Hematopoietic Stem Cell Transplantation (HSCT): Practice Essentials, Historical Background, Indications for HSCT (original) (raw)
Practice Essentials
Hematopoietic stem cell transplantation (HSCT) involves the intravenous infusion of hematopoietic stem cells in order to reestablish blood cell production in patients whose bone marrow or immune system is damaged or defective. Over the past half century, this technique has been used with increasing frequency to treat numerous malignant and nonmalignant diseases.
Cells for HSCT may be obtained from the patient himself or herself (autologous transplant) or from another person, such as a sibling or unrelated donor (allogeneic transplant) or an identical twin (syngeneic transplant). Cell sources include bone marrow; peripheral blood; umbilical cord blood; or, rarely, fetal liver. Each of those cell sources has specific advantages and disadvantages, and each has found particular clinical applications.
Allogeneic donor types include the following:
- Matched related donors
- Matched unrelated donors
- Mismatched related donors
- Haploidentical donors
- Umbilical cord blood donors
The list of diseases for which HSCT is being used is rapidly increasing, and currently numbers more than 70. [1] More than half of autologous transplantations are performed for multiple myeloma and non-Hodgkin lymphoma, and the vast majority of allogeneic transplants are performed for hematologic and lymphoid cancers.
Worldwide, approximately 90,000 first HSCTs—53% autologous and 47% allogeneic—are performed every year, according to the World Wide Network of Blood and Marrow Transplantation. [1] The number continues to increase by 10-20% annually, and reductions in organ damage, infection, and severe, acute graft versus host disease (GVHD) seem to be contributing to improved outcomes. [2]
An important barrier to HSCT has been the inability to secure a suitable donors. The National Marrow Donor Program (NMDP), founded in 1986, and the World Marrow Donor Association (WMDA), founded in 1988, were established to (1) locate and secure appropriate unrelated-donor HSCT sources for patients by promoting volunteer donation of bone marrow and peripheral blood stem cells in the community and (2) promote ethical practices of sharing stem cell sources by need, rather than by geographic location of the donor. This, along with the development of unrelated cord blood transplantation and familial haploidentical transplantation methods, have improved the likelihood of finding an appropriate HSCT source in a timely manner.
The transplant process
The transplant process generally is divided into the following 5 phases:
- Conditioning
- Stem cell infusion
- Neutropenic phase
- Engraftment phase
- Postengraftment period
The conditioning period typically lasts 7-14 days. The purpose is to deliver chemotherapy, immunotherapy, and/or radiation to eliminate malignancy, prevent rejection of new stem cells, and create space for the new cells. Conditioning regimens may be classified as follows:
- Myeloablative regimens – These are designed to kill all residual cancer cells in autologous or allogeneic transplantation and to cause immunosuppression for engraftment in allogeneic transplantation; they may be further subclassified as radiation-containing or non–radiation-containing
- Nonmyeloablative regimens – These are immunosuppressive but not myeloablative and rely on the graft-versus-tumor effect to kill tumor cells with donor T cells
Stem cell infusion is a relatively simple process that is performed at the bedside. Minimal toxicity is observed in most cases.
During the neutropenic phase (2-4 wk) the patient essentially has no effective immune system. Healing is poor, and susceptibility to infection is high. Supportive care and empiric antibiotic therapy are the mainstays of successful passage through this phase.
During the engraftment phase, which lasts several weeks, the healing process begins with resolution of mucositis and other acquired lesions. In addition, fever begins to subside, and infections often begin to clear. The greatest challenges at this time include management of GVHD and prevention of viral infections, especially cytomegalovirus (CMV).
Hallmarks of the postengraftment period, which lasts for months to years, include the gradual development of tolerance, weaning off of immunosuppression, management of chronic GVHD, and documentation of immune reconstitution. Patients who receive transplants using T-cell depletion or from mismatched or haplotypic sources often have delayed or incomplete immune reconstitution, and patients who received total-body irradiation (TBI) as part of their conditioning regimen often have significant splenic dysfunction.
Antibody titers to vaccine-preventable diseases decline after autologous or allogeneic HSCT. Revaccination schedules must be tailored to the individual patient scenario (eg, transplant type, GVHD status or immunosuppression).
Patient education
For patient education information, see Stem Cell Transplants for Blood Cancers. Patient education information is also available at Be The Match, operated by the National Marrow Donor Program and Stem Cell or Bone Marrow Transplant from the American Cancer Society.
![]()
Historical Background
The starting point of HSCT may be traced to a 1939 report describing a patient who received 18 mL of bone marrow intravenously from his brother as treatment of aplastic anemia. Following this report, major advances in transplantation science occurred in the 1950s. [3] The following fundamental concepts of HSCT were discovered:
- Mice could be protected from lethal effects of whole-body irradiation by shielding the spleen or by intravenous bone marrow infusions
- Barnes and Loutit’s observation of antileukemic effects and graft versus host disease effects of transplanted spleen cells in murine models
- Thomas’ bone marrow transplantation in dogs using high-dose irradiation as conditioning
- Dausset and Van Rood’s unraveling of the human leukocyte antigen (HLA) system in humans
In the 1950s and 1960s, almost 200 allogeneic marrow transplants were performed in humans, with no long-term successes. However, during this time, transplantation using identical twin donors brought a fair amount of success and provided a crucial foundation to continued clinical research in the field. [1, 2]
These advances culminated in Thomas’ 1959 syngeneic bone marrow transplantation in 2 patients with acute lymphoblastic leukemia resulting in successful grafts. For his contribution in the development of bone marrow transplantation, Thomas shared the Nobel Prize in physiology in medicine with Joseph Murray, a surgeon instrumental in the development of kidney transplantation. [3]
Since the 1950s, many advances have been made in improving the success of bone marrow transplantation. Among the most important have been in selecting hematopoietic stem cell donors and the tissue source, optimizing transplantation conditioning, reducing the morbidity and mortality from transplantation conditioning, and preventing and treating graft versus host disease.
Bone marrow
In 1968, the first major landmark in bone marrow transplantation occurred with successful allogeneic transplantations performed for an infant with X-linked lymphopenic immune deficiency and for another with Wiskott-Aldrich syndrome. [4, 3] These successes were followed by reports of effective transplantation for aplastic anemia and, later, for leukemia. [5] Advances in histocompatibility testing and development of marrow donor registries, such as the National Marrow Donor Program (NMDP), have facilitated the use of unrelated donors, thus expanding the number of patients who can receive transplants.
Cord blood
Transplantation of umbilical cord blood was successfully performed for the first time in 1988, in a young boy with Fanconi anemia using umbilical cord blood collected at the birth of his sibling. [6] The patient remains alive and well to this date. In 1992, a patient was successfully transplanted with cord blood instead of bone marrow for the treatment of leukemia. Over the subsequent decades, the use of cord blood has rapidly expanded; several thousand transplants have been performed using cord blood as a stem cell source. [7] Cord blood transplants have been used for any disease state for which bone marrow can be used.
The 5-year leukemia-free survival rate in 503 children with acute lymphoblastic leukemia (ALL) who received a transplant of umbilical cord blood that was mismatched for either one or two human leukocyte antigens (HLA) was similar when compared with the survival rate of 282 children who received bone marrow transplants. [8]
The limited number of cells in cord blood traditionally restricted its use to children and small adults. Techniques studied for making this source available to a wider range of patients have included transfusion of multiple units, ex vivo expansion of the cord blood unit before transplantation, and exposure of the cord blood graft to compounds intended to improve homing and engraftment. [9]
Peripheral blood stem cells
Peripheral blood stem cells (PBSCs) have gained popularity as a source of stem cells since their initial introduction in the 1980s. Over time, peripheral blood stem cells—mobilized from the bone marrow by cytokine treatment and collected by apheresis—have come to account for the majority of HSCTs, as this process results in a much higher content of stem cells than in bone marrow. [10]
![]()
Indications for HSCT
HSCT is often performed as part of therapy to eliminate a bone marrow infiltrative process, such as leukemia, or to correct congenital immunodeficiency disorders. In addition, HSCT is used to allow patients with cancer to receive higher doses of chemotherapy than bone marrow can usually tolerate; bone marrow function is then salvaged by replacing the marrow with previously harvested stem cells. Examples of emerging indications for HSCT include replacement of marrow progenitors for the purpose of making normal red cells (eg, in hemoglobinopathies), making corrective enzymes (eg, in storage disorders), and mediating tissue repair (eg, in epidermolysis bullosa).
The indications for HSCT vary according to disease categories and are influenced by factors such as the following:
- Cytogenetic abnormalities
- Response to prior therapy
- Patient age and performance status
- Disease status (remission vs relapse)
- Disease-specific prognostic factors
- Most importantly, availability of a suitable graft source, time of referral, and time to transplant
Autologous HSCT is used to treat the following conditions:
- Germ cell tumors
Allogeneic HSCT is used to treat the following disorders:
- Wiskott-Aldrich syndrome
- Hemophagocytic lymphohistiocytosis (HLH)
- Inborn errors of metabolism - Eg, adrenoleukodystrophy, Gaucher disease, Hurler syndrome, metachromatic leukodystrophy, mucopolysaccharidosis, osteopetrosis
- Severe congenital neutropenia
- Shwachman-Diamond syndrome
- Diamond-Blackfan anemia
- Leukocyte adhesion deficiency
Indications for HSCT in specific diseases
Acute myeloid leukemia
Allogeneic HSCT is the treatment of choice for most children with AML in their first complete remission (CR1) who have a human leukocyte antigen (HLA) ̶ matched sibling, except for those with favorable cytogenetic characteristics or for Down syndrome. In adults, HSCT is reserved for those with high-risk features who are in their CR1 and for those with standard or good risk features, in their second complete remission (CR2). HSCT is the only curative option for patients with primary refractory or relapsed AML.
Acute lymphoblastic leukemia
Indications for stem cell transplantation in adults with ALL are similar to those for persons with AML.Recommendations for considering HSCT in pediatric patients with ALL vary according to cytogenetics, minimimal residual disease state, and response to pharmacologic therapy. [12]
Chronic myeloid leukemia
Allogeneic transplantation for many years was the standard of care for patients with CML who had donors, and even now it offers the potential for cure for advanced stages of the disease. However, the advent of tyrosine kinase inhibitor (TKI) therapy, starting with imatinib, has altered therapy significantly for patients with CML. In current treatment models, TKIs are considered first-line therapy for CML and can produce lasting cytogenetic remission. HSCT is used when TKI therapy fails or for patients who have not achieved an optimal response. [13]
Myelodysplastic syndrome
Allogeneic HSCT should be considered for patients with myelodysplastic syndrome who are younger than 60 years and who have an HLA-matched sibling donor.
Chronic lymphocytic leukemia
Autologous and allogeneic HSCT have been used with success in young patients with CLL.
Non-Hodgkin lymphoma
A combination of high-dose chemotherapy and autologous or allogeneic HSCT has produced complete remissions in patients with relapsed disease and in patients who have not achieved complete remission with primary therapy. [6]
Hodgkin lymphoma
High-dose chemotherapy and autologous HSCT are the treatments of choice for patients with poor prognoses and early relapse after initial chemotherapy or induction failure (ie, resistant disease), a second relapse after conventional treatment for the first relapse, or a generalized systemic relapse after initial chemotherapy. [14]
Multiple myeloma
Although HSCT does not cure multiple myeloma, high-dose chemotherapy with early autologous HSCT is recommended for these patients, as it has demonstrated a significant survival advantage compared with conventional chemotherapy. Whether double autologous HSCT (tandem transplants) provides an advantage remains uncertain. [15]
Neuroblastoma
Autologous transplantation (typically tandem transplants) is the current backbone of therapy for patients with high-risk neuroblastoma in CR1. [16, 17, 18] Allogeneic transplantation has been reported for patients with relapsing or refractory disease, but no standard guidelines for its use are available.
