Current Understanding of Molecular Pathology and Treatment of Cardiomyopathy in Duchenne Muscular Dystrophy (original) (raw)
Author / Affiliation / Email
![]()
Article Menu
/ajax/scifeed/subscribe
Font Type:
Arial Georgia Verdana
Open AccessReview
by
Tirsa L. E. Van Westering
,
Corinne A. Betts
and
Matthew J. A. Wood
*
Department of Physiology, Anatomy and Genetics, University of Oxford, South Parks Road, Oxford, OX1 3QX, UK
*
Author to whom correspondence should be addressed.
Submission received: 31 March 2015 /Revised: 8 May 2015 /Accepted: 11 May 2015 /Published: 15 May 2015
Abstract
:
Duchenne muscular dystrophy (DMD) is a genetic muscle disorder caused by mutations in the Dmd gene resulting in the loss of the protein dystrophin. Patients do not only experience skeletal muscle degeneration, but also develop severe cardiomyopathy by their second decade, one of the main causes of death. The absence of dystrophin in the heart renders cardiomyocytes more sensitive to stretch-induced damage. Moreover, it pathologically alters intracellular calcium (Ca2+) concentration, neuronal nitric oxide synthase (nNOS) localization and mitochondrial function and leads to inflammation and necrosis, all contributing to the development of cardiomyopathy. Current therapies only treat symptoms and therefore the need for targeting the genetic defect is immense. Several preclinical therapies are undergoing development, including utrophin up-regulation, stop codon read-through therapy, viral gene therapy, cell-based therapy and exon skipping. Some of these therapies are undergoing clinical trials, but these have predominantly focused on skeletal muscle correction. However, improving skeletal muscle function without addressing cardiac aspects of the disease may aggravate cardiomyopathy and therefore it is essential that preclinical and clinical focus include improving heart function. This review consolidates what is known regarding molecular pathology of the DMD heart, specifically focusing on intracellular Ca2+, nNOS and mitochondrial dysregulation. It briefly discusses the current treatment options and then elaborates on the preclinical therapeutic approaches currently under development to restore dystrophin thereby improving pathology, with a focus on the heart.
Graphical Abstract
1. Introduction
Duchenne muscular dystrophy is a severe muscle wasting X-linked genetic disease which affects 1 in 3500 children [1]. It is characterized by loss in muscle function leading to decreased ambulation and most patients succumb to cardiac or respiratory failure [2,3]. The disease is caused by the almost complete absence of the dystrophin protein due to out-of-frame mutations in the DMD gene [4]. Dystrophin is an important protein for cytoskeletal structure and normal muscle function and plays a vital role in membrane stability and signaling [5].
Patients with DMD suffer from severe cardiomyopathy. Although the degree of cardiac involvement varies between patients, cardiomyopathy generally manifests at about 10 years of age and is prevalent in most patients by 20 years of age [6]. These patients first exhibit left ventricle (LV) dilation and hypertrophy, which progresses to a stage known as dilated cardiomyopathy (DCM). Additional cardiomyopathic features include decreased fractional shortening and electrocardiogram (ECG) abnormalities [7,8,9,10]. Approximately 26% of patients also display tachycardia, and 51% exhibit an increase in pathological heart rate variability [7]. These ECG abnormalities are associated with morphological changes to cardiac muscle (thin and thick filaments of heart) [11], myocardial fibrosis and conduction defects [12]. Indeed, deep Q wave abnormalities have been associated with lateral wall scarring [13].
The cardiac phenotype continues to deteriorate as dystrophic hearts undergo hypertrophy in an effort to cope with the increase in wall stress imposed by pressure/volume overload. The heart further undergoes maladaptive remodeling, developing DCM [6]. Approximately 25% of patients under the age of 6 present with DCM, which escalates to 59% by 10 years of age and is prevalent in all patients by adulthood [8]. Unfortunately, severe scoliosis hampers the ability to accurately measure cardiac function which results in late diagnosis and late employment of treatment [6,14,15].
Marked decline in cardiac function correlates with increased fibrotic deposition, impeding normal heart contraction. Fibrotic involvement is prevalent from a young age (17% of patients under 10 years), and escalates with age (34% between 10–15 years and 59% older than 15 years of age) [16].
The most widely used mouse model of DMD is the mdx mouse which has a point mutation in exon 23, thus preventing the production of dystrophin protein [17,18]. This model exhibits an obvious skeletal phenotype including elevated creatine kinase (CK) plasma levels, irregular muscle histology [17], muscle necrosis [19], and a decline in specific force and power with age [20]. Additionally, respiratory function deteriorates and correlates with degeneration of the diaphragm as the disease progresses with age.
Mdx mice also display a cardiac phenotype. Increased right ventricle (RV) systolic volume and subsequent reduction in RV ejection fraction (EF) is seen at 3 months [21]. An increase in myocardial fibrosis is apparent from 6 months of age, whilst LV cardiac output (CO) deteriorates. By 9 months of age, stroke volume (SV) is reduced followed by a reduction in LV EF at 12 months. Although this model has been criticized for the late onset cardiac phenotype, it should be noted that, at 1 month of age, mdx mice exhibit early intolerance to dobutamine stress [21]. As with DMD patients, ECG recordings in conscious mdx mice revealed tachycardia, decreased heart rate variability [22], deep Q waves, diminished S:R ratios, polyphasic R-waves and shorter QT and PR intervals [23] compared to controls.
The major disparity between mdx mice and DMD patients is that RV involvement does not occur in all DMD patients and cardiac complications contribute significantly to early mortality in patients. Interestingly BMD patients also exhibit RV changes prior to LV, analogous to mdx cardiac progression. DMD patients generally receive early ventilatory intervention due to the rapid deterioration in respiratory function. This intervention would be likely to improve pulmonary hypertension and thus may compensate for RV dysfunction.
It is thus evident that the heart is an important system affected in DMD. More recently, research has focused on characterizing heart pathology and therefore this review focuses on the cardiac aspects of DMD. First the result of the absence of dystrophin on molecular heart pathology will be discussed, particularly concentrating on the role of intracellular calcium (Ca2+) increase, the perturbed nitric oxide (NO) signaling and neuronal NO synthase (nNOS) function and the role of mitochondria within DMD. It should be noted that many other cellular pathways are important in DMD pathology (e.g., inflammation, matrix metalloproteinase function, autophagy and apoptosis), but will not be discussed here (see e.g., Shin et al. [24] and Mosquiera et al. [25]). Additionally, current therapies involving corticosteroids, respiratory support, ACE-inhibitors, beta-blockers and cardiac resynchronization techniques are reviewed. Lastly, preclinical therapeutic approaches that are presently investigated, such as exon skipping and viral gene therapy, will be explored within the context of improving heart pathology.
2. Molecular Pathology of DMD
It is clear that dystrophin plays an important role in the cell. By connecting with laminin at the C-terminus through the dystroglycan complex, actin at the N-terminus, and spectrin-like repeat 11–17 in the rod domain, dystrophin provides stability to the cell and prevents damage from muscle contraction [5,26,27]. It is also involved in signaling through its association to the dystroglycan complex (DGC) at the cysteine-rich domain and C-terminus and neuronal nitric oxide synthase (nNOS) at its rod domain (see Figure 1) [5]. The location of mutation/deletion within the dystrophin gene correlates with the severity of cardiomyopathy. Deletions which affect the amino-terminal domain (muscle promoter, exon 1 or intronic regions) are associated with early-onset DCM, whereas deletions in the rod domain and hinge 3 region result in later onset DCM (mid-40’s) [8]. Absence of this protein, as is the case in DMD, renders both skeletal and cardiac cells more susceptible to damage upon muscle contraction. Increased permeability of the cell membrane has also been observed, possibly due to lipid peroxidation by phospholipase A2 or reactive oxygen species (ROS). This allows larger proteins, such as CK, to traverse the cell membrane [28]. Furthermore, many signaling pathways within the cell are affected and these factors lead to an imbalance in the intracellular environment, further resulting in cell muscle damage and eventually necrosis. The ensuing muscle pathology is characterized by cell degeneration and regeneration, whereby muscle cells are eventually replaced by fibrotic tissue [19].
2.1. Intracellular Ca2+ Increase
One of the first events thought to influence molecular pathology is the increase in intracellular Ca2+. Given the importance of Ca2+ in excitation-contraction (E-C) coupling and as a signaling molecule, this has a detrimental effect on heart function and Ca2+ downstream pathways. The heart exhibits a smaller Ca2+ influx than skeletal muscle, but this smaller influx has a greater influence on cardiac cells, suggesting perhaps a more sensitive Ca2+-induced Ca2+ release [29]. Elevated intracellular Ca2+ in the heart leads to mitochondrial deregulation, protease calpain-mediated necrosis and NF-κB activation, which induces transcription of inducible NOS (iNOS)-mediated inflammation [24,30]. In addition, it activates Ca2+/calmodulin (CaM) and CaM kinase II (CaMKII) and protein kinase A (PKA) which hyper-phosphorylate Ca2+ channels [31,32,33]. Seval calcium binding proteins are known to be affected in the skeletal muscle phenotype. However, only two have been discussed in heart pathology in DMD, namely calsequestrin and sarcalumenin, which are involved in sequestration and SERCA Ca2+ uptake, respectively, and are both decreased in mdx hearts [34].
As of yet it is unclear how the increase in Ca2+ commences, particularly the initial entry of Ca2+. Many studies have focused on this topic and membrane tears and stretch-activated channels (SACs) have been proposed as possible mechanisms. More indirect entry of Ca2+ from the extracellular space is thought to be contributed by the Na+-Ca2+-exchanger (NCX) and voltage-gated Ca2+ channels (VGCC). The initial intracellular Ca2+ increase brings about the activation of store-operated channels (SOCs) such as sarcoplasmic/endoplasmic reticulum Ca2+ ATPase (SERCA), the ryanodine receptor (RyR) and the inositol 1,4,5-triphosphate (IP3) receptor (IP3R), thereby further increasing the intracellular Ca2+ concentration ([Ca2+]i) (see Figure 1).
2.1.1. Membrane Tears
Membrane tears were suggested to arise from a fragile membrane due to the absence of dystrophin [35]. This would make the membrane leaky, allowing the influx and efflux of ions and proteins [28,36] (see Figure 1). Within the heart, a membrane sealant lowered, but not abolished, increased Ca2+ influx and improved left ventricular end diastolic volume in mdx mice [37,38]. More recently, the contribution of membrane tears to increased Ca2+ influx has been debated. In an extensive review, Allen and Whitehead indicated that the time required for Ca2+ to increase (10–20 min after contraction) did not correlate with membrane tears [39]. The leakage of ions and proteins through the membrane can also be explained by SACs and by lipid peroxidation [28]. Given these opposing results, more research is needed to elucidate these findings.
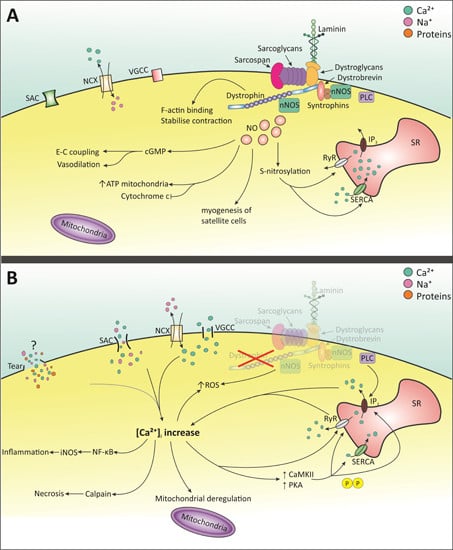
Figure 1. Molecular pathways involving Ca2+ and NO signaling in healthy and dystrophic cell. (A) Dystrophin present: Ca2+ is balanced by SACs, NCX and VGCC. nNOS is activated by Ca2+-dependent calmodulin [40,41,42]. NO is produced which is involved in many processes including normal E-C coupling, and S-nitrosylation of SERCA and RyR [25,40,42,43]. Upon E-C coupling, [Ca2+]i rises via opening of RyR and IP3 receptor. When the stimulus stops, intracellular Ca2+ returns to normal via SERCA and other cell membrane Ca2+-permeable channels. (B) Dystrophin absent: membrane tears, SACs, NCX and VGCC increase intracellular Ca2+ [28,35,36,38,44,45]. Elevated Ca2+ increases CaMKII and PKA which hyper-phosphorylate Ca2+ channels on the SR [31,32,33]. SERCA levels may decrease and RyR becomes sensitive to Ca2+ [31,32,46,47,48]. Mis-localization of nNOS disrupts NO signaling and function [49,50]. This combined with raised Ca2+ increases ROS production [28,51]. Increased [Ca2+]i elicits multiple detrimental events [24,30]. SACs: stretched-activated channels; NCX: Na+-Ca2+ exchanger; VGCC: Voltage-gated Ca2+ channels; nNOS: neuronal nitric oxide synthase; SERCA: Sarcoplasmic/endoplasmic reticulum Ca2+-ATPase; RyR: ryanodine receptor; NO: nitric oxide; IP3: inositol 1,4,5-triphosphate; SR: sarcoplasmic reticulum; CaMKII: calmodulin-kinase II; PKA: protein kinase A.
Figure 1. Molecular pathways involving Ca2+ and NO signaling in healthy and dystrophic cell. (A) Dystrophin present: Ca2+ is balanced by SACs, NCX and VGCC. nNOS is activated by Ca2+-dependent calmodulin [40,41,42]. NO is produced which is involved in many processes including normal E-C coupling, and S-nitrosylation of SERCA and RyR [25,40,42,43]. Upon E-C coupling, [Ca2+]i rises via opening of RyR and IP3 receptor. When the stimulus stops, intracellular Ca2+ returns to normal via SERCA and other cell membrane Ca2+-permeable channels. (B) Dystrophin absent: membrane tears, SACs, NCX and VGCC increase intracellular Ca2+ [28,35,36,38,44,45]. Elevated Ca2+ increases CaMKII and PKA which hyper-phosphorylate Ca2+ channels on the SR [31,32,33]. SERCA levels may decrease and RyR becomes sensitive to Ca2+ [31,32,46,47,48]. Mis-localization of nNOS disrupts NO signaling and function [49,50]. This combined with raised Ca2+ increases ROS production [28,51]. Increased [Ca2+]i elicits multiple detrimental events [24,30]. SACs: stretched-activated channels; NCX: Na+-Ca2+ exchanger; VGCC: Voltage-gated Ca2+ channels; nNOS: neuronal nitric oxide synthase; SERCA: Sarcoplasmic/endoplasmic reticulum Ca2+-ATPase; RyR: ryanodine receptor; NO: nitric oxide; IP3: inositol 1,4,5-triphosphate; SR: sarcoplasmic reticulum; CaMKII: calmodulin-kinase II; PKA: protein kinase A.

