Kinetin Improves IKBKAP mRNA Splicing in Patients With Familial Dysautonomia (original) (raw)
Main
Familial dysautonomia (FD), also known as Riley Day syndrome or hereditary sensory and autonomic neuropathy type III, is a rare fatal autosomal recessive disease, which impairs the development of sensory and autonomic nerves (1). Affected patients have characteristic alacrima, depressed tendon reflexes, decreased pain and temperature perception, gastrointestinal dysmotility (1,2), and afferent baroreflex failure resulting in extreme blood pressure volatility (3). The leading causes of death include respiratory infections, renal failure, and unexpected sleep death (4). Current treatments are limited to supportive measures and are not always effective.
In 2001, we discovered that the disease was caused by mutations in I-κ-B kinase complex-associated protein (IKBKAP) gene, leading to a deficiency of I-κ-B kinase complex-associated protein (IKAP)/elongation protein 1 (ELP-1) (5). IKAP, also known as ELP-1, is an essential component of the human elongator complex, which promotes the transcriptional elongation of a number of target genes (6). Ninety-nine percent of patients with FD are homozygous for the IVS20+6T>C mutation (5), which weakens the splice site, causing variable, tissue-specific skipping of exon 20 in the transcribed RNA message, resulting in reduced protein expression of IKAP. Interestingly, the splicing abnormality primarily affects neuronal tissue, which produces mostly the shortened or mutant IKBKAP mRNA and a minimal amount of functional protein product (5,7). Other cells, like fibroblasts, produce almost equal levels of the normally spliced or WT IKBKAP mRNA and mutant IKBKAP mRNA.
As a result of a National Institute of Neurological Disorders and Stroke-sponsored drug screen (8), kinetin (6-furfurylaminopurine), a dietary supplement, was shown to rescue the splicing abnormality and restore normal IKAP protein levels in FD fibroblasts and transformed lymphoblast lines after only 1 wk in culture (9,10). Recently, the ability of kinetin to correct the splicing defect was also demonstrated in induced pluripotent FD stem cell-derived neural crest precursors from fibroblasts (11) and olfactory stem cells (12). Animal studies showed that kinetin was well absorbed orally, followed first-order elimination, was distributed into plasma and into the CNS, and was not a clastogenic agent (Sitek studies No. 0849-M208, No. 0849-1721.4, No. 0849-1731.C, SITEK Research Laboratories, Rockville, MD, 2005–2007).
Initial clinical studies of kinetin were performed in carriers of the IVS20+6T>C mutation, which despite being asymptomatic have some degree of mis-splicing and decreased levels of WT IKBKAP mRNA (13). Encouragingly, after receiving kinetin for 8 d, an increase in WT IKBKAP mRNA was found in circulating leukocytes. Here, we report the result of a pharmacokinetics study and 1 mo trial of kinetin in patients with FD.
METHODS
Study population.
Eight individuals with FD (mean age 36 ± 9 y; 2 males; Table 1), who were homozygous for the IVS20+6T>C mutation, were enrolled in the study. Subjects were recruited from the FD registry database at the Dysautonomia Center of the New York University School of Medicine. NYU School of Medicine Institutional Research Board approved the study and informed consent was obtained from all participants.
Table 1 Demographics and pharmacokinetics
Study preparation.
Patients were admitted to the General Clinical Research Center (Bellevue Hospital, New York, NY). An indwelling venous catheter was inserted. Vital signs were measured and baseline bloods obtained to establish pretreatment IKBKAP mRNA levels and safety parameters. The daily dose of kinetin for each patient was calculated based on weight as 23.5 mg/kg. This dose was identical to the dose used in the carrier study (13), which was extrapolated from cell culture data (9,10). The initial kinetin dose was administered in the morning, 2 h after eating. Doses ranged from 1000 to 1500 mg (Table 1). The kinetin was given in clear capsules containing 250 mg kinetin and 120 mg of maltodextrin (Nature's Best, Inc., Hauppauge, NY). Dosages were administered either via the gastrostomy tube or by mouth. Standard meals were served throughout the day.
Kinetin pharmacokinetics study.
