The protective effects of granulocyte-macrophage colony-stimulating factor against radiation-induced lung injury (original) (raw)

Original Article
Introduction
Radiation therapy is an established modality for the treatment of various malignancies (1,2). Radiation-induced lung injury (RILI) is one of the most typical side effects experienced by patients with thoracic or breast cancer who undergo therapeutic radiation (3,4). As many as 15% of these patients suffer from pneumonitis and subsequently pulmonary fibrosis (5,6). The progression of RILI and radiation-induced lung fibrosis (RILF) involves a multi-factorial process characterized by persistent lung inflammation and extracellular matrix (ECM) production. RILI starts with an obvious acute inflammatory reaction which occurs 6–12 weeks after radiation (7).
Pulmonary fibrosis is a late-onset adverse event that manifests as alveolar edema, stromal fibroblast activation, proinflammatory and profibrotic cytokine release, destruction of the vasculature and lung parenchyma, and disordered progressive deposition of the ECM. Deposition of ECM in the distal vacuum can lead to respiratory failure (3,8). RILI is associated with chronic inflammation, this injury is perpetuated by the stimulation of chemokines and proinflammatory cytokines. The production of these cytokines and chemokines by inflammatory cells initiates the irreversible progression of fibrosis. This response can induce repetitive alveolar epithelial cell injury, fibroblast hyperplasia, and extreme accumulation of ECM, finally resulting in lung structure distortion and respiratory failure (9). RILF may occur several weeks, or even years, after the completion of radiotherapy (10). Immunosuppressive cytokines and other anti-inflammatory methods have been used to control acute lung inflammation and delay the progression of RILF (11). However, no effective strategy is currently available for the prevention or treatment of RILI.
Granulocyte-macrophage colony-stimulating factor (GM-CSF) belongs to the hematopoietic cytokine family (12), has been used clinically as a strategy for granulocytopenia after myelotoxic radiotherapy or chemotherapy in cancer patients. GM-CSF has previously been shown to induce the differentiation and proliferation of distinct subsets of granulocytes and macrophages from bone marrow progenitor cells (13,14). Recent research suggests that GM-CSF plays pivotal roles in the progression of autoimmune and inflammatory diseases (15). GM-CSF is essential for multiple fundamental biological immunity processes, and it has been demonstrated to be necessary for the maintenance of normal pulmonic homeostasis (16,17). Accumulating evidences show that GM-CSF has a crucial part in the process of lung injury due to hyperoxic stress and it is also play a role in the wound repair (18-21). Furthermore, GM-CSF has been reported to have remarkable protective effects in bleomycin-induced pulmonary fibrosis (22). However, the role of GM-CSF in RILI is not well understood.
Interestingly, the application of GM-CSF in patients with granulocytopenia has been shown to also alleviate RILI. An analysis of the clinicopathological characteristics of 41 patients demonstrated that RILI remission was significantly correlated with GM-CSF treatment (P=0.011, Table 1). Therefore, we hypothesized that GM-CSF may act as a potential protective agent against RILI. In the present study, we aimed to explore the effect of GM-CSF in a mouse model of RILI and investigate the potential mechanism. Our study revealed that GM-CSF–/– mice were susceptible to RILI and that GM-CSF alleviated RILI through regulating inflammatory cytokines, such as interleukin (IL)-6, IL-1β, and tumor necrosis factor (TNF)-α, as well as epithelial-mesenchymal transition (EMT), thus providing a feasible strategy for the prevention and treatment of RILI.
We present the following article in accordance with the ARRIVE reporting checklist (available at http://dx.doi.org/10.21037/tlcr-20-1272).
Methods
Animals
As described in a previous study (23), 8-week-old male C57BL/6 mice [wild-type (WT) mice, Vitalriver Company, Beijing, China] and GM-CSF–/– mice (Jackson Laboratory, Bar Harbor, ME, USA), weighing approximately 16–20 g, were used to establish a mouse model of RILI. The mice were bred in The First Affiliated Hospital of Shandong First Medical University & Shandong Provincial Qianfoshan Hospital Laboratory Animal Medicine facilities under specific-pathogen-free conditions. All animal protocols were approved by The First Affiliated Hospital of Shandong First Medical University & Shandong Provincial Qianfoshan Hospital Animal Ethics Committee in accordance with the Guide for Care and Use of Laboratory Animals published by the US National Institute of Health (publication No. 96-01). Both 90 WT and 90 GM-CSF–/– mice were randomly divided into non-radiated control, radiation + saline, and radiation + GM-CSF groups [n=30 per group, 10 µg/kg/twice weekly (24)]. The mice were observed daily and weighed weekly to ensure that the interventions were well tolerated. At 1, 3, 6, 10, and 20 weeks after radiation, the mice were killed by isoflurane in halation and decapitation. The lung tissues of the mice were collected for further analysis.
Radiation
The mice were anesthetized with an intraperitoneal injection of 10% chloral hydrate (400 mg/kg; Yongda Chemical Reagent Co. Ltd., Tianjin, China) 5 minutes before radiation. The mice were radiated at a dose rate of 400 cGy/min with a single fraction of 16 Gy photons (6 MV linac, Siemens, Munich, Germany) targeted at the entire thorax (25). During the radiation procedure, the brain, neck, and body of the mice were protected with customized lead shielding. Radiation was performed at a source-to-skin distance (SSD) of 100 cm. At 1, 3, 6, 10, and 20 weeks after radiation, six mice from each group were chosen randomly to undergo examination.
Hydroxyproline (Hyp) assay
Hyp content in lung tissues from the mice was determined by colorimetric assay. At 3, 6, 10, and 20 weeks, the lung tissues (n=6 mice per group) were weighed, mechanically homogenized, and snap-frozen. Hydrolysate was analyzed with a Hyp assay kit (Jiancheng, China) in accordance with the manufacturer’s instructions. The Hyp content was calculated according to the total lung weight and was expressed as micrograms.
Histology
At 3, 6, 10, and 20 weeks after radiation, mice selected from the groups were sacrificed and their lung tissues were harvested for histological examination. The lung samples were fixed in 4% paraformaldehyde and embedded in paraffin. The samples were sectioned for staining with hematoxylin and eosin (H&E) or Masson’s trichrome stain to assess the fibrosis score according to standard protocols. The histopathological evaluation of pulmonary fibrosis was scored according to the density of Masson’s staining (Jiancheng, China) using an image J analysis program. Ten microscopic fields were randomly selected for analysis. Assessment was performed in a double-blind fashion. All images were acquired using an Olympus LCX100 Imaging System and analyzed using Imagepro plus software (Olympus Corporation, Tokyo, Japan).
RNA isolation and quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA was extracted from lung tissues cleared of blood using TRIzol reagent (Invitrogen). The concentration and purity of the RNA was determined with a spectrophotometer. Total RNA was reversed transcribed to cDNA using PrimeScript RT kit (Takara) in line with the manufacturer’s instructions. qRT-PCR was performed using SYBR Green Master Mix Kit (Takara) on the ABI ViiA 7 Real-Time PCR system (Applied Biosystems, Foster City, CA, USA). The sequences of specific primer pairs are listed in Table 2. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as the internal control for determining ΔCT values (26). The 2–ΔΔCT method was used to normalize fold increases in expression to the saline control group.