Sickle cell disease
Allogeneic HSCT may cure sickle cell disease. Lack of matched sibling donors has limited its use, but better conditioning regimens and GVHD prophylaxis, as well as the use of unrelated donors and related haploidentical donors, has expanded its use. [19, 20]
Other disorders
HSCT may also be effective in the treatment of the following conditions:
- Breast cancer - HSCT in breast cancer patients is controversial and currently has limited application.
- Testicular cancer - Autologous HSCT is used in refractory or recurrent cases [21]
- Thalassemia - An 80% disease-free survival rate is achieved after allogeneic HSCT [22]
- Genetic disorders - Many genetic immunologic or hematopoietic disorders are potentially curable with allogeneic HSCT
Results of HSCT in patients with acquired immunodeficiency syndrome (AIDS) have not been very encouraging. However, transfecting the hematopoietic stem cells with retroviral vectors that cleave the ribonucleic acid (RNA) of the human immunodeficiency virus (HIV) is in the experimental stage. In addition, HSCT transplantation using donors who have a mutation in the receptor CCR5 has led to successful HSCT in situations in which the lymphocytes generated from transplantation are resistant to HIV infection.
Table 1 (below) summarizes the common indications for hematopoietic stem cell transplantation. Cord blood transplants are being used for many of the allogeneic transplant indications whenever a suitable HLA-matched donor is unavailable or whenever time for identifying, typing, and harvesting a transplant from an unrelated donor is limited.
Table 1. Common Indications for HSCT [8] (Open Table in a new window)
![]()
Prognosis
HSCT has led to the cure of diverse forms of cancer, bone marrow failure, hereditary metabolic disorders, hemoglobinopathies, and severe congenital immunodeficiencies that would otherwise have been fatal. Transplantation-related mortality and morbidity rates have considerably decreased because of improved conditioning regimens, human leukocyte antigen (HLA) typing, supportive care, and prevention and treatment of serious infections. Currently, overall and event-free survival rates are based on the individual's disease pathology and on the stage of disease.
However, interpretation of the results of HSCT trials is always complicated by the problem of patient selection. The efficacy of transplantation can be underestimated if only patients with the worst prognoses are studied, or it can be overestimated if only those with the best prognoses are studied.
Table 2 (below) lists survival rates for patients with different diseases after HSCT. Patients undergoing HLA-matched sibling allogeneic transplantation have the best 5-year survival rate of all treated patients. These data need to be interpreted carefully because methods of data collection, the way in which survival is counted, and the length of follow-up can be important.
Table 2. Five-Year Survival Data by Disease* (Open Table in a new window)
| Disease | Stage | Survival Rate (%) | ||
|---|---|---|---|---|
| Autologous Transplantation | Allogeneic Transplantation | |||
| Sibling Donor | Unrelated Donor | |||
| Acute lymphoblastic leukemia (ALL) | Complete response (CR) 1 | N/A | 65 | 45 |
| CR2 | N/A | 55 | 35 | |
| Acute myeloid leukemia (AML) | CR1 | N/A | 70 | 50 |
| CR2 | N/A | 50 | 40 | |
| No remission | N/A | N/A | 25 | |
| Chronic myeloid leukemia (CML) | Chronic phase < 1 y | N/A | 70 | 55 |
| Chronic phase >1 y | N/A | 60 | 50 | |
| Hodgkin lymphoma | CR1 | 80 | N/A | N/A |
| CR2 | 70 | N/A | N/A | |
| No remission | 45 | N/A | N/A | |
| Diffuse large-cell lymphoma | CR1 | 65 | 25 | 30 |
| 50 | 25 | N/A | ||
| 45 | 20 | N/A | ||
| Neuroblastoma | 60 | N/A | N/A | |
| * Based on Kaplan-Meier curves of data from the Center for International Blood and Marrow Transplant Research (CIBMTR) and National Marrow Donor Program (NMDP) data. |
![]()
Donation for HSCT
Donors for hematopoietic stem cell transplantation (HSCT) must be in generally good health, without other comorbid conditions, and should in general have the same qualifications as a blood donor. The donor must have a performance status that permits safe collection of cells, be able to tolerate anesthesia (general or regional), and have adequate cardiac, pulmonary, hepatic, and renal function. Pediatric donors are used only for autologous collection or donation to siblings.
Donors with ongoing malignancies or a history of a malignant condition other than minor skin cancers (eg, basal cell carcinomas) are generally excluded from further consideration.
The following studies are routinely performed on hematopoietic cell donors:
- History and physical examination to detect health and behavioral problems, such as signs of intravenous drug use, mental health problems, and signs of significant illness
- Complete blood count (CBC) and platelet count
- Serum creatinine, electrolyte, and liver function studies
- Tests for infection: Serologic studies for cytomegalovirus (CMV), herpes viruses, HIV RNA, anti-HIV antibodies, hepatitis B and C viruses (including HCV nucleic acid amplification testing [NAT]), human T-cell lymphotropic virus-1/2 (HTLV-I/II), and syphilis (using the Venereal Disease Research Laboratory [VDRL] test); in autologous donations, CMV and VDRL testing is not required
- ABO blood typing
- Human leukocyte antigen (HLA) typing
- Chest radiography
- Electrocardiography
Qualification of donors of cellular therapy products in the United States is regulated by the US Food and Drug Administration (FDA). The regulations are contained within the Code of Federal Regulations. Donors must provide written consent and must be provided complete information about donation, including a clear description of the procedure risks and alternatives. [23]
Donors are kept anonymous from the recipient until 1 year after transplant. At that time, identifying information can be exchanged if mutually desired.
Cord blood
Cord blood transplantation refers to the use of hematopoietic stem cells collected from the umbilical cord and placenta. About 50-200 mL of cord blood is collected immediately after the cord is clamped and cut. These units are cryopreserved and stored in private and public cord blood banks worldwide for future use. This type of collection has no risk to the donor if the cord is appropriately clamped.
The American Academy of Pediatrics has published a policy statement that provides information to guide physicians in responding to parents' questions about cord blood donation and banking. [24] Directed donation of cord blood should be encouraged when there is a specific diagnosis of a disease within a family known to be amenable to stem cell transplantation.
![]()
Donor Sources
Once transplantation is considered as a possible therapy, an appropriate donor must be identified. The best possible match results in the least complications. For allogeneic transplants, HLA histocompatibility typing is performed for immediate family members, initially using intermediate-resolution typing.
Human leukocyte antigen matching
HLAs are expressed on the surface of various cells, in particular white blood cells (WBCs). These antigens are also known as the major histocompatibility complex (MHC) and occupy the short arm of chromosome 6. [25] This genetic region has been divided into chromosomal regions, called classes. Classes I, II, and III have been defined, although class III information is still too sparse to include here.
Class I is made up of HLA-A, HLA-B, and HLA-C, as well as genes that are less frequently discussed (eg, HLA-E, HLA-F, and HLA-G). Class II is made up of HLA-DR, HLA-DP, and HLA-DQ, as well as variations on these genes. Traditionally, the loci critical for matching are HLA-A, HLA-B, and HLA-DR. HLA-C and HLA-DQ are also now considered when determining the appropriateness of a donor.
A completely matched sibling—that is, one in whom both alleles of HLA-A, HLA-B, and HLA-DR are the same as in the recipient, a so-called 6-of-6 match—is generally considered the ideal donor. Fully matched family members provide the most compatible matches because they often share minor HLA antigens not usually included in testing. If the donor and recipient are not a 6-of-6 match, they are said to be mismatched. When only half the alleles match, the term haplotypic donor applies. Unrelated-donor searches generally look for 10-of-10 (all but HLA-DP) or 12-of-12 matches. For unrelated donors, a complete match or a single mismatch is considered acceptable for most transplantations, although in certain circumstances a greater mismatch is tolerated.
Umbilical cord blood has historically been thought of as “immunologically naive” and matched for HLA-A, HLA-B, and HLA-DRB1, with consideration of HLA-C. More recent data suggest that mismatch of HLA-C is an independent risk factor for transplant-related mortality. [26]
Other genetic loci currently being studied include the ligands for natural killer cells known as killer immunoglobulinlike receptors (KIRs). A KIR, along with its HLA-C ligand, is part of the biological process to prime natural killer cells to attack non–self-intruders such as leukemia cells. KIR and HLA-C mismatching between donor and HSCT recipient has been associated with reduced posttransplantation relapse of leukemia. [27]
With syngeneic transplantation, in which the donor is an identical twin sibling of the patient, the graft would be considered a perfect match. Graft rejection is less of an issue for such transplants when compared with other allogeneic transplants, but there is also limited to no graft-versus-tumor effect when treating malignancy.
Autologous transplantation
Generally, candidates for autologous transplantation have no demonstrable malignancy in the blood or bone marrow. Treatment-related morbidity and mortality rates are lowest with autografts, with the major problem being tumor relapse. This finding relates to the absence of a graft-versus-tumor effect (ie, immunologic attack on the tumor by immunocompetent T cells and natural killer cells in the donor graft) and concern over the reinfusion of occult tumor cells in the graft.
Autologous transplantation is typically used as a method of returning the patient's own stem cells as a rescue therapy after high-dose myeloablative therapy. This transplantation technique is generally used in chemosensitive hematopoietic and solid tumors to eliminate malignant cells. Prior to transplant, the patient receives a higher dose of chemotherapy than can normally be tolerated by the bone marrow, in order to increase the chances of killing remaining tumor cells.
The high-dose chemotherapy is followed by rescue of the host's bone marrow with the previously collected autologous stem cells. Immunosuppression is not required after autologous transplantation, because the immune system that is reconstituted is that of the original host. Because the native immune system returns after autologous transplant, this technique is not used for correction of immunodeficiencies.
Identical twin donors
In rare instances, patients who are candidates for HSCT have an identical twin who can serve as a donor. These patients do not require posttransplantation immunosuppressive therapy and do not develop graft versus host disease (GVHD), although they are at a higher risk of recurrence of the underlying malignant disease than are nonidentical sibling donors with similar HLAs. The reason for this apparent disparity, although uncertain, is presumably related to the greater ability of lymphocytes from nonidentical sibling donors to recognize recipient tumor cells as foreign (ie, graft-vs-leukemia effect).
Interestingly, survival rates are similar with identical twin and nonidentical sibling transplants, because the increased frequency of leukemia relapse with identical twin donors is counterbalanced by lower treatment-related mortality rates.
Allogeneic transplantation
Allogeneic transplantation refers to the use of stem cells from a donor source other than the subject. The source of donated stem cells (the donor) may be genetically related or unrelated to the recipient. This type of transplant is used in the context of many malignant and nonmalignant disorders to replace a defective host marrow or immune system with a normal donor marrow and immune system.
The degree of HLA match between the donor and the recipient is perhaps the most important factor in these transplants; well-matched transplants decrease the risk of graft rejection and GVHD, both of which are among the most serious sequelae of transplantation. Moreover, because of the graft-versus-tumor effect, allogeneic transplants are associated with lower relapse rates than are autologous transplants.
Patients older than 50 years, however, experience higher transplant-related morbidity and mortality rates with allogeneic grafts. This phenomenon relates to the need for continuing immunosuppression after the transplantation to prevent the development of GVHD. [28]
Matched, related donors
Related donors are usually siblings, because they have the opportunity to inherit the same HLA genes located on chromosome 6. A given sibling has a 25% chance of being HLA matched at the A, B, and DRB1 loci.
ABO red blood cell (RBC) antigens are not expressed on stem cells. Although hemolysis, delayed erythropoietic engraftment, and pure RBC aplasia may complicate HSCT if the patient and donor are ABO incompatible, these complications are uncommon; therefore, ABO incompatibility is not a barrier to successful nonmyeloablative transplantation.