To determine the pharmacokinetics of kinetin, blood was drawn 2 h before the single dose of kinetin was given. Blood samples were repeated after 0.5, 1, 1.5, 2, 3, 4, 6, 9, 12, and 24 h. All blood samples were taken after 10 min in the seated position. At the 8-d follow-up visit, blood was obtained 2 h after the morning kinetin dose to measure plasma kinetin levels.
One-month safety and tolerability study.
After completing the pharmacokinetics study, patients were discharged from the General Clinical Research Center and instructed to continuing taking the same kinetin dose once daily for 28 consecutive days. Participants returned weekly for repeat safety bloods and clinical assessment.
Measurement of plasma kinetin levels.
Whole blood was collected in heparinized tubes. After centrifugation (10 min at 2000 × g), the plasma was extracted and stored in aliquots at −20°C. Kinetin levels were determined using a validated solid-phase extraction method using caffeine (2.5 ng) as an internal standard and Varian Bond Elut 200 mg C18 extraction columns (Varian, Lake Forest, CA). One milliliter plasma was mixed with 70 μL of 1N acetic acid. The extraction columns were activated with 3 mL of acetonitrile and washed with 3 mL of distilled H2O. The kinetin containing plasma sample (1 mL) was applied to the extraction column followed by 2 mL of distilled H2O. The kinetin was eluted with the addition of 1 mL methanol. The samples were taken to dryness by vacuum centrifugation. Before analysis, the samples were reconstituted with 250 μL of the A component of the mobile-phase buffer (5% acetonitrile/H2O pH = 3.0); 100 μL was injected into the analytical system via a WISP 717 autosampler (Waters, Milford, MA). The analysis used a 15-min linear gradient to 90% acetonitrile/H2O, (B component) followed by a 5-min isocratic B, and then a 5-min linear return to A mobile phase using Knauer type 42 pumps (Sonntek, Saddle River, NJ) controlled by Pyramid software (Axxiom Chromatography, Moorepark, CA). The reverse phase HPLC used a Luna C18, 3 μ analytical column (4.6 × 150 mm; Phenomenex, Torrance, CA) with ultraviolet detection at 280 nm. The assay for kinetin was linear over the range of 0.05 to 5.0 μg/mL. We used nonlinear models available in Winnonlin 5.1 (Pharsight, Sunnyvale, CA) to calculate values of maximum plasma concentration (_C_max) and times of maximal concentration (_T_max), the area under curve (AUC), elimination rate constant (_K_el), and time of half-life (_T_1/2).
Measurement of IKBKAP mRNA levels.
Blood samples for IKBKAP mRNA were obtained before administration of the first kinetin dose and then at 8 d and at 28 d. Five milliliter of blood was collected into two vacutainers using the PAXgene Blood RNA System containing the PreAnalytiX reagent (PreAnalytiX, Switzerland) to stabilize intracellular RNA.
As previously described (13,14), RNA extraction and quantification were performed, and the relative amount of WT IKBKAP mRNA was measured using semi-quantitative RT-PCR. Briefly, the relative amount of spliced transcripts was determined using an Alpha 2000 Imager Analyzer (BioRad, Hercules, CA) and Image Quant QL (Amersham, Piscataway, NJ) software, using the integrated density value for each band. These values were then expressed as percent exon inclusion i.e. the percent of IKBKAP transcript produced that contains exon 20 (10).
Statistics.
A repeated-measure ANOVA was used to compare IKBKAP mRNA exon inclusion levels at baseline, d 8 and d 28. Statistical analyses were performed using SPSS (version 14.0). Data are shown as mean ± SD, unless otherwise stated. The p values of 0.05 or less were considered statistically significant.
RESULTS
Kinetin absorption.
Seven of the eight subjects completed the trial; one subject (female, 37 y old) developed a community-acquired respiratory infection after 21-d of receiving kinetin and was withdrawn from the study. Based on the extrapolation of efficacy data obtained in cell culture studies (9), the target kinetin level was a plasma concentration (_C_max) of 2150 ng/mL. Only subjects 2 and 4 did not meet this target. The remaining participants either met or exceeded the target (Table 1). The mean _C_max was 2866 ± 1889 ng/mL and _T_max = 2.1 h. The mean AUC was 20,291 ± 15,203 ng/mL·h. Kinetin was rapidly absorbed and then cleared according to first-order elimination kinetics (Fig. 1). Eight days after receiving kinetin, repeated plasma kinetin levels 2 h after dose administration were 2994 ± 3168 ng/mL and did not show accumulation or slower metabolism.
