheral blood mononuclear cells have increased proinflammatory cytokine secretion (TNF-alpha, IFN-gamma, and IL-12) after stimulation with specific agonists (5,32).
However, the absence of hematopoietic Stat3 in mice is also associated with impaired B cell numbers in the bone marrow and the periphery (34). Mice with Stat3 deficient bone marrow have both poor B cell differentiation and enhanced apoptosis of B-cell progenitors. The correlation of these murine findings with human disease is not clear. Patients with HIES usually have normal B cell numbers, as well as normal IgG, IgM, and IgA, but the number of B-memory cells is diminished (27). In one study, there was no correlation between the ability to mount specific antibodies and the relative number of memory B cells, making the clinical significance of the lack of memory B cells unclear.
In mice and humans, STAT3 is integral to Th17 cell differentiation and IL-17 production. One of the most consistent immunologic findings in HIES is the near total absence of Th17 cells and severely impaired IL-17 production (28,29,35,36). Th17 CD4 cells are thought to be important in host defense through recruitment of neutrophils well as up-regulation of antimicrobial peptides (37). The significance of diminished IL-17 production in humans is still being elucidated, but mice that are deficient in IL-17 are prone to Candida and Klebsiella infections (38,39). Th17 cells are also important for IL-22 secretion, which is critical for beta-defensin production (40). Lack of beta-defensins has been implicated in susceptibility to S. aureus in individuals with eczema (41). Therefore, the S. aureus abscesses of HIES may in part reflect decreased defensins secondary to the lack of Th17 cells. Beta-defensins are also expressed in the lung and might in part explain the susceptibility to pneumonias (42).
Further evidence of impaired immunity is the greatly diminished monocyte chemoattractant protein-1 levels in HIES patient peripheral blood mononuclear cells after IL-6 stimulation (5,6). Monocyte chemoattractant protein-1 is a chemokine involved in chemotaxis and the simulation of several cytokines. In addition, C-reactive protein, an acute phase reactant produced in response to IL-6, is often diminished in acute infections.
The extreme elevation of IgE in HIES remains poorly understood. There were initial hypotheses that the elevated IgE might reflect impaired IL-21 signaling, because IL-21 receptor knockout mice have elevated IgE (43). However, human B cells differ from those of mice in that IL-21 actually acts synergistically with IL-4 to increase IgE production (44). Therefore, lack of IL-21 might be associated with less IgE, not more. Perhaps, the increased IgE reflects a lack of suppression of IL-21 or lack of IL-4 inhibition by IL-10 or IFN-gamma. It is noteworthy that this cardinal feature of HIES has not yet been clearly explained.
Somatic Features of HIES
Homozygous Stat3 knockout mice die in utero, but many tissue specific Stat3 knockouts are viable. It is through these conditional knockout animals that much of the function of Stat3 has been deduced (45). Poor wound healing, eosinophilia, osteoporosis, cardiomyopathy, lung, and brain abnormalities are seen in these tissue-specific deletants.
In pulmonary epithelium-specific Stat3 knockout models, hyperoxia caused excessive inflammation and pulmonary injury with associated alveolar enlargement (46). This may relate to the pneumatocoeles of HIES that are thought to be related to an exuberant inflammatory response.
In hematopoietic Stat3 knockouts, mice have increased TNF-alpha and IFN-gamma levels as well as increased osteoclastogenesis and decreased bone density (47). The bone findings may relate to the minimal trauma fractures and osteopenia observed in HIES. This may relate to the inhibitory effect on osteoblasts of TNF-alpha, or to abnormal IL-10 signaling, because IL-10 typically inhibits osteoclast activity (48).
The hyperintensities seen on brain MRI in HIES may relate to increased inflammation, demyelination, and astrocytosis after neurologic injury, as seen in astrocyte-specific Stat3 knockout mice (49). As IL-6 is involved in astrocyte differentiation, these findings may relate to disrupted IL-6 signaling.