Finding matched, related donors other than siblings is unlikely unless the patient’s parents happen to have very common haplotypes or intermarrying among families has occurred such that first cousins are fully HLA matched.
Mismatched, related donors
Although most centers require a complete match at the HLA-A, HLA-B, and HLA-DRB1 loci for an individual to be used as a transplant donor, some centers consider the use of single antigen–mismatched siblings. As expected, transplants from such donors pose a higher risk of GVHD, although the overall survival rate may not differ significantly from that observed with fully matched siblings.
Unrelated donors
When a related donor cannot be identified, a search for an unrelated donor is often initiated. The process begins with high-resolution HLA typing of the recipient, which defines the specific DNA sequence of the HLA molecule's antigen binding site and is a more accurate method of typing than the serologic method. [29] This is followed by a preliminary search of registry databases, which can be performed relatively quickly, yielding general information as per number of potential marrow donors and cord blood units within 24 hours.
The largest donor registry is the National Marrow Donor Program (NMDP), a repository for HLA-typing information that permits matching of recipients with unrelated donors. At present, more than 3 million donors, all of whom have undergone HLA-A and HLA-B serologic typing, are registered in the program’s data bank with over 325,000 added in 2020 alone. The World Marrow Donor Association (WMDA is a collective database of more than 59 registries in 43 countries and 37 cord blood registries from 21 countries. Almost 39 million potential donors and over 800,000 cord units were available as of May 2021.
When a potential donor is identified, a formal search is begun, including high-resolution typing of potential donors and subsequent confirmatory typing of selected matches. This incurs a cost to the recipient or the insurer.
High-resolution HLA typing is done because if a donor and recipient are not related, serologic typing alone does not ensure that the individuals share the same HLA genes. This is evident clinically by the higher risk of GVHD in recipients of unrelated donor grafts. DNA-based techniques for molecular typing have demonstrated that only 55% of serologically identical donor and recipient pairs (ie, antigen matched) are highly matched by molecular typing (ie, allele matched).
Patients who are truly highly matched appear to have better outcomes. As a result, most transplantation centers now require complete serologic and molecular matching at the class II region before using a donor for a given transplantation procedure.
A study by Horan et al indicated that when nonmalignant diseases are treated with HSCT using unrelated donors, HLA mismatches are associated with graft failure but not with GVHD. Reviewing 663 HSCTs that used bone marrow or peripheral blood stem cells from donors who were not related to the transplant recipients, the investigators found a link between patient mortality and HLA-A, HLA-B, HLA-C, and HLA-DRB1, but not HLA-DQB1 or HLA-DPB1, mismatches. [30]
When donors are requested, they undergo a workup that includes laboratory testing and infectious disease screening, physical examination, and an informational session. (See Donatioon for HSCT, above.)
Haploidentical donors
When well-matched unrelated donor and cord blood are not available, then a haploidentical donor may be considered. As the name implies, the donor and recipient in these cases match on only half or more HLA alleles. The advantage of this donor source is availability, as multiple individuals are usually available who could serve as potential donors within a given family, including parents, siblings, and children. Another advantage is an equal availability of donors for all ethnic and racial groups, in contrast to the availability of matched, unrelated donors
In haploidentical transplantations, mismatching of maternal antigens, rather than paternal antigens, seems to be better tolerated, presumably because of exposure to maternal HLA antigens during the prenatal and perinatal period.
Although initial studies reported a high incidence of acute GVHD with haploidential HSCT, strategies including T-cell depletion of the graft and post-transplant cyclophosphamide have greatly limited this problem. With the success of those strategies, haploidentical donors have been utilized more frequently in both myeloid and lymphoid malignancies, as well as nonmalignant diseases. [31]
Cord blood transplantation
Cord blood can be used to transplant in any disease state for which bone marrow can be used. The small number of cells collected in each unit typically limits the size of the recipient, although successful transplantations in patients weighing more than 100 kg have been reported.
Cord blood is increasingly used in the pediatric setting when a suitable donor is not available, either due to small family size or the patient’s status as a member of an ethnic minority. (Ethnic minorities represent only a small proportion of donors to the National Marrow Donor Program.)
In 2011, the US Food and Drug Administration (FDA) approved the first umbilical cord blood product for use in stem cell transplantation. The product contains hematopoietic progenitor cells from human cord blood (HemaCord, New York Blood Center). [32, 33]
More than 800,000 cord blood units are currently stored in banks worldwide. The New York Cord Blood Bank was the first and remains the largest cord blood bank in the world. Several additional cord blood banks are organized throughout the United States, Europe, and Asia; these collect units much like a blood bank solicits volunteer blood donations.
In addition, private for-profit cord blood banks often solicit funds from expectant mothers to pay for the collection and storage of cord blood following delivery. Typically, an upfront processing fee and an ongoing storage fee are charged.
To date, autologous stored cord blood from private banks has rarely been used. Calculations suggest that, of the individuals who store cord blood, fewer than 1 in 20,000 will use the blood as an autologous transplant. When faced with the choice of private storage versus donation to a public bank, an individual must consider the cost of long-term storage, potential for current and future use, as well as the availability to the public at large.
Owing to the relative immaturity of the fetal immune system, stem cells from umbilical cord blood allow the crossing of immunologic barriers that would otherwise be prohibitive. As a result, the degree of tolerable HLA disparity is much greater in cord blood transplants. A match of 3-4 of the 6 HLA-A, HLA-B and HLA-DRB1 antigens is sufficient for transplantation. For the same reason, the degree and severity of GVHD are low following cord blood transplants.
In addition, cord blood contains relatively high numbers of hematopoietic stem cells with superior proliferative capacity, compared with hematopoietic stem cells from marrow and blood in adults. Other advantages of cord blood are that it carries less risk of transmission of blood-borne infections and is readily available. [34]
A major limitation is the relatively small volume obtained from cord blood collections. In adults, the small volume results in delayed engraftment and increased risk of infections and mortality. The use of multiple cord units for a single patient is one strategy for overcoming this limitation. Studies have shown the feasibility, safety, and efficacy of double umbilical cord blood transplantation. [35] The possibility of expanding cord blood stem cells in vitro is an area of active investigation. [36]
However, a randomized study of cord blood transplantation in 224 patients 1 to 21 years of age with hematologic cancer found that survival rates were similar after single-unit and double-unit cord-blood transplantation, but single-unit transplants were associated with better platelet recovery and a lower risk of GVHD. [37]
Another disadvantage is that engrafting with a cord blood unit can be attempted only once, whereas bone marrow donors can give again if the first transplant fails to engraft. Cord blood is associated with a higher rate of nonengraftment. This is especially evident in bone marrow failure diseases, especially aplastic anemia.
The median time to neutrophil recovery after cord blood transplantation is 4 weeks, compared with 10-14 d for bone marrow and 7-12 d with peripheral blood stem cell transplantation.
In a study of 434 consecutive unrelated transplants performed from 1997 to 2009, Parody and colleagues found that cord blood transplantation was associated with a higher risk of invasive aspergillosis and cytomegalovirus infection compared with bone marrow and peripheral blood transplants, but rates of infection-related mortality and non-relapse mortality were similar between groups. [38]
![]()
Donor Selection
The selection of the donor HSCT graft is multifactorial and includes the following considerations:
- Availability of a matched sibling donor
- Survival and disease control statistics for specific illnesses with different stem cell sources
- The urgency of the transplantation (An urgent transplantation may preclude use of a matched unrelated donor, which can take time to identify and procure stem cells from.)
- Speed of engraftment
- Risk of graft versus host disease (GVHD)
- Potential need for a subsequent graft
- Transplant center preference
Additional considerations include the following [39, 40] :
- Donor age (younger is better)
- Sex (female stem cells given to a male is less favorable)
- Cytomegalovirus (CMV) serology (CMV-negative has better outcome)
- Pregnancy and transfusion history
- Body weight
Below is an algorithm for typical selection of a graft for most indications for HSCT, based on multifactorial analysis.
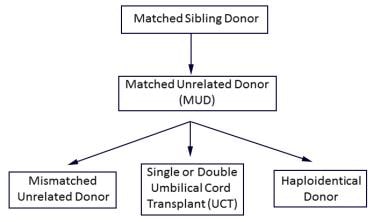
An algorithm for typically preferred hematopoietic stem cell transplantation cell source for treatment of malignancy: If a matched sibling donor is not available, then a MUD is selected; if a MUD is not available, then choices include a mismatched unrelated donor, umbilical cord donor(s), and a haploidentical donor.
Table 6 (below) lists the cellular characteristics of the commonly used stem cell sources, and Table 7 lists the clinical characteristics of these sources.
Table 6. Cellular Characteristics of Various Sources of Stem Cells (Open Table in a new window)
| Cellular Characteristics | Source | ||
|---|---|---|---|
| Bone Marrow | Peripheral Blood | Cord Blood | |
| Stem cell content | Adequate | Good | Low |
| Progenitor cell content | Adequate | High | Low |
| T-cell content | Low | High | Low, functionally immature |
| Risk of tumor cell contamination | High* | Low* | Not applicable† |
| *Risk of tumor cell contamination from an autologous bone marrow source would be high; an allogeneic source should have negligible risk.†Risk of tumor cell contamination from cord blood from an allogeneic source should be negligible. |
Table 7. Clinical Characteristics of Various Sources of Stem Cells (Open Table in a new window)
| Cellular Characteristics | Source | ||
|---|---|---|---|
| Peripheral Blood | Bone Marrow | Cord Blood | |
| HLA matching | Close matching required | Close matching required | Less restrictive than others |
| Engraftment | Fastest | Faster than cord blood but slower than peripheral blood | Slowest |
| Risk of acute GVHD | Same as in bone marrow | Same as in peripheral blood | Lowest |
| Risk of chronic GVHD | Highest | Lower than peripheral blood | Lowest |
Peripheral blood HSCT versus bone marrow HSCT
Bone marrow has been the traditional source of hematopoietic stem cells for use in autologous and allogeneic transplantations. However, peripheral blood has replaced bone marrow as a source of these cells for most autologous transplantations and a significant proportion of allogeneic transplantations. [41]
Advantages of peripheral blood stem cell transplantation (PBSCT) include the nonsurgical method of collection and the far larger number of stem cells that can be collected with apheresis. [41] Moreover, peripheral blood contains higher numbers of T cells, and PBSCT is associated with higher graft-versus-tumor or graft-versus-leukemia effect and decreased relapse rates. It is also associated with rapid engraftment (because a higher number of committed progenitor cells are collected), which translates into decreased mortality and early hospital discharges.
The disadvantage with PBSCT is the increased incidence of GVHD, which is due to a higher T-cell load in peripheral blood stem cells in comparison with bone marrow stem cells. Bone marrow transplantation is considered superior to PBSCT for nonmalignant conditions (hemoglobinopathies), in which rapid engraftment is not crucial and a graft-versus-tumor effect is not required. and graft availability. , and faster engraftment time.