Figure 1
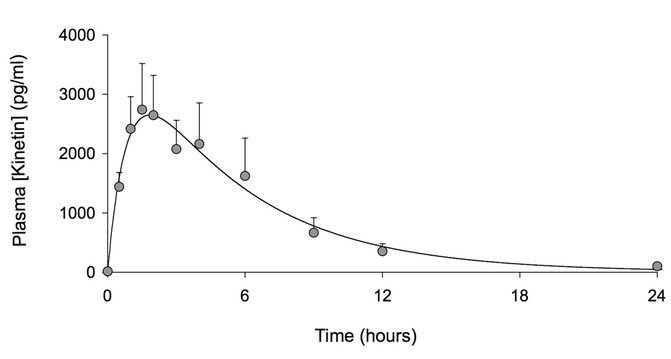
Venous plasma levels of kinetin after administration (n = 8, data are mean ± SEM). Time zero represents the time of drug administration.
Effect on IKBKAP mRNA.
All patients with FD increased the amount of normal WT IKBKAP mRNA produced in white blood cells after taking kinetin. At baseline, the mean percentage exon inclusion (i.e. the percentage of correctly spliced IKBKAP mRNA) was 54 ± 10%. This increased to 57 ± 10% after receiving kinetin for 8 d and increased further to 71 ± 9% after receiving kinetin for 28 d (p = 0.002, Fig. 2). There was no relationship between the kinetin dosages and peak concentration levels achieved or the effect on WT IKBKAP mRNA production.
Figure 2
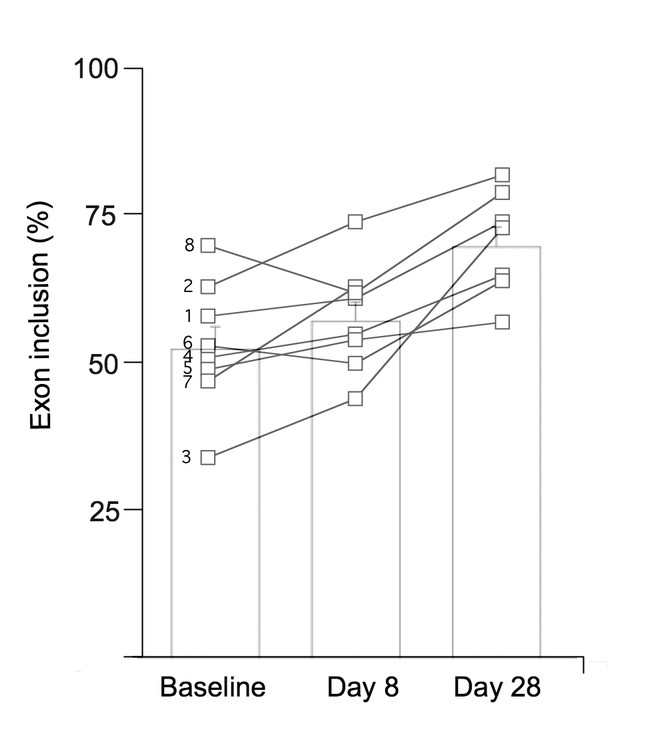
The percent of IKBKAP transcript produced that include exon 20 at baseline, after 8 d of receiving kinetin and after 28 d of receiving kinetin. Open squares show individual data. Subject number is to the left of the baseline values. Bar charts show mean ± SEM; p = 0.002.
Tolerability.
Overall, kinetin was well tolerated with only minimal side effects reported over the 28-d period. Diarrhea occurred in two subjects. Mild to moderate nausea was reported in three participants. These side effects were anticipated because they were common complaints in all subjects before they began taking kinetin. Three subjects complained of headaches. The side effects were not severe enough to cause any of the subjects to drop out of the study over the 28 d.