2.1.2. Stretched-Activated Channels (SACs)
More recently, SACs have been investigated as a possible reason for the initial Ca2+ influx [36]. These channels open upon stretched muscle contraction and allow the influx of cationic ions [52]. Inhibition of SACs following exposure to mechanical stress significantly diminished the rise of [Ca2+]i in ventricular cardiomyocytes and restored protein kinase C (PKC) and phospholipase C (PLC) signaling [53,54]. It should be noted that the non-specificity of the blockers used means they may have also been blocking Na+ channels [53]. Therefore, the increase in [Ca2+]i could be mediated by both direct entry of Ca2+ ions through SACs, and indirect entry through the Na+/Ca2+-exchanger (NCX) or voltage-gated Ca2+ channels (VGCC) that open due to Na+-induced membrane depolarization (see Figure 1) [38,53,55,56,57]. SACs are composed of multiple channels and subunits, some of which have been implicated in the composition of these SACs and DMD pathology. These channels include the mechano-sensitive transient receptor potential cation (TRPC) channels, namely TRPC1, TRPC3, TRPC6 and TRPV2.
TRPC1 channels are activated by the ROS-dependent Src kinase leading to an influx of Ca2+. Both the absence of dystrophin-associated nNOS [51] as well as the increase in [Ca2+]i [28] can induce increased ROS levels. The role of TRPC1 in the heart remains elusive, but the expression levels of this channel increase with age in the mdx mouse [53]. Interestingly, a more recent experiment did not see an involvement of TRPC1 channels in skeletal myotubes [54]. Whether this is also the case in cardiomyocytes is unclear. TRPV2 channels have also been implicated in DMD and upon activation migrate to the plasma membrane. Cultured mdx cardiomyocytes exhibit increased TRPV2 expression at the plasma membrane, and inhibition of these channels with non-specific blockers and targeted siRNAs reduced cellular Ca2+ influx upon osmotic stress [52]. TRPC6 channels are also sensitive to stretch contraction [58] and inhibition of this channel in the mdx heart reduced contractions and arrhythmias [59]. Finally, relevance of TRPC3 in the heart remains to be established as TRPC3 knock-out (KO) mice exhibit a dystrophic phenotype, but stretch-induced contraction of TRPC3-/- cardiomyocytes does not induce a Ca2+ response [59,60].
2.1.4. Store-Operated Ca2+ Release
Other intracellular channels on the sarcoplasmic reticulum (SR) involved in regulating [Ca2+]i are SERCA, RyR and IP3 channels (see Figure 1). Being a major store of Ca2+, SERCA pumps Ca2+ back into the SR when [Ca2+]i increases [65]. Interestingly, expression levels of SERCA in the mdx heart appear to be similar to that of wild-type (WT), though other reports have shown down-regulation [46,47]. This may be attributed to age, where older mice would exhibit a reduction in SERCA channels. RyRs are activated by Ca2+ binding, thereby leading to Ca2+-induced Ca2+ release stimulating excitation-contraction (E-C) coupling [65,66]. Hyper-phosphorylation of the channel, possibly by protein kinase A (PKA) or CaMKII, could make it more sensitive to Ca2+ [31,48,66]. Persistent hyper-phosphorylation of RyR leads to dissociation from calstabin 2, a stabilizing protein [67]. This in turn disrupts Ca2+ regulation whereby the SR becomes leaky, ultimately resulting in heart failure [33]. Indirect evidence of RyR involvement in the mdx heart is seen, whereby increased SR Ca2+ leak was observed in 3–4 and 12–15 month old mice, with a worsened pathology in the older mice [68]. These events are also observed in arrhythmias and heart failure [69]. Lastly, IP3 receptors are activated by IP3, a downstream product of PLC. Application of PLC inhibitors reduced the Ca2+ concentration in the heart of mdx mice compared to WT. Older mdx animals (9 and 12 months) showed a larger diastolic Ca2+ decrease upon inhibition compared to WT. These results indicate the involvement of PLC and its downstream effects on IP3 and diastolic Ca2+ concentration in the heart [69]. Moreover, it also indicates that age plays an important role in the increases of intracellular Ca2+ concentration.
Much work has been done to elucidate the role of Ca2+ in DMD pathology, which suggests the interaction of multiple mechanisms. It is possible other uncovered avenues play an important role in addition to the mechanisms proposed so far.
2.2. NO and NOS
An important second messenger in cell physiology is the signaling molecule nitric oxide (NO). Its production is regulated by NO synthase (NOS) of which there are three isoforms: neuronal NOS (nNOS), endothelial NOS (eNOS) and inducible NOS (iNOS). nNOS and eNOS are both constitutively expressed in muscle and heart and activated by the Ca2+-dependent calmodulin. eNOS is associated with caveolin 3 on the sarcolemma and nNOS is associated with the DGC and is in close proximity to SERCA and RyR [40,41,42,70]. Conversely, iNOS is only expressed in the cell during inflammation and partly responsible for the inflammatory response seen in DMD [40,41]. The localization of NOS enzymes within the cell is crucial to their function as NO is only active for several seconds and thus needs to reach its target quickly [42]. However, in DMD nNOS is displaced, with considerably lower nNOS activity levels in patients and mdx mice [49,50].
The function of nNOS in the heart is currently still being investigated. Several knock-out studies have implicated a role in LV systolic function and shortening of myocytes, but this remains elusive due to opposing results [42,71,72]. Overproduction of NO by nNOS is associated with cardiac depression, which increases the chances of heart failure [49]. Absence of nNOS impacts several downstream pathways, including the mitochondrial respiratory chain, RyR regulation and reactive oxygen species (ROS) control, which induces cell damage and further increases Ca2+ [25,40,41,72,73]. Transgenic up-regulation of nNOS in the mdx mouse restored heart phenotype, with a decrease in inflammatory markers, fibrosis and apoptosis. Some aspects of heart function also improved, such as stroke volume and ejection fraction [74]. Together these studies underscore the importance of nNOS and NO function and their interaction with [Ca2+]i in the heart and loss of these molecules results in worsening of heart function, though further research is needed to elucidate its exact function.
2.3. Mitochondrial Dysfunction
In any cell, mitochondria are an important part of energy metabolism and healthy muscle contraction [65]. In addition to this role, it also functions as a Ca2+ store, supplying and taking up Ca2+ to and from the cell [75,76]. Furthermore, it is involved in ROS production and plays an important role in cell death through necrosis and apoptosis [77]. Mdx mice display disturbed mitochondrial function and thus these organelles are thought to play a role in DMD.
Altered mitochondrial energy production is one of the first pathophysiological changes known to occur in mdx heart. Mdx mice were assessed at 10–12 weeks of age, considered the pre-cardiomyopathic stage, and revealed a slight shift in energy consumption, from the normally utilized fatty acids to a higher usage of carbohydrates [78]. The functional oxidative capacity of the mitochondria is also disturbed in the mdx mouse model. It is suggested that mild oxidative stress induces a reduction in oxidative phosphorylation, thereby reducing the production of ATP. Due to the energy demand of the cell and the inability of the mitochondria to supply the cell, cardiomyocytes are severely affected [43]. Mitochondria are also a target of NO signaling and its downstream effectors. Overexpression of guanylyl cyclase (GC; activated by cGMP), in 12 and 20 week old mdx mice improved muscle function and cardiac endurance and led to reduced heart production of lactate dehydrogenase, a marker for tissue damage. The GC transgenic mdx mouse heart still displayed a slight shift in energy metabolism from fatty acids to carbohydrate substrates, but mitochondrial function was enhanced as higher levels of the compounds for the citric acid cycle were observed [79]. Given this data, the lack of dystrophin seems to disrupt metabolism in the heart. However, it remains unclear what brings about this change in metabolism and whether this is a compensatory or detrimental mechanism [78].
In addition to the perturbed energy metabolism exhibited by mitochondria in dystrophin-deficient cells, the increased intracellular Ca2+ levels have a detrimental effect on the mitochondria. The rising [Ca2+]i is thought to induce a sudden increase in mitochondrial membrane permeability through the mitochondrial permeability transition pore (PTP). The PTP is a voltage-sensitive channel and an increase in the mitochondrial Ca2+ concentration induces the opening of this channel, leading to swelling. Short-term opening of these channels is thought to be beneficial as the mitochondria are able to regulate intracellular Ca2+ and ROS balance [80,81]. However, long-term opening of this channel is thought to be detrimental to the cell and induces necrosis of the mitochondria [82]. The ruptured mitochondria contributes to myofiber necrosis [56].
3. Current Clinical Disease Management and Application of Commercially Available Drugs
There is currently no cure for DMD, and therefore therapy is limited to the management of symptoms. The three predominant treatment approaches include corticosteroids in order to ameliorate skeletal muscle phenotype, mechanical respiratory support and cardiac assessment and treatment. Corticosteroids have been shown to markedly improve muscle strength and function [83]. More recent studies have implicated corticosteroids in the stabilization of pulmonary function, prolonging ambulation and reducing the prevalence of scoliosis [84]. In addition they have been associated with delaying the onset of cardiomyopathy by 4% for each year of corticosteroid treatment [85]. However, the unfortunate side effects including Cushing’s syndrome, short stature, hypertension, hyperglycemia, cataracts and osteoporosis, hampers the use of corticosteroids [86].
Deterioration in respiratory muscles results in respiratory insufficiency and pulmonary hypertension. An increase in pulmonary hypertension compounds cardiac function leading to DCM [87]. Thus mechanical support is required as soon as nocturnal hypoventilation is apparent, in order to maintain adequate ventilation and coughing reflex [88]. A non-invasive positive-pressure ventilation (NIPPV) machine with a mechanical insufflation-exsufflation device is the preferred respiratory support device and has substantially reduced mortality in DMD patients [89].
Lastly, the approaches designed to specifically target cardiac involvement include angiotensin-converting enzyme (ACE) inhibitors, beta-blockers (β-blockers) and diuretics [90]. ACE inhibitors are generally implemented when LV systolic function declines [3]. These inhibitors prevent the conversion of angiotensin-I to angiotensin-II (Ang-II), thereby reducing circulating levels of the Ang-II. In addition, ACE inhibitors reduce kinase-II and increase bradykinin levels, which have been shown to reduce peripheral vascular resistance, improve endothelial function and prevent fibrosis. The use of ACE inhibitors for the treatment of DMD patients with an ejection fraction (EF) below 55%, showed a significant improvement in cardiac function (relative to the pre-therapy results; p < 0.0001) and delayed the progression of cardiomyopathy [91].
Beta-Blockers prevent the binding of catecholamines by interfering with β-receptor binding, thus reducing sympathetic nervous system activity. They are therefore prescribed for arrhythmic patients, and those with symptomatic but stable systolic dysfunction. Clinicians often prescribe a cocktail of ACE inhibitors, β-blockers and diuretics which significantly improves cardiac function and survival in DMD patients [91,92]. However, regular cardiac assessments are imperative to ensure the correct treatment regimen, as it has been observed that administration of the β-blocker metoprolol was detrimental to RV function when administered at early stages of cardiomyopathic progression [93].
In DMD patients whom are unresponsive to pharmacological treatment, cardiac resynchronization therapy may be considered, i.e., patients with low LV EF, accompanied by ventricular dyssynchrony (QRS > 120 ms) [94]. These patients have a bi-ventricular pacemaker surgically installed, which allows the pacing of the septal and lateral walls of the LV. Other measures such as left ventricular assist devices HeartMate II and HeartWare, have also been employed in an effort to reduce heart failure [95]. In both cases these devices markedly improve cardiac function and quality of life, thus demonstrating their potential for DMD patients who do not respond to conventional interventional measures.
Several drugs that are already commercially available may benefit DMD patients and have therefore been investigated in animal models of DMD to explore their therapeutic potential. The synthetic copolymer poloxamer P188, which stabilizes the sarcolemma, has been shown to improve the cardiac phenotype and even prevent isoproterenol/dobutamine-induced cardiomyopathy/death [37,96]. The phosphodiesterase 5 (PDE5) inhibitor, sildenafil, acts on the cGMP pathway, where it enhances NO-cGMP signaling. This signaling is impaired in mdx mice and administration of sildenafil improved contractile performance, cardiac metabolism and sarcolemmal integrity [97]. Furthermore, it reversed pathological cardiac dysfunction in mdx hearts [98], and improved diaphragmatic muscle strength thereby enhancing respiratory function [99]. Unfortunately, when translated to clinic, no cardiac function improvement was observed in patients and the clinical trial was terminated early [100,101]. It is important to note, however, that a relatively small cohort of patients was used in this study and higher numbers may better elucidate the underlying mechanisms. In addition, the drugs losartan and pirfenidone have been investigated for their anti-fibrotic properties. These drugs inhibit TGF-β expression, thereby reducing fibrosis and they were seen to improve cardiac pathology and function [102,103]. Pharmacological measures to target the NO signaling pathway have also been utilized and shown to slower the muscular dystrophy progression. Indeed, histone deacteylase activity (HDAC), which is perturbed in DMD may be inhibited with the use of NO donors, and has also been shown to benefit heart pathology [104]. In addition the combination of a non-steroidal anti-inflammatory drug, ibuprofen, and an NO donor, isosorbide dinitrate, were capable of preventing morphological changes in the heart of mdx mice [105]. A recent phase 1 clinical trial using this combinatory therapy, demonstrated an excellent safety profile in healthy patients [106]. This provides a solid basis for further trials in muscular dystrophy patients.
These drugs have demonstrated encouraging results in animal models of DMD and it is hoped that they will infer some clinical benefit in patients. Furthermore, these drugs may present viable combinatory therapies which may be administered in conjunction with treatments specifically aimed at correcting/compensating for the dystrophin mutation.
4. Preclinical Therapeutic Approaches
Given that current treatment strategies are limited to reducing the symptoms of the disease, it is vital that new therapies are developed that directly target the underlying mutation. Several very promising strategies currently under investigation for treating DMD also have potential for treating the heart. This review will discuss some of these most promising therapies, namely; utrophin up-regulation, ribosomal read-through therapy, stem cell therapy, viral gene therapy delivering mini/micro-dystrophin and exon skipping antisense oligonucleotides. Not all experiments regarding the different approaches are discussed in this review, but a summary of selected references are cited in Table 1.
Table 1. DMD therapies under development. Several strategies have been employed to further develop the different types of therapies (specific strategy), which are in different stages of research, either clinical or pre-clinical (research stage and selected models). A brief summary of the results of these strategies is mentioned (results of therapy) with selected references. IV: Intravenous; IM: intramuscular; IP: intraperitoneal.