Cardiac myocyte-specific Stat3 deficiency in mice leads to cardiac dysfunction with early cardiac failure and increased infarct size after ischemic insult (50). Cardiac myocytes stimulated with LPS had increased TNF-alpha levels and subsequent inflammation and dysfunction (51). Cardiomyopathy has not been a reported feature of HIES, but has not been examined systematically. Coronary artery tortuosity and aneurysms have been found in HIES, as well as decreased atherosclerosis (21–23). The pathogenesis and implications of these findings remain to be determined. Some mouse models of atherosclerosis have increased Stat3 expression, suggesting that a lack of Stat3 functioning may be associated with decreased atherosclerosis, but this has not been evaluated (52).
Poor wound healing after skin biopsy has been associated with mouse keratinocytes deficient in Stat3 (53). These mice have normal initial hair growth, but then abnormal hair follicles form in subsequent growth cycles with resultant epidermal hyperplasia and keratosis. Wound healing is thought to be normal in HIES, but the skin is often porous with a doughy texture. The oral mucosa variants may reflect aberrant responses to infection, such as Candida, or injury. The pathogenesis of the eczema of HIES is not well understood.
Therapy of AD-HIES
Therapy of HIES remains largely supportive, but will likely be refined greatly in the next several years as the pathogenesis of HIES is delineated and animal models created. Treatment of eczema and prevention of S. aureus abscesses are most successfully accomplished with antiseptics such as bathing in bleach (120 mL of bleach in tub of water for 15 min three times weekly) or swimming in chlorinated pools. Antimicrobial prophylaxis prevents recurrent sinopulmonary infections, with trimethoprim-sulfamethoxazole a frequent choice. Pneumonias should be treated aggressively to try to prevent parenchymal damage; bronchoscopy may help isolate causative pathogens and clear pus. Active suspicion of pneumonia is necessary because systemic symptoms of illness are often lacking.
If pneumatocoeles and bronchiectasis are present, antimicrobial prophylaxis often needs to be broadened to cover Gram-negative bacteria and fungi. Management of pneumatocoeles is complex, as these cysts when secondarily infected carry significant risk for morbidity and mortality; however, surgery is not without risk, as HIES patients may have trouble re-expanding their lungs and soilage of the pleural space can occur.
Immunomodulatory agents in HIES have not been well studied. Intravenous immunoglobulin (IVIG) may decrease the number of infections for some individuals and is the most frequent immunomodulator used (54,55). Levamisole, an antihelminthic that stimulates T- and natural killer cell function, was found to be inferior to placebo (56). IFN-gamma in vitro showed promise, but clinically had mixed results (57). Histamine-2 antagonists and cyclosporine may be helpful (58,59). Omalizumab, the MAb against IgE, has not been studied. Bone marrow transplantation has been performed but is likely not fully corrective (60).
Autosomal Recessive Hyper-IgE Syndrome
A similar but distinct syndrome was reported by Renner et al. (9), characterized by extremely elevated serum IgE, severe eczema, recurrent skin bacterial and viral infections as well as sinopulmonary infection. Compared with AD-HIES, these individuals lack the somatic features, such as the characteristic facies, scoliosis, and the failure of baby teeth to exfoliate. In addition, although pneumonias occur in AR-HIES, pneumatocoeles do not form.
AR-HIES has a much higher rate of cutaneous viral infections such as Molluscum contagiosum, Herpes simplex, and varicella infections. They also have frequent neurologic disease, ranging from facial paralysis to hemiplegia, in some cases due to CNS vasculitis. Mortality is high at a young age in AR-HIES with sepsis more frequent than in AD-HIES.
Eosinophilia and elevated serum IgE are the most consistent laboratory findings and may be more dramatic than in AD-HIES. Autoimmune cytopenias may occur. Other immunologic studies, such as lymphocyte phenotyping, do not have characteristic findings.
The etiology of most cases of AR-HIES is unknown, but autosomal recessive inheritance is assumed because of consanguinity and multiple affected siblings. One patient has been found to have a homozygous mutation of Tyk2 with a four-nucleotide deletion resulting in a premature stop codon (61). This patient had the common features of AR-HIES such as eczema, viral infections, and recurrent sinopulmonary infections, but he also had Bacille Calmette–Guerin and Salmonella infections, infections classic for IL-12/IFN-gamma defects. Mutations of Tyk2 have been absent in the other reported cases of AR-HIES (62).