A 2005 meta-analysis of research articles evaluating matched sibling bone marrow versus matched sibling peripheral blood HSCT for treatment of leukemia showed that peripheral blood HSCT was associated with decreased leukemia relapse rates and better overall survival but increased chronic GVHD. [42] A long-term follow-up of matched related HSCT for hematological malignancy showed both 2- and 10-year overall survival was superior with PBSCT compared with bone marrow HSCT. [43] When unrelated PBSCT was compared with unrelated bone marrow transplants in patients with leukemia and myelodysplastic syndrome, an equivalence in 3-year survival was confirmed; however, the rate of chronic GVHD was higher with unrelated PBSCT grafts. [44]
In summary, those data would suggest that for treatment of leukemia, matched sibling PBSCT may be superior to matched sibling bone marrow HSCT. In matched unrelated donor transplants, there does not seem to be an antileukemic benefit for PBSCT compared with bone marrow HSCT. In fact, a multicenter phase III trial (Blood and Marrow Transplant Clinical Trials Network [BMT CTN] 0902) showed no benefit and increased GVHD in matched unrelated peripheral blood versus bone marrow HSCT. [10]
Other comparison studies
A study of allogeneic HSCT in 2223 adult patients with acute myeloid leukemia reported comparable survival in matched sibling transplantation and matched unrelated transplantation. The 7/8 matched unrelated donor HSCTs showed higher early mortality, but comparable long-term survival compared with matched related donors and matched unrelated donors. It was noted that matched related HSCTs have the lowest frequency of GVHD. [45]
In a retrospective analysis of 1525 adults with leukemia treated with primarily 4/6 matched umbilical cord HSCT versus peripheral blood or bone marrow 8/8 or 7/8 matched unrelated HSCT, no difference was shown in leukemia-free survival in all arms. Umbilical cord transplants were associated with later engraftment and higher treatment-related mortality, despite a lower incidence of GVHD. Surprisingly, rates of GVHD resulting in mortality did not diffe between umbilical cord HSCT and peripheral blood HSCT. [46] Of note, most of the umbilical cord transplants were 4/6 matches, and there is evidence that closer HLA matching in umbilical cord transplants improves outcomes. [47]
![]()
Procurement of Stem Cells
When donor approval has been obtained, a date is set for the collection. Bone marrow or peripheral blood stem cells are harvested on the same day of transplantation, with the intent to infuse the recipient within 24 hours of the harvest. Generally, cord blood is shipped to the transplant center prior to the onset of the recipient's conditioning regimen.
Bone marrow
Bone marrow harvesting has become a relatively routine procedure. Bone marrow is generally aspirated from the posterior iliac crests while the donor is under either regional or general anesthesia. This can be a difficult procedure in donors who are smaller than the recipient, such as sibling donors, and several aspirations may be required to obtain an adequate cell dose.
Additional bone marrow can be obtained from the anterior iliac crest; however, the amounts available are relatively limited, and the marrow from this site is generally used only for diagnostic purposes.
Because only a small percentage of the total bone marrow is removed during a harvesting procedure, peripheral blood leukocyte counts do not significantly decrease. Bone marrow is a highly vascular organ, however, so harvesting can result in substantial blood loss, depending on the volume of marow removed. Guidelines established by the National Marrow Donor Program limit the volume of bone marrow removed to 15 mL/kg of donor weight. A minimum dose of 1 × 108 and 2 × 108 marrow mononuclear cells per kilogram is required to establish engraftment in autologous and allogeneic marrow transplants, respectively.
Complications related to bone marrow harvesting are rare and involve anesthetic, infectious, and bleeding problems.
Bone marrow primed with granulocyte colony-stimulating factor (G-CSF; filgrastim) has been used in pediatric and adult patients to increase the stem cell count and, thus, to reduce the number of aspirations from the donor and speed engraftment in the recipient. [48] Filgrastim and chemotherapy can be used alone or in combination to mobilize stem cells. Interleukin-2 increases T-cell function but is not a stem cell mobilizer.
A randomized trial suggested that the risk of GVHD is not increased by the use of G-CSF–primed marrow and that such marrow does not affect engraftment. [49] The potential risks of G-CSF use include increased bone pain, rare events (eg, splenic rupture), and the theoretical risk of leukemia.
Studies have suggested that the risk of chronic GVHD from G-CSF–primed bone marrow may be less than that from G-CSF–primed peripheral blood stem cells.
Peripheral blood
Hematopoietic stem cells circulate in blood, albeit in very low concentrations. Administration of recombinant hematopoietic growth factors (ie, the cytokines G-CSF and granulocyte-macrophage colony-stimulating factor [GM-CSF]) to patients or donors down-regulates the adhesion molecules on these cells, so they are released into the peripheral blood. The cells can then be collected by apheresis and can be identified and quantified using flow cytometry (hematopoietic stem cells express the CD34 antigen). The efficacy of mobilization of stem cells from the bone marrow by cytokines has led to the widespread adoption of peripheral blood as a cell source for HSCT.
Stem cell mobilization
Cytokine treatment for mobilization is initiated at least 4 days before apheresis. The dose of G-CSF used for mobilization is 10 mcg/kg/day. In autologous donors who are heavily pretreated, however, doses of up to 40 mcg/kg/day can be given. Clinical trials have shown that mobilization with G-CSF is better than with GM-CSF.
Common adverse effects of G-CSF include bone pain, malaise, headaches, chills, and (sometimes) fever. Filgrastim induces a hypercoagulable state, and in rare cases it causes vascular thrombosis. Moreover, G-CSF can exacerbate autoimmune disease, and some cases of ophthalmologic events have been reported in healthy donors. So far, however, there has been no evidence linking the development of myelodysplasia or hematologic malignancies with G-CSF administration.
GM-CSF leads to the same somatic complaints as G-CSF. In addition, it can cause abnormal findings on liver function tests (LFT), fluid retention, serositis, and “first dose reaction,” characterized by hypoxia and hypotension within 3 hours of administration.
CXCR4 inhibitors
Motixafortide and plerixafor are inhibitors of C-X-C motif chemokine receptor 4 (CXCR4) and block binding of its cognate ligand, stromal-derived factor-1α (SDF-1α)/CXCL12. SDF-1α and CXCR4 play a role in trafficking and homing of hematopoietic stem cells to the marrow compartment.
Motixafortide (Aphexda) is indicated in combination with G-CSF/ to mobilize hematopoietic stem cells to the peripheral blood for collection and subsequent autologous transplantation in patients with multiple myeloma.
Approval was supported by the phase 3 GENESIS trial. The primary endpoint was the collection of ≥6 × 106 CD34+ cells/kg within 2 apheresis procedures; the secondary endpoint was to achieve this goal in 1 apheresis. The American Society for Transplantation and Cellular Therapy (ASTCT) guidelines recommend a collection target of 3-5 × 106 CD34+ cells/kg. Motixafortide plus G-CSF enabled 92.5% of patients to successfully meet the primary endpoint versus 26.2% with placebo plus G-CSF (P < 0.0001). Motixafortide plus G-CSF also enabled 88.8% to meet the secondary endpoint versus 9.5% with placebo plus G-CSF (P < 0.0001). [50]
Plerixafor (Mozobil) is used in conjunction with G-CSF to mobilize hematopoietic stem cells to peripheral blood for collection and subsequent autologous transplant in non-Hodgkin lymphoma and myeloma. Plerixafor is administered at a dose of 0.24 mg/kg subcutaneously. The dose has to be adjusted in kidney failure, and leukocyte and platelet counts have to be monitored. Common adverse effects include nausea, vomiting, diarrhea, flatulence, arthralgias, dizziness, headache, and injection-site sequelae. Other adverse effects include myalgia, hyperhidrosis, xerostomia, dyspnea, hypoxia, orthostatic hypotension, leukocytosis, and thrombocytopenia.
Plerixafor and motixafortide are not indicated in leukemias, due to the possibility of mobilization of leukemic cells and contamination of apheresis product.
Apheresis
Apheresis instruments, similar to those used for collecting platelet concentrates from volunteer donors for transplantation, are used in an ambulatory setting for collecting CD34+ stem cells. For a typical donor, approximately 24 L of whole blood can be processed over 2 days to collect approximately 500 million CD34+ cells, a cell progenitor population enriched for hematopoietic stem cells. Mobilization of stem cells may be enhanced approximately 10-fold for patients with cancer who receive chemotherapy (eg, cyclophosphamide) along with hematopoietic growth factors (eg, G-CSF), compared with mobilization with G-CSF alone.
The minimum dose required for engraftment is 1-2 × 106 CD34+ cells/kg body weight for autologous and allogeneic transplants. Higher doses would result in better engraftment, but doses in the range of 8 × 106 are associated with increased risk of extensive GVHD.
Two issues in the collection of peripheral blood stem cells—priming and anticoagulation—require special consideration in children.
Priming
Although devices to minimize extracorporeal volume are used, priming of the apheresis machine with RBCs may be required for children based on their total blood volume. This step prevents unacceptable dilutional anemia during the procedure, as well as fluid overload associated with the return of red cells from the centrifuge chamber at the end of the procedure.
Anticoagulation
In older patients, anticoagulation required for the apheresis procedure is accomplished using anticoagulant citrate dextrose (ACD). Although ACD does not result in systemic anticoagulation, its citrate component increases the risk of symptomatic hypocalcemia in young patients. In addition, citrate toxicity often limits the rate of blood processing, prolonging the procedure.
However, pediatric patients can be treated with a combination of ACD and heparin. Heparin can allow the use of decreased amounts of ACD, making symptomatic hypocalcemia rare. Even so, the patient treated with heparin and ACD may be fully anticoagulated by the end of the procedure, slightly increasing the bleeding risk and possibly requiring reversal of heparinization after the procedure has been completed.
Poor mobilizers
Up to 20% of normal donors can be poor mobilizers with G-CSF, showing less than 5 CD34+ cells/µL after a full course of medication administration. The management of poor mobilizers to improve stem cell collection yield is on a case-by-case basis and can involve the use of additional mobilizing drugs such as plerixafor.
Contraindications
Contraindications to therapeutic hematopoietic stem cell collection includes donor assessment to determine whether the donor can tolerate the mobilization drug or procedure, based on size, health, and hematologic parameters. In addition, donors with sickle cell disease or sickle cell trait have been reported to have serious adverse consequences, including sickle crisis, multiorgan failure, and death, following administration of G-CSF and apheresis.
![]()
Manipulation of Stem Cell Grafts
These techniques often require sophisticated laboratories and highly trained technical personnel, increasing the price of the product.
ABO-incompatible allogeneic transplants
ABO-mismatched donors are acceptable donors for bone marrow transplantation, but they may have ABO-mismatch–related complications.
In a major mismatch, the recipient’s immune system has antibodies against the donor’s red blood cells. In this situation, acute hemolysis may occur with infusion of the product. Red blood cell depletion of the product is used to help prevent this. Delayed red blood cell engraftment may also occur, owing to the recipient’s residual immune system targeting the donor's developing red blood cells. This is often temporary and requires supportive care with transfusions.
In a minor mismatch, the donor’s immune system is implicated. Donor isohemagglutinins from a product’s plasma component can bind and destroy the recipient’s red blood cells soon after the product is given. This can be prevented by plasma depletion of the product. In addition, the donor lymphocytes from the product can engraft and temporarily produce antibodies against the recipient red blood cells. This is known as donor lymphocyte syndrome. This is often clinically transient, and it mandates carefully monitoring and supportive care. The removal of isoagglutinins or red blood cells from the donor graft prevents hemolysis in the recipient.
T-cell depletion in the allogeneic transplantation setting
Immunocompetent donor T cells may be removed using a variety of methods to reduce or eliminate the possibility that graft versus host disease (GVHD) will develop. Although such a strategy is often effective in lowering the morbidity and mortality associated with GVHD, removal of these accessory cells may be associated with an increase in engraftment failure in up to 10% of transplantations (due to the loss of T-helper cells, which facilitate engraftment). Furthermore, T-cell depletion results in higher relapse rates than do T-cell–replete grafts, because the removal of cytotoxic T cells eliminates the potential for a graft-versus-malignancy effect. To counter this, a T-cell add-back is often given to achieve a certain optimal number of T cells to obtain engraftment yet minimize GVHD. More specific T-cell selection, such as removing the majority of alpha/beta T cells and leaving the gamma/delta T cells, has shown increasing promise in non-malignant diseases and may minimize the need for prophylactic immunosuppression.
In vitro purging in autografts
Tumor cells can be detected by using sophisticated means such as tumor clonogenic assays, flow cytometry, or polymerase chain reaction assay. Tumor cell removal methods include chemical and immunologic methods, positive selection of CD34+, and negative selection.