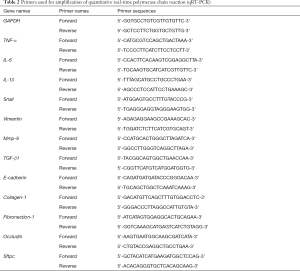
Table 2 Primers used for amplification of quantitative real-time polymerase chain reaction (qRT-PCR)
Western blot
Proteins were extracted from mouse lung tissues as previously described. RIPA buffer (Beyotime) containing protease inhibitors (Beyotime) and phosphatase inhibitors (Roche, Basel, Switzerland) was used for cell lysis. All of the lung tissues used in this analysis were cleared of blood. Total protein was estimated using a bicinchoninic acid protein assay kit (Pierce; Thermo Fisher Scientific, Inc.). The primary antibodies used included Collagen-I (cat. no. NBP2-29651; 1:500; Novus); E-cadherin (cat. no. 3195S; 1:1,000; CST), vimentin (cat. no. 5741S; 1:1,000, CST), Snail (cat. no. ab180714; 1:1,000; Abcam), and GAPDH (cat. no. ab8245; 1:3,000; Abcam). Enhanced chemiluminescence images were captured with the FluorChem E instrument (Cell Biosciences, Inc., Santa Clara, CA, USA). Quantification of the band intensity relative to GADPH was performed using Image J software (version 1.62; National Institutes of Health, Bethesda, MD, USA).
Immunohistochemistry (IHC)
Immunohistochemical characterization of the tissue was performed on 4-µm thick sections of formalin-fixed, paraffin-embedded lung tissue. The sections were blocked with goat serum and incubated with primary antibody solutions including E-cadherin (1:200), vimentin (1:200), and TGF-β1 (1:150) overnight at 4 °C. Following that, the sections were incubated with goat anti-rabbit biotinylated secondary antibody (SP-9001; Zhongshan Biotechnology, Beijing, China). Immunoactivity was detected with 3,3'-diaminobenzidine (DAB). Finally, the sections were dehydrated with ethanol gradient and mounted with rhamsan gum. Five equal-sized fields were randomly chosen. The results of IHC were analyzed blindly by two independent pathologists.
Enzyme-linked immunosorbent assay (ELISA)
After the body weight of the mice had been measured, 1 mL blood was obtained via cardiac puncture. The serum was collected and stored at –80 °C for later analysis by ELISA. Serum levels of IL-6, IL-1β, and TNF-α were determined using a double-antibody sandwich ELISA kit (DG Biotechnology, Wuhan, China). All of the procedures were performed following the manufacturer’s instructions. The sensitivity of this assay is 1 pg/mL. OD values were read at a wavelength of 450 nm by using a microplate ELISA reader. Each treatment was performed three times. The mean values of IL-6, IL-1β, and TNF-α in the different groups were compared statistically.
Patients
A total of 41 patients (28 males and 13 females) with lung or esophageal primary malignant tumors who received definitive thoracic radiation therapy between May 2015 and January 2018 were enrolled. The patients were divided into the lung cancer (LC) group (21 patients, 51.22%) and the esophageal cancer (EC) group (20 patients, 48.78%). The median age was 64 (range, 50–88) years in the LC group, and 67 (range, 48–82) years in the EC group. The median age for the entire study population was 65.5 (range, 48–88) years. This study was performed in compliance with the Declaration of Helsinki (as revised in 2013) and was approved by the Ethics Committee of The First Affiliated Hospital of Shandong First Medical University & Shandong Provincial Qianfoshan Hospital. Written informed consent was obtained from all study participants. Clinical information retrieved from the patients’ medical records included current smoking status and smoking history, alcohol consumption, and pathologic tumor node metastasis (pTNM) stage. Patients were also dichotomized according to GM-CSF or granulocyte colony-stimulating factor (G-CSF) treatment.
Statistical analysis
All quantitative experiments were performed at least three times with consistent results. All data were analyzed using SPSS software (version 22.0; SPSS, Inc., Chicago, IL, USA). All data were expressed as the mean ± standard deviation. Unpaired Student’s _t_-tests and one-way ANOVA were performed for comparisons between groups. P<0.05 was considered to indicate a statistically significant difference.
Results
GM-CSF alleviated RILI
To determine the side effects of radiation and the biological outcomes of GM-CSF on RILI, we established an experimental thoracic radiation mouse model (27,28). Phosphate-buffered saline or 10 µg/kg GM-CSF was administered to the mice twice weekly after thoracic radiation (Figure 1A). Between 8 and 9 weeks after radiation, the hair in the exposed area turned gray. In the radiation + saline group, this gray band was extremely distinct compared to that of the radiation + GM-CSF group. The radiation + GM-CSF mice exhibited a similar hair color to the non-radiated control mice (Figure 1B). At 20 weeks, lung bleeding sites in the GM-CSF-treated mice were attenuated compared to those in the vehicle-treated animals (Figure 1C).
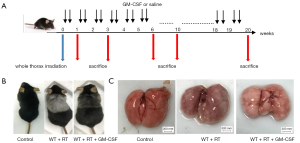
Figure 1 Establishment of a thoracic radiation mouse model. (A) Intervention with GM-CSF in the experimental mouse model of whole thorax radiation. C57BL/6 WT mice were intraperitoneally injected with GM-CSF or vehicle (n=6 per group) treatment twice weekly after radiation. Lung tissues were harvested at the indicated time points for the following analysis. (B) Pattern of color change in the hair of the radiated thoracic region at 20 weeks after radiation. (C) The lung phenotype among the control, vehicle, and GM-CSF intervention groups at 20 weeks post radiation. Scale bar represents 300 mm. GM-CSF, granulocyte-macrophage colony-stimulating factor; WT, wild-type; RT, radiation.
Next, we examined whether GM-CSF treatment could exhibit anti-fibrotic effect in RILI in mice after radiation. Pulmonary structure damage and disordered collagen deposition were found in the radiated mice. After radiation, GM-CSF treatment group displayed less alveolitis and fibrosis than the vehicle-treated mice (Figure 2A). In the GM-CSF-treated group, radiated animals showed significantly less alveolar septa damage and collagen deposition, especially at 20 weeks after radiation (Figure 2B). We also examined the content of Hyp, which is the major constituent of collagen. The Hyp content was significantly increased in saline-treated mice at 10 and 20 weeks after radiation. It was significantly decreased in GM-CSF-treated animals compared to the control mice (Figure 2C). Therefore, we demonstrated that GM-CSF treatment had a significant inhibitory effect on the occurrence both pneumonia and pulmonary fibrosis.
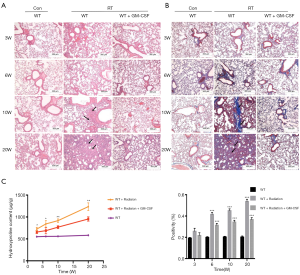
Figure 2 Pulmonary fibrosis caused by radiation was attenuated by intraperitoneal administration of GM-CSF. (A) Lung sections of WT mice from different groups were subjected to H&E staining at 3, 6, 10, and 20 weeks after radiation. Lung bleeding sites and alveolitis were showed with arrows. Representative images are shown (n=6 per group). Scale bar represents 300 μm. (B) Masson trichrome staining of collagen in lung sections from WT mice in the normal non-radiated, radiation only, and GM-CSF treatment groups at 3, 6, 10, and 20 weeks after radiation. Abnormal collagen deposition were showed with arrows. Representative images are shown (n=6 per group). Bar represents 300 μm. The down panel is quantification of Masson’s trichrome staining for collagen in lung sections from different groups. The fibrotic area is presented as percentage numbers. (C) Hyp content was measured between different groups. Error bar indicates mean ± standard deviation (n=6; *, P<0.05; **, P<0.01; ***, P<0.001, WT + radiation vs. WT + radiation + GM-CSF). GM-CSF, granulocyte-macrophage colony-stimulating factor; WT, wild-type; H&E, hematoxylin and eosin; Hyp, hydroxyproline; RT, radiation.