Conclusion
HIES was the last major primary immunodeficiency for which the genetic etiology was unknown. STAT3 mutations account for most cases of this multifaceted disease. Understanding the pathogenesis of the immunologic and the somatic features has already been greatly aided by genetic dissection of this disease. It is to be hoped that understanding the pathophysiology of these rare conditions will shed light on the more common problems in the general population, such as eczema, scoliosis, osteoporosis, and S. aureus infection.
References
- Davis SD, Schaller J, Wedgwood RJ 1966 Job's syndrome. Recurrent, “cold” staphylococcal abscesses. Lancet 1: 1013–1015
Article CAS PubMed Google Scholar - Buckley RH, Wray BB, Belmaker EZ 1972 Extreme hyperimmunoglobulinemia E and undue susceptibility to infection. Pediatrics 49: 59–70
CAS PubMed Google Scholar - Buckley RH 2001 The hyper IgE syndrome. Clin Rev Allergy Immunol 20: 139–154
Article CAS PubMed Google Scholar - Grimbacher B, Holland SM, Gallin JI, Greenberg F, Hill SC, Malech HL, Miller JA, O'Connell AC, Puck JM 1999 Hyper-IgE syndrome with recurrent infections—an autosomal dominant multisystem disorder. N Engl J Med 340: 692–702
Article CAS PubMed Google Scholar - Holland SM, DeLeo FR, Elloumi HZ, Hsu AP, Uzel G, Brodsky N, Freeman AF, Demidowich A, Davis J, Turner ML, Anderson VL, Darnell DN, Welch PA, Kuhns DB, Frucht DM, Malech HL, Gallin JI, Kobayashi SD, Whiteny AR, Voyich JM, Musser JM, Woellner C, Schaffer AA, Puck JM, Grimbacher B 2007 STAT3 mutations in they hyper-IgE syndrome. N Engl J Med 357: 1608–1619
Article CAS PubMed Google Scholar - Jiao H, Toth B, Fransson I, Rakoczi E, Balogh I, Magyarics Z, Derfalvi B, Csorba G, Szaflarska A, Megarbane A, Akatcherian C, Dbaibo G, Rajnavolgyi E, Hammarstrom L, Kere J, Lefranc G, Marodi L 2008 Novel and recurrent STAT3 mutations in hyper-IgE syndrome patients from different ethnic groups. Mol Immunol 46: 202–206
Article CAS PubMed Google Scholar - Minegishi Y, Saito M, Tsuchiya S 2007 Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature 448: 1058–1062
Article CAS PubMed Google Scholar - Renner ED, Ryalaarsdam S, Anover-Sombke S, Rack AL, Reichenbach J, Carey JC, Zhu Q, Jansson AF, Barboza J, Schimke LF, Leppert MF, Getz MM, Seger RA, Hill HR, Belohradsky BH, Torgerson TR, Ochs HD 2008 Novel signal transducer and activator of transcription 3 (STAT3) mutations, reduced T(H)17 cell numbers, and STAT3 phosphorylation in hyper-IgE syndrome. J Allergy Clin Immunol 122: 181–187
Article CAS PubMed PubMed Central Google Scholar - Renner ED, Puck JM, Holland SM, Schmitt M, Weiss M, Frosch M, Bergmann M, Davis J, Belohradsky BH, Grimbacher B 2004 Autosomal recessive hyperimmunoglobulin E syndrome: a distinct disease entity. J Pediatr 144: 93–99
Article CAS PubMed Google Scholar - Chamlin SL, McCalmont TH, Cunningham BB, Esterly NB, Lai CH, Mallory SB, Mancini AJ, Tamburro J, Frieden IJ 2002 Cutaneous manifestations of hyper-IgE syndrome in infants and children. J Pediatr 141: 572–575
Article PubMed Google Scholar - Eberting CL, Davis J, Puck JM, Holland SM 2004 Dermatitis and the newborn rash of hyper-IgE syndrome. Arch Dermatol 140: 1119–1125
Article PubMed Google Scholar - Freeman A, Kleiner D, Nadiminti H, Davis J, Quezado M, Anderson V, Puck JM, Holland SM 2007 Causes of death in hyper IgE syndrome. J Allergy Clin Immunol 119: 1234–1240