With regard to chemical and immunologic methods, in vitro chemotherapy with 4-hydroperoxycyclophosphamide or mafosfamide (cyclophosphamide derivatives) kills residual autologous tumor cells. However, normal stem cells are injured, resulting in slower or incomplete engraftment.
For positive selection of CD34+ cells, commercial instruments can be employed, using solid-phase, anti-CD34 monoclonal antibodies. With negative selection, anticancer monoclonal antibodies can be used to remove tumor cells, leaving stem cells in the graft.
Although in vitro purging has been attempted for many diseases, most notably neuroblastoma, outcomes have not significantly improved with this practice.
![]()
Preparative Regimens for HSCT
The preparative or conditioning regimen is a critical element in hematopoietic stem cell transplantation (HSCT). The purpose of the preparative regimen is to provide immunoablation and cytoreductioin sufficient to prevent rejection of the transplanted graft and to eradicate the disease for which the transplantation is being performed. These goals have traditionally been achieved by delivering maximally tolerated doses of multiple chemotherapeutic agents and immunologic agents with nonoverlapping toxicities (with or without radiation).
Several novel approaches have been evaluated in an attempt to minimize toxicity. For example, nonmyeloablative preparative regimens have been used to induce a state of mixed chimerism (defined as the concurrent presence of donor and recipient hematopoietic cells); this can be followed by cellular therapy via the administration of donor lymphocyte infusions. Another alternative is targeted therapy in the form of monoclonal antibodies with or without radiolabeling.
Infusion of hematopoietic cells circumvents the problem of prolonged myelosuppression from chemotherapy, permitting escalation to considerably higher dose levels. However, marrow recovery still takes weeks and requires sophisticated supportive care until the effects of chemotherapy have lessened.
In addition to myelotoxicity, common adverse effects of preparative regimens include the following:
- Mucositis
- Nausea
- Vomiting
- Alopecia
- Diarrhea
- Rash
- Peripheral neuropathies
- Pulmonary and hepatic toxicity
In addition, infertility is an almost universal occurrence when myeloablative regimens are used. This problem can be addressed with sperm cryopreservation for male patients, assuming that they have adequate sperm number and function. Oocyte cryopreservation in female patients has generally been less successful.
Long-term complications following total body irradiation include asymptomatic alterations in pulmonary function, cataracts, sicca syndrome, hypothyroidism, and thyroiditis.
Myeloablative preparative regimens
Myeloablative regimens are designed to kill all residual cancer cells in autologous or allogeneic transplantation and to cause immunosuppression for engraftment in allogeneic transplantation.
Myeloablative regimens can be classified as radiation-containing or non–radiation-containing regimens, therapies that were developed by escalating the dose of radiation or of a particular drug to the maximally tolerated dose. Drugs with nonoverlapping toxicities have been used in an effort to avoid synergistic injury to a particular organ.
Total-body irradiation and cyclophosphamide or busulfan and cyclophosphamide are the commonly used myeloablative therapies. These regimens are especially used in aggressive malignancies, such as leukemias.
Radiation-containing preparative regimens
Total-body irradiation has been the mainstay of preparative regimens since the inception of HSCT. Total-body irradiation ̶ based regimens typically fractionate the radiation and administer the total dose over several days (a strategy termed fractionated total-body irradiation [FTBI]), which helps to decrease toxicity and increase tolerability. Partial lung shielding is included in an effort to reduce the potential for irreversible lung injury.
The maximally tolerated dose of total-body irradiation is approximately 1500 cGy. Higher doses produce excessive nonhematologic toxicity, primarily to the lungs, but also to other organs, including the heart.
Commonly used radiation-containing preparative regimens are as follows:
- FTBI at 1200 cGy and cyclophosphamide at 120 mg/kg
- FTBI at 1320 cGy and etoposide at 60 mg/kg
- FTBI at 1320 cGy, etoposide at 60 mg/kg, and cyclophosphamide at 60 mg/kg
- FTBI at 1200 cGy and melphalan at 200 mg/kg
- FTBI at 1200 cGy, etoposide at 60 mg/kg, and cyclophosphamide (for autologous) at 100 mg/kg
Non-radiation-containing preparative regimens
Regimens have been developed in which total-body irradiation is replaced with additional chemotherapeutic agents. These approaches have primarily been developed for autologous transplantation, but they have also been used in the allogeneic setting. The primary advantage of regimens that lack total-body irradiation is reduced toxicity. Additionally, the cost is lower, the regimen is easier to administer, and radiation can still be given to sites of prior disease following transplantation.
Commonly used non–radiation-containing preparative regimens are as follows:
- Busulfan at 16 mg/kg and cyclophosphamide at 120 mg/kg
- Busulfan at 16 mg/m2 and etoposide at 60 mg/m2
- Cyclophosphamide at 6-7.2 g/m2, carmustine at 300-500 mg/m2, and etoposide at 600-2400 mg/m2
- Cyclophosphamide at 7.2 g/m2, carmustine at 600 mg/m2, etoposide at 1200 mg/m2, and cisplatin at 150 mg/m2
- Carmustine at 300 mg/m2, etoposide at 400-800 mg/m2, cytarabine at 800-1600 mg/m2, and melphalan at 140 mg/m2
Multiple cycles of high-dose chemotherapy
With the introduction of mobilized peripheral blood progenitor cell (PBPC) collection techniques, the collection of doses of stem cells that far exceed the thresholds required for engraftment can be readily achieved. This permits an approach consisting of multiple cycles of high-dose chemotherapy, with PBPCs infused after each cycle in an attempt to increase the intensity of anticancer therapy beyond that achievable with a standard autologous transplantation.
Each cycle is at or near myeloablative levels of chemotherapy, followed by rescue with previously collected PBPCs. At present, however, little evidence indicates that this approach increases the long-term disease-free survival rate over that achieved with standard transplantation.
Radiolabeled monoclonal antibodies
The maximally tolerated dose of total-body irradiation is approximately 1500 cGy. Randomized trials comparing 1220- and 1575-cGy doses found that the higher dose was associated with a lower relapse rate but, because of a higher rate of complications, no improvement in overall survival rates. These observations suggest that improvements in disease-free survival rates can be attained if the radiation dose can be increased without excessive toxicity.
One approach to achieving this goal has been the administration of monoclonal antibodies radiolabeled with high-energy–emitting radioisotopes. This would permit targeting of the radiation dose to the tumor cells and marrow, with a potential reduction in dose to other organs, such as the liver, lungs, and kidneys. One such monoclonal antibody is directed against CD45, which is highly expressed in hematopoietic cells, thereby allowing targeting to the marrow space.
Nonmyeloablative preparative regimens
For patients with leukemia, an important contributing factor to effective treatment is a graft-versus-tumor effect mediated by the donor cells. This effect requires the engraftment of donor-type immunocompetent cells, which does not necessarily require a toxic myeloablative preparative regimen. As a result, the possibility of achieving mixed chimerism—which, as previously stated, is the concurrent presence of donor and recipient hematopoietic cells—using nonmyeloablative regimens (ie, mini-transplants) is being explored.
Nonmyeloablative regimens use doses of chemotherapeutic drugs and radiation that are substantially lower than those of myeloablative regimens. These regimens are immunosuppressive but not myeloablative. Reduced-intensity regimens involve drugs such as fludarabine, melphalan, antithymocyte globulin, and busulfan.
The chimeric approach, which relies more on donor cellular immune effects and less on the cytotoxic effects of the preparative regimen to control the underlying disease, has less acute and chronic toxicity compared with myeloablative regimens. This permits transplantation in older patients (aged 55 years or older), patients at high risk due to comorbidities, and heavily pretreated patients.
The major use of mini-transplants is in treating patients with immunologically responsive disorders such as myeloma, and for slow-growing tumors such as those of chronic lymphocytic leukemia or chronic myeloid leukemia; they are also beneficial for various nonmalignant disorders, such as thalassemia and autoimmune disorders.
The advantage of this kind of transplantation is that patients in whom the stem cells do not engraft still have an autologous recovery due to the nonmyeloablative preparatory regimen. However, most nonmyeloablative transplants require donor lymphocyte infusions for maximum graft-versus-tumor effect, with attendant risk of GVHD. The incidence of GVHD is comparable with that of myeloablative regimens; however, the onset of GVHD is delayed with reduced-intensity regimens compared with other types.
![]()
Infusion of Stem Cells and Engraftment
The infusion of hematopoietic stem cells is a relatively simple process that is performed at the bedside, through a central venous catheter. The patient is premedicated with acetaminophen and diphenhydramine. Allogeneic or syngeneic transplants are generally used immediately after harvesting and processing, and are infused over a period of several hours. Autologous products are almost always cryopreserved; they are thawed at the bedside and infused rapidly over a period of several minutes.
The hematopoietic stem cells home in on the bone marrow cavity and engraft, by mechanisms that have not yet been fully elucidated. Vascular cell adhesion molecule–1, heparan sulphate, and stromal cell–derived factor–1 and its receptor (CXCR4) appear to play roles in this process.
Minimal toxicity has been observed in most cases. ABO-mismatched bone marrow infusions could occasionally lead to hemolytic reactions. Dimethylsulfoxide (DMSO), which is used for the cryopreservation of stem cells, may give rise to facial flushing, tickling sensation in the throat, and strong (garlic) taste in the mouth (the taste of garlic). Rarely, it could cause bradycardia, abdominal pain, encephalopathy/seizures, and renal failure. To avoid the risk of encephalopathy, which occurs with doses above 2 g/kg/day of DMSO, stem cell infusions exceeding 500 mL are infused over 2 days and the rate of infusion is limited to 20 mL/min.
![]()
Infection Prophylaxis for HSCT Patients
All hospitalized HSCT recipients are kept in high-efficiency particulate air (HEPA)–filtered, positive-air-pressure–sealed rooms, and strict hand hygiene is practiced. Autologous HSCT recipients may be managed in an outpatient setting, as they have only a brief period of neutropenia. Most patients receive antibacterial prophylaxis with a fluoroquinolone; antifungal prophylaxis is given with fluconazole, amphotericin B, or voriconazole until day 75-100 posttransplantation.
Herpes simplex–positive patients receive acyclovir prophylaxis (5 mg/kg IV q12h). Cytomegalovirus (CMV)–seronegative patients receive immunosuppression with ganciclovir and intravenous immunoglobulin (CytoGam) and with CMV-negative blood products.
Letermovir (Prevymis) is an antiviral drug against CMV that is approved by the US Food & Drug Administration (FDA) for CMV prophylaxis in adult CMV-seropositive recipients of an allogeneic (HSCT). Letermovir inhibits the CMV DNA terminase complex (pUL51, pUL56, and pUL89), which is required for viral DNA processing and packaging, by affecting the production of proper unit length genomes and interfering with virion maturation. Approval of letermovir was based on a phase III clinical trial in which rates of clinically significant CMV infection (38% versus 61%, respectively) and all-cause mortality (12% versus 17%, respectively) were lower in signficantly fewer patients in the letermovir group than in the placebo group. [51]
All patients should receive Pneumocystis jiroveci prophylaxis with trimethoprim/sulfamethoxazole (double-strength tablet) twice weekly or pentamidine 300 mg once monthly for 1 year posttransplant.
Patients with graft versus host disease (GVHD) on immunosuppression should be on prophylaxis for P jiroveci and fungal infections for 1 month after discontinuation of immunosuppression. They should also receive prophylaxis with penicillin, erythromycin, or extended-spectrum fluoroquinolones for pneumococcal bacteremia.
Patients with documented hypogammaglobulinemia receive intravenous immunoglobulin. Patients with acute GVHD should receive gut decontamination with metronidazole or fluoroquinolones. All patients who are negative for hepatitis B surface antigen (HBsAg) should receive hepatitis B virus (HBV) vaccine before HSCT.