Table 1. DMD therapies under development. Several strategies have been employed to further develop the different types of therapies (specific strategy), which are in different stages of research, either clinical or pre-clinical (research stage and selected models). A brief summary of the results of these strategies is mentioned (results of therapy) with selected references. IV: Intravenous; IM: intramuscular; IP: intraperitoneal.
| Therapy | Specific Strategy | Research Stage and Selected Models | Results of Therapy | Selected References |
|---|---|---|---|---|
| Utrophin up-regulation | Utrophin transgene | Preclinical—mdx/utrn−/− | Transgenic utrophin expression which improved pathology in skeletal muscle, but not heart. | [107,108,109] |
| Zinc fingers | Preclinical—cultured cells, mdx muscle | Successful activation of utrophin improved muscle function and reduced pathology in TA. No heart data. | [110,111,112] | |
| Biglycan | Preclinical—mdx | Localizes utrophin to sarcolemma. Treatment reduced pathology in quadriceps and diaphragm and improved physiology in EDL. No heart data. | [113] | |
| SMT C1100 | Preclinical—mdx;Clinical trials—Phase Ia and Ib | Preclinically: increased RNA and protein of utrophin in skeletal and cardiac muscle. Reduced pathology and improved muscle function in skeletal muscle. Phase Ia: mild side-effects at higher dose. Phase Ib: no data. | [114,115] | |
| Read-through therapy | Gentamicin | Preclinical—mdx; Clinical trials—Phase I | Preclinically: Low levels of dystrophin expression, including in heart, protection against muscle damage in EDL. Clinical trials: inconclusive. | [116,117] |
| Negamycin | Preclinical—mdx | Antibiotic drug to reduce side effects seen in gentamycin. Subcutaneous injections negamycin safer than gentamycin, but induced low dystrophin expression in skeletal muscle and heart. | [118] | |
| PTC124 | Preclinical—HEK293 cells and mdx;Clinical trials—Phase I, 2a/b | Preclinically: 20%–25% increase in dystrophin in TA, diaphragm and heart. Improved physiological function of EDL. Clinical trials: Generally well tolerated. Overall no significant improvement, but certain subgroups responded well to treatment. | [119,120,121,122] | |
| RTC13/RTC14 | Preclinical—mdx | RTC13 demonstrated better efficacy (restored dystrophin in skeletal muscle and heart) than gentamicin, PTC124 and RTC14. Improved muscle function and decreased serum CK. | [123] | |
| Viral gene therapy | Lentivirus | Preclinical—myotubes, primary myoblasts and mdx | Transfection with mini- or microdystrophin: 20%–25% dystrophin expression in TA muscles (for 2 year period). Less central nucleation, but no protection from muscle injury. Able to transfect TA myogenic progenitor cells. | [124,125,126] |
| ‘Gutted’ adenovirus | Preclinical—mdx | IM with full dystrophin cDNA displayed dystrophin expression, improved muscle force and protected against muscle damage. | [127] | |
| rAAV2/AAV8 | Preclinical—mdx | Chimeric vector containing codon-optimized micro-dystrophin. IV injection resulted in almost 100% transfection, effective dystrophin expression in skeletal muscle and heart and improved muscle function. No immunological response was observed. | [128] | |
| rAAV6 | Preclinical—mdx/utrn−/− and mdx | Microdystrophin rAAV6 administered in 1 month old mdx/utrn−/− increased life span, improved pathology and dystrophin (1 year post-injection). Dystrophin restored in heart and heart mass normal, but function not recovered. 20 mo mdx (4 months after injection) showed dystrophin expression in skeletal muscle and heart and improved pathology. | [129,130] | |
| AAV9 | Preclinical—GRMD and mdx | IV mini-dystrophin administration to GRMD revealed varied dystrophin expression, also in heart. Micro-dystrophin administration in young mdx induced dystrophin expression and slowed progression of cardiac phenotype. 10 mo mice expressed dystrophin and cardiac function improved. | [131,132,133] | |
| Cell-based therapy | Myoblasts | Preclinical—mdxClinical trials | Ability to differentiate into myotubes.Preclinically: partial dystrophin expression in mdx mice. No heart data. Clinical trial: no beneficial effects | [134,135] |
| Fibroblasts | Preclinical—mdx | Ability to differentiate into myotubes.Effective transfection with dystrophin expression in immunocompromised mice. No heart data. | [136] | |
| Bone marrow-derived stem cells | Preclinical—mdx and GRMD | Migrate to damaged muscle areas, differentiate into myogenic cells and aid regeneration. Substantial dystrophin restoration in skeletal muscle of mdx, but no restoration in GRMD dogs. No heart data. | [137,138] | |
| Cd133+ stem cells | Preclinical—scid/mdx;Clinical trial Phase I | Ability to differentiate into myocytes.Preclinically: effective dystrophin restoration in scid/mdx. No heart data.Clinical trial demonstrated safety. | [139,140] | |
| Mesangio-blasts | Preclinical—GRMD | Improved functional mobility and partial dystrophin restoration in skeletal muscle. No heart data. | [141] | |
| iPS cells | Preclinical—immuno-compromised mdx | Differentiating iPS cells into muscle precursor cells followed by injection into TA induced dystrophin expression. Cells integrated with muscle cells and settled in satellite cell population. Improved TA function. No heart data. | [142,143,144] | |
| Antisense oligonucleo-tides | 2′O MePS | Preclinical—mdx;Clinical trial Phase III | Preclinical: IM revealed low dystrophin restoration, even with multiple high doses. Clinical: 6 mg/kg was maximal tolerated dose in patients. Phase III trial did not meet 6MWD endpoint. | [145,146,147,148] |
| PMO | Preclinical—mdx;Clinical trial Phase IIb | Preclinical: repeat IV administrations of high dose restored dystrophin in multiple skeletal muscles of the mdx mouse, <2% in heart. Clinically: well tolerated and dystrophin present after 48 weeks. At 84 weeks stabilization in the 6MWD; 120 weeks stabilized pulmonary function. | [149,150,151,152,153] (Sarepta press release, February 2014) | |
| Tricyclo-DNA | Preclinical- mdx | Multiple IV administrations and very high doses (200 mg/kg per week) resulted in dystrophin in skeletal muscle and heart, with low levels in the brain and improvements in cardiac and pulmonary function. | [154] | |
| Octa-guanidium conjugated PMO | Preclinical- mdx and GRMD | Capable of restoring dystrophin in skeletal muscle and hearts of mdx mice. This has further been demonstrated in dystrophic dogs. High doses led to adverse events in GRMD. | [155,156] | |
| CPP-AOs- Arginine rich | Preclinical—mdx and mdx/utrn−/− | (RXR)4 multiple IP produced ~100% dystrophin in diaphragm and low levels in skeletal muscles. Single IV restored dystrophin in skeletal muscle and diaphragm, ~50% in the heart. Improved mortality rate and corrected kyphosis in mdx/utrn−/−. (RXRRBR)2: Less toxic, repeat and high dose IV illustrated impressive exon skipping notably in heart (72%). Improvements in cardiac function, with preserved diastolic function after 6 months | [157,158,159,160,161,162,163,164] | |
| CPP-AOs- Pips | Preclinical—mdx | Pip2a and Pip2b: strong exon skipping following IM. Following IV, Pip5e induced high dystrophin restoration body wide including heart. Pip6-PMO series: Pip6a, Pip6b and Pip6f exhibited best dystrophin expression in heart. Long-term IV administration prevented deterioration in heart function in the event of exercise. | [165,166,167] | |
| CPP-AOs- Phage Peptides | Preclinical—mdx | MSP enhanced in vivo skeletal and cardiac muscle binding capacity. B-MSP-PMO showed 2–5 fold improvement in skeletal muscle compared to B-PMO (no dystrophin in heart). T-9 (SKTFNTHPQSTP) specificity in mdx quad and improved specificity over MSP. 12-mer phage resulted in ~25% dystrophin expression in skeletal muscle (75 mg/kg).A 7-mer phage conjugated to 2′OMePS resulted in exon skipping in multiple tissues including heart and diaphragm. | [168,169,170,171,172]. |
4.1. Utrophin Up-Regulation
Utrophin up-regulation was one of the first therapeutic strategies developed to compensate for the lack of dystrophin in the cell. In the adult cell, utrophin is mainly found in regenerating cells and at the neuromuscular and myotendinous junctions [173]. It is thought that utrophin could be important for determining actin filament length during development [174]. Utrophin is a homologue of dystrophin with a molecular weight of 395 kDa and a similar amino acid composition, although the rod domain is significantly shorter [175]. Despite functional differences, utrophin appears to have many of the same binding proteins, including certain components of the DAPC and F-actin [173,174,176]. Given these characteristics, it is believed that utrophin could partially compensate for dystrophin function and as such it has been considered a viable option for therapeutic purposes.
Consequently, up-regulation of utrophin to ameliorate DMD pathology and improve the functional deficit is being investigated. Evidence supporting this concept comes from the dystrophin/utrophin double knockout (mdx/utrn-/−) mouse model. These animals exhibit worsened skeletal muscle and cardiac pathology and have a life span of only 2–3 months [162,177,178,179], indicating that utrophin compensates for the absence of dystrophin in these animals. The positive effects of utrophin up-regulation are also seen in mdx skeletal muscle, though it should be noted that the heart is not corrected [107,108,109,110,111,112]. Another strategy called biglycan localizes utrophin to the sarcolemma, though no new utrophin is generated [113] (Tivorsan Pharmaceuticals: www.trivorsan.com).
The most promising utrophin targeting drug is SMT C1100, a drug discovered from a small molecule screen, currently undergoing clinical trials. Application of this drug in mdx mice increased utrophin RNA and protein levels, and the mice exhibited a reduced pathology and improved muscle function. The heart also displayed the presence of utrophin, but no functional tests were performed [114]. Phase Ia and Ib clinical trials have been performed, where the phase Ia trials showed that mild side-effects only occurred at higher doses [115]. The phase Ib trials have been completed, but results have yet to be published (clinicaltrials.gov, identifier: NCT02056808) (see Table 1).
It should be noted that utrophin compensation is unable to completely restore pathology. For instance, utrophin is unable to bind nNOS, a key factor in DMD pathophysiology [180]. Therefore, given the structural differences compared to dystrophin, it is not yet clear if utrophin can fully compensate for the absence of dystrophin. Nevertheless, utrophin up-regulation remains a very valuable therapeutic option as it could benefit all patients, irrespective of the underlying mutation causing the disease.
4.2. Stop Codon Read-Through Therapy
A small number of patients have a nonsense mutation which leads to a premature stop-codon (about 10%–15%) thereby disrupting the open reading frame [181,182]. Antibacterial drugs called aminoglycosides are able to ‘read-through’ these premature stop codons without affecting normal stop codons, thereby restoring the reading frame and generating a functional dystrophin protein [119,181,183]. Both gentamicin and negamycin have been examined in mdx mice, whereby low levels of dystrophin expression are observed. Gentamycin has been tested in DMD patients, where earlier clinical trials saw contradictory and inconclusive results and later trials saw only low levels of dystrophin expression in 50% of patients [117].
Subsequently, a high-throughput screen has identified PTC124, a small-molecule able to induce read-through of premature stop codons, which has shown significant promise in both in vitro and in vivo studies [119]. A major concern of this drug is the possibility of off-target effects [183], though there is no indication of this as of yet [119]. Nonetheless, positive preclinical results have led to PTC124, or ataluren, undergoing clinical trials, whereby the drug was well tolerated and most importantly, read-through of normal stop codons did not occur [120]. Further trials showed no significant functional outcome, though interestingly, several subgroups were positively affected by treatment. There was no mention of the heart [121,122] (clinicaltrials.gov, identifier: NCT01826487) (see Table 1). The results from this clinical trial clearly show that a better understanding regarding subgroups of patients and their functional outcomes is needed.
Other studies have aimed at identifying more effective drugs to induce read-through. A sensitive small-molecule screen was performed, which identified compounds RTC13 and RTC14. These compounds were subsequently administered intramuscularly and intravenously in mdx mice, and RTC13 demonstrated better efficacy (restoring dystrophin in skeletal muscle and especially the heart) than gentamicin, PTC124 and RTC14. In addition, muscle function was improved and serum CK levels were decreased in mdx mice treated with RTC13 [123]. RTC13 provides a promising new therapeutic molecule for patients with a premature stop codon.
4.3. Viral Gene Therapy
Another therapeutic strategy undergoing development is the use of viral vectors to deliver a dystrophin transgene to the cell. Given dystrophin’s enormous size, multiple studies have endeavored to distinguish the integral components of the dystrophin gene to develop a shortened truncated version that may be inserted into the viral capsid (mini- or microdystrophin) [184]. Several viruses have been considered as a potential delivery vector, including lentivirus, adenovirus and adeno-associated virus (AAV; see Table 1).
In order to safely apply the AAV virus, it has been modified to exclude all viral genes, but leave essential repeats for replication (recombinant AAV, rAAV) [182]. The AAV vector requires co-transfection of a vector carrying components necessary for replication. Different AAV serotypes exhibit different tropism [185], of which rAAV2/AAV8, rAAV6 and rAAV9 have shown particular promise in the mdx heart when using mini- or micro-dystrophin. rAAV6 microdystrophin was successful at transfection and induction of dystrophin in TA, diaphragm and heart in young mdx mice. Interestingly, however, heart function did not recover even though dystrophin was restored in the heart and heart mass was normal [129]. Injection of old mdx mice also produced positive results [130], suggesting that even though mdx muscles had been significantly damaged by the disease, treatment was still capable of improving some muscle pathology [130].
rAAV9 has proven to be particularly efficient in transfecting the heart [132]. Mini-dystrophin rAAV9 administered intravenously in golden retriever muscular dystrophy (GRMD) dogs revealed varied dystrophin expression (between 15% and 100% depending on the muscle analyzed) and was also present in the heart [131]. Micro-dystrophin rAAV9 administration in young and adult mdx mice also induced dystrophin expression and improved cardiac function [132,133]. Despite these positive results, immunological responses to the AAV virus have still been observed in larger animals and humans [180,186]. Moreover, high titers of virus are necessary to induce effective dystrophin expression in muscle cells. In an effort to reduce this, chimeric vectors such as rAAV2/8 vector containing a codon-optimized microdystrophin have been generated and intravenously injected in 10 week old mdx mice. Lower titer levels of the virus resulted in almost 100% transfection, effective dystrophin expression in both skeletal muscle and heart and improved muscle function. Moreover, no immunological response was observed [128].
Overall, viral vectors are promising candidates for the delivery of the dystrophin gene. However, special care should be taken regarding immunogenicity as the human population carries antibodies towards many prevalent viruses, such as AAV [182,187], though immunosuppression may be implemented to reduce this risk [188]. Moreover, incorrect insertions of the virus into the host genome may occur with vectors such as lentiviruses [182]. Lastly, repeat administration is problematic due to the presence of neutralizing antibodies from the first administration [180]. Further research is needed to eliminate these concerns.