Article CAS PubMed Google Scholar - Freeman AF, Davis J, Anderson VL, Barson W, Darnell DN, Puck JM, Holland SM 2006 Pneumocystis jiroveci infection in patients with hyper-immunoglobulin E syndrome. Pediatrics 118: e1271–e1275
Article PubMed Google Scholar - Hutto JO, Bryan CS, Greene FL, White CJ, Gallin JL 1988 Cryptococcosis of the colon resembling Crohn's disease in a patient with the hyperimmunoglobulinemia E syndrome. Gastroenterology 94: 808–812
Article CAS PubMed Google Scholar - Jacobs DH, Macher AM, Handler R, Bennett JE, Collen MJ, Gallin JI 1984 Esophageal cryptococcosis in a patient with the hyperimmunoglobulin E-recurrent infection (Job's) syndrome. Gastroenterology 87: 201–203
Article CAS PubMed Google Scholar - Freeman AF, Collura-Burke CJ, Patronas NJ, Ilcus LS, Darnell D, Davis J, Puck JM, Holland SM 2007 Brain abnormalities in patients with hyperimmunoglobulin E syndrome. Pediatrics 119: e1121–e1125
Article PubMed Google Scholar - Hoger PH, Boltshauser E, Hitzig WH 1985 Craniosynostosis in hyper-IgE syndrome. Eur J Pediatr 144: 414–417
Article CAS PubMed Google Scholar - Smithwick EM, Finelt M, Pahwa S, Good RA, Naspitz CK, Mendes NF, Kopersztyk S, Spira TJ, Nahmias AJ 1978 Cranial synostosis in job's syndrome. Lancet 1: 826
Article CAS PubMed Google Scholar - O'Connell AC, Puck JM, Grimbacher B, Facchetti F, Majorana A, Gallin JI, Malech HL, Holland SM 2000 Delayed eruption of permanent teeth in hyperimmunoglobulinemia E recurrent infection syndrome. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 89: 177–185
Article CAS PubMed Google Scholar - Domingo DL, Freeman AF, Davis J, Puck JM, Tianxia W, Holland SM, Hart TC 2008 Novel intraoral phenotypes in hyper immunoglobulin E syndrome. Oral Dis 14: 73–81
CAS PubMed Google Scholar - Alomar-Melero E, Martin TD, Janelle GM, Peng YG 2008 An unusual giant right coronary artery aneurysm resembles an intracardiac mass. Anesth Analg 107: 1161–1162
Article PubMed Google Scholar - Ling JC, Freeman AF, Gharib AM, Arai AE, Lederman RJ, Rosing DR, Holland SM 2007 Coronary artery aneurysms in patients with hyper IgE recurrent infection syndrome. Clin Immunol 122: 255–258
Article CAS PubMed Google Scholar - Young TY, Jerome D, Gupta S 2007 Hyperimmunoglobulinemia E syndrome associated with coronary artery aneurysms: deficiency of central memory CD4+ T cells and expansion of effector memory CD4+ T cells. Ann Allergy Asthma Immunol 98: 389–392
Article CAS PubMed Google Scholar - Leonard GD, Posadas E, Herrmann PC, Anderson VL, Jaffe ES, Holland SM, Wilson WH 2004 Non-Hodgkin's lymphoma in Job's syndrome: a case report and review of the literature. Leuk Lymphoma 45: 2521–2525
Article PubMed Google Scholar - Gorin LJ, Jeha SC, Sullivan MP, Rosenblatt HM, Shearer WT 1989 Burkitt's lymphoma developing in a 7 year old body with hyper IgE syndrome. J Allergy Clin Immunol 83: 5–10
Article CAS PubMed Google Scholar - Oztop I, Demirkan B, Tarhan O, Kayahan H, Yilmaz U, Kargi A, Alakavuklar M 2004 The development of a pulmonary adenocarcinoma in a patient with Job's syndrome. Tumori 90: 132–135
Article PubMed Google Scholar - Speckmann C, Enders A, Woellner C 2008 Reduced memory B cells in patients with hyper IgE syndrome. Clin Immunol 129: 448–454