Antifungal prophylaxis
A guideline on the use of primary antifungal prophylaxis in pediatric cancer patients who are undergoing HSCT includes the following recommendations [52] :
- For children aged 1 month to less than 19 years undergoing allogeneic HSCT, administer fluconazole 6–12 mg/kg/day (maximum, 400 mg/day) orally or IV from the beginning of conditioning until engraftment; when fluconazole is contraindicated, an echinocandin can be used
- For children aged 13 years or older undergoing allogeneic HSCT with acute grade II–IV or chronic extensive graft-versus-host-disease (GVHD), administer prophylaxis with posaconazole 200 mg PO 3 times daily from the time of GVHD diagnosis until its resolution; when posaconazole is contraindicated, fluconazole can be used; for children aged 1 month to less than 13 years, fluconazole is recommended
- Patients aged 1 month to less than 19 years undergoing autologous HSCT with anticipated neutropenia for more than 7 days should be given fluconazole 6–12 mg/kg/day (maximum, 400 mg/day) IV or orally from the start of conditioning until engraftment
- Children aged 1 month to less than 19 years with acute myeloid leukemia (AML) or myelodysplastic syndrome (MDS) should receive fluconazole 6–12 mg/kg/day (maximum, 400 mg/day) IV or orally during chemotherapy-associated neutropenia; children aged 13 years or older may also be treated with posaconazole 200 mg PO 3 times daily instead of fluconazole in centers with a high local incidence of mold infections or in cases where fluconazole is not available
- Antifungal prophylaxis should not be administered routinely to children with malignancy and neutropenia anticipated to persist for more than 7 days, outside of those undergoing HSCT or those with AML or MDS
Vaccination and revaccination
Antibody titers to vaccine-preventable diseases (eg, tetanus, polio, measles, mumps, rubella, infection from encapsulated bacteria) decrease 1-4 years after autologous or allogeneic HSCT, so revaccination is routinely recommended for all HSCT recipients. [53] Guidelines for vaccination in HSCT recipients have been published by the American Society of Blood and Marrow Transplantation, the European Group of Blood and Marrow Transplantation, the Infectious Disease Society of America, and the American Academy of Pediatrics. [53, 54, 55, 56]
Most killed vaccines are considered safe. Revaccination with killed vaccines typically begins 6 months after transplantation, although this period may need to be individualized based on the patient's immune function, especially in the presence of GVHD. In patients who have undergone HSCT because of primary immune deficiency, routine posttransplant vaccination (except for influenza vaccination) is not considered until at least 1 year after transplantation, and there is robust evidence of functional B-cell recovery. [54]
Inactivated influenza vaccine can be given 6 months after HSCT, regardless of conditioning regimen or donor type, and should continue annually lifelong. Children aged 6 months to 8 years who have never had flu vaccine before should receive a second dose ≥4 weeks after the first. In the event of a community outbreak, HSCT recipients may receive the vaccine at 3 to 4 months posttransplant; these patients also receive a second dose, 1 month later.
Other recommended killed vaccines include the following:
- Inactivated polio vaccine (IPV)
- Diphtheria, tetanus, acellular pertussis (DTap)
- Haemophilus influenzae type B (HIB) conjugate
- Meningococcal conjugated quadrivalent vaccine (MCV4)
- Pneumococcal vaccine: 13-valent pneumococcal conjugate vaccine (PCV13), 3 doses 1-2 months apart, then 1 dose of 23-valent pneumococcal polysaccharide vaccine (PPSV23) 6-12 months after the last PCV13
- Hepatitis A and B
The use of live-virus vaccines, such as measles-mumps-rubella (MMR) is generally contraindicated until at least 24 months posttransplant. In addition, patients must be free from GVHD and have immunologic response; they must have been off all systemic immmunosuppressive therapy for at least 1 year, and have not received intravenous immunoglobulin for at least 8 months. [54, 57]
![]()
Complications
Complications associated with transplantation consist of both early and late effects. Early-onset problems include the following:
- Mucositis
- Hemorrhagic cystitis
- Prolonged, severe pancytopenia
- Infection
- Graft-versus-host disease (GVHD)
- Graft failure
- Pulmonary complications
- Hepatic veno-occlusive disease
- Thrombotic microangiopathy
Mucositis
Mucositis is the most common short-term complication of myeloablative preparatory regimens (common with etoposide-containing regimens) and methotrexate used to prevent GVHD. Oropharyngeal mucositis is a painful condition and may require intubation when it involves the supraglottic area. Intestinal mucositis results in nausea, abdominal cramping, and diarrhea and requires total parenteral nutrition (TPN) to maintain caloric requirements. [58]
Topical pain medications and systemic opioids are required to manage pain adequately. A recombinant human keratinocyte growth factor, palifermin, reduces the incidence of mucositis after autologous transplant. Amifostine reduces the risk of mucositis following radiation and alkylating agents.
Hemorrhagic cystitis
Hemorrhagic cystitis is a disorder that manifests as dysuria and hematuria, with the hematuria being microscopic or gross. Clots may form within the bladder, occluding the urethral exit of urine, and may be severe. This disorder may occur in the immediate posttransplant period or may appear much later.
Medications used in conditioning (especially cyclophosphamide) are well known to be associated with hemorrhagic cystitis, especially earlier in the transplant timeline. A bladder protectant (mesna) is often used to help prevent this disorder, but it may still occur. Later onset is associated with infections such as adenovirus or BK virus. [59] Therapy is largely supportive, with hyperhydration, bladder irrigation, pain medications, and, in the most severe cases, surgery. Cidofovir may be helpful in viral infection with BK virus. [60]
Prolonged and severe pancytopenia
Severe (< 500/µL but often < 100/µL), prolonged (up to 4 wk) neutropenia is common after transplantation and invariably requires the use of empiric broad-spectrum antimicrobials until recovery of the neutrophils.
Empiric antifungal therapy with amphotericin B, fluconazole, or other agents is often administered if unexplained fever persists despite the use of broad-spectrum antibacterials. Antiviral therapy is usually given as prophylaxis (acyclovir for autografts; ganciclovir for allografts).
Serious infections (eg, pneumonia, bacteremia, fungemia, viremia) may occur in up to 50% of patients after transplantation and may develop more frequently with grafts from matched, unrelated donors (MUDs) than with autografts and sibling-matched allografts. Such infections are the major contributors to the mortality associated with these procedures. Recombinant hematopoietic growth factors (eg, filgrastim 5-10 mcg/kg/day SC, sargramostim 250 mcg/m2/day SC) started 24-72 hours after stem cell infusion reduce the time to blood neutrophil recovery.
Severe thrombocytopenia requires prophylactic transfusions for platelet counts of less than 10,000/µL, but for bleeding episodes or surgical procedures, the target may exceed 50,000/µL. Bleeding may occur despite platelet transfusions, as a result of visceral organ injury from chemotherapy (eg, gastritis, pneumonitis), acute GVHD, or infection (eg, adenovirus-induced hematuria due to cystitis).
Severe anemia requires frequent red blood cell (RBC) transfusions; recombinant erythropoietin (30,000-60,000 U/wk SC) is sometimes used after stem cell infusion to enhance erythroid recovery.
Infections
Many factors predispose to the development of infections in a transplant patient. Damage to the mucosal surfaces and skin from preparatory regimens and catheters, neutropenia and immunodeficiency from immunosuppressive medication, GVHD, and T-cell depletion of the graft all contribute to this.
The risk of infection with transplantation varies based on the underlying disease process, conditioning regimen, type of stem cells used as donor cells, posttransplantation immunosuppression, and the complications associated with transplantation.
Early after transplantation (0-30 days), mucosal and skin injury, as well as neutropenia, contribute to infections with aerobic bacteria (particularly coagulase-negative Staphylococcus species and viridans streptococci, gram-negative bacilli), Candida species, and herpes simplex. Colonizing yeasts invade the mucosa and cause systemic mycotic infections in 10-15% of patients.
One to 3 months after transplantation, T-cell dysfunction, hypogammaglobulinemia, and acute GVHD predispose to infections with the following:
- Encapsulated bacteria (eg, pneumococcus, Haemophilus influenzae)
- Viruses (eg, cytomegalovirus [CMV])
- Pneumocystis jiroveci
- Molds (eg, Aspergillus, Zygomycetes)
- Candida species.
Between 3 and 12 months after transplantation, slow T-cell reconstitution and chronic GVHD predispose patients to infections with encapsulated bacteria, CMV, P jiroveci, and herpes zoster virus. CMV, Epstein-Barr virus (EBV), and hepatitis viruses are particularly important.
Risk for specific bacterial and viral infections, as well as antibiotic/antifungal/antiviral susceptibilities, is often location and season dependent. The primary methods for prevention of infections are careful handwashing, sterile methods for accessing central lines, and limiting contact with the sick. In addition, hospital transplantation units typically test air and water purity to ensure a minimal threshold of exposure to concerning airborne and waterborne infections.
CMV and EBV
CMV infections or reactivation can be related to impaired viral immunity during the first year following transplantation, acute GVHD, and chronic GVHD. CMV-positive patients carry a higher peritransplantation mortality rate than do CMV-negative patients. Many patients, even though they have received a CMV-negative graft, become seropositive with time. Posttransplant patients are monitored for CMV infection and are treated prophylactically and preemptively to prevent pneumonitis and CMV viremia. However, CMV infections still occur late posttransplant and carry high mortality.
EBV infections are most common in patients who are EBV naive, patients receiving T-cell–depleted grafts, or those receiving antithymocyte globulin for in vivo T-cell depletion. EBV causes posttransplant lymphoproliferative disorder (PTLD) in HSCT patients and is more common in patients receiving T-cell depleted grafts and intensive treatment for GVHD.
Viral hepatitis
Viral hepatitis is the third most common cause of liver disease in transplant patients. (Liver GVHD and drug-induced hepatotoxicity are the first and second most common causes of liver disease in HSCT patients.)
In patients with prior exposure to hepatitis B virus (HBV), the impaired cellular immunity in the first 3-6 months after transplantation can cause reactivation of latent virus and lead to fulminant hepatic failure. Hepatitis B surface antigen (HBsAg)–positive patients should begin prophylactic antiviral therapy with nucleoside analogues before chemotherapy and continue for at least 3 months after chemotherapy. HBsAg-negative patients should receive HBV vaccination before HSCT.
Unlike HBV, infection with hepatitis C virus (HCV) appears to have little impact on short-term survival after HSCT. However, in the long term, it is a risk factor for hepatic venoocclusive disease and GVHD.
In long-term survivors with HCV, fibrosis progresses rapidly in the presence of HSCT and leads to cirrhosis, decompensation, and malignancy; liver-related mortality is the third leading cause of late deaths after transplant. Therefore, selected long-term survivors who have been off immunosuppression for more than 6 months and who have no evidence of GVHD should be considered for therapy with pegylated interferon and ribavirin.
Graft versus host disease
GVHD occurs when immunocompetent T cells and natural killer cells in the donor graft recognize host antigens as foreign targets and mediate a reaction. GVHD occurs very frequently in the allograft setting but rarely occurs in the autologous or syngeneic setting. The disease may cause significant morbidity and mortality and has been divided into acute and chronic forms.
The severity of GVHD is inversely related to the risk of relapse, because GVHD and the graft-versus-leukemia effect are interrelated. Therefore, strategies that reduce GVHD may increase relapse rates. New strategies are being developed to separate these effects in order to decrease the incidence and severity of GVHD without increasing the risk of relapse.
Strategies to reduce GVHD involve selection of the optimal available donor and graft type, along with posttransplantation immunosuppression. It appears the relative risk for GVHD increases from peripheral blood HSCT to bone marrow–derived HSCT to umbilical cord HSCT. The HLA mismatching increases the incidence of GVHD, with the relative importance of various HLA genes under investigation.