GM-CSF reduced inflammatory cytokine production in the RILI mouse model
Next, we investigated whether GM-CSF could exert an anti-inflammatory effect against RILI. We found that radiation significantly impaired the production of key proinflammatory cytokines, such as IL-6 and TNF-α. Profibrotic cytokines in serum such as IL-1β closely correlated with the occurrence of lung fibrosis. GM-CSF treatment significantly decreased proinflammatory and profibrotic levels in the serum of radiated C57BL/6 mice compared with those in the vehicle group (Figure 3A). GM-CSF treatment also remarkably reduced the expression of TNF-α, IL-1β, and IL-6 from 3 weeks after radiation (Figure 3B).
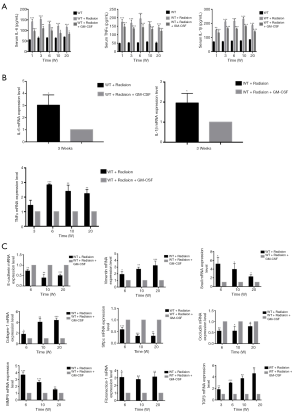
Figure 3 Effect of GM-CSF on inflammatory cytokines in the radiation mouse model. (A) ELISA analysis of the serum levels of IL-6, TNF-α, and IL-1β in mice from different groups in the radiation-induced injury mouse model at 1, 3, 6, 10, and 20 weeks post radiation. (B) qRT-PCR analysis of IL-6, TNF-α, and IL-1β mRNA levels in the lung tissues of mice from different groups at different time points (n=6 per group). (C) qRT-PCR analysis of EMT-related markers. The target gene mRNA levels were normalized to GAPDH (n=6 per group). *, P<0.05; **, P<0.01; ***, P<0.001. GM-CSF, granulocyte-macrophage colony-stimulating factor; ELISA, enzyme-linked immunosorbent assay; qRT-PCR, quantitative real-time polymerase chain reaction; EMT, epithelial-mesenchymal transition; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; WT, wild-type.
GM-CSF decreased EMT in the radiated mouse model
The messenger RNA (mRNA) levels of interstitial markers (Vimentin, Snail) and fibrosis markers (collagen-1 and fibronectin-1) were increased in the radiated mouse model. The epithelial marker (E-cadherin), surfactant protein C (Sftpc) and a tight junction marker (Occludin) were progressively down-regulated at 6, 10, and 20 weeks after radiation. In the GM-CSF treatment group, the upregulation of mesenchymal markers and downregulation of epithelial markers were significantly inhibited (Figure 3C). To further verify whether GM-CSF treatment affects the EMT pathway, the expression of key molecules in the EMT pathway was detected in mouse lung tissues. The results showed that compared with the control group, radiation significantly upregulated the expression of collagen-1 as well as that of vimentin and Snail, but downregulated the expression of E-cadherin. In contrast, GM-CSF treatment decreased the expression of collagen-1 as well as that of vimentin and Snail, but increased E-cadherin expression. We confirmed that GM-CSF inhibited the EMT process at the protein level at 20 weeks after radiation (Figure 4A). In this study, TGF-β1 mRNA expression was found in the lungs of mice by qRT-PCR (Figure 3C) and was further confirmed by IHC staining. The protein levels of TGF-β and the interstitial marker Vimentin were up-regulated after 20 weeks after radiation but were significantly inhibited by GM-CSF treatment. However, E-cadherin expression showed contrary tendency (Figure 4B). These results indicated that GM-CSF treatment significantly reduced the expression levels of markers related to EMT and fibrosis.
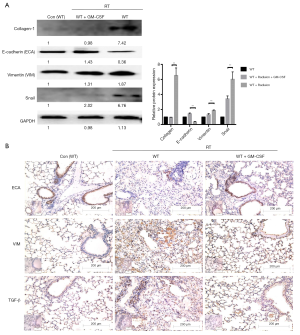
Figure 4 Effect of GM-CSF on mesenchymal markers. (A) Lung tissues were subjected to western blot analysis to detect the expression of collagen-1, ECA, VIM, and Snail at 20 weeks after radiation. GAPDH was used as an internal reference for relative quantification. (B) Relative protein expression of ECA, VIM, and TGF-β was measured by IHC. Scale bars represent 200 μm (20×) and 100 μm (40×). Error bar indicates mean ± standard deviation (n=6; **, P<0.01). GM-CSF, granulocyte-macrophage colony-stimulating factor; ECA, E-cadherin; VIM, vimentin; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; IHC, immunohistochemistry; WT, wild-type; con, control.
GM-CSF–/– mice had higher susceptibility to RILI than WT mice
In order to further examine the role of GM-CSF in RILI, we subjected GM-CSF knockout mice to radiation (Figure 5A). At 10 weeks after radiation, the radiated lungs of GM-CSF–/– mice with vehicle treatment exhibited severe pulmonary congestion compared with those of mice in other groups (Figure 5B). Damage to the pulmonary alveolar architecture and abnormal collagen deposition were observed in the radiated GM-CSF–/– mice. The GM-CSF–/– mice in the vehicle treatment groups had a more severe phenotype, whereas the RILI was significantly relieved in the GM-CSF treatment groups (Figure 5C). The results of Masson staining suggested that GM-CSF–/– mice not treated with GM-CSF were more susceptible to radiation-induced lung fibrosis (Figure 5D). In accordance with these observations, the Hyp content in the vehicle group was also remarkably increased at 10 weeks, whereas GM-CSF treatment significantly reduced the Hyp levels in the lungs of radiated GM-CSF–/– mice (Figure 5E).
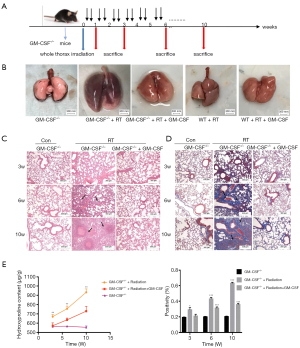
Figure 5 Effect of GM-CSF on radiation-induced pneumonia and collagen deposition in GM-CSF–/– mice. (A) GM-CSF–/– mice were radiated with a single fraction of 16-Gy photons targeted at the entire thorax. GM-CSF–/– mice were intraperitoneally injected with 10 μg/kg GM-CSF (n=6 per group) twice weekly after radiation therapy, while the matched group received the same volume of vehicle instead. (B) GM-CSF reduced radiation-induced hyperemia and edema in pulmonary tissue at 10 weeks post-radiation. Representative images of GM-CSF–/– mouse lung tissues are shown from the control group, single radiation group, and GM-CSF-treated group. (C,D) GM-CSF mitigated radiation-induced pneumonia and collagen deposition in lung tissues. The arrows showed the damage to the pulmonary alveolar architecture and abnormal collagen deposition. Representative images of H&E and Masson staining of lung tissue sections at 3–10 weeks post radiation (bar represents 300 μm). The down panel of (D) is quantification of collagen deposition in slides from lung tissues. Values are given as mean ± standard deviation (n=6), *, P<0.05 and ***, P<0.001 versus single radiation group. (E) Lung tissues of GM-CSF–/– mice from 3, 6, 10 weeks after radiation was analyzed for Hyp content. Error bar indicates mean ± standard deviation (n=6; *, P<0.05; **, P<0.01; ***, P<0.001, GM-CSF–/– + radiation vs. GM-CSF–/– + radiation + GM-CSF). GM-CSF, granulocyte-macrophage colony-stimulating factor; H&E, hematoxylin and eosin; Hyp, hydroxyproline.