Article CAS PubMed Google Scholar - de Beaucoudrey L, Puel A, Filipe-Santos O, Cobat A, Ghandil P, Chrabieh M, Feinberg J, von Bernuth H, Samarina A, Jannière L, Fieschi C, Stéphan JL, Boileau C, Lyonnet S, Jondeau G, Cormier-Daire V, Le Merrer M, Hoarau C, Lebranchu Y, Lortholary O, Chandesris MO, Tron F, Gambineri E, Bianchi L, Rodriguez-Gallego C, Zitnik SE, Vasconcelos J, Guedes M, Vitor AB, Marodi L, Chapel H, Reid B, Roifman C, Nadal D, Reichenbach J, Caragol I, Garty BZ, Dogu F, Camcioglu Y, Gülle S, Sanal O, Fischer A, Abel L, Stockinger B, Picard C, Casanova JL 2008 Mutations in STAT3 and IL12RB1 impair the development of human IL-17 producing T cells. J Exp Med 205: 1543–1550
Article CAS PubMed PubMed Central Google Scholar - Ma CS, Chew GY, Simpson N, Priyadarshi A, Wong M, Grimbacher B, Fulcher DA, Tangye SG, Cook MC 2008 Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. J Exp Med 205: 1551–1557
Article CAS PubMed PubMed Central Google Scholar - Levy DE, Loomis CA 2007 STAT3 signaling and the hyper IgE syndrome. N Engl J Med 357: 1655–1658
Article CAS PubMed Google Scholar - Murray PJ 2007 The JAK-STAT signaling pathway: input and output integration. J Immunol 178: 2623–2629
Article CAS PubMed Google Scholar - Yeganeh M, Henneke P, Rezaei N, Ehl S, Thiel D, Matamoros N, Pietrogrande C, Espanol T, Litzman J, Franco JL, Sanal O, Kilic SS, Breborowicz A, Plebani A, Renner E, Rothenfusser S, Hawn TR, Woellner C, Grimbacher B 2008 Toll-like receptor stimulation induces higher TNF-a secretion in peripheral blood mononuclear cells from patients with hyper IgE syndrome. Int Arch Allergy Immunol 146: 190–194
Article CAS PubMed Google Scholar - Takeda K, Clausen BE, Kaisho T, Tsujimura T, Terada N, Forster I, Akira S 1999 Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity 10: 39–49
Article CAS PubMed Google Scholar - Chou WC, Levy DE, Lee CK 2006 STAT3 positively regulates an early step in B cell development. Blood 108: 3005–3011
Article CAS PubMed PubMed Central Google Scholar - Harris TJ, Grosso JF, Yen HR, Xin H, Kortylewski M, Albesiano E, Hipkiss EL, Getnet D, Goldberg MV, Maris CH, Housseau F, Yu H, Pardoll DM, Drake CG 2007 An in vivo requirement for STAT3 signaling in Th17 Development and Th17 dependant autoimmunity. J Immunol 179: 4313–4317
Article CAS PubMed Google Scholar - Milner JD, Brenchley JM, Laurence A, Freeman AF, Hill BJ, Elias KM, Kanno Y, Spalding C, Elloumi HZ, Paulson ML, Davis J, Hsu A, Asher AI, O'Shea J, Holland SM, Paul WE, Douek DC 2008 Impaired Th17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature 452: 773–776
Article CAS PubMed PubMed Central Google Scholar - Dong C 2008 Th17 cells in development: an updated view of their molecular identity and genetic programming. Nat Rev Immunol 8: 337–348
Article CAS PubMed Google Scholar - Aujla SJ, Chan YR, Zheng M, Fei M, Askew DJ, Pociask DA, Reinhart TA, McAllister F, Edeal J, Gaus K, Husain S, Kreindler JL, Dubin PJ, Pilewski JM, Myerburn MM, Mason CA, Iwakura Y, Kolls JK 2008 IL-22 medicates mucosal host defense against gram-negative bacterial pneumonia. Nat Med 14: 275–281
Article CAS PubMed PubMed Central Google Scholar - Huang W, Na L, Fidel PL, Schwarzenberger P 2004 Requirement of interleukin-17A for systemic anti-Candida albicans host defense in mice. J Infect Dis 190: 624–631
Article CAS PubMed Google Scholar - Wolk K, Kunz S, Witte E, Friedrich M, Asadullah K, Sabat R 2004 IL-22 increases the innate immunity of tissues. Immunity 21: 241–254