Immunosuppression, to optimize the balance between stable engraftment and reduction of risk and severity of GVHD, can be achieved with total body irradiation. Pharmacologic agents commonly used for immunosuppression are as follows:
- Gene expression regulators - Corticosteroids, vitamin D
- DNA alkylating agent - Cyclophosphamide
- Inhibitors of de novo pyrimidine synthesis - Leflunomide
Acute GVHD
Acute GVHD is a common complication of allogeneic transplantation; it occurs within the first 100 days after the procedure.
Acute GVHD involves skin, mucosal surfaces, gut, and liver. It starts as an erythematous, macular skin rash, and as it progresses, blistering of the skin similar to severe burns, severe abdominal pain, profound diarrhea, and hyperbilirubinemia develop. Acute GVHD is graded as per Glucksberg criteria. Stage I disease is confined to the skin and is mild; stage II-IV have systemic involvement. Stage III and IV acute GVHD carry a grave prognosis. [61, 62]
Risk factors for acute GVHD include HLA-mismatched grafts, MUD grafts, grafts from a parous female donor, and advanced patient age.
Pathogenesis of acute GVHD is considered to be a cytokine storm. The cytokines tumor-necrosis factor (TNF) and interleukin (IL)-1, which are released due to damage to host tissues from the preparative regimen, provoke increased major histocompatibility complex (MHC) expression and amplify the recognition of minor HLA differences by the donor T cells (which proliferate and release cytokines). These cytokines recruit more T cells and macrophages, which in turn release TNF and IL-1, setting up a vicious cycle of inflammation and tissue damage with the clinical manifestations of acute GVHD.
Prophylaxis of GVHD, which is more successful than treatment, can be achieved either by T-cell depletion of the graft or by using immunosuppressive agents against donor cytotoxic lymphocytes. T-cell depletion results in a significant reduction in GVHD but is accompanied by an increased risk of engraftment failure and rate of relapse due to the loss of the graft-versus-tumor effect.
A common regimen used to prevent acute GVHD consists of cyclosporine or tacrolimus along with a few days of methotrexate. However, cyclosporine and tacrolimus are each associated with renal toxicity, and methotrexate is associated with severe mucositis. Sirolimus (Rapamycin) and mycophenolate mofetil are alternatives with lower toxicity. Other measures to decrease acute GVHD include gut decontamination with metronidazole, the administration of intravenous immunoglobulin, and the use of a less intense preparative regimen.
The treatment of acute GVHD, which consists of high-dose steroids and antithymocyte globulin (ATG), remains disappointing. Experimental therapies with monoclonal antibodies to TNF (infliximab) or IL-2, extracorporeal photopheresis using apheresis machines, and drugs such as pentostatin are being offered to patients. [28, 63] The mortality rate is increased in patients who do not respond to treatment due to the increased risk of infection (particularly invasive fungal infections) and chronic GVHD.
Chronic GVHD
Approximately 40-80% of long-term survivors of hematopoietic cell transplantation experience this complication. The incidence of chronic GVHD is rising as an increasing number of transplants are being performed in older patients. Other risk factors include peripheral blood stem cell transplants, mismatched or unrelated donors, second transplant, and donor leukocyte infusions (DLIs). The greatest risk for chronic GVHD is acute GVHD.
Chronic GVHD develops 2-12 months after HSCT and involves the skin, eyes, mouth, liver, fascia, and almost any organ in the body. Patients with chronic GVHD present with chronic lichenoid skin changes, dryness of the eyes and mouth, and lichenoid skin changes in the oral mucosa, with ulceration and oral pain. Impaired range of motion occurs from fibrosis of the dermis and fascia. Hyperbilirubinemia and elevated alkaline phosphatase can occur. Although the clinical presentation of chronic GVHD mostly resembles scleroderma, it can mimic any other autoimmune disease.
The pathogenesis of chronic GVHD is not well studied, as most of the patients with this complication are at home and a good animal model for chronic GVHD is lacking. Some believe that it represents the consequence of an old acute GVHD, while others believe that it results from dysfunctional immunologic recovery after transplant.
Immunosuppression with corticosteroids, tacrolimus, and mycophenolate mofetil are the mainstays of treatment. Hydroxychloroquine, an antimalarial drug, is effective in several autoimmune disorders, including chronic GVHD. Some studies have suggested that the use of keratinocyte growth factors prevents GVHD, [64] presumably by preventing host thymic injury. [65, 66] Other studies have not confirmed this finding. [67]
Therapies under evaluation include extracorporeal phototherapy, pentostatin, acitretin, psoralen plus ultraviolet A (PUVA) therapy, thalidomide, and total-body irradiation. [63]
A major cause of death in chronic GVHD is infection from profound immunodeficiency associated with the disease. All patients require prophylaxis against encapsulated organisms, and patients with frequent infections and low immunoglobulin levels should receive intravenous immunoglobulin replacement.
Graft failure
Primary graft failure results from failure to establish hematologic engraftment after transplantation; late graft failure results from loss of an established graft. Graft failure is associated with increased risk of infection and peritransplant mortality.
Graft failure occurs in approximately 1-5% of sibling-matched allografts, as compared with 10-15% of MUD grafts. The greater the degree of HLA mismatch, the greater the risk of graft rejection. Other risk factors for failure include the following:
- Aplastic anemia
- T-cell depletion of the donor graft (loss of helper T cells, which help in engraftment)
- Infusion of lower number of hematopoietic stem cells - As in cord blood transplants
- Nonmyeloablative transplants
- GVHD
- Splenomegaly
- Use of methotrexate, mycophenolate mofetil, antithymocyte globulin, and ganciclovir
Poor graft function after transplant can be improved by growth factors such as granulocyte colony-stimulating factor (G-CSF), granulocyte-macrophage colony-stimulating factor (GM-CSF), and erythropoietin. In the case of graft failure, a second stem cell infusion can be useful.
Pulmonary complications
Transplantation-related lung injury (TRLI) is an acute inflammatory response that leads to severe lung injury. TRLI is seen in allogeneic transplants. Early treatment with corticosteroids and etanercept, an anti-TNF agent, can reduce the extent of this injury.
In patients receiving allografts, interstitial pneumonitis is frequently fatal. Often caused by viral infections (eg, cytomegalovirus infection), it is characterized by fever, infiltrates, hypoxemia, and acute respiratory distress syndrome. However, the prevalence has been lowered owing to the use of anti-infective prophylaxis and the selection of CMV-negative blood products (either leukoreduced blood or blood drawn from CMV-seronegative donors). Treatment with ganciclovir or foscarnet plus intravenous immunoglobulin is often effective.
A diffuse alveolar hemorrhage is sometimes observed in the autograft setting. Lung injury can also be due to total-body irradiation or pulmonary toxins (carmustine/methotrexate).
Hepatic veno-occlusive disease
Hepatic veno-occlusive disease (VOD), more accurately termed sinusoidal obstruction syndrome, is a common and potentially lethal complication that can develop after HSCT. It occurs in 10-60% of patients receiving HSCT and accounts for 50% of deaths after the procedure. The incidence in children ranges from 27% to 40%. [60] Severe VOD develops in less than 2% of patients after HSCT but has been fatal in up to 80% of cases. [68]
Clinically, hepatic VOD is characterized by weight gain and fluid retention, tender hepatomegaly, jaundice, and ascites and can progress to fulminant hepatic failure, respiratory failure, and renal failure. It usually starts 8-10 days after starting the preparatory regimen. Risk factors include the following:
- Prior hepatocellular damage
- High levels of busulphan
- Total-body irradiation of more than 10-12 Gy
- Heavy pretreatment prior to stem cell transplantation
- Presence of the C282Y allele of the hemochromatosis gene
Pathologically, hepatic VOD is characterized by damage to the sinusoidal endothelium, which sloughs off and obstructs the hepatic circulation, leading to centrilobular hepatic injury and portal hypertension. Perivenular fibrosis and cholestasis also occur. Elevated levels of tumor necrosis factor alpha (TNF-alpha) precede the development of hepatic VOD.
Prevention is the best approach to this disorder. Substitution of fludarabine for cyclophosphamide and the use of nonmyeloablative regimens decrease the incidence of hepatic VOD. Ursodiol (ursodeoxycholic acid) given in doses of 12 mg/kg in 2 divided doses starting a day before the preparatory regimen has been shown to reduce the risk of VOD and of grade III and IV GVHD. Jiang et al reported a significant reduction in the rate of VOD in patients receiving ursodiol plus pravastatin. [69]
Treatment is largely supportive. Recombinant tissue plasminogen activator (tPA) and heparin use has been associated with a significant risk of hemorrhage. Defibrotide (Defitelio) is approved for the treatment of adult and pediatric patients with severe hepatic VOD following HSCT. Approval was based on results of three studies in which 38% to 45% of patients treated with defibrotide were alive 100 days after HSCT, compared with expected survival rates of 21-31% for patients with severe hepatic VOD who received only supportive care or interventions. [68, 70] Defibrotide is a single-stranded polydeoxyribonucleotide derived from porcine tissue that possesses antithrombotic, thrombolytic, anti-inflammatory, and anti-ischemic properties. [71]
In 2013, the British Committee for Standards in Haematology (BCSH) and the British Society for Blood and Marrow Transplantation (BSBMT) issued the following guidelines for the diagnosis and management of hepatic VOD after HSCT [72] :
- Diagnosis can be based primarily on established clinical criteria; ultrasonography may help exclude other disorders; liver biopsy should be reserved for patients in whom the diagnosis is unclear.
- For prophylaxis, defibrotide is recommended at a dosage of 6.25 mg/kg IV every 6 hours in children and adults undergoing allogeneic HSCT who have certain risk factors.
- Defibrotide is also recommended for the treatment of VOD in adults and children.
For full discussion of this topic, see Veno-occlusive Hepatic Disease.
Transplantation-associated thrombotic microangiopathy
Transplantation-associated thrombotic microangiopathy (TA-TMA) is a microangiopathic hemolytic anemia that has been described in the early posttransplant period, and it is typically characterized by microangiopathic hemolytic anemia, platelet consumption, and fibrin deposition/thrombosis in the microcirculation, most commonly in the kidney. [73] The etiology of TA-TMA is not obviously related to ADAM-TS13 levels (von-Willebrand multimer–cleaving enzyme), as has been shown in thrombotic thrombocytopenic purpura (TTP). Investigational use of plasmapheresis or eculizumab (a monoclonal antibody to complement) is currently being studied.
Late-onset problems
Late-onset problems following HSCT are related to organ toxicity from large doses of chemotherapy received in the treatment of cancer and/or conditioning for transplantation, transplant-related GVHD, or posttransplantation immunosuppression. Since the field of modern transplantation is only about 50 years old and is rapidly changing, it can be difficult to determine the precise risks for late effects.
Late-onset problems include the following:
- Chronic GVHD
- Ocular effects
- Endocrine effects
- Pulmonary effects
- Musculoskeletal effects
- Neurologic effects
- Immune effects
- Infection
- Congestive heart failure
- Subsequent malignancy
Late malignancy, most often occurring many years following the transplantation procedure. Secondary acute leukemias, solid tumors, and myelodysplastic syndromes have been described. These conditions are disease and regimen dependent, with increased prevalence after total-body irradiation.
Late-onset infections can occur months after transplantation. These infections usually occur after allograft procedures in association with GVHD or GVHD therapy. Occasionally, they occur in autograft procedures after posttransplantation immunotherapy. Vaccinations are strongly recommended (ie, pneumococcus, H influenzae b, hepatitis B, poliovirus, diphtheria/tetanus, influenza). All patients receiving HSCT should be carefully followed by a physician, preferably a late-effects specialist.