Radiation considerably increased the serum levels of TNF-α and IL-6 in GM-CSF–/– mice as compared to the controls. The level of IL-6 was elevated at 1, 3 weeks but was decreased at 6 weeks after radiation in the GM-CSF–/– mice. GM-CSF treatment significantly reduced the levels of these inflammatory cytokines in the GM-CSF–/– mice. At 10 weeks, the level of IL-1β in the serum of radiated mice without GM-CSF treatment was consistently higher than that in other groups. GM-CSF treatment also significantly decreased profibrotic cytokine production in the GM-CSF–/– mice (Figure 6A). Furthermore, GM-CSF treatment reduced the expression of cytokine-related genes, which is consistent with our previous finding and is shown in Figure 6B.
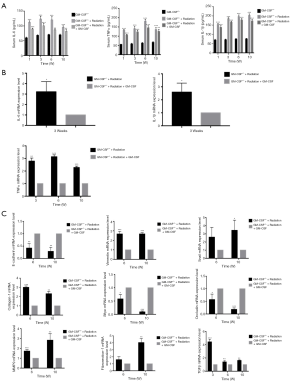
Figure 6 Effect of GM-CSF on lung inflammatory cytokines in radiated GM-CSF–/– mice. (A) GM-CSF reduced the levels of profibrotic cytokines in GM-CSF–/– mice with lung injury after radiation. Cytokine concentration in the serum of radiated mice with different treatments. (B) Relative mRNA levels of inflammatory markers including IL-6, IL-1β, and TNF-α in the lungs were calculated in different groups (n=6 per group). (C) Relative mRNA levels of EMT-related markers including TGF-β1, MMP-9, epithelial cell markers including Occuludin, E-cadherin, and SFTPC, and mesenchymal cell markers including vimentin, were calculated in different groups (n=6 per group). *, P<0.05; **, P<0.01; ***, P<0.001. GM-CSF, granulocyte-macrophage colony-stimulating factor; EMT, epithelial-mesenchymal transition.
To evaluate whether EMT could also be a potential mechanism in the attenuation of RILI through GM-CSF treatment we detected the expression of epithelial and mesenchymal markers in GM-CSF–/– mice at 10 weeks after radiation. As shown in Figure 6C, the radiation + saline groups showed significantly enhanced expression of the mesenchymal markers (vimentin and Snail), and the fibrosis markers (collagen-1 and fibronectin-1). However, the mRNA expression of those markers was remarkably reduced in the GM-CSF treatment group. At the same time, E-cadherin was significantly repressed in the vehicle group after radiation. Matrix metallopeptidase-9 (MMP-9) has been identified as a hallmark of EMT and fibrosis (29). Our data showed that the mice that received radiation with vehicle treatment showed a similar significant increase in MMP-9 mRNA expression, while treatment with GM-CSF inhibited the expression of MMP-9 in GM-CSF–/– mice.
Furthermore, radiation significantly up-regulated the protein levels of vimentin, while markedly down-regulating E-cadherin expression in GM-CSF–/– mice. Snail has also been reported to suppress E-cadherin expression and drive EMT (30). Radiation significantly increased the expression of Snail in the groups without GM-CSF treatment. We also found that the fibrosis marker collagen-1 was significantly over-expressed in the lungs of the vehicle treatment groups after radiation. In contrast, a low expression of collagen-1 was observed in WT mice in the GM-CSF treatment groups. Interestingly, its expression was hardly detected in the radiated GM-CSF–/– mice treated with GM-CSF (Figure 7A).
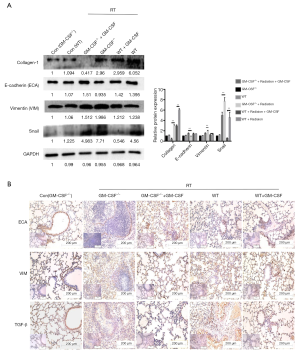
Figure 7 Effect of GM-CSF on EMT in radiated pulmonary fibrosis. (A) Western blot analysis of EMT markers in radiated lung tissues with/without GM-CSF treatment at 10 weeks. Quantification analysis of raw density of Western blot figures are measured in different groups. (B) GM-CSF reduced the expression levels of ECA, VIM and TGF-β1. Representative images of IHC staining of ECA, VIM, and TGF-β1 are assessed in lung tissues from different groups at 10 weeks. Error bar indicates mean ± standard deviation (n=6; **, P<0.01). GM-CSF, granulocyte-macrophage colony-stimulating factor; EMT, epithelial-mesenchymal transition; ECA, E-cadherin; VIM, vimentin; IHC, immunohistochemistry.
At 10 weeks post radiation, the level of E-cadherin was found to be reduced in the radiation + saline group compared to the radiation + GM-CSF groups in GM-CSF–/– mice. In contrast, the radiation-induced increase in vimentin expression was further augmented in the vehicle group compared to in the GM-CSF groups. IHC revealed that TGF-β expression was stronger in the lungs of the radiated group with vehicle treatment than in the lungs of the controls. GM-CSF treatment significantly decreased the expression of TGF-β in 16 Gy-radiated lung tissues (Figure 7B). These results revealed that GM-CSF treatment after radiation administration could dramatically mitigate radiation-induced lung fibrosis via inhibition of EMT.
GM-CSF administration alleviated RILI in several patients
In the LC group, the number of patients with squamous cell carcinoma, adenocarcinoma, and small cell LC was 8, 3, and 9, respectively. Almost all of the EC patients were the squamous cell carcinoma histologic type. Eleven patients in the LC group and 11 patients in the EC group had stage III cancer, and 10 LC patients and 9 EC patients had stage IV cancer. In the LC group, 14 patients accepted concurrent chemoradiotherapy and 7 patients accepted radiotherapy only; in the EC group, these numbers were 13 and 7, respectively. Overall, 38 patients suffered from cough, fever, tachycardia, cyanosis, and exercise intolerance during the 5 months of follow-up after radiotherapy. In terms of severe adverse effects, three patients developed multiple bacterial infections and received antibiotic therapy. Computed tomographic images showed regions of high-density consolidation that expanded gradually during post-intensity modulated radiation therapy (IMRT) follow-up. The morbidity of RILI was graded according to the National Cancer Institute’s Common Terminology Criteria for Adverse Events version 4 (CTCAE v4) for pneumonitis. In RILI patients, remission means that the patients developed clinical radiation pneumonitis of Grade ≥2 gradually decline until Grades 0–1. Further analysis of the clinicopathological characteristics of the 41 patients showed that RILI remission was significantly correlated with GM-CSF treatment (P=0.011, Table 1).