Article CAS PubMed Google Scholar - Ong PY, Ohtake T, Brandt C 2002 Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N Engl J Med 347: 1151–1160
Article CAS PubMed Google Scholar - Kao CY, Chen Y, Thai P, Wachi S, Kim C, Tam L, Wu R 2004 IL-17 markedly up-regulates beta-defensin-2 expression in human airway epithelium via JAK and NF-kB signaling pathways. J Immunol 173: 3482–3491
Article CAS PubMed Google Scholar - Ozaki K, Spolski R, Feng CG, Qi CF, Cheng J, Sher A, Morse HC 3rd, Liu C, Schwartzberg PL, Leonard WJ 2002 A critical role for IL-21 in regulating immunoglobulin production. Science 298: 1630–1634
Article CAS PubMed Google Scholar - Avery DT, Ma CS, Bryant VL, Santner-Nanan B, Nanan R, Wong M, Fulcher DA, Cook MC, Tangye SG 2008 STAT3 is required for IL-21 induced secretion of IgE from human naïve B cells. Blood 112: 1784–1793
Article CAS PubMed Google Scholar - Takeda K, Noguchi K, Shi W, Tanaka T, Matsumoto M, Yoshida N, Kishimoto T, Akira S 1997 Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc Natl Acad Sci U S A 94: 3801–3804
Article CAS PubMed PubMed Central Google Scholar - Hokuto I, Ikegami M, Yoshida M, Takeda K, Akira S, Perl AK, Hull WM, Wert SE, Whitsett JA 2004 Stat-3 is required for pulmonary homeostasis during hyperoxia. J Clin Invest 113: 28–37
Article CAS PubMed PubMed Central Google Scholar - Zhang Z, Welte T, Troiano N, Maher SE, Fu XY, Bothwell AL 2005 Osteoporosis with increased osteoclastogenesis in hematopoietic cell-specific STAT3-deficient mice. Biochem Biophys Res Commun 328: 800–807
Article CAS PubMed Google Scholar - Datta HK, Ng WF, Walker JA, Tuck SP, Varanasi SS 2008 The cell biology of bone metabolism. J Clin Pathol 61: 577–587
Article CAS PubMed Google Scholar - Okada S, Nakamura M, Katoh H, Miyao T, Shimazaki T, Ishii K, Yamane J, Yoshimura A, Iwamoto Y, Toyama Y, Okano H 2006 Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat Med 12: 829–834
Article CAS PubMed Google Scholar - Hilfiker-Kleiner D, Hilfiker A, Fuchs M, Kaminski K, Schaefer A, Schieffer B, Hillmer A, Schmiedl A, Ding Z, Podewski E, Podewski E, Poli V, Schneider MD, Schulz R, Park JK, Wollert KC, Drexler H 2004 Signal transducer and activator of transcription 3 is required for myocardial capillary growth, control of interstitial matrix deposition and heart protection from ischemic injury. Circ Res 95: 187–195
Article CAS PubMed Google Scholar - Scarabelli TM, Townsend PA, Chen Scarabelli C, Yuan Z, McCauley RB, Di Rezze J, Patel D, Putt J, Allebban Z, Abboud J, Chilukuri K, Gardin J, Saravolatz L, Knight RA, Latchman DS, Stephanou A 2008 Amino acid supplementation differentially modulates STAT1 and STAT3 activation in the myocardium exposed to ischemia/reperfusion injury. Am J Cardiol 101: 63E–68E
Article CAS PubMed Google Scholar - Olive M, Mellad JA, Beltran LE, Ma M, Cimato T, Noguchi AC, San H, Childs R, Kovacic JC, Boehm M 2008 p21Cip1 modulates arterial wound repair through the stromal cell-derived factor-1/CXCR4 axis in mice. J Clin Invest 118: 2050–2061
CAS PubMed PubMed Central Google Scholar - Sano S, Itami S, Takeda K, Tarutani M, Yamaguchi Y, Miura H, Yoshikawa K, Akira S, Takeda J 1999 Keratinocyte-specific ablation of Stat3 exhibits impaired skin remodeling, but does not affect skin morphogenesis. EMBO J 18: 4657–4668