4.4. Cell-Based Therapy
With cell-based therapy, healthy dystrophin-expressing cells are transplanted into DMD patient tissue which not only integrate into healthy adult muscle cells, but ideally also co-populate the satellite cells. These cells can be derived either from healthy precursor cells or from patient cells that are genetically modified in vitro [189]. Several different muscle precursor cells have been explored for therapeutic purposes in DMD, namely myoblasts, fibroblasts, bone-marrow derived stem cells, CD133+ stem cells and mesangioblasts (see Table 1) [134,135,136,137,138,139,140,141,190,191].
A more recent shift to the use of induced pluripotent stem (iPS) cells has been observed. Indeed, human iPS cells have been differentiated into muscle precursor cells and injected into TA muscles of immunocompromised mdx mouse models [143]. Following injection, dystrophin protein was restored, regeneration led to higher dystrophin levels and functional tests showed improvement [143,144]. These experiments illustrated that iPS cells were able to integrate with muscle cells and settle in the satellite cell population.
Several groups have aimed at utilizing genetically modified patient precursor cells as treatment. This would circumvent immunological suppression needed when introducing foreign material into the body. The genome of the cells are modified in vitro with the use of nucleases [192,193,194,195], whereby more recently, TALENs and CRISPR-cas9 have been used for splicing correction of the dystrophin gene. They were utilized in iPS cells in vitro to induce exon skipping, shift the reading frame or insert the missing exon with successful results. Subsequently, these iPS cells were differentiated into skeletal muscle cells and dystrophin expression was observed. The knock-in of the missing exon was most effective [193]. Given the novelty of the work, the study focused on skeletal cells and did not encompass the cardiomyocytes, but the results offer interesting prospects for dystrophin heart restoration.
Overall, utilizing genetically modified cells from patients would be a more valuable treatment when considering immunological responses. However, the ability to induce and sustain adequate dystrophin levels remains difficult. Currently, delivery of progenitor cells is most successful if done intramuscularly [182], yet this would only allow the treatment of one muscle at a time and muscles like the diaphragm and the heart would not be restored. Additionally, high titers of modified cells are needed to significantly alter the phenotype and functionality. Several obstacles are yet to overcome, but this technique appears promising and may prove to be very effective.
4.5. Antisense Oligonucleotides
Antisense oligonucleotides (AONs) are another viable approach for treating DMD patients, and are specifically utilized in the skipping of exons with mutations or deletions in the dystrophin gene in order to correct the aberrant reading frame [196]. AONs are short, single stranded DNA sequences which are complementary to a target pre-mRNA splice site, thereby sterically blocking splice enhancers and skipping the desired exon which produces a functional, albeit shorter, dystrophin protein [197]. This therapy is capable of treating ~83% of DMD patients with dystrophin mutations, specifically 79% of patients with deletions, 91% with small mutations and 73% with duplications [198]. A significant shortfall of this therapy is that AONs can only specifically target one exon at a time and therefore current efforts are focused on exons which would assist the greatest numbers of patients [198].
The predominant AON chemistries which are currently undergoing clinical evaluation are phosphorodiamidate morpholino oligomers (PMOs) [149] and 2′O methyl phosphorothioate (2′OMePS) [146] and both have shown promising results in the clinic (see Table 1) [146,149,153,199]. Although these chemistries demonstrate potential for treating DMD, the confounding issues are their relatively poor systemic delivery and their inability to restore dystrophin in the heart [146,149,200,201]. More recently, a new AON chemistry, tricyclo-DNA, has demonstrated marked preclinical success and was capable of restoring dystrophin in skeletal muscle and heart, with low levels in the brain of DMD mouse models. However, it should be noted that these positive findings required multiple dose administration and very high doses (200 mg/kg per week) [154].
In an effort to improve the delivery capacity of AONs and reduce the dose required, short peptide sequences, ‘cell penetrating peptides’ (CPPs), have been chemically conjugated to neutral AONs such as PMO to facilitate their delivery across the plasma membrane. A multitude of studies have investigated peptide conjugated AONs in the context of DMD and can broadly be classified into 3 distinct groups namely arginine rich, Pip (PNA/PMO internalization peptide), and phage and chimeric peptides.
Most importantly the arginine rich and Pip peptides have demonstrated cardiac dystrophin expression. Arginine rich CPPS conjugated to PMO were the first to show activity in the heart [163] and indeed further showed that sustained treatment improved cardiac function and resistance to dobutamine stress [158]. More recently, the Pip6 series has demonstrated dystrophin expression in the heart at very low doses (12.5 mg/kg) [166] and prevented exercise-induced cardiomyopathy following long-term treatment [167]. The phage peptides have been less successful in restoring cardiac dystrophin and function, with the exception of a 7-mer phage (conjugated to 2′OMePS), which resulted in exon skipping in multiple tissues including heart [172].
Another method to improve delivery of charged AONs is to encapsulate them in liposomes [202] or nanoparticles. More recent advancements have given rise to ZM4 nanoparticles bound to AONs which demonstrated widespread distribution to skeletal muscle and heart, thus highlighting the potential of this delivery method. Other new and innovative technologies being investigated include bispecific splice oligonucleotides targeting dystrophin and myostatin regulation [203], and knocking-in of deleted exons using meganucleases [195]. Given the current developments of AON therapy in the DMD field, it is no surprise that such a variety of novel approaches are being developed for the treatment of DMD.
To summarize, with the exception of tricyclo-DNA, naked AONs have demonstrated limited systemic delivery and an inability to restore dystrophin in the heart, whereas conjugation to CPPs or the use of encapsulation technologies have significantly improved upon this. From a CPP perspective, both poly-arginine and Pip CPPs have displayed widespread dystrophin restoration in multiple skeletal muscle tissues and the heart. Nanoparticles are another promising delivery strategy, particularly for charged AONs such as 2′OMePS. This field is rapidly evolving and it is hoped that one or more of these technologies is advanced to clinical trial in the near future.
Whilst these experimental therapies have inspired new hope for patients and families affected by DMD, the feasibility of clinically translating some of these therapies has been brought into question. Not only should species differences be considered in terms of cellular processes and pathways, but also concerning immunological responses. Mouse models of DMD are somewhat resilient, and studies in dog models have highlighted inevitable obstacles particularly due to our complex immune system. This is impossible to avoid given the requirement of a systemic approach to treat a disease which is devastating to the entire body. Not only is preventing an immunological response particularly relevant for cell-based therapies and viral vectors, but also for peptide-conjugated AONs which may cause complications. Certainly, long term administration of ‘naked’ PMO did not elicit an immunological response. Other therapies such as aminoglycosides may induce off-target or other toxicological affects. Ultimately, in order to substantially ameliorate or abrogate this disease, targeting the underlying defect, specifically the lack of dystrophin protein, is required. Current pharmacological interventions can only delay or partially ameliorate the symptoms of this disease. Therefore it is imperative that one or more, or indeed a new therapy altogether, is able to overcome these obstacles and by-pass our innate immune system.
5. Conclusions
Our current understanding of Duchenne muscular dystrophy has increased significantly over the last few decades. The absence of the dystrophin protein gives rise to a complex pathology with the involvement of many different processes in the cell all playing an intricate part in further disease progression. Much research has already been performed which indicate the importance of Ca2+, NO, mitochondria and other second messengers and organelles in the cell. However, given the gaps that still remain in our knowledge, it seems likely that other unknown proteins or pathways also affect disease pathology. Furthermore, as dystrophin is expressed body-wide, it is important that individual systems such as the heart are considered, as specific mechanistic findings from skeletal muscle pathology may not necessarily be applied to the heart. Moreover, the comparison of molecular aspects of dystrophin function between different cell types could provide valuable information.
The knowledge gained from improved understanding of molecular pathological pathways provides more ways of developing and assessing the effectiveness of treatment. With the availability of steroids and other drugs, improvements have been made to ameliorate the skeletal muscle phenotype. Whilst pharmacological treatments to stabilize cardiac function, such as ACE inhibitors and beta-blockers, are in use, mortality from cardiorespiratory complications in DMD patients remains high. Moreover, whilst some experimental therapies are under development, these may not benefit cardiac aspects of the disease and most clinical trials to date have focused exclusively on the beneficial effects of drug treatment on skeletal muscle function, without considering the impact on heart function. Therefore, an increased focus on the heart is essential, both in terms of existing treatments and the effects on heart function, but also more critically on novel treatments that could be developed to specifically restore dystrophin or upregulate utrophin in cardiac muscle, resulting in clinical benefit for patients.
Acknowledgments
This work was supported by the MD UK grant and the BHF grant.
Author Contributions
All authors contributed to the planning of the manuscript. TLEW and CAB wrote the manuscript and all authors edited and approved the final manuscript.
Conflicts of Interest
MJAW has worked as a consultant for Sarepta Therapeutics.
References
- Finsterer, J.; Stöllberger, C. The heart in human dystrophinopathies. Cardiology 2003, 99, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Province, M.A.; Moxley, R.T., 3rd; Griggs, R.C.; Brooke, M.H.; Fenichel, G.M.; Miller, J.P.; Kaiser, K.K.; King, W.; Robison, J.; et al. Clinical investigation of Duchenne muscular dystrophy. A methodology for therapeutic trials based on natural history controls. Arch. Neurol. 1987, 44, 808–811. [Google Scholar] [CrossRef] [PubMed]
- Fayssoil, A.; Nardi, O.; Orlikowski, D.; Annane, D. Cardiomyopathy in Duchenne muscular dystrophy: Pathogenesis and therapeutics. Heart Fail. Rev. 2010, 15, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Douglas, A.G.L.; Wood, M.J. A splicing therapy for neuromuscular disease. Mol. Cell. Neurosci. 2013, 56, 169–185. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.E.; Nowak, K.J. Molecular mechanisms of muscular dystrophies: Old and new players. Nat. Rev. Mol. Cell Biol. 2006, 7, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Mazur, W.; Hor, K.N.; Germann, J.T.; Fleck, R.J.; Al-Khalidi, H.R.; Wansapura, J.P.; Chung, E.S.; Taylor, M.D.; Jefferies, J.L.; Benson, D.W.; et al. Patterns of left ventricular remodeling in patients with Duchenne Muscular Dystrophy: A cardiac MRI study of ventricular geometry, global function, and strain. Int. J. Cardiovasc. Imag. 2012, 28, 99–107. [Google Scholar] [CrossRef]
- Kirchmann, C.; Kececioglu, D.; Korinthenberg, R.; Dittrich, S. Echocardiographic and electrocardiographic findings of cardiomyopathy in Duchenne and Becker-Kiener muscular dystrophies. Pediatr. Cardiol. 2005, 26, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Nigro, G.; Comi, L.I.; Politano, L.; Bain, R.J.I. The incidence and evolution of cardiomyopathy in Duchenne muscular dystrophy. Int. J. Cardiol. 1990, 26, 271–277. [Google Scholar] [CrossRef]
- Bushby, K.; Muntoni, F.; Bourke, J.P. 107th ENMC International Workshop: The management of cardiac involvement in muscular dystrophy and myotonic dystrophy. 7–9 June 2002, Naarden, the Netherlands. Neuromuscular Disord. 2003, 13, 166–172. [Google Scholar] [CrossRef]