Safety.
Mild transitory changes in laboratory values were noted. Three patients with FD (no. 3, 6, and 8) had modest increases in liver enzymes. Subject no. 3 had the highest serum glutamic oxaloacetic transaminase and serum glutamic pyruvic transaminase values at 110 U/L and 169 U/L, respectively. Values in subjects 6 and 8 never exceeded 82 U/L for either transaminase, and bilirubin and alkaline phosphatase values remained normal for all three subjects. One subject (2) had a decrease in platelet count from his baseline 168,000/mL to 47,000/mL accompanied by a slight decrease in white blood count from baseline of 5300/mL to 3300/mL after 28 d of receiving kinetin but had no overt signs of viral illness. The possibility of a laboratory error could not be excluded. All laboratory changes were reversible and normalized after discontinuation of kinetin. Blood pressure and heart rate in supine and upright positions were unaffected by kinetin administration. There were no changes in weight or differences noted in the neurological examination during the 28-d period.
DISCUSSION
FD is caused by an intronic splice mutation in the IKBKAP gene that leads to partial skipping of exon 20 and tissue-specific reduction in IKAP/ELP-1 protein expression. Because kinetin has been shown to improve splicing and increase WT IKBKAP mRNA and IKAP protein expression in FD cell lines, we administered kinetin to eight FD individuals who are homozygous for the splice mutation and were able to demonstrate in vivo splicing changes.
To date, four substances have been identified that potentially increase total IKAP protein levels in FD cell lines, by either increasing total IKBKAP gene expression or modifying mRNA splicing and increasing WT IKBKAP mRNA production (9,15–17). These compounds have been postulated as therapeutic agents for patients with FD. Although in vitro administration of tocotrienols was reported to increase total IKBKAP gene expression (16), its effectiveness could not be replicated by other recent studies (11,17). Epigallocatechin gallate, like kinetin, reportedly modifies mRNA splicing (15), but its effectiveness in cell lines is modest when compared with kinetin (9,11). Most recently, phosphatidylserine was shown to also increase total IKBKAP expression, however, its effects on IKAP levels in patients with FD are still unknown. To date, kinetin is the first of these compounds to be subjected to an objective assessment in humans regarding pharmacokinetics, impact on mRNA splicing and safety.
Our results address several fundamental issues relevant to kinetin's possible role as a therapeutic agent for individuals affected with FD. First, we confirm that kinetin results in plasma levels equivalent to cell culture concentrations reported to affect IKBKAP splicing (9). Consistent with results from a previous study of oral administration of kinetin, kinetin elimination is almost complete by 12 h after ingestion (13).
Second, our data show that kinetin corrects the FD splicing defect in affected individuals and raises the level of normal IKBKAP mRNA in vivo. After 8 d of single daily dosing, normal splicing of IKBKAP had increased in six of eight subjects. Even more intriguing was the finding that the magnitude of splicing continued to improve without dosage change, because the levels of WT IKBKAP mRNA were greater in all subjects on d 28 than on d 8. It is possible that this is the result of a cumulative effect, similar to that seen in FD cell lines (10). Furthermore, the kinetin was relatively well tolerated and safe over this 28-d period. Given the severity of FD and the possibility that kinetin may alter the natural history of the disease, some tolerance for minor side effects might be warranted.
Our data clearly demonstrates that kinetin can modify splicing in peripheral leukocytes. We anticipate that IKBKAP mRNA expression may be similarly affected in neural tissues where mis-splicing is most profound causing the deficits characteristic to this disorder. Gene expression studies in HeLa cells after reduction of ELP-1 by RNAi has shown that a subset of Elongator target genes play a role in cell migration (6). This is consistent with previously described neuropathology in FD, such as arrested small fiber neuronal development resulting in variable degrees of disease severity (1). Because of its small molecular size (215.21), kinetin crosses the blood brain barrier quickly (Sitek study No. 0849-M208; SITEK Research Laboratories, Rockville, MD, 2005). Therefore, we project that target levels of kinetin will be achieved in human neuronal tissue to improve splicing efficiency so that there is increased production of WT IKBKAP mRNA transcript and thus increased amounts of normal functioning IKAP. IKAP is an integral component of the human Elongator complex, a transcriptional regulator that impacts on a host of other target genes (6) some of which may be involved in maintenance of sensory and autonomic nervous systems. Thus by modifying activity of these target genes, the natural history of FD may be altered and relentless progression attenuated. Long-term clinical trials of kinetin are warranted.