Discussion
RILI is a common and serious complication of radiotherapy for chest tumors (31). Approximately 13–37% of LC patients who receive radiation therapy are likely to develop RILI, and some patients develop late-phase fibrosis (32). Currently, there is no effective treatment for RILI. In the present study, we performed a detailed analysis of the pathological phenotype underlying RILI by establishing a mouse model of thoracic radiation, to mimic human chronic pneumonitis after radiation therapy. Radiation-induced pneumonitis and fibrosis have been repeatedly documented in humans and experimental animals (3,33,34). Zhou et al. (35) evaluated mice 24 weeks after they were administered single-dose or fractionated thoracic radiation by quantitatively assessing lung variations using computed tomography. They found that 14.55±0.34 Gy induced lung fibrosis in 50% of the mice (ED50). C57BL/6 mice were chosen for the model due to their proneness to fibrosis compared with other strains, such as BALB/c mice, which express various cytokines or proteases/anti-proteases and are considered to be fibrosis resistant (36,37). Therefore, we used 16 Gy-radiated C57BL/6 mice for our fibrosis model.
A number of therapeutic strategies that effectively reduce inflammatory damage and suppress the fibrotic process have been explored in previous studies. Jiang et al. (38) reported that adipose-derived mesenchymal stromal cells had a therapeutic benefit for acute RILI and mediated anti-inflammatory and anti-fibrotic effects. The TGF-β receptor I inhibitor LY2109761 was shown to suppress radiation-induced inflammation and achieved its anti-fibrotic effect by suppressing radiation-induced proinflammatory, proangiogenic, and profibrotic signals (39). However, no effective method exists for the clinical treatment of RILI. In the present research, our data provide compelling evidence to support the application of GM-CSF as a therapeutic strategy for RILI patients by revealing several novel findings. First, RILI was alleviated in several patients following treatment with GM-CSF. Secondly, thoracic radiation triggered the upregulation of inflammatory cytokine expression in pulmonary tissue and progressive accumulation of inflammatory cytokines in the serum. Radiation led to lung fibrosis in the mouse model, which was accompanied by an increased expression of profibrotic factors such as TGF-β and mesenchymal markers such as vimentin. Thirdly, GM-CSF–/– mice were sensitive to lung injury; compared to WT mice, the GM-CSF–/– mice exhibited more aggravated lung fibrosis after exposure to radiation. The knockout mice showed progressive increases in the levels of inflammatory cytokines, as well as the accumulation of TGF-β1 and vimentin. Fourthly, and most importantly, all GM-CSF-treated groups demonstrated benefits, with RILI attenuated or prevented (as assessed by histology). However, prospective, controlled, large-sample size clinical trials are needed to further confirm our findings. Furthermore, in clinical practice, radiotherapy is not used to radiate both lungs simultaneously. It should also be pointed out that the difference in radiation volume between the thoracic radiation mouse model and clinical patients in this study was a limitation. Herein, we provide results that further illustrate the effect of GM-CSF in fibrogenesis. We indicated that the anti-fibrotic role of GM-CSF treatment may be a result of anti-inflammatory responses and the dysregulation of EMT.
GM-CSF, which is a recombinant growth factor, has recently been approved by the U.S. Food and Drug Administration as a therapy for granulocytopenia after radiation- or chemotherapy-induced myelosuppression (40). Previous studies indicated that GM-CSF could be abundantly secreted by alveolar epithelial cells, interstitial cells, and macrophages after inflammatory stimulation (41). GM-CSF is very importment for the maturation of alveolar macrophage and mononuclear phagocytes (42,43). Many studies have shown that alveolar macrophages are essential for initial defense against pathogens and the surfactant homeostasis in lungs (17,44). GM-CSF was found to be over-expressed in radiated lungs and reduced pulmonary injury by stimulating the protective granulocytes and monocytes in situ, which might be sites of the widespread infection (45). GM-CSF is also related to a rare lung pathology characteristic of pulmonary alveolar proteinosis (46,47), which is a characteristic of extensive pulmonary fibrosis (48,49). Recent studies using GM-CSF-deficient mice have clearly demonstrated that GM-CSF enhances the pulmonary host defense and is critically involved in the maintenance of normal macrophage function (50). Moore et al. (22) reported that GM-CSF-deficient mice showed genetically caused fibrogenesis in bleomycin-related lung fibrosis. Another study revealed that alveolar GM-CSF acts as a local mediator of macrophage maturation (50). However, exogenous expression of GM-CSF in rats via adenovirus-mediated gene transfection was found to result in pulmonary fibrosis (51). Recent reports have described that GM-CSF–/– mice exhibit enhanced susceptibility to pneumonia from two specific pathogenic agents, Pneumocystis carinii (52) and group B streptococcus (53). However, in a randomized phase II trial, GM-CSF did not lower the rate of ventilator usage among patients with acute pulmonary injury/acute respiratory distress syndrome (54). These inconsistent outcomes could be explained by the different sample sizes, drug doses, methods of drug administration. In particular, the observation window for treatment in the phase II trial emphasizes the first 7 days after the onset of acute lung injury/acute respiratory distress syndrome, while our research emphasizes the role of GM-CSF in RILI after 7 days. Further research in the inflammatory or fibrotic activities of lung macrophages is necessary to better evaluate if there are other unusual factors in GM-CSF-deficient mice conduce to the severity of RILI.
It is now clear that innate and acquired immune responses are integral parts of the differentiation and activation of fibroblasts, which can lead to a local environment enriched in chemokines as well as proinflammatory cytokines, fibrogenic cytokines, and growth factors (55). Radiation of the lungs can induce the accumulation of innate inflammatory cells, including macrophages, neutrophils, mast cells, and eosinophils, and elevate the secretion of cytokines including TNF-α, IL-1β, IL-6, and TGF-β, during the innate immune stage (9,56). Previous studies have revealed that overexpression of TNF-α or IL-1β in the lungs results in procedural lung fibrosis (57,58). Similar to TNF-α, IL-1β also has several biological functions that might facilitate fibrosis. IL-1β and TNF-α can also increase the expression of IL-6, which has critical activity in fibroblasts (59-61). These functions may include leukocyte infiltration, the expression of proinflammatory regulators that exacerbate parenchymal cell injury, and the production of the key profibrotic growth factor TGF-β. Wang et al. (62) showed that combination of mean lung dose (MLD), IL-8 level before radiation, and the ratio of TGF-β1 level two weeks vs before radiation provided a more accurate model to be correlated with an increased risk of RILI2 (Grade ≥2 RILI) compared with MLD alone. In our research, we choosed those representative and classical cytokines to prove that GM-CSF alleviated RILI and inflammation in both WT and GM-CSF–/– mice. So far, our existing data did not show that our cytokine profiling could predict symptomatic radiation lung injury as predict markers. Furthermore, recent studies have proposed that EMT might play a key role in lung fibrosis (63,64), possibly through a TGF-β-mediated mechanism (65-67). During lung fibrosis, up to 20% cells underwent EMT in the mouse model or human samples (68,69). TGF-β is a multifunctional regulator of cell growth, EMT, and differentiation in response to injuries (70). The expression of TGF-β induced by radiation has been reported as part of RILI (71-73). EMT is characterized by a reduction in E-cadherin expression and an increase in vimentin expression. Snail, another critical factor involved in EMT, has also been discovered to repress the transcription of the CDH1 gene (encoding E-cadherin) (74,75). Choi et al. showed that transcription factor Hypoxia-inducible factor-1α (HIF-1α) could influence TGF-β-R1/Smad3 signaling pathway altering EMT (76). Si-HIF-1α could inhibit radiation-induced EMT through downregulating TGF-β-R1/Smad3 signaling (77,78). Moreover, transcription factors e.g. Snail and Twist are considered as pivotal regulators of EMT, which repressing E-cadherin and increasing the mesenchymal transcriptomes (79). We discovered that radiated mice treated with GM-CSF had decreased lung inflammation and reduced mRNA levels of TGF-β, TNF-α, IL-1β, and IL-6. This study is the first to confirm that GM-CSF participates in TGF-β-induced EMT during radiation-induced pulmonary fibrosis. In this study, treatment with GM-CSF dramatically elevated the expression of epithelial markers while reducing that of interstitial markers in GM-CSF-deficient mice compared to WT mice. We also found that the expression of Snail was much higher in the GM-CSF–/– mice than in the WT mice after exposure to radiation, which shows that GM-CSF can affect the expression of Snail at the transcriptional and translational levels. These findings demonstrate that GM-CSF exerts a protective effect against pulmonary fibrosis by decreasing the incidence of EMT. Recently, it has been discovered that fibroblasts may derive from multiple formats, such as resident mesenchymal cells, bone marrow stem cells and epithelial cells (80). After exposure to fibrosis-inducing doses of radiation, a time- and dose-dependent senescence and pneumocyte depletion of airway epithelial cell II (AEC II) cells has frequently been observed (81). AEC II senescence is hypothesized to be a major contributor to less or ineffective ventilation, leading to fibroproliferation and epithelial stress. Myofibroblasts are likely originated from bone marrow (BM)—those fibroblast progenitor cells that are crucial in the fibrogenesis (82). Ligand stromal cell-derived factor 1 (SDF-1/CXCL12) could active the chemokine receptor CXCR4, which is expressed by the BM- originated fibrocytes. The activation of CXCR4 may be important in the development of RILF. Therefore, the CXCR4/CXCL12 axis play a critical role in recruiting BM-derived precursors that differentiate into the fibroblasts that cause RILF. Furthermore, thorax radiation not only affects the function of macrophages in the lung tissue but also leads to various immune cells recruitment, including basophils, lymphocytes, monocytic cells and neutrophils (83). The recruitment of immune cells is related to characteristic changes in local and systemic cytokine and chemokine expression. In the future, additional studies are necessary to define how the various immune cells interact and coordinate in multiple formats to reduce the side effects of LC radiotherapy.