- Sultan, A.; Fayaz, M. Prevalence of cardiomyopathy in Duchenne and Becker’s muscular dystrophy. J. Ayub Med. Coll. Abbottabad 2008, 20, 7–13. [Google Scholar] [PubMed]
- Sanyal, S.K.; Johnson, W.W.; Thapar, M.K.; Pitner, S.E. An ultrastructural basis for electrocardiographic alterations associated with Duchenne’s progressive muscular dystrophy. Circulation 1978, 57, 1122–1129. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, S.K.; Johnson, W.W. Cardiac conduction abnormalities in children with Duchenne’s progressive muscular dystrophy: Electrocardiographic features and morphologic correlates. Circulation 1982, 66, 853–863. [Google Scholar] [CrossRef] [PubMed]
- Perloff, J.K.; Roberts, W.C.; de Leon, A.C., Jr.; O’Doherty, D. The distinctive electrocardiogram of Duchenne’s progressive muscular dystrophy. An electrocardiographic-pathologic correlative study. Am. J. Med. 1967, 42, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Hor, K.N.; Wansapura, J.; Markham, L.W.; Mazur, W.; Cripe, L.H.; Fleck, R.; Benson, D.W.; Gottliebson, W.M. Circumferential Strain Analysis Identifies Strata of Cardiomyopathy in Duchenne Muscular Dystrophy. A Cardiac Magnetic Resonance Tagging Study. J. Am. Coll. Cardiol. 2009, 53, 1204–1210. [Google Scholar] [CrossRef] [PubMed]
- Hagenbuch, S.C.; Gottliebson, W.M.; Wansapura, J.; Mazur, W.; Fleck, R.; Benson, D.W.; Hor, K.N. Detection of Progressive Cardiac Dysfunction by Serial Evaluation of Circumferential Strain in Patients With Duchenne Muscular Dystrophy. Am. J. Cardiol. 2010, 105, 1451–1455. [Google Scholar] [CrossRef] [PubMed]
- Hor, K.N.; Taylor, M.D.; Al-Khalidi, H.R.; Cripe, L.H.; Raman, S.V.; Jefferies, J.L.; O’Donnell, R.; Benson, D.W.; Mazur, W. Prevalence and distribution of late gadolinium enhancement in a large population of patients with Duchenne muscular dystrophy: Effect of age and left ventricular systolic function. J. Cardiovasc. Magn. Reson. 2013, 15, 107. [Google Scholar] [CrossRef] [PubMed]
- Bulfield, G.; Siller, W.G.; Wight, P.A.; Moore, K.J. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc. Natl. Acad. Sci. USA 1984, 81, 1189–1192. [Google Scholar] [CrossRef] [PubMed]
- Sicinski, P.; Geng, Y.; Ryder-Cook, A.S.; Barnard, E.A.; Darlison, M.G.; Barnard, P.J. The molecular basis of muscular dystrophy in the mdx mouse: A point mutation. Science 1989, 244, 1578–1580. [Google Scholar] [CrossRef] [PubMed]
- Pastoret, C.; Sebille, A. mdx mice show progressive weakness and muscle deterioration with age. J. Neurol. Sci. 1995, 129, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Lynch, G.S.; Hinkle, R.T.; Chamberlain, J.S.; Brooks, S.V.; Faulkner, J.A. Force and power output of fast and slow skeletal muscles from mdx mice 6–28 months old. J. Physiol. 2001, 535, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Stuckey, D.J.; Carr, C.A.; Camelliti, P.; Tyler, D.J.; Davies, K.E.; Clarke, K. In vivo MRI characterization of progressive cardiac dysfunction in the mdx mouse model of muscular dystrophy. PLoS ONE 2012, 7, e28569. [Google Scholar] [CrossRef] [PubMed]
- Chu, V.; Otero, J.M.; Lopez, O.; Sullivan, M.F.; Morgan, J.P.; Amende, I.; Hampton, T.G. Electrocardiographic findings in mdx mice: A cardiac phenotype of Duchenne muscular dystrophy. Muscle Nerve 2002, 26, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Wehling-Henricks, M.; Jordan, M.C.; Roos, K.P.; Deng, B.; Tidball, J.G. Cardiomyopathy in dystrophin-deficient hearts is prevented by expression of a neuronal nitric oxide synthase transgene in the myocardium. Hum. Mol. Genet. 2005, 14, 1921–1933. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Tajrishi, M.M.; Ogura, Y.; Kumar, A. Wasting mechanisms in muscular dystrophy. Int. J. Biochem. Cell Biol. 2013, 45, 2266–2279. [Google Scholar] [CrossRef] [PubMed]
- Mosqueira, M.; Baby, S.M.; Lahiri, S.; Khurana, T.S. Ventilatory Chemosensory Drive Is Blunted in the mdx Mouse Model of Duchenne Muscular Dystrophy (DMD). PLoS ONE 2013, 8, e69567. [Google Scholar] [CrossRef] [PubMed]
- Amann, K.J.; Guo, A.W.X.; Ervasti, J.M. Utrophin lacks the rod domain actin binding activity of dystrophin. J. Biol. Chem. 1999, 274, 35375–35380. [Google Scholar] [CrossRef] [PubMed]
- Blake, D.J.; Weir, A.; Newey, S.E.; Davies, K.E. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol. Rev. 2002, 82, 291–329. [Google Scholar] [PubMed]
- Allen, D.G.; Whitehead, N.P. Duchenne muscular dystrophy—What causes the increased membrane permeability in skeletal muscle? Int. J. Biochem. Cell Biol. 2011, 43, 290–294. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.; Martins, A.S.; Niggli, E.; Shirokova, N. Dystrophic cardiomyopathy: Amplification of cellular damage by Ca2+ signalling and reactive oxygen species-generating pathways. Cardiovasc. Res. 2008, 77, 766–773. [Google Scholar] [CrossRef] [PubMed]
- Altamirano, F.; López, J.R.; Henríquez, C.; Molinski, T.; Allen, P.D.; Jaimovich, E. Increased resting intracellular calcium modulates NF-κB-dependent inducible nitric-oxide synthase gene expression in dystrophic mdx skeletal myotubes. J. Biol. Chem. 2012, 287, 20876–20887. [Google Scholar] [CrossRef] [PubMed]
- Wehrens, X.H.T.; Lehnart, S.E.; Reiken, S.R.; Marks, A.R. Ca2+/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ. Res. 2004, 94, e61–e70. [Google Scholar] [CrossRef] [PubMed]
- Shirokova, N.; Niggli, E. Cardiac phenotype of Duchenne Muscular Dystrophy: Insights from cellular studies. J. Mol. Cell. Cardiol. 2013, 58, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Shan, J.; Betzenhauser, M.J.; Kushnir, A.; Reiken, S.; Meli, A.C.; Wronska, A.; Dura, M.; Chen, B.X.; Marks, A.R. Role of chronic ryanodine receptor phosphorylation in heart failure and β-adrenergic receptor blockade in mice. J. Clin. Investig. 2010, 120, 4375. [Google Scholar] [CrossRef] [PubMed]
- Lohan, J.; Ohlendieck, K. Drastic reduction in the luminal Ca2+-binding proteins calsequestrin and sarcalumenin in dystrophin-deficient cardiac muscle. BBA-Mol. Basis Dis. 2004, 1689, 252–258. [Google Scholar] [CrossRef]
- Petrof, B.J.; Shrager, J.B.; Stedman, H.H.; Kelly, A.M.; Sweeney, H.L. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl. Acad. Sci. USA 1993, 90, 3710–3714. [Google Scholar] [CrossRef] [PubMed]
- Yeung, E.W.; Whitehead, N.P.; Suchyna, T.M.; Gottlieb, P.A.; Sachs, F.; Allen, D.G. Effects of stretch-activated channel blockers on [Ca2+]i and muscle damage in the mdx mouse. J. Physiol. 2005, 562, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, S.; Townsend, D.; Michele, D.E.; Favre, E.G.; Day, S.M.; Metzger, J.M. Dystrophic heart failure blocked by membrane sealant poloxamer. Nature 2005, 436, 1025–1029. [Google Scholar] [CrossRef] [PubMed]
- Fanchaouy, M.; Polakova, E.; Jung, C.; Ogrodnik, J.; Shirokova, N.; Niggli, E. Pathways of abnormal stress-induced Ca2+ influx into dystrophic mdx cardiomyocytes. Cell Calcium 2009, 46, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M.S.; Khakee, R.; McNeil, P.L. Loss of cytoplasmic basic fibroblast growth factor from physiologically wounded myofibers of normal and dystrophic muscle. J. Cell Sci. 1993, 106, 121–133. [Google Scholar] [PubMed]
- De Palma, C.; Clementi, E. Nitric oxide in myogenesis and therapeutic muscle repair. Mol. Neurobiol. 2012, 46, 682–692. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.N.; Duglan, D.; Casadei, B.; Carnicer, R. Nitric oxide synthase regulation of cardiac excitation-contraction coupling in health and disease. J. Mol. Cell. Cardiol. 2014, 73, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.H.; Casadei, B. Sub-cellular targeting of constitutive NOS in health and disease. J. Mol. Cell. Cardiol. 2012, 52, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Percival, J.M.; Siegel, M.P.; Knowels, G.; Marcinek, D.J. Defects in mitochondrial localization and ATP synthesis in the mdx mouse model of duchenne muscular dystrophy are not alleviated by PDE5 inhibition. Hum. Mol. Genet. 2013, 22, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Viola, H.M.; Arthur, P.G.; Hool, L.C. Evidence for regulation of mitochondrial function by the l-type Ca2+ channel in ventricular myocytes. J. Mol. Cell. Cardiol. 2009, 46, 1016–1026. [Google Scholar] [CrossRef] [PubMed]
- Viola, H.M.; Adams, A.M.; Davies, S.M.K.; Fletcher, S.; Filipovska, A.; Hool, L.C. Impaired functional communication between the l-type calcium channel and mitochondria contributes to metabolic inhibition in the mdx heart. Proc. Natl. Acad. Sci. USA 2014, E2905–E2914. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, S.; Zhang, X.; Li, J.; Ai, X.; Zhang, L.; Yu, D.; Ge, S.; Peng, Y.; Chen, X. Blunted cardiac beta-adrenergic response as an early indication of cardiac dysfunction in Duchenne muscular dystrophy. Cardiovasc. Res. 2014, 103, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Rohman, M.S.; Emoto, N.; Takeshima, Y.; Yokoyama, M.; Matsuo, M. Decreased mAKAP, ryanodine receptor, and SERCA2a gene expression in mdx hearts. Biochem. Biophys. Res. Commun. 2003, 310, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Respress, J.L.; van Oort, R.J.; Li, N.; Rolim, N.; Dixit, S.; DeAlmeida, A.; Voigt, N.; Lawrence, W.S.; Skapura, D.G.; Skardal, K.; et al. Role of RyR2 Phosphorylation at S2814 during Heart Failure Progression. Circ. Res. 2012, 110, 1474–1483. [Google Scholar] [CrossRef] [PubMed]
- Damy, T.; Ratajczak, P.; Shah, A.M.; Camors, E.; Marty, I.; Hasenfuss, G.; Marotte, F.; Samuel, J.L.; Heymes, C. Increased neuronal nitric oxide synthase-derived NO production in the failing human heart. Lancet 2004, 363, 1365–1367. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.J.; Iannaccone, S.T.; Lau, K.S.; Masters, B.S.; McCabe, T.J.; McMillan, K.; Padre, R.C.; Spencer, M.J.; Tidball, J.G.; Stull, J.T. Neuronal nitric oxide synthase and dystrophin-deficient muscular dystrophy. Proc. Natl. Acad. Sci. USA 1996, 93, 9142–9147. [Google Scholar] [CrossRef] [PubMed]
- Wallace, G.Q.; McNally, E.M. Mechanisms of muscle degeneration, regeneration, and repair in the muscular dystrophies. Annu. Rev. Physiol. 2009, 71, 37–57. [Google Scholar] [CrossRef] [PubMed]
- Lorin, C.; Vogeli, I.; Niggli, E. Dystrophic cardiomyopathy: Role of TRPV2 channels in stretch-induced cell damage. Cardiovasc. Res. 2015, 106, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Ward, M.L.; Williams, I.A.; Chu, Y.; Cooper, P.J.; Ju, Y.K.; Allen, D.G. Stretch-activated channels in the heart: Contributions to length-dependence and to cardiomyopathy. Prog. Biophys. Mol. Biol. 2008, 97, 232–249. [Google Scholar] [CrossRef] [PubMed]
- Harisseh, R.; Chatelier, A.; Magaud, C.; Déliot, N.; Constantin, B. Involvement of TRPV2 and SOCE in calcium influx disorder in DMD primary human myotubes with a specific contribution of α1-syntrophin and PLC/PKC in SOCE regulation. Am. J. Physiol. Cell Physiol. 2013, 304, C881–C894. [Google Scholar] [CrossRef] [PubMed]
- Blaustein, M.P.; Lederer, W.J. Sodium/calcium exchange: Its physiological implications. Physiol. Rev. 1999, 79, 763–854. [Google Scholar] [PubMed]
- Burr, A.R.; Millay, D.P.; Goonasekera, S.A.; Park, K.H.; Sargent, M.A.; Collins, J.; Altamirano, F.; Philipson, K.D.; Allen, P.D.; Ma, J.; et al. Na+ dysregulation coupled with Ca2+ entry through NCX1 promotes muscular dystrophy in mice. Mol. Cell. Biol. 2014, 34, 1991–2002. [Google Scholar] [CrossRef] [PubMed]
- Gavillet, B.; Rougier, J.S.; Domenighetti, A.A.; Behar, R.; Boixel, C.; Ruchat, P.; Lehr, H.A.; Pedrazzini, T.; Abriel, H. Cardiac sodium channel Nav1.5 is regulated by a multiprotein complex composed of syntrophins and dystrophin. Circ. Res. 2006, 99, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Dyachenko, V.; Husse, B.; Rueckschloss, U.; Isenberg, G. Mechanical deformation of ventricular myocytes modulates both TRPC6 and Kir2.3 channels. Cell Calcium 2009, 45, 38–54. [Google Scholar] [CrossRef] [PubMed]
- Seo, K.; Rainer, P.P.; Lee, D.I.; Hao, S.; Bedja, D.; Birnbaumer, L.; Cingolani, O.H.; Kass, D.A. Hyperactive adverse mechanical stress responses in dystrophic heart are coupled to transient receptor potential canonical 6 and blocked by cgmp-protein kinase g modulation. Circ. Res. 2014, 114, 823–832. [Google Scholar] [CrossRef] [PubMed]
- Millay, D.P.; Goonasekera, S.A.; Sargent, M.A.; Maillet, M.; Aronow, B.J.; Molkentin, J.D. Calcium influx is sufficient to induce muscular dystrophy through a TRPC-dependent mechanism. Proc. Natl. Acad. Sci. USA 2009, 106, 19023–19028. [Google Scholar] [CrossRef] [PubMed]
- Hohaus, A.; Person, V.; Behlke, J.; Schaper, J.; Morano, I.; Haase, H. The carboxyl-terminal region of ahnak provides a link between cardiac l-type Ca2+ channels and the actin-based cytoskeleton. FASEB J. 2002, 16, 1205–1216. [Google Scholar] [CrossRef] [PubMed]
- Sadeghi, A.; Doyle, A.D.; Johnson, B.D. Regulation of the cardiac l-type Ca2+ channel by the actin-binding proteins alpha-actinin and dystrophin. Am. J. Physiol. Cell Physiol. 2002, 282, C1502–C1511. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, T.; Beam, K.G.; Adams, B.A.; Niidome, T.; Numa, S. Regions of the skeletal muscle dihydropyridine receptor critical for excitation-contraction coupling. Nature 1990, 346, 567–569. [Google Scholar] [CrossRef] [PubMed]