Abbreviations
AUC:
area under curve
C max :
maximum plasma concentration
ELP-1:
elongation protein 1
FD:
familial dysautonomia
IKBKAP :
I-κ-B kinase complex-associated protein gene
IKAP:
I-κ-B kinase complex-associated protein
T max :
time of maximal concentration
References
- Axelrod FB 2004 Familial dysautonomia. Muscle Nerve 29: 352–363
Article Google Scholar - Axelrod FB, Gold-von Simson G 2007 Hereditary sensory and autonomic neuropathies: types II, III, and IV. Orphanet J Rare Dis 2: 39
Article Google Scholar - Norcliffe-Kaufmann L, Axelrod F, Kaufmann H 2010 Afferent baroreflex failure in familial dysautonomia. Neurology 75: 1904–1911
Article Google Scholar - Axelrod FB, Goldberg JD, Ye XY, Maayan C 2002 Survival in familial dysautonomia: impact of early intervention. J Pediatr 141: 518–523
Article Google Scholar - Slaugenhaupt SA, Blumenfeld A, Gill SP, Leyne M, Mull J, Cuajungco MP, Liebert CB, Chadwick B, Idelson M, Reznik L, Robbins C, Makalowska I, Brownstein M, Krappmann D, Scheidereit C, Maayan C, Axelrod FB, Gusella JF 2001 Tissue-specific expression of a splicing mutation in the IKBKAP gene causes familial dysautonomia. Am J Hum Genet 68: 598–605
Article CAS Google Scholar - Close P, Hawkes N, Cornez I, Creppe C, Lambert CA, Rogister B, Siebenlist U, Merville MP, Slaugenhaupt SA, Bours V, Svejstrup JQ, Chariot A 2006 Transcription impairment and cell migration defects in elongator-depleted cells: implication for familial dysautonomia. Mol Cell 22: 521–531
Article CAS Google Scholar - Cuajungco MP, Leyne M, Mull J, Gill SP, Lu W, Zagzag D, Axelrod FB, Maayan C, Gusella JF, Slaugenhaupt SA 2003 Tissue-specific reduction in splicing efficiency of IKBKAP due to the major mutation associated with familial dysautonomia. Am J Hum Genet 72: 749–758
Article CAS Google Scholar - Heemskerk J, Tobin AJ, Bain LJ 2002 Teaching old drugs new tricks. Meeting of the Neurodegeneration Drug Screening Consortium, 7–8 April 2002, Washington, DC, USA. Trends Neurosci 25: 494–496
Article Google Scholar - Slaugenhaupt SA, Mull J, Leyne M, Cuajungco MP, Gill SP, Hims MM, Quintero F, Axelrod FB, Gusella JF 2004 Rescue of a human mRNA splicing defect by the plant cytokinin kinetin. Hum Mol Genet 13: 429–436
Article CAS Google Scholar - Hims MM, Ibrahim EC, Leyne M, Mull J, Liu L, Lazaro C, Shetty RS, Gill S, Gusella JF, Reed R, Slaugenhaupt SA 2007 Therapeutic potential and mechanism of kinetin as a treatment for the human splicing disease familial dysautonomia. J Mol Med 85: 149–161
Article CAS Google Scholar - Lee G, Papapetrou EP, Kim H, Chambers SM, Tomishima MJ, Fasano CA, Ganat YM, Menon J, Shimizu F, Viale A, Tabar V, Sadelain M, Studer L 2009 Modelling pathogenesis and treatment of familial dysautonomia using patient-specific iPSCs. Nature 461: 402–406
Article CAS Google Scholar - Boone N, Loriod B, Bergon A, Sbai O, Formisano-Tréziny C, Gabert J, Khrestchatisky M, Nguyen C, Féron F, Axelrod FB, Ibrahim el C 2010 Olfactory stem cells, a new cellular model for studying molecular mechanisms underlying familial dysautonomia. PLoS One 5: e15590
Article CAS Google Scholar - Gold-von Simson G, Goldberg JD, Rolnitzky LM, Mull J, Leyne M, Voustianiouk A, Slaugenhaupt SA, Axelrod FB 2009 Kinetin in familial dysautonomia carriers: implications for a new therapeutic strategy targeting mRNA splicing. Pediatr Res 65: 341–346