In conclusion, our study provides evidence that GM-CSF protects against the development of RILI. Thus, GM-CSF may be a potential therapeutic reagent for preventing and treating RILI.
Acknowledgments
Funding: This study was supported by grants from the National Natural Science Foundation of China (81703033 and 81672974) and the Fundamental Research Funds of Shandong University.
Reporting Checklist: The authors have completed the ARRIVE reporting checklist. Available at http://dx.doi.org/10.21037/tlcr-20-1272
Data Sharing Statement: Available at http://dx.doi.org/10.21037/tlcr-20-1272
Conflicts of Interest: All authors have completed the ICMJE uniform disclosure form (available at http://dx.doi.org/10.21037/tlcr-20-1272). The authors have no conflicts of interest to declare.
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All animal protocols were approved by The First Affiliated Hospital of Shandong First Medical University & Shandong Provincial Qianfoshan Hospital Animal Ethics Committee in accordance with the Guide for Care and Use of Laboratory Animals published by the US National Institute of Health (publication No. 96-01). This study was performed in compliance with the Declaration of Helsinki (as revised in 2013) and was approved by the Ethics Committee of The First Affiliated Hospital of Shandong First Medical University & Shandong Provincial Qianfoshan Hospital. Written informed consent was obtained from all study participants.
Open Access Statement: This is an Open Access article distributed in accordance with the Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License (CC BY-NC-ND 4.0), which permits the non-commercial replication and distribution of the article with the strict proviso that no changes or edits are made and the original work is properly cited (including links to both the formal publication through the relevant DOI and the license). See: https://creativecommons.org/licenses/by-nc-nd/4.0/.
References
- Killock D. Lung cancer: thoracic radiotherapy improves survival in small-cell lung cancer. Nat Rev Clin Oncol 2014;11:623. [Crossref] [PubMed]
- Choi JI, Simone CB 2nd. The current and future faces of stereotactic body radiation therapy for thoracic malignancies. Transl Lung Cancer Res 2019;8:1-4. [Crossref] [PubMed]
- Ding NH, Li JJ, Sun LQ. Molecular mechanisms and treatment of radiation-induced lung fibrosis. Curr Drug Targets 2013;14:1347-56. [Crossref] [PubMed]
- Graves PR, Siddiqui F, Anscher MS, et al. Radiation pulmonary toxicity: from mechanisms to management. Semin Radiat Oncol 2010;20:201-7. [Crossref] [PubMed]
- Kong FM, Zhao L, Hayman JA. The role of radiation therapy in thoracic tumors. Hematol Oncol Clin North Am 2006;20:363-400. [Crossref] [PubMed]
- Tian S, Switchenko JM, Cassidy RJ, et al. Predictors of pneumonitis-free survival following lung stereotactic body radiation therapy. Transl Lung Cancer Res 2019;8:15-23. [Crossref] [PubMed]
- Zheng L, Zhu Q, Xu C, et al. Glycyrrhizin mitigates radiation-induced acute lung injury by inhibiting the HMGB1/TLR4 signalling pathway. J Cell Mol Med 2020;24:214-26. [Crossref] [PubMed]
- Yarnold J, Brotons MC. Pathogenetic mechanisms in radiation fibrosis. Radiother Oncol 2010;97:149-61. [Crossref] [PubMed]
- Bickelhaupt S, Erbel C, Timke C, et al. Effects of CTGF blockade on attenuation and reversal of radiation-induced pulmonary fibrosis. J Natl Cancer Inst 2017. [Crossref] [PubMed]
- Marks LB, Yu X, Vujaskovic Z, et al. Radiation-induced lung injury. Semin Radiat Oncol 2003;13:333-45. [Crossref] [PubMed]
- Koukourakis MI. Radiation damage and radioprotectants: new concepts in the era of molecular medicine. Br J Radiol 2012;85:313-30. [Crossref] [PubMed]
- Arellano M, Lonial S. Clinical uses of GM-CSF, a critical appraisal and update. Biologics 2008;2:13-27. [Crossref] [PubMed]
- Hamilton JA, Achuthan A. Colony stimulating factors and myeloid cell biology in health and disease. Trends Immunol 2013;34:81-9. [Crossref] [PubMed]
- Metcalf D. Hematopoietic cytokines. Blood 2008;111:485-91. [Crossref] [PubMed]
- Shiomi A, Usui T. Pivotal roles of GM-CSF in autoimmunity and inflammation. Mediators Inflamm 2015;2015:568543. [Crossref] [PubMed]
- Steinwede K, Tempelhof O, Bolte K, et al. Local delivery of GM-CSF protects mice from lethal pneumococcal pneumonia. J Immunol 2011;187:5346-56. [Crossref] [PubMed]
- Trapnell BC, Whitsett JA. Gm-CSF regulates pulmonary surfactant homeostasis and alveolar macrophage-mediated innate host defense. Annu Rev Physiol 2002;64:775-802. [Crossref] [PubMed]
- Mann A, Breuhahn K, Schirmacher P, et al. Keratinocyte-derived granulocyte-macrophage colony stimulating factor accelerates wound healing: Stimulation of keratinocyte proliferation, granulation tissue formation, and vascularization. J Invest Dermatol 2001;117:1382-90. [Crossref] [PubMed]
- Yuan L, Minghua C, Feifei D, et al. Study of the use of recombinant human granulocyte-macrophage colony-stimulating factor hydrogel externally to treat residual wounds of extensive deep partial-thickness burn. Burns 2015;41:1086-91. [Crossref] [PubMed]
- Baleeiro CE, Christensen PJ, Morris SB, et al. GM-CSF and the impaired pulmonary innate immune response following hyperoxic stress. Am J Physiol Lung Cell Mol Physiol 2006;291:L1246-55. [Crossref] [PubMed]
- Fang Y, Shen J, Yao M, et al. Granulocyte-macrophage colony-stimulating factor enhances wound healing in diabetes via upregulation of proinflammatory cytokines. Br J Dermatol 2010;162:478-86. [Crossref] [PubMed]
- Moore BB, Coffey MJ, Christensen P, et al. GM-CSF regulates bleomycin-induced pulmonary fibrosis via a prostaglandin-dependent mechanism. J Immunol 2000;165:4032-9. [Crossref] [PubMed]
- Huffman JA, Hull WM, Dranoff G, et al. Pulmonary epithelial cell expression of GM-CSF corrects the alveolar proteinosis in GM-CSF-deficient mice. J Clin Invest 1996;97:649-55. [Crossref] [PubMed]