- Koenig, X.; Rubi, L.; Obermair, G.J.; Cervenka, R.; Dang, X.B.; Lukacs, P.; Kummer, S.; Bittner, R.E.; Kubista, H.; Todt, H.; Hilber, K. Enhanced currents through l-type calcium channels in cardiomyocytes disturb the electrophysiology of the dystrophic heart. Am. J. Physiol. Heart Circ. Physiol. 2013, 306, H564–H573. [Google Scholar] [CrossRef] [PubMed]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell, 5th ed.; Garland Science: New York, NY, USA, 2007. [Google Scholar]
- Niggli, E.; Ullrich, N.D.; Gutierrez, D.; Kyrychenko, S.; Poláková, E.; Shirokova, N. Posttranslational modifications of cardiac ryanodine receptors: Ca2+ signaling and EC-coupling. BBA-Mol. Cell Res. 2013, 1833, 866–875. [Google Scholar]
- Marx, S.O.; Reiken, S.; Hisamatsu, Y.; Jayaraman, T.; Burkhoff, D.; Rosemblit, N.; Marks, A.R. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): Defective regulation in failing hearts. Cell 2000, 101, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Kyrychenko, S.; Poláková, E.; Kang, C.; Pocsai, K.; Ullrich, N.D.; Niggli, E.; Shirokova, N. Hierarchical accumulation of RyR post-translational modifications drives disease progression in dystrophic cardiomyopathy. Cardiovasc. Res. 2013, 97, 666–675. [Google Scholar] [CrossRef] [PubMed]
- Mijares, A.; Altamirano, F.; Kolster, J.; Adams, J.A.; López, J.R. Biochemical and Biophysical Research Communications Age-dependent changes in diastolic Ca2+ and Na+ concentrations in dystrophic cardiomyopathy: Role of Ca2+ entry and IP 3. Biochem. Biophys. Res. Commun. 2014, 452, 1054–1059. [Google Scholar] [CrossRef] [PubMed]
- Gervásio, O.L.; Whitehead, N.P.; Yeung, E.W.; Phillips, W.D.; Allen, D.G. TRPC1 binds to caveolin-3 and is regulated by Src kinase—role in Duchenne muscular dystrophy. J. Cell Sci. 2008, 121, 2246–2255. [Google Scholar] [CrossRef] [PubMed]
- Ashley, E.A.; Sears, C.E.; Bryant, S.M.; Watkins, H.C.; Casadei, B. Cardiac nitric oxide synthase 1 regulates basal and beta-adrenergic contractility in murine ventricular myocytes. Circulation 2002, 105, 3011–3016. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Kohr, M.J.; Traynham, C.J.; Wheeler, D.G.; Janssen, P.M.L.; Ziolo, M.T. Neuronal nitric oxide synthase signaling within cardiac myocytes targets phospholamban. Am. J. Physiol. Cell Physiol. 2008, 294, C1566–C1575. [Google Scholar] [CrossRef] [PubMed]
- Nisoli, E.; Falcone, S.; Tonello, C.; Cozzi, V.; Palomba, L.; Fiorani, M.; Pisconti, A.; Brunelli, S.; Cardile, A.; Francolini, M.; et al. Mitochondrial biogenesis by NO yields functionally active mitochondria in mammals. Proc. Natl. Acad. Sci. USA 2004, 101, 16507–16512. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.; Zhao, J.; Yue, Y.; Wasala, N.B.; Duan, D. Partial restoration of cardiac function with ΔPDZ nNOS in aged mdx model of Duchenne cardiomyopathy. Hum. Mol. Genet. 2014, 23, 3189–3199. [Google Scholar] [CrossRef] [PubMed]
- Alonso, M.T.; Villalobos, C.; Chamero, P.; Alvarez, J.; García-Sancho, J. Calcium microdomains in mitochondria and nucleus. Cell Calcium 2006, 40, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Ichas, F.; Jouaville, L.S.; Mazat, J.P. Mitochondria are excitable organelles capable of generating and conveying electrical and calcium signals. Cell 1997, 89, 1145–1153. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed]
- Khairallah, M.; Khairallah, R.; Young, M.E.; Dyck, J.R.B.; Petrof, B.J.; Des Rosiers, C. Metabolic and signaling alterations in dystrophin-deficient hearts precede overt cardiomyopathy. J. Mol. Cell. Cardiol. 2007, 43, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Khairallah, R.J.; Khairallah, M.; Gélinas, R.; Bouchard, B.; Young, M.E.; Allen, B.G.; Lopaschuk, G.D.; Deschepper, C.F.; Des Rosiers, C. Cyclic GMP signaling in cardiomyocytes modulates fatty acid trafficking and prevents triglyceride accumulation. J. Mol. Cell. Cardiol. 2008, 45, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P.; di Lisa, F. The mitochondrial permeability transition pore: Molecular nature and role as a target in cardioprotection. J. Mol. Cell. Cardiol. 2015, 78, 100–106. [Google Scholar] [PubMed]
- Pan, X.; Liu, J.; Nguyen, T.; Liu, C.; Sun, J.; Teng, Y.; Fergusson, M.M.; Rovira, I.I.; Allen, M.; Springer, D.A.; et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat. Cell Biol. 2013, 15, 1464–1472. [Google Scholar] [CrossRef] [PubMed]
- Millay, D.P.; Sargent, M.A.; Osinska, H.; Baines, C.P.; Barton, E.R.; Vuagniaux, G.; Sweeney, H.L.; Robbins, J.; Molkentin, J.D. Genetic and pharmacologic inhibition of mitochondrial-dependent necrosis attenuates muscular dystrophy. Nat. Med. 2008, 14, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Griggs, R.C.; Moxley, R.T.; Mendell, J.R.; Fenichel, G.M.; Brooke, M.H.; Pestronk, A.; Miller, J.P. Prednisone in Duchenne dystrophy. A randomized, controlled trial defining the time course and dose response. Clinical Investigation of Duchenne Dystrophy Group. Arch. Neurol. 1991, 48, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Biggar, W.D.; Harris, V.A.; Eliasoph, L.; Alman, B. Long-term benefits of deflazacort treatment for boys with Duchenne muscular dystrophy in their second decade. Neuromuscul. Disord. 2006, 16, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Barber, B.J.; Andrews, J.G.; Lu, Z.; West, N.A.; Meaney, F.J.; Price, E.T.; Gray, A.; Sheehan, D.W.; Pandya, S.; Yang, M.; et al. Oral corticosteroids and onset of cardiomyopathy in Duchenne muscular dystrophy. J. Pediatr. 2013, 163, 1080–1084. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.R.; Lechtzin, N.; Judge, D.P. Current treatment of adult Duchenne muscular dystrophy. Biochim. Biophys. Acta 2007, 1772, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Melacini, P.; Vianello, A.; Villanova, C.; Fanin, M.; Miorin, M.; Angelini, C.; Dalla Volta, S. Cardiac and respiratory involvement in advanced stage Duchenne muscular dystrophy. Neuromuscul. Disord. 1996, 6, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Finsterer, J. Cardiopulmonary support in Duchenne muscular dystrophy. Lung 2006, 184, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Toussaint, M.; Chatwin, M.; Soudon, P. Mechanical ventilation in Duchenne patients with chronic respiratory insufficiency: Clinical implications of 20 years published experience. Chron. Respir. Dis. 2007, 4, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Goemans, N.; Buyse, G. Current treatment and management of dystrophinopathies. Curr. Treat. Options Neurol. 2014, 16, 1–13. [Google Scholar] [CrossRef]
- Viollet, L.; Thrush, P.T.; Flanigan, K.M.; Mendell, J.R.; Allen, H.D. Effects of angiotensin-converting enzyme inhibitors and/or beta blockers on the cardiomyopathy in duchenne muscular dystrophy. Am. J. Cardiol. 2012, 110, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Ogata, H.; Ishikawa, Y.; Minami, R. Beneficial effects of beta-blockers and angiotensin-converting enzyme inhibitors in Duchenne muscular dystrophy. [Erratum appears in J Cardiol. 2009 Apr;53(2):316]. J. Cardiol. 2009, 53, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Blain, A.; Greally, E.; Laval, S.; Blamire, A.; Straub, V.; MacGowan, G.A. Beta-Blockers, Left and Right Ventricular Function, and in-Vivo Calcium Influx in Muscular Dystrophy Cardiomyopathy. PLoS ONE 2013, 8, e57260. [Google Scholar] [CrossRef] [PubMed]
- Fayssoil, A.; Nardi, O.; Annane, D.; Orlikowski, D. Successful cardiac resynchronisation therapy in Duchenne muscular dystrophy: A 5-year follow-up. Presse Med. 2014, 43, 330–331. [Google Scholar] [CrossRef] [PubMed]
- Ryan, T.D.; Jefferies, J.L.; Sawnani, H.; Wong, B.L.; Gardner, A.; Del Corral, M.; Lorts, A.; Morales, D.L.S. Implantation of the HeartMate II and HeartWare left ventricular assist devices in patients with duchenne muscular dystrophy: Lessons learned from the first applications. ASAIO J. 2014, 60, 246–248. [Google Scholar] [CrossRef] [PubMed]
- Spurney, C.F.; Guerron, A.D.; Yu, Q.; Sali, A.; van der Meulen, J.H.; Hoffman, E.P.; Nagaraju, K. Membrane sealant Poloxamer P188 protects against isoproterenol induced cardiomyopathy in dystrophin deficient mice. BMC Cardiovasc. Disord. 2011, 11, 20. [Google Scholar] [CrossRef] [PubMed]
- Khairallah, M.; Khairallah, R.J.; Young, M.E.; Allen, B.G.; Gillis, M.A.; Danialou, G.; Deschepper, C.F.; Petrof, B.J.; Des Rosiers, C. Sildenafil and cardiomyocyte-specific cGMP signaling prevent cardiomyopathic changes associated with dystrophin deficiency. Proc. Natl. Acad. Sci. USA 2008, 105, 7028–7033. [Google Scholar] [CrossRef] [PubMed]
- Adamo, C.M.; Dai, D.-F.; Percival, J.M.; Minami, E.; Willis, M.S.; Patrucco, E.; Froehner, S.C.; Beavo, J.A. Sildenafil reverses cardiac dysfunction in the mdx mouse model of Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 2010, 107, 19079–19083. [Google Scholar] [CrossRef] [PubMed]
- Percival, J.M.; Whitehead, N.P.; Adams, M.E.; Adamo, C.M.; Beavo, J.A.; Froehner, S.C. Sildenafil reduces respiratory muscle weakness and fibrosis in the mdx mouse model of Duchenne muscular dystrophy. J. Pathol. 2012, 228, 77–87. [Google Scholar] [PubMed]
- Leung, D.G.; Herzka, D.A.; Thompson, W.R.; He, B.; Bibat, G.; Tennekoon, G.; Russell, S.D.; Schuleri, K.H.; Lardo, A.C.; Kass, D.A.; et al. Sildenafil does not improve cardiomyopathy in Duchenne/Becker muscular dystrophy. Ann. Neurol. 2014, 76, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Witting, N.; Kruuse, C.; Nyhuus, B.; Prahm, K.; Citirak, G.; Lundgaard, S.; von Huth, S.; Vejlstrup, N.; Lindberg, U.; Krag, T.; Vissing, J. Effect of sildenafil on skeletal and cardiac muscle in Becker muscular dystrophy. Ann. Neurol. 2014, 76, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Spurney, C.F.; Sali, A.; Guerron, A.D.; Iantorno, M.; Yu, Q.; Gordish-Dressman, H.; Rayavarapu, S.; van der Meulen, J.; Hoffman, E.P.; Nagaraju, K. Losartan decreases cardiac muscle fibrosis and improves cardiac function in dystrophin-deficient mdx mice. J. Cardiovasc. Pharmacol. Ther. 2011, 16, 87–95. [Google Scholar] [CrossRef]
- Van Erp, C.; Irwin, N.G.; Hoey, A.J. Long-term administration of pirfenidone improves cardiac function in mdx mice. Muscle Nerve 2006, 34, 327–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vianello, S.; Consolaro, F.; Bich, C.; Cancela, J.M.; Roulot, M.; Lanchec, E.; Touboul, D.; Brunelle, A.; Israël, M.; Benoit, E.; et al. Low doses of arginine butyrate derivatives improve dystrophic phenotype and restore membrane integrity in DMD models. FASEB J. 2014, 28, 2603–2619. [Google Scholar] [CrossRef] [PubMed]
- Sciorati, C.; Staszewsky, L.; Zambelli, V.; Russo, I.; Salio, M.; Novelli, D.; Di Grigoli, G.; Moresco, R.M.; Clementi, E.; Latini, R. Ibuprofen plus isosorbide dinitrate treatment in the mdx mice ameliorates dystrophic heart structure. Pharmacol. Res. 2013, 73, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Cossu, M.V.; Cattaneo, D.; Fucile, S.; Pellegrino, P.; Baldelli, S.; Cozzi, V.; Capetti, A.; Clementi, E. Combined isosorbide dinitrate and ibuprofen as a novel therapy for muscular dystrophies: Evidence from Phase I studies in healthy volunteers. Drug Des. Devel. Ther. 2014, 8, 411–419. [Google Scholar] [PubMed]
- Tinsley, J.M.; Potter, A.C.; Phelps, S.R.; Fisher, R.; Trickett, J.I.; Davies, K.E. Amelioration of the dystrophic phenotype of mdx mice using a truncated utrophin transgene. Nature 1996, 384, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Tinsley, J.; Deconinck, N.; Fisher, R.; Kahn, D.; Phelps, S.; Gillis, J.M.; Davies, K. Expression of full-length utrophin prevents muscular dystrophy in mdx mice. Nat. Med. 1998, 4, 1441–1444. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, R.; Nalbanoglu, J.; Tinsley, J.M.; Massie, B.; Davies, K.E.; Karpati, G. Efficient utrophin expression following adenovirus gene transfer in dystrophic muscle. Biochem. Biophys. Res. Commun. 1998, 242, 244–247. [Google Scholar] [CrossRef] [PubMed]
- Corbi, N.; Libri, V.; Fanciulli, M.; Tinsley, J.M.; Davies, K.E.; Passananti, C. The artificial zinc finger coding gene “Jazz” binds the utrophin promoter and activates transcription. Gene Ther. 2000, 7, 1076–1083. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Tian, C.; Danialou, G.; Gilbert, R.; Petrof, B.J.; Karpati, G.; Nalbantoglu, J. Targeting artificial transcription factors to the utrophin A promoter: Effects on dystrophic pathology and muscle function. J. Biol. Chem. 2008, 283, 34720–34727. [Google Scholar] [CrossRef] [PubMed]
- Onori, A.; Pisani, C.; Strimpakos, G.; Monaco, L.; Mattei, E.; Passananti, C.; Corbi, N. UtroUp is a novel six zinc finger artificial transcription factor that recognises 18 base pairs of the utrophin promoter and efficiently drives utrophin upregulation. BMC Mol. Biol. 2013, 14, 3. [Google Scholar] [CrossRef] [PubMed]
- Amenta, A.R.; Yilmaz, A.; Bogdanovich, S.; McKechnie, B.A.; Abedi, M.; Khurana, T.S.; Fallon, J.R. Biglycan recruits utrophin to the sarcolemma and counters dystrophic pathology in mdx mice. Proc. Natl. Acad. Sci. USA 2011, 108, 762–767. [Google Scholar] [CrossRef] [PubMed]