Article CAS Google Scholar - Gold-von Simson G, Leyne M, Mull J, Rolnitzky LM, Goldberg JD, Berlin D, Axelrod FB, Slaugenhaupt SA 2008 IKBKAP mRNA in peripheral blood leukocytes: a molecular marker of gene expression and splicing in familial dysautonomia. Pediatr Res 63: 186–190
Article CAS Google Scholar - Anderson SL, Qiu J, Rubin BY 2003 EGCG corrects aberrant splicing of IKAP mRNA in cells from patients with familial dysautonomia. Biochem Biophys Res Commun 310: 627–633
Article CAS Google Scholar - Anderson SL, Qiu J, Rubin BY 2003 Tocotrienols induce IKBKAP expression: a possible therapy for familial dysautonomia. Biochem Biophys Res Commun 306: 303–309
Article CAS Google Scholar - Keren H, Donyo M, Zeevi D, Maayan C, Pupko T, Ast G 2010 Phosphatidylserine increases IKBKAP levels in familial dysautonomia cells. PLoS ONE 5: e15884
Article CAS Google Scholar
Acknowledgements
We thank the staff of the General Clinical Research Center at Bellevue Hospital for their diligence in obtaining and preparing samples. We thank the participants and their families for their continued support.
Author information
Authors and Affiliations
- Department of Pediatrics, New York University School of Medicine, New York, 10016, New York
Felicia B Axelrod & Gabrielle Gold-von Simson - Department of Neurology, New York University School of Medicine, New York, 10016, New York
Felicia B Axelrod & Horacio Kaufmann - Department of Medicine, New York University School of Medicine, New York, 10016, New York
Leonard Liebes & Sandra Mendoza - Department of Physiology and Neuroscience, New York University School of Medicine, New York, 10016, New York
Lucy Norcliffe-Kaufmann - Department of Neurology (Genetics), Center for Human Genetic Research, Massachusetts General Hospital, Boston, 02114, Massachusetts
James Mull, Maire Leyne & Susan A Slaugenhaupt
Authors
- Felicia B Axelrod
You can also search for this author inPubMed Google Scholar - Leonard Liebes
You can also search for this author inPubMed Google Scholar - Gabrielle Gold-von Simson
You can also search for this author inPubMed Google Scholar - Sandra Mendoza
You can also search for this author inPubMed Google Scholar - James Mull
You can also search for this author inPubMed Google Scholar - Maire Leyne
You can also search for this author inPubMed Google Scholar - Lucy Norcliffe-Kaufmann
You can also search for this author inPubMed Google Scholar - Horacio Kaufmann
You can also search for this author inPubMed Google Scholar - Susan A Slaugenhaupt
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toFelicia B Axelrod.
Additional information
Supported by CTSI Grant 1UL1RR029893 and grants from the NIH General Clinical Research Centers Program of the National Center for Research Resources M01 RR-00096: NIH NS058318, NS036326, U54NS065736: FDA FD-R-3731-01, and awards from the Dysautonomia Foundation, Inc, New York, NY.
The authors report no conflicts of interest.
Rights and permissions
About this article
Cite this article
Axelrod, F., Liebes, L., Gold-von Simson, G. et al. Kinetin Improves IKBKAP mRNA Splicing in Patients With Familial Dysautonomia.Pediatr Res 70, 480–483 (2011). https://doi.org/10.1203/PDR.0b013e31822e1825
- Received: 28 February 2011
- Accepted: 10 May 2011
- Issue Date: November 2011
- DOI: https://doi.org/10.1203/PDR.0b013e31822e1825