- Romero F, Shah D, Duong M, et al. A pneumocyte-macrophage paracrine lipid axis drives the lung toward fibrosis. Am J Respir Cell Mol Biol 2015;53:74-86. [Crossref] [PubMed]
- Dabjan MB, Buck CM, Jackson IL, et al. A survey of changing trends in modelling radiation lung injury in mice: bringing out the good, the bad, and the uncertain. Lab Invest 2016;96:936-49. [Crossref] [PubMed]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 2001;29:e45. [Crossref] [PubMed]
- Haston CK, Travis EL. Murine susceptibility to radiation-induced pulmonary fibrosis is influenced by a genetic factor implicated in susceptibility to bleomycin-induced pulmonary fibrosis. Cancer Res 1997;57:5286-91. [PubMed]
- Tucker SL, Travis EL. Time course for the hazard of radiation-induced pneumonitis death in mice. Int J Radiat Biol 1992;62:627-39. [Crossref] [PubMed]
- Zucker S, Hymowitz M, Conner C, et al. Measurement of matrix metalloproteinases and tissue inhibitors of metalloproteinases in blood and tissues. Clinical and experimental applications. Ann N Y Acad Sci 1999;878:212-27. [Crossref] [PubMed]
- Yanagawa J, Walser TC, Zhu LX, et al. Snail promotes CXCR2 ligand-dependent tumor progression in non-small cell lung carcinoma. Clin Cancer Res 2009;15:6820-9. [Crossref] [PubMed]
- Vujaskovic Z, Marks LB, Anscher MS. The physical parameters and molecular events associated with radiation-induced lung toxicity. Semin Radiat Oncol 2000;10:296-307. [Crossref] [PubMed]
- Rodrigues G, Lock M, D'Souza D, et al. Prediction of radiation pneumonitis by dose - volume histogram parameters in lung cancer--a systematic review. Radiother Oncol 2004;71:127-38. [Crossref] [PubMed]
- Benveniste MF, Welsh J, Godoy MC, et al. New era of radiotherapy: an update in radiation-induced lung disease. Clin Radiol 2013;68:e275-90. [Crossref] [PubMed]
- Lee SJ, Yi CO, Heo RW, et al. Clarithromycin attenuates radiation-induced lung injury in mice. PLoS One 2015;10:e0131671. [Crossref] [PubMed]
- Zhou C, Jones B, Moustafa M, et al. Quantitative assessment of radiation dose and fractionation effects on normal tissue by utilizing a novel lung fibrosis index model. Radiat Oncol 2017;12:172. [Crossref] [PubMed]
- Phan SH, Kunkel SL. Lung cytokine production in bleomycin-induced pulmonary fibrosis. Exp Lung Res 1992;18:29-43. [Crossref] [PubMed]
- Schrier DJ, Kunkel RG, Phan SH. The role of strain variation in murine bleomycin-induced pulmonary fibrosis. Am Rev Respir Dis 1983;127:63-6. [Crossref] [PubMed]
- Jiang X, Jiang X, Qu C, et al. Intravenous delivery of adipose-derived mesenchymal stromal cells attenuates acute radiation-induced lung injury in rats. Cytotherapy 2015;17:560-70. [Crossref] [PubMed]
- Flechsig P, Dadrich M, Bickelhaupt S, et al. LY2109761 attenuates radiation-induced pulmonary murine fibrosis via reversal of TGF-beta and BMP-associated proinflammatory and proangiogenic signals. Clin Cancer Res 2012;18:3616-27. [Crossref] [PubMed]
- Singh VK, Seed TM. A review of radiation countermeasures focusing on injury-specific medicinals and regulatory approval status: part I. Radiation sub-syndromes, animal models and FDA-approved countermeasures. Int J Radiat Biol 2017;93:851-69. [Crossref] [PubMed]
- Burgess AW, Camakaris J, Metcalf D. Purification and properties of colony-stimulating factor from mouse lung-conditioned medium. J Biol Chem 1977;252:1998-2003. [PubMed]
- Shi Y, Liu CH, Roberts AI, et al. Granulocyte-macrophage colony-stimulating factor (GM-CSF) and T-cell responses: what we do and don’t know. Cell Res 2006;16:126-33. [Crossref] [PubMed]
- Min L, Mohammad Isa SA, Shuai W, et al. Cutting edge: granulocyte-macrophage colony-stimulating factor is the major CD8+ T cell-derived licensing factor for dendritic cell activation. J Immunol 2010;184:4625-9. [Crossref] [PubMed]
- Fels AO, Cohn ZA. The alveolar macrophage. J Appl Physiol (1985) 1986;60:353-69. [Crossref] [PubMed]
- Fedorocko P, Egyed A, Vacek A. Irradiation induces increased production of haemopoietic and proinflammatory cytokines in the mouse lung. Int J Radiat Biol 2002;78:305-13. [Crossref] [PubMed]
- Reed JA, Whitsett JA. Granulocyte-macrophage colony-stimulating factor and pulmonary surfactant homeostasis. Proc Assoc Am Physicians 1998;110:321-32. [PubMed]
- Luisetti M. Role for granulocyte/macrophage colony-stimulating factor in pulmonary surfactant homeostasis. Monaldi Arch Chest Dis 1997;52:176-7. [PubMed]
- Ono M, Saito R, Tominaga J, et al. Pathological features of explant lungs with fibrosis in autoimmune pulmonary alveolar proteinosis. Respirol Case Rep 2017;5:e00255. [Crossref] [PubMed]
- Holbert JM, Costello P, Li W, et al. CT features of pulmonary alveolar proteinosis. AJR Am J Roentgenol 2001;176:1287-94. [Crossref] [PubMed]
- Paine R 3rd, Morris SB, Jin H, et al. Impaired functional activity of alveolar macrophages from GM-CSF-deficient mice. Am J Physiol Lung Cell Mol Physiol 2001;281:L1210-8. [Crossref] [PubMed]
- Xing Z, Ohkawara Y, Jordana M, et al. Transfer of granulocyte-macrophage colony-stimulating factor gene to rat lung induces eosinophilia, monocytosis, and fibrotic reactions. J Clin Invest 1996;97:1102-10. [Crossref] [PubMed]
- Paine R 3rd, Preston AM, Wilcoxen S, et al. Granulocyte-macrophage colony-stimulating factor in the innate immune response to Pneumocystis carinii pneumonia in mice. J Immunol 2000;164:2602-9. [Crossref] [PubMed]
- LeVine AM, Reed JA, Kurak KE, et al. GM-CSF-deficient mice are susceptible to pulmonary group B streptococcal infection. J Clin Invest 1999;103:563-9. [Crossref] [PubMed]
- Paine R 3rd, Standiford TJ, Dechert RE, et al. A randomized trial of recombinant human granulocyte-macrophage colony stimulating factor for patients with acute lung injury. Crit Care Med 2012;40:90-7. [Crossref] [PubMed]