- Tinsley, J.M.; Fairclough, R.J.; Storer, R.; Wilkes, F.J.; Potter, A.C.; Squire, S.E.; Powell, D.S.; Cozzoli, A.; Capogrosso, R.F.; Lambert, A.; et al. Daily treatment with SMTC1100, a novel small molecule utrophin upregulator, dramatically reduces the dystrophic symptoms in the mdx mouse. PLoS ONE 2011, 6, e19189. [Google Scholar] [CrossRef] [PubMed]
- Tinsley, J.; Robinson, N.; Davies, K.E. Safety, Tolerability, and Pharmacokinetics of SMT C1100, a 2-Arylbenzoxazole Utrophin Modulator, Following Single- and Multiple-Dose Administration to Healthy Male Adult Volunteers † Jon Tinsley, PhD. J. Clin. Pharmacol. 2015, 55, 698–707. [Google Scholar] [CrossRef] [PubMed]
- Barton-Davis, E.R.; Cordier, L.; Shoturma, D.I.; Leland, S.E.; Sweeney, H.L. Aminoglycoside antibiotics restore dystrophin function to skeletal muscles of mdx mice. J. Clin. Investig. 1999, 104, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Malik, V.; Rodino-Klapac, L.R.; Viollet, L.; Wall, C.; King, W.; Al-Dahhak, R.; Lewis, S.; Shilling, C.J.; Kota, J.; Serrano-Munuera, C.; et al. Gentamicin-induced readthrough of stop codons in Duchenne muscular dystrophy. Ann. Neurol. 2010, 67, 771–780. [Google Scholar] [PubMed]
- Arakawa, M.; Shiozuka, M.; Nakayama, Y.; Hara, T.; Hamada, M.; Kondo, S.; Ikeda, D.; Takahashi, Y.; Sawa, R.; Nonomura, Y.; et al. Negamycin Restores Dystrophin Expression in Skeletal and Cardiac Muscles of mdx Mice. J. Biochem. 2003, 134, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Welch, E.M.; Barton, E.R.; Zhuo, J.; Tomizawa, Y.; Friesen, W.J.; Trifillis, P.; Paushkin, S.; Patel, M.; Trotta, C.R.; Hwang, S.; et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature 2007, 447, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Hirawat, S.; Welch, E.M.; Elfring, G.L.; Northcutt, V.J.; Paushkin, S.; Hwang, S.; Leonard, E.M.; Almstead, N.G.; Ju, W.; Peltz, S.W.; et al. Safety, tolerability, and pharmacokinetics of PTC124, a nonaminoglycoside nonsense mutation suppressor, following single- and multiple-dose administration to healthy male and female adult volunteers. J. Clin. Pharmacol. 2007, 47, 430–444. [Google Scholar] [CrossRef] [PubMed]
- Finkel, R.S.; Flanigan, K.M.; Wong, B.; Bönnemann, C.; Sampson, J.; Sweeney, H.L.; Reha, A.; Northcutt, V.J.; Elfring, G.; Barth, J.; et al. Phase 2a study of ataluren-mediated dystrophin production in patients with nonsense mutation Duchenne muscular dystrophy. PLoS ONE 2013, 8, e81302. [Google Scholar] [CrossRef] [PubMed]
- Bushby, K.; Finkel, R.; Wong, B.; Barohn, R.; Campbell, C.; Comi, G.P.; Connolly, M.; Day, J.W.; Flanigan, K.M. Ataluren Treatment of Patients with Nonsense Mutation Dystrophinopathy. Muscle Nerve 2014, 50, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Kayali, R.; Ku, J.M.; Khitrov, G.; Jung, M.E.; Prikhodko, O.; Bertoni, C. Read-through compound 13 restores dystrophin expression and improves muscle function in the MDX mouse model for duchenne muscular dystrophy. Hum. Mol. Genet. 2012, 21, 4007–4020. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Kimura, E.; Fall, B.M.; Reyes, M.; Angello, J.C.; Welikson, R.; Hauschka, S.D.; Chamberlain, J.S. Stable transduction of myogenic cells with lentiviral vectors expressing a minidystrophin. Gene Ther. 2005, 12, 1099–1108. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ha, T.; Liu, L.; Zou, J.; Zhang, X.; Kalbfleisch, J.; Gao, X.; Williams, D.; Li, C. Increased expression of microRNA-146a decreases myocardial ischaemia/reperfusion injury. Cardiovasc. Res. 2013, 97, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Kimura, E.; Li, S.; Gregorevic, P.; Fall, B.M.; Chamberlain, J.S. Dystrophin delivery to muscles of mdx mice using lentiviral vectors leads to myogenic progenitor targeting and stable gene expression. Mol. Ther. 2010, 18, 206–213. [Google Scholar] [CrossRef] [PubMed]
- DelloRusso, C.; Scott, J.M.; Hartigan-O’Connor, D.; Salvatori, G.; Barjot, C.; Robinson, A.S.; Crawford, R.W.; Brooks, S.V.; Chamberlain, J.S. Functional correction of adult mdx mouse muscle using gutted adenoviral vectors expressing full-length dystrophin. Proc. Natl. Acad. Sci. USA 2002, 99, 12979–12984. [Google Scholar] [CrossRef] [PubMed]
- Foster, H.; Sharp, P.S.; Athanasopoulos, T.; Trollet, C.; Graham, I.R.; Foster, K.; Wells, D.J.; Dickson, G. Codon and mRNA sequence optimization of microdystrophin transgenes improves expression and physiological outcome in dystrophic mdx mice following AAV2/8 gene transfer. Mol. Ther. 2008, 16, 1825–1832. [Google Scholar] [CrossRef] [PubMed]
- Gregorevic, P.; Allen, J.M.; Minami, E.; Blankinship, M.J.; Haraguchi, M.; Meuse, L.; Finn, E.; Adams, M.E.; Froehner, S.C.; Murry, C.E.; et al. rAAV6-microdystrophin preserves muscle function and extends lifespan in severely dystrophic mice. Nat. Med. 2006, 12, 787–789. [Google Scholar] [CrossRef] [PubMed]
- Gregorevic, P.; Blankinship, M.J.; Allen, J.M.; Chamberlain, J.S. Systemic microdystrophin gene delivery improves skeletal muscle structure and function in old dystrophic mdx mice. Mol. Ther. 2008, 16, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Kornegay, J.N.; Li, J.; Bogan, J.R.; Bogan, D.J.; Chen, C.; Zheng, H.; Wang, B.; Qiao, C.; Howard, J.F.; Xiao, X. Widespread muscle expression of an AAV9 human mini-dystrophin vector after intravenous injection in neonatal dystrophin-deficient dogs. Mol. Ther. 2010, 18, 1501–1508. [Google Scholar] [CrossRef] [PubMed]
- Bostick, B.; Yue, Y.; Lai, Y.; Long, C.; Li, D.; Duan, D. Adeno-associated virus serotype-9 microdystrophin gene therapy ameliorates electrocardiographic abnormalities in mdx mice. Hum. Gene Ther. 2008, 19, 851–856. [Google Scholar] [CrossRef] [PubMed]
- Schinkel, S.; Bauer, R.; Bekeredjian, R.; Stucka, R.; Rutschow, D.; Lochmüller, H.; Kleinschmidt, J.A.; Katus, H.A.; Müller, O.J. Long-Term Preservation of Cardiac Structure and Function After Adeno-Associated Virus Serotype 9-Mediated Microdystrophin Gene Transfer in mdx Mice. Hum. Gene Ther. 2012, 23, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Partridge, T.A.; Morgan, J.E.; Coulton, G.R.; Hoffman, E.P.; Kunkel, L.M. Conversion of mdx myofibres from dystrophin-negative to -positive by injection of normal myoblasts. Nature 1989, 337, 176–179. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Kissel, J.T.; Amato, A.A.; King, W.; Signore, L.; Prior, T.W.; Sahenk, Z.; Benson, S.; McAndrew, P.E.; Rice, R. Myoblast transfer in the treatment of Duchenne’s muscular dystrophy. N. Engl. J. Med. 1995, 333, 832–838. [Google Scholar] [CrossRef] [PubMed]
- Muir, L.A.; Nguyen, Q.G.; Hauschka, S.D.; Chamberlain, J.S. Engraftment potential of dermal fibroblasts following in vivo myogenic conversion in immunocompetent dystrophic skeletal muscle. Mol. Ther. Methods Clin. Dev. 2014, 1, 14025. [Google Scholar] [CrossRef] [PubMed]
- Gussoni, E.; Soneoka, Y.; Strickland, C.D.; Buzney, E.A.; Khan, M.K.; Flint, A.F.; Kunkel, L.M.; Mulligan, R.C. Dystrophin expression in the mdx mouse restored by stem cell transplantation. Nature 1999, 401, 390–394. [Google Scholar] [PubMed]
- Dell’Agnola, C.; Wang, Z.; Storb, R.; Tapscott, S.J.; Kuhr, C.S.; Hauschka, S.D.; Lee, R.S.; Sale, G.E.; Zellmer, E.; Gisburne, S.; et al. Hematopoietic stem cell transplantation does not restore dystrophin expression in Duchenne muscular dystrophy dogs. Blood 2004, 104, 4311–4318. [Google Scholar] [CrossRef] [PubMed]
- Benchaouir, R.; Meregalli, M.; Farini, A.; D’Antona, G.; Belicchi, M.; Goyenvalle, A.; Battistelli, M.; Bresolin, N.; Bottinelli, R.; Garcia, L.; et al. Restoration of Human Dystrophin Following Transplantation of Exon-Skipping-Engineered DMD Patient Stem Cells into Dystrophic Mice. Cell Stem Cell 2007, 1, 646–657. [Google Scholar] [CrossRef] [PubMed]
- Torrente, Y.; Belicchi, M.; Marchesi, C.; D’Antona, G.; Cogiamanian, F.; Pisati, F.; Gavina, M.; Giordano, R.; Tonlorenzi, R.; Fagiolari, G.; et al. Autologous transplantation of muscle-derived CD133+ stem cells in Duchenne muscle patients. Cell Transplant. 2007, 16, 563–577. [Google Scholar] [CrossRef] [PubMed]
- Sampaolesi, M.; Blot, S.; D’Antona, G.; Granger, N.; Tonlorenzi, R.; Innocenzi, A.; Mognol, P.; Thibaud, J.-L.; Galvez, B.G.; Barthélémy, I.; et al. Mesoangioblast stem cells ameliorate muscle function in dystrophic dogs. Nature 2006, 444, 574–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goudenege, S.; Lamarre, Y.; Dumont, N.; Rousseau, J.; Frenette, J.; Skuk, D.; Tremblay, J.P. Laminin-111: A potential therapeutic agent for Duchenne muscular dystrophy. Mol. Ther. 2010, 18, 2155–2163. [Google Scholar] [CrossRef] [PubMed]
- Goudenege, S.; Lebel, C.; Huot, N.B.; Dufour, C.; Fujii, I.; Gekas, J.; Rousseau, J.; Tremblay, J.P. Myoblasts Derived From Normal hESCs and Dystrophic hiPSCs Efficiently Fuse with Existing Muscle Fibers Following Transplantation. Mol. Ther. 2012, 20, 2153–2167. [Google Scholar] [CrossRef] [PubMed]
- Darabi, R.; Arpke, R.W.; Irion, S.; Dimos, J.T.; Grskovic, M.; Kyba, M.; Perlingeiro, R.C.R. Human ES- and iPS-derived myogenic progenitors restore dystrophin and improve contractility upon transplantation in dystrophic mice. 2012, 10, 610–619. [Google Scholar]
- Lu, Q.L.; Mann, C.J.; Lou, F.; Bou-Gharios, G.; Morris, G.E.; Xue, S.; Fletcher, S.; Partridge, T.A.; Wilton, S.D. Functional amounts of dystrophin produced by skipping the mutated exon in the mdx dystrophic mouse. Nat. Med. 2003, 9, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.L.; Rabinowitz, A.; Chen, Y.C.; Yokota, T.; Yin, H.; Alter, J.; Jadoon, A.; Bou-Gharios, G.; Partridge, T. Systemic delivery of antisense oligoribonucleotide restores dystrophin expression in body-wide skeletal muscles. Proc. Natl. Acad. Sci. USA 2005, 102, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Flanigan, K.M.; Voit, T.; Rosales, X.Q.; Servais, L.; Kraus, J.E.; Wardell, C.; Morgan, A.; Dorricott, S.; Nakielny, J.; Quarcoo, N.; et al. Pharmacokinetics and safety of single doses of drisapersen in non-ambulant subjects with Duchenne muscular dystrophy: results of a double-blind randomized clinical trial. Neuromuscul. Disord. 2014, 24, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Goemans, N.M.; Tulinius, M.; van den Akker, J.T.; Burm, B.E.; Ekhart, P.F.; Heuvelmans, N.; Holling, T.; Janson, A.A.; Platenburg, G.J.; Sipkens, J.A.; et al. Systemic Administration of PRO051 in Duchenne’s Muscular Dystrophy. N. Engl. J. Med. 2011, 364, 1513–1522. [Google Scholar] [CrossRef] [PubMed]
- lter, J.; Lou, F.; Rabinowitz, A.; Yin, H.; Rosenfeld, J.; Wilton, S.D.; Partridge, T.A.; Lu, Q.L. Systemic delivery of morpholino oligonucleotide restores dystrophin expression bodywide and improves dystrophic pathology. Nat. Med. 2006, 12, 175–177. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Xiao, B.; Cloer, C.; Shaban, M.; Sali, A.; Lu, P.; Li, J.; Nagaraju, K.; Xiao, X.; Lu, Q.L. One-year treatment of morpholino antisense oligomer improves skeletal and cardiac muscle functions in dystrophic mdx mice. Mol. Ther. 2011, 19, 576–583. [Google Scholar] [CrossRef] [PubMed]
- Kinali, M.; Arechavala-Gomeza, V.; Feng, L.; Cirak, S.; Hunt, D.; Adkin, C.; Guglieri, M.; Ashton, E.; Abbs, S.; Nihoyannopoulos, P.; et al. Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: A single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol. 2009, 8, 918–928. [Google Scholar] [CrossRef] [PubMed]
- Cirak, S.; Arechavala-Gomeza, V.; Guglieri, M.; Feng, L.; Torelli, S.; Anthony, K.; Abbs, S.; Garralda, M.E.; Bourke, J.; Wells, D.J.; et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: An open-label, phase 2, dose-escalation study. Lancet 2011, 378, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Rodino-Klapac, L.R.; Sahenk, Z.; Roush, K.; Bird, L.; Lowes, L.P.; Alfano, L.; Gomez, A.M.; Lewis, S.; Kota, J.; et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann. Neurol. 2013, 74, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Goyenvalle, A.; Griffith, G.; Babbs, A.; El Andaloussi, S.; Ezzat, K.; Avril, A.; Dugovic, B.; Chaussenot, R.; Ferry, A.; Voit, T.; et al. Functional correction in mouse models of muscular dystrophy using exon-skipping tricyclo-DNA oligomers. Nat. Med. 2015, 21, 270–275. [Google Scholar] [PubMed]
- Wu, B.; Li, Y.; Morcos, P.A.; Doran, T.J.; Lu, P.; Lu, Q.L. Octa-guanidine morpholino restores dystrophin expression in cardiac and skeletal muscles and ameliorates pathology in dystrophic mdx mice. Mol. Ther. 2009, 17, 864–871. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Nakamura, A.; Nagata, T.; Saito, T.; Kobayashi, M.; Aoki, Y.; Echigoya, Y.; Partridge, T.; Hoffman, E.P.; Takeda, S. Extensive and prolonged restoration of dystrophin expression with vivo-morpholino-mediated multiple exon skipping in dystrophic dogs. Nucleic Acid Ther. 2012, 22, 306–315. [Google Scholar] [PubMed]
- Jearawiriyapaisarn, N.; Moulton, H.M.; Buckley, B.; Roberts, J.; Sazani, P.; Fucharoen, S.; Iversen, P.L.; Kole, R. Sustained dystrophin expression induced by peptide-conjugated morpholino oligomers in the muscles of mdx mice. Mol. Ther. 2008, 16, 1624–1629. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Moulton, H.M.; Iversen, P.L.; Jiang, J.; Li, J.; Li, J.; Spurney, C.F.; Sali, A.; Guerron, A.D.; Nagaraju, K.; et al. Effective rescue of dystrophin improves cardiac function in dystrophin-deficient mice by a modified morpholino oligomer. Proc. Natl. Acad. Sci. USA 2008, 105, 14814–14819. [Google Scholar] [CrossRef] [PubMed]