Article CAS PubMed PubMed Central Google Scholar - Kimata H 1995 High dose intravenous gammaglobulin treatment for hyperimmunoglobulinemia E syndrome. J Allergy Clin Immunol 95: 771–774
Article CAS PubMed Google Scholar - Wakim M, Alazard M, Yajima A, Speights D, Saxon A, Stiehm ER 1998 High dose intravenous immunoglobulin in atopic dermatitis and hyper-IgE syndrome. Ann Allergy Asthma Immunol 81: 153–158
Article CAS PubMed Google Scholar - Donabedian H, Alling DW, Gallin JI 1982 Levamisole is inferior to placebo in the hyperimmunoglobulin E recurrent infection (Job's) syndrome. N Engl J Med 307: 290–292
Article CAS PubMed Google Scholar - King CL, Gallin JI, Malech HL, Abramson SL, Nutman TB 1989 Regulation of immunoglobulin production in hyperimmunoglobulin E recurrent infection syndrome by IFN-gamma. Proc Natl Acad Sci U S A 86: 10085–10089
Article CAS PubMed PubMed Central Google Scholar - Thompson RA, Kumararatne DS 1989 Hyper-IgE syndrome and H2 receptor blockade. Lancet 2: 630
Article CAS PubMed Google Scholar - Etzioni A, Shehadeh N, Brecher A, Yorman S, Pollack S 1997 Cyclosporin A in hyperimmunoglobulin E syndrome. Ann Allergy Asthma Immunol 78: 413–414
Article CAS PubMed Google Scholar - Gennery AR, Flood TJ, Abinun M, Cant AJ 2000 Bone marrow transplantation does not correct the hyper-IgE syndrome. Bone Marrow Transplant 25: 1303–1305
Article CAS PubMed Google Scholar - Minegishi Y, Saito M, Morio T, Watanabe K, Agematsu K, Tsuchiya S, Takada H, Hara T, Kawamaura N, Ariga T, Kaneko H, Kondo N, Tsuge I, Yachie A, Sakiyama Y, Iwata T, Bessho F, Ohishi T, Joh K, Imai K, Kogawa K, Shinohara M, Fujieda M, Wakiguchi H, Pasic S, Abinun M, Ochs HD, Renner ED, Jansson A, Belohradsky BH, Metin A, Shimizu N, Mizutani S, Miyawaki T, Nonoyama S, Karasuyama H 2006 Human tyrosine kinase 2 deficiency reveals its requisite roles in multiple cytokine signals involved in innate and acquired immunity. Immunity 25: 745–755
Article CAS PubMed Google Scholar - Woellner C, Schaffer AA, Puck JM, Renner ED, Knebel C, Holland SM, Plebani A, Grimbacher B 2007 The hyper IgE syndrome and mutations in Tyk2. Immunity 26: 535
Article CAS PubMed Google Scholar
Author information
Authors and Affiliations
- Laboratory of Clinical Infectious Diseases, SAIC-Frederick, Inc., Frederick, 27102, Maryland
Alexandra F Freeman - Laboratory of Clinical Infectious Diseases, National Institutes of Health, Bethesda, 20892, Maryland
Steven M Holland
Authors
- Alexandra F Freeman
You can also search for this author inPubMed Google Scholar - Steven M Holland
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toSteven M Holland.
Additional information
This research was supported by the National Institute of Allergy and Infectious Diseases. This project has been funded, in part, with federal funds from the National Cancer Institute, National Institutes of Health, under contract HHSN261200800001E.
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government.
Rights and permissions
About this article
Cite this article
Freeman, A., Holland, S. Clinical Manifestations, Etiology, and Pathogenesis of the Hyper-IgE Syndromes.Pediatr Res 65, 32–37 (2009). https://doi.org/10.1203/PDR.0b013e31819dc8c5
- Received: 19 November 2008
- Accepted: 01 December 2008
- Issue Date: May 2009
- DOI: https://doi.org/10.1203/PDR.0b013e31819dc8c5