- Wynn TA. Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J Clin Invest 2007;117:524-9. [Crossref] [PubMed]
- Meziani L, Mondini M, Petit B, et al. CSF1R inhibition prevents radiation pulmonary fibrosis by depletion of interstitial macrophages. Eur Respir J 2018;51:1702120. [Crossref] [PubMed]
- Miyazaki Y, Araki K, Vesin C, et al. Expression of a tumor necrosis factor-alpha transgene in murine lung causes lymphocytic and fibrosing alveolitis. A mouse model of progressive pulmonary fibrosis. J Clin Invest 1995;96:250-9. [Crossref] [PubMed]
- Kolb M, Margetts PJ, Anthony DC, et al. Transient expression of IL-1beta induces acute lung injury and chronic repair leading to pulmonary fibrosis. J Clin Invest 2001;107:1529-36. [Crossref] [PubMed]
- Yang J, Ding C, Dai X, et al. Soluble dietary fiber ameliorates radiation-induced intestinal epithelial-to-mesenchymal transition and fibrosis. JPEN J Parenter Enteral Nutr 2017;41:1399-410. [Crossref] [PubMed]
- Choi YJ, Baek GY, Park HR, et al. Smad2/3-regulated expression of DLX2 is associated with radiation-induced epithelial-mesenchymal transition and radioresistance of A549 and MDA-MB-231 human cancer cell lines. PLoS One 2016;11:e0147343. [Crossref] [PubMed]
- Liu W, Huang YJ, Liu C, et al. Inhibition of TBK1 attenuates radiation-induced epithelial-mesenchymal transition of A549 human lung cancer cells via activation of GSK-3beta and repression of ZEB1. Lab Invest 2014;94:362-70. [Crossref] [PubMed]
- Wang S, Campbell J, Stenmark MH, et al. Plasma levels of IL-8 and TGF-beta1 predict radiation-induced lung toxicity in non-small cell lung cancer: a validation study. Int J Radiat Oncol Biol Phys 2017;98:615-21. [Crossref] [PubMed]
- Grabias BM, Konstantopoulos K. Epithelial-mesenchymal transition and fibrosis are mutually exclusive reponses in shear-activated proximal tubular epithelial cells. FASEB J 2012;26:4131-41. [Crossref] [PubMed]
- Zhou J, Wu P, Sun H, et al. Lung tissue extracellular matrix-derived hydrogels protect against radiation-induced lung injury by suppressing epithelial-mesenchymal transition. J Cell Physiol 2020;235:2377-88. [Crossref] [PubMed]
- Fan JM, Huang XR, Ng YY, et al. Interleukin-1 induces tubular epithelial-myofibroblast transdifferentiation through a transforming growth factor-beta1-dependent mechanism in vitro. Am J Kidney Dis 2001;37:820-31. [Crossref] [PubMed]
- Lei X, Ma N, Liang Y, et al. Glucosamine protects against radiation-induced lung injury via inhibition of epithelial-mesenchymal transition. J Cell Mol Med 2020;24:11018-23. [Crossref] [PubMed]
- Park HR, Jo SK, Jung U. Ionizing radiation promotes epithelial-to-mesenchymal transition in lung epithelial cells by TGF-beta-producing M2 macrophages. In Vivo 2019;33:1773-84. [Crossref] [PubMed]
- Tanjore H, Xu XC, Polosukhin VV, et al. Contribution of epithelial-derived fibroblasts to bleomycin-induced lung fibrosis. Am J Respir Crit Care Med 2009;180:657-65. [Crossref] [PubMed]
- Marmai C, Sutherland RE, Kim KK, et al. Alveolar epithelial cells express mesenchymal proteins in patients with idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 2011;301:L71-8. [Crossref] [PubMed]
- Chapman HA. Epithelial-mesenchymal interactions in pulmonary fibrosis. Annu Rev Physiol 2011;73:413-35. [Crossref] [PubMed]
- Rube CE, Uthe D, Schmid KW, et al. Dose-dependent induction of transforming growth factor beta (TGF-beta) in the lung tissue of fibrosis-prone mice after thoracic irradiation. Int J Radiat Oncol Biol Phys 2000;47:1033-42. [Crossref] [PubMed]
- Tsoutsou PG, Gourgoulianis KI, Petinaki E, et al. ICAM-1, ICAM-2 and ICAM-3 in the sera of patients with idiopathic pulmonary fibrosis. Inflammation 2004;28:359-64. [Crossref] [PubMed]
- Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 2003;425:577-84. [Crossref] [PubMed]
- Saitoh M. Epithelial-mesenchymal transition is regulated at post-transcriptional levels by transforming growth factor-beta signaling during tumor progression. Cancer Sci 2015;106:481-8. [Crossref] [PubMed]
- Batlle E, Sancho E, Franci C, et al. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol 2000;2:84-9. [Crossref] [PubMed]
- Choi SH, Hong ZY, Nam JK, et al. A hypoxia-induced vascular endothelial-to-mesenchymal transition in development of radiation-induced pulmonary fibrosis. Clin Cancer Res 2015;21:3716-26. [Crossref] [PubMed]
- Sanders KA, Hoidal JR. The NOX on pulmonary hypertension. Circ Res 2007;101:224-6. [Crossref] [PubMed]
- Basu RK, Hubchak S, Hayashida T, et al. Interdependence of HIF-1alpha and TGF-beta/Smad3 signaling in normoxic and hypoxic renal epithelial cell collagen expression. Am J Physiol Renal Physiol 2011;300:F898-905. [Crossref] [PubMed]
- Das V, Bhattacharya S, Chikkaputtaiah C, et al. The basics of epithelial-mesenchymal transition (EMT): a study from a structure, dynamics, and functional perspective. J Cell Physiol 2019. Epub ahead of print. [Crossref] [PubMed]
- Kisseleva T, Brenner D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat Rev Gastroenterol Hepatol 2020. Epub ahead of print. [Crossref] [PubMed]
- Citrin DE, Shankavaram U, Horton JA, et al. Role of type II pneumocyte senescence in radiation-induced lung fibrosis. J Natl Cancer Inst 2013;105:1474-84. [Crossref] [PubMed]
- Hashimoto N, Jin H, Liu T, et al. Bone marrow-derived progenitor cells in pulmonary fibrosis. J Clin Invest 2004;113:243-52. [Crossref] [PubMed]
- Cappuccini F, Eldh T, Bruder D, et al. New insights into the molecular pathology of radiation-induced pneumopathy. Radiother Oncol 2011;101:86-92. [Crossref] [PubMed]
Cite this article as: Hu D, Zhang Y, Cao R, Hao Y, Yang X, Tian T, Zhang J. The protective effects of granulocyte-macrophage colony-stimulating factor against radiation-induced lung injury. Transl Lung Cancer Res 2020;9(6):2440-2459. doi: 10.21037/tlcr-20-1272