- Moulton, H.M.; Fletcher, S.; Neuman, B.W.; McClorey, G.; Stein, D.A.; Abes, S.; Wilton, S.D.; Buchmeier, M.J.; Lebleu, B.; Iversen, P.L. Cell-penetrating peptide-morpholino conjugates alter pre-mRNA splicing of DMD (Duchenne muscular dystrophy) and inhibit murine coronavirus replication in vivo. Biochem. Soc. Trans. 2007, 35, 826–828. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, S.; Honeyman, K.; Fall, A.M.; Harding, P.L.; Johnsen, R.D.; Steinhaus, J.P.; Moulton, H.M.; Iversen, P.L.; Wilton, S.D. Morpholino oligomer-mediated exon skipping averts the onset of dystrophic pathology in the mdx mouse. Mol. Ther. 2007, 15, 1587–1592. [Google Scholar] [CrossRef] [PubMed]
- Goyenvalle, A.; Babbs, A.; Powell, D.; Kole, R.; Fletcher, S.; Wilton, S.D.; Davies, K.E. Prevention of dystrophic pathology in severely affected dystrophin/utrophin-deficient mice by morpholino-oligomer-mediated exon-skipping. Mol. Ther. 2010, 18, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Deconinck, A.E.; Rafael, J.A.; Skinner, J.A.; Brown, S.C.; Potter, A.C.; Metzinger, L.; Watt, D.J.; Dickson, J.G.; Tinsley, J.M.; Davies, K.E. Utrophin-dystrophin-deficient mice as a model for Duchenne muscular dystrophy. Cell 1997, 90, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Moulton, H.M.; Seow, Y.; Boyd, C.; Boutilier, J.; Iverson, P.; Wood, M.J.A. Cell-penetrating peptide-conjugated antisense oligonucleotides restore systemic muscle and cardiac dystrophin expression and function. Hum. Mol. Genet. 2008, 17, 3909–3918. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.P.; Youngblood, D.S.; Hassinger, J.N.; Lovejoy, C.E.; Nelson, M.H.; Iversen, P.L.; Moulton, H.M. Cell-penetrating peptides as transporters for morpholino oligomers: Effects of amino acid composition on intracellular delivery and cytotoxicity. Nucleic Acids Res. 2007, 35, 5182–5191. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Saleh, A.F.; Betts, C.; Camelliti, P.; Seow, Y.; Ashraf, S.; Arzumanov, A.; Hammond, S.; Merritt, T.; Gait, M.J.; et al. Pip5 transduction peptides direct high efficiency oligonucleotide-mediated dystrophin exon skipping in heart and phenotypic correction in mdx mice. Mol. Ther. 2011, 19, 1295–1303. [Google Scholar] [CrossRef] [PubMed]
- Betts, C.; Saleh, A.F.; Arzumanov, A.A.; Hammond, S.M.; Godfrey, C.; Coursindel, T.; Gait, M.J.; Wood, M.J. Pip6-PMO, A New Generation of Peptide-oligonucleotide Conjugates With Improved Cardiac Exon Skipping Activity for DMD Treatment. Mol. Ther. Nucleic Acids 2012, 1, e38. [Google Scholar] [CrossRef] [PubMed]
- Betts, C.A.; Saleh, A.F.; Carr, C.A.; Hammond, S.M.; Coenen-Stass, A.M.L.; Godfrey, C.; McClorey, G.; Varela, M.A.; Roberts, T.C.; Clarke, K.; et al. Prevention of exercise induced cardiomyopathy following Pip-PMO treatment in dystrophic mdx mice. Sci. Rep. 2015, 5, 8986. [Google Scholar] [CrossRef] [PubMed]
- Samoylova, T.I.; Smith, B.F. Elucidation of muscle-binding peptides by phage display screening. Muscle Nerve 1999, 22, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Lu, Q.; Wood, M. Effective exon skipping and restoration of dystrophin expression by peptide nucleic acid antisense oligonucleotides in mdx mice. Mol. Ther. 2008, 16, 38–45. [Google Scholar] [CrossRef]
- Yin, H.; Moulton, H.M.; Betts, C.; Seow, Y.; Boutilier, J.; Iverson, P.L.; Wood, M.J.A. A fusion peptide directs enhanced systemic dystrophin exon skipping and functional restoration in dystrophin-deficient mdx mice. Hum. Mol. Genet. 2009, 18, 4405–4414. [Google Scholar] [CrossRef] [PubMed]
- Seow, Y.; Yin, H.; Wood, M.J.A. Identification of a novel muscle targeting peptide in mdx mice. Peptides 2010, 31, 1873–1877. [Google Scholar] [CrossRef] [PubMed]
- Jirka, S.M.G.; Heemskerk, H.; Tanganyika-de Winter, C.L.; Muilwijk, D.; Pang, K.H.; de Visser, P.C.; Janson, A.; Karnaoukh, T.G.; Vermue, R.; ’t Hoen, P.A.C.; et al. Peptide Conjugation of 2′-O-methyl Phosphorothioate Antisense Oligonucleotides Enhances Cardiac Uptake and Exon Skipping in mdx Mice. Nucleic Acid Ther. 2014, 24, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Rybakova, I.N.; Ervasti, J.M. Identification of spectrin-like repeats required for high affinity utrophin-actin interaction. J. Biol. Chem. 2005, 280, 23018–23023. [Google Scholar] [CrossRef] [PubMed]
- Fairclough, R.J.; Bareja, A.; Davies, K.E. Progress in therapy for Duchenne muscular dystrophy. Exp. Physiol. 2011, 96, 1101–1113. [Google Scholar] [CrossRef] [PubMed]
- Tinsley, J.M.; Blake, D.J.; Roche, A.; Fairbrother, U.; Riss, J.; Byth, B.C.; Knight, A.E.; Kendrick-Jones, J.; Suthers, G.K.; Love, D.R. Primary structure of dystrophin-related protein. Nature 1992, 360, 591–593. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa-Sakurai, M.; Yoshida, M.; Imamura, M.; Davies, K.E.; Ozawa, E. ZZ domain is essentially required for the physiological binding of dystrophin and utrophin to beta-dystroglycan. Hum. Mol. Genet. 2004, 13, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Deconinck, N.; Rafael, J.A.; Beckers-Bleukx, G.; Kahn, D.; Deconinck, A.E.; Davies, K.E.; Gillis, J.M. Consequences of the combined deficiency in dystrophin and utrophin on the mechanical properties and myosin composition of some limb and respiratory muscles of the mouse. Neuromuscul. Disord. 1998, 8, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Verhaart, I.E.C.; van Duijn, R.J.M.; den Adel, B.; Roest, A.A.W.; Verschuuren, J.J.G.M.; Aartsma-Rus, A.; van der Weerd, L. Assessment of cardiac function in three mouse dystrophinopathies by magnetic resonance imaging. Neuromuscul. Disord. 2012, 22, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Chun, J.L.; O’Brien, R.; Berry, S.E. Cardiac dysfunction and pathology in the dystrophin and utrophin-deficient mouse during development of dilated cardiomyopathy. Neuromuscul. Disord. 2012, 22, 368–379. [Google Scholar] [CrossRef] [PubMed]
- Fairclough, R.J.; Wood, M.J.; Davies, K.E. Therapy for Duchenne muscular dystrophy: Renewed optimism from genetic approaches. Nat. Rev. Genet. 2013, 14, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Finkel, R.S. Read-through strategies for suppression of nonsense mutations in Duchenne/Becker muscular dystrophy: aminoglycosides and ataluren (PTC124). J. Child Neurol. 2010, 25, 1158–1164. [Google Scholar] [CrossRef] [PubMed]
- Konieczny, P.; Swiderski, K.; Chamberlain, J.S. Gene and cell-mediated therapies for muscular dystrophy. Muscle Nerve 2013, 47, 649–663. [Google Scholar] [CrossRef] [PubMed]
- Muntoni, F.; Wood, M.J. A Targeting RNA to treat neuromuscular disease. Nat. Rev. Drug Discov. 2011, 10, 621–637. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.M.; Li, S.; Harper, S.Q.; Welikson, R.; Bourque, D.; DelloRusso, C.; Hauschka, S.D.; Chamberlain, J.S. Viral vectors for gene transfer of micro-, mini-, or full-length dystrophin. Neuromuscul. Disord. 2002, 12, 23–29. [Google Scholar] [CrossRef]
- Asokan, A.; Schaffer, D.V.; Samulski, R.J. The AAV Vector Toolkit: Poised at the Clinical Crossroads. Mol. Ther. 2012, 20, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Rodino-Klapac, L.; Sahenk, Z.; Malik, V.; Kaspar, B.K.; Walker, C.M.; Clark, K.R. Gene therapy for muscular dystrophy: Lessons learned and path forward. Neurosci. Lett. 2012, 527, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Foster, H.; Popplewell, L.; Dickson, G. Genetic Therapeutic Approaches for Duchenne Muscular Dystrophy. Hum. Gene Ther. 2012, 23, 676–687. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Kuhr, C.S.; Allen, J.M.; Blankinship, M.; Gregorevic, P.; Chamberlain, J.S.; Tapscott, S.J.; Storb, R. Sustained AAV-mediated dystrophin expression in a canine model of Duchenne muscular dystrophy with a brief course of immunosuppression. Mol. Ther. 2007, 15, 1160–1166. [Google Scholar] [PubMed]
- Skuk, D.; Goulet, M.; Roy, B.; Tremblay, J.P. Efficacy of myoblast transplantation in nonhuman primates following simple intramuscular cell injections: toward defining strategies applicable to humans. Exp. Neurol. 2002, 175, 112–126. [Google Scholar] [CrossRef] [PubMed]
- Huard, C.; Moisset, P.A.; Dicaire, A.; Merly, F.; Tardif, F.; Asselin, I.; Tremblay, J.P. Transplantation of dermal fibroblasts expressing MyoD1 in mouse muscles. Biochem. Biophys. Res. Commun. 1998, 248, 648–654. [Google Scholar] [CrossRef] [PubMed]
- Goodell, M.A.; Brose, K.; Paradis, G.; Conner, A.S.; Mulligan, R.C. Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. J. Exp. Med. 1996, 183, 1797–1806. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, J.; Chapdelaine, P.; Boisvert, S.; Almeida, L.P.; Corbeil, J.; Montpetit, A.; Tremblay, J.P. Endonucleases: Tools to correct the dystrophin gene. J. Gene Med. 2011, 13, 522–537. [Google Scholar] [CrossRef] [PubMed]
- Li, H.L.; Fujimoto, N.; Sasakawa, N.; Shirai, S.; Ohkame, T.; Sakuma, T.; Tanaka, M.; Amano, N.; Watanabe, A.; Sakurai, H.; et al. Precise Correction of the Dystrophin Gene in Duchenne Muscular Dystrophy Patient Induced Pluripotent Stem Cells by TALEN and CRISPR-Cas9. Stem Cell Rep. 2015, 4, 143–154. [Google Scholar] [CrossRef] [Green Version]
- Alberts, B.; Johnson, A.; Lewis, J.; Morgan, D.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell, 6th ed.; Garland Science: New York, NY, USA, 2014. [Google Scholar]
- Popplewell, L.; Koo, T.; Leclerc, X.; Duclert, A.; Mamchaoui, K.; Gouble, A.; Mouly, V.; Voit, T.; Pâques, F.; Cédrone, F.; et al. Gene correction of a duchenne muscular dystrophy mutation by meganuclease-enhanced exon knock-in. Hum. Gene Ther. 2013, 24, 692–701. [Google Scholar] [CrossRef] [PubMed]
- Wilton, S.D.; Lloyd, F.; Carville, K.; Fletcher, S.; Honeyman, K.; Agrawal, S.; Kole, R. Specific removal of the nonsense mutation from the mdx dystrophin mRNA using antisense oligonucleotides. Neuromuscul. Disord. 1999, 9, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Cartegni, L.; Chew, S.L.; Krainer, A.R. Listening to silence and understanding nonsense: Exonic mutations that affect splicing. Nat. Rev. Genet. 2002, 3, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; Fokkema, I.; Verschuuren, J.; Ginjaar, I.; van Deutekom, J.; van Ommen, G.J.; den Dunnen, J.T. Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Hum. Mutat. 2009, 30, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Voit, T.; Topaloglu, H.; Straub, V.; Muntoni, F.; Deconinck, N.; Campion, G.; de Kimpe, S.J.; Eagle, M.; Guglieri, M.; Hood, S.; et al. Safety and efficacy of drisapersen for the treatment of Duchenne muscular dystrophy (DEMAND II): an exploratory, randomised, placebo-controlled phase 2 study. Lancet Neurol. 2014, 13, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Malerba, A.; Thorogood, F.C.; Dickson, G.; Graham, I.R. Dosing regimen has a significant impact on the efficiency of morpholino oligomer-induced exon skipping in mdx mice. Hum. Gene Ther. 2009, 20, 955–965. [Google Scholar] [CrossRef] [PubMed]
- Malerba, A.; Sharp, P.S.; Graham, I.R.; Arechavala-Gomeza, V.; Foster, K.; Muntoni, F.; Wells, D.J.; Dickson, G. Chronic systemic therapy with low-dose morpholino oligomers ameliorates the pathology and normalizes locomotor behavior in mdx mice. Mol. Ther. 2011, 19, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Negishi, Y.; Hamano, N.; Shiono, H.; Akiyama, S.; Endo-Takahashi, Y.; Suzuki, R.; Maruyama, K.; Aramaki, Y. The development of an ultrasound-mediated nucleic acid delivery system for treating muscular dystrophies. Yakugaku Zasshi 2012, 132, 1383–1388. [Google Scholar] [CrossRef] [PubMed]
- Bestas, B.; McClorey, G.; Tedebark, U.; Moreno, P.M.D.; Roberts, T.C.; Hammond, S.M.; Smith, C.I.E.; Wood, M.J.A.; Andaloussi, S. El Design and application of bispecific splice-switching oligonucleotides. Nucleic Acid Ther. 2014, 24, 13–24. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Van Westering, T.L.E.; Betts, C.A.; Wood, M.J.A. Current Understanding of Molecular Pathology and Treatment of Cardiomyopathy in Duchenne Muscular Dystrophy. Molecules 2015, 20, 8823-8855. https://doi.org/10.3390/molecules20058823
AMA Style
Van Westering TLE, Betts CA, Wood MJA. Current Understanding of Molecular Pathology and Treatment of Cardiomyopathy in Duchenne Muscular Dystrophy. Molecules. 2015; 20(5):8823-8855. https://doi.org/10.3390/molecules20058823
Chicago/Turabian Style
Van Westering, Tirsa L. E., Corinne A. Betts, and Matthew J. A. Wood. 2015. "Current Understanding of Molecular Pathology and Treatment of Cardiomyopathy in Duchenne Muscular Dystrophy" Molecules 20, no. 5: 8823-8855. https://doi.org/10.3390/molecules20058823
Article Metrics
Article Access Statistics
For more information on the journal statistics, click here.
Multiple requests from the same IP address are counted as one view.
We use cookies on our website to ensure you get the best experience.
Read more about our cookies here.