Ocular effects
Posterior subcapsular cataract formation is common in HSCT recipients. Total-body irradiation is the predisposing risk factor, but fractionation of the dose substantially decreases the risk. Keratoconjunctivitis sicca, or dry eyes, is part of chronic GVHD syndrome. Other adverse effects include retinopathy, infectious retinitis, and hemorrhage. Treatment includes the use of topical lubricants and steroids. [74]
Endocrine effects
With improved HSCT outcomes, the number of long-term survivors has increased, including those who would like to have children. Although fertility can be preserved following HSCT, infertility is common in males and females. Young adults (aged 15-30 y) and prepubertal patients may be the most protected.
Pregnancies have been reported in patients receiving alkylating agents and total body irradiation. Although pregnancies following transplantation are currently considered high risk, the frequency of congenital malformations in children born posttransplantation does not appear to be increased, and increased risk of preterm birth has not been conclusively shown. Nonmyeloablative/reduced-intensity conditioning regimens may have higher posttransplantation fertility rates. [75]
Options to preserve fertility are available to patients, including sperm cryopreservation for men. In women, the options are investigational and include oocyte stimulation and retrieval, followed by in vitro fertilization and embryo cryopreservation, unfertilized oocyte cryopreservation, and ovarian cryopreservation.
In children, growth and development are impaired; these children may require growth hormone supplements. Hypothyroidism, overt and subclinical, is also common in these patients; they should be screened for low levels of thyroid hormone. [74]
Pulmonary effects
Pulmonary infiltrates in the setting of HSCT can be infectious or noninfectious and can be categorized by focality and infectious versus noninfectious, and early versus late. Infectious complications include respiratory viruses, CMV, P jiroveci, and adenovirus, typically producing diffuse infiltrates, and other bacterial, fungal (eg, Aspergillus), and nocardial pneumonia, typically producing focal infiltrates. Noninfectious etiologies of pulmonary infiltrates include acute respiratory distress syndrome, bronchiolitis obliterans, congestive heart failure, and hemorrhagic alveolitis, producing diffuse infiltrates, and aspiration, pulmonary embolism, and chemotherapy-related micronodules, producing focal infiltrates. [76]
Pulmonary effects include restrictive and chronic obstructive lung disease. Conditioning regimens, infections, and GVHD are important risk factors. [77]
Bronchiolitis obliterans is a specific form of obstructive lung disease seen in HSCT recipients and has a fatality rate of 50%. Corticosteroids are generally not helpful. Some patients respond to azathioprine and mycophenolate mofetil. [74] The French ALLOZITHRO trial, which was investigating the efficacy of long-term azithromycin in preventing bronchiolitis obliterans after allogeneic HSCT, was terminated early after an increased risk for cancer relapse was seen in patients taking azithromycin compared with placebo, and in August 2018 the FDA issued a safety warning advising against the use of azithromycin for prevention of bronchiolitis obliterans in patients undergoing HSCT for hematologic malignancies. [78]
Musculoskeletal effects
Osteopenia, osteoporosis, and avascular necrosis are common adverse effects in HSCT recipients. [79] Bisphosphonate therapy may be able to reverse some of the effects of this early-onset osteoporosis. [80]
Neurocognitive and neuropsychological effects
Lower intelligence quotient (IQ) scores, sleep disorders, fatigue, memory problems, and developmental delays and declines have all been reported. The greatest declines in these functional areas occur in patients who have received cranial radiation either as part of their oncologic therapy or as part of their HSCT conditioning. These issues must be addressed appropriately to improve the person's overall quality of life. [81]
Immune effects
Host immunity is suppressed for months to years after HSCT. This effect is more pronounced following allogeneic transplantation than it is after the autologous procedure. The factors responsible for depressed immunity include severe myelosuppression due to the myeloablative conditioning of the host, acute GVHD that further suppresses host immunity, and the use of immunosuppressants to prevent or treat GVHD. [82]
In allogeneic transplant recipients, complete immune reconstitution takes a few years and depends on the ability of naïve prethymic donor T cells to mature in the host's thymus and to become host tolerant and antigen specific. This process is most efficient in children and young adults because they have an active thymus. Older patients may never completely recover their immunity, because their thymic tissue may not be fully functional.
![]()
Future Directions for HSCT
Improved patient selection
Patient selection can be improved by identifying and treating malignancies at high risk for recurrence using newer prognostic guides (eg, International Age-Adjusted Index in non-Hodgkin lymphoma, International Performance Scoring in myelodysplastic syndrome, Hasenclever-Diehl Classification in Hodgkin lymphoma, poor-risk cytogenetics in leukemia) and by excluding high-risk patients or those unlikely to benefit from transplantation.
In addition, identification and matching or mismatching of genetic factors such as HLA-C and killer immunoglobulinlike receptors (KIRs) improves engraftment and reduces graft versus host disease (GVHD), along with optimizing graft-versus-leukemia effect in different hematopoietic graft types. Understanding donor characteristics that confer protection against disease may allow for a disease-specific effective HSCT. A good illustration is the successful transplantation of and treatment of HIV disease with donors who have CCR5 mutations that confer lymphocyte resistance to HIV infection.
Improved preparative regimens
Laboratory and clinical studies evaluating reduced-intensity conditioning regimens to improve graft stability; preserve graft-versus-leukemia effect; and reduce treatment-related morbidity, mortality, and late effects will likely result in improved outcomes for HSCT recipients. Reduced-intensity therapies have been pioneered in the young and old patients, who may have the worst toxicity from classic conditioning regimens, and have been shown to be effective. New conditioning regimens may also include targeted therapies to enhance engraftment or graft-versus-tumor effect, while minimizing the off-target effects of chemotherapy and radiation.
Stem cell mobilization and engraftment
The next generation of stem cell mobilization involves new pathways for release of hematopoietic stem cells from their specialized niche, for the purpose of collecting them or making space for newly infused stem cells to optimally engraft.
Along with enhancement of the disruption of CXCR4 ligand/CXCL12 receptor by small molecules that can reduce levels of CXCL12 or CXCR4, alternative target integrin ligand/receptor interactions such as VLA-4 and VCAM-1 are being explored. Downstream cell signaling pathways such as Rac1, which can be targeted by small molecules, such as NSC23766, have shown the ability to mobilize hematopoietic progenitors in murine models.
An alternative approach is to enhance hematopoietic stem cell homing to the niche of a patient to enhance long-term engraftment. [83] Potentially, this could be accomplished by dislodging present stem cells from the niche by targeted therapy such as C-kit antibodies or by enhancing homing by agents such as prostaglandin E2. [84]
GVHD and graft-versus-tumor effect
Prevention and treatment of transplant-associated GVHD has been a clinical challenge with an unpredictable clinical course, particularly in the setting of steroid-refractory GVHD. Three current strategies under investigation are targeting antigen-presenting cells, lymphocytes, and GVHD neovascularization in order to slow down or shut off pathogenic lymphocyte alloreactivity.
Adoptive T-regulatory cell therapy has entered clinical trials. Phase I/II studies have shown possible benefit in preventing GHVD. [85]
In vivo T regulatory expansion by mTOR inhibition as single or combination therapy has shown response in steroid-refractory GVHD. Natural killer cell immune down-regulation via interleukin 4–mediated pathways and mesenchymal stem cells as immunoregulators are currently being investigated. Research efforts in elucidating the pathways of lymphocyte dysregulation involved in GVHD are critical to the development effective treatments. [86]
For graft-versus-tumor effect, donor lymphocyte infusion was an effective tool in HSCT for chronic myeloid leukemia, but it has not translated well to other hematopoietic malignancies. Ex vivo or in vivo methods of activation of donor lymphocytes and natural killer cells are currently being investigated. [87]
Novel uses of HSCT
The use of HSCT to mediate tissue repair has been a longstanding controversy with scientists in disagreement about the ability of hematopoietic stem cells to differentiate into cells that could impact tissue angiogenesis and regeneration.
Clinical trials with peripheral or intracoronary infusion of hematopoietic progenitors have not shown homing of donor cells to the site of injury.
Clinical trials using intramuscular injection of bone marrow mononuclear cells or CD34+ cells for myocardial injury have shown to be safe, but show mixed efficacy. The PROTECT-CAD trial showed the most promising results, in which bone marrow mononuclear cells were injected directly into ischemic myocardium in patients with refractory myocardial ischemia. The 6-month follow-up showed improvement in exercise time and left ventricular function. Follow-up studies will be critical to determine efficacy. [88]
Another novel use of HSCT has been the treatment of recessive dystrophic epidermolysis bullosa, a genetic defect in type 7 collagen. Following bone marrow transplantation, there was evidence of type 7 collagen deposition in the skin dermoepidermal junction and detectable donor hematopoietic cells in the skin. There was also significant improvement in skin integrity. The mechanism of hematopoietic progenitor homing and clinical improvement is unclear, but it is suspected that donor cells may secrete replacement enzyme, which is taken up by recipient skin progenitors. [11]
These studies suggest the possibility of a therapeutic role for HSCT in tissue repair in specific disease processes.
Ex vivo expansion of hematopoietic stem cellOs
Ex vivo expansion of hematopoietic progenitor cells from a single umbilical cord blood unit may reduce the time of engraftment for a patient if co-infused with a second, unmanipulated umbilical cord unit. This may result from rapid engraftment and “burn out” of the expanded cord blood, resulting in transient hematopoiesis until the unmanipulated graft is functional. In April 2023 the US Food and Drug Administration (FDA) approved omidubicel, which uses this process. Omidubical is indicated for adult and pediatric patients 12 years and older with hematologic malignancies who are planned for umbilical cord blood transplantation following myeloablative conditioning, to reduce the time to neutrophil recovery and the incidence of infection. [89]
Pluripotent progenitors cells have the capability of long-term ex-vivo expansion, and multilineage differentiation has been a working alternative to expansion of hematopoietic progenitors. In theory, a few pluripotent progenitors could be expanded into many pluripotent progenitors and then differentiated into hematopoietic progenitors. Embryonic stem cells have been the longest studied pluripotent stem cells, but restricted access to tissue has resulted in relatively slow progress.
Discovery of the Yamanaka factors (transcription factors that can reprogram terminally differentiated cells into expandable pluripotent progenitors) has resulted in the broad availability of “inducible pluripotent stem cells,” termed iPS cells, for study. These iPS cells are extremely close in phenotype to embryonic stem cells.
In concept, a human can donate a small amount of blood or tissue, which can be transformed into his or her own iPS cells. These iPS cells can then be differentiated into any tissue in the body, including hematopoietic progenitors. Since the iPS cells can expand in tissue culture, they can also be efficiently modified by techniques such as gene therapy.
In a murine model, murine iPS cells have been expanded and differentiated into hematopoietic progenitors, followed by syngeneic transplantation to cure murine sickle cell disease. The differentiation of these murine iPS cells to hematopoietic progenitors required genetic integration of an oncogene, and some of the hematopoietic progenitors produced developed into tumors in some mice. A critical barrier for functional use of human iPS cells in therapeutics as a hematopoietic progenitor stem cell source is the ability to reliably differentiate these cells into a purified nontumorigenic hematopoietic stem cell population in vitro.
Gene therapy and autologous HSCT
Gene therapy for immunodeficiency has had a renaissance in the last decade, and it has been shown to be effective in generating an effective, albeit not completely normal, immune system in X-linked severe combined immune deficiency (X-SCID), adenine deaminase deficiency SCID (ADA-SCID), and chronic granulomatous disease (CGD).
These clinical trials used retroviral ex vivo additive gene transfer, during which an extra functional gene was added into the genome of hematopoietic progenitors, followed by autologous HSCT. The ADA-SCID trial results suggested that “some” compared with “no” conditioning prior to reinfusion of the gene-corrected cells improved engraftment of gene-transduced progenitors. The X-SCID and CGD trials showed evidence of insertional mutagenesis, wherein a proto-oncogene was activated, resulting in cancer in some of the patients who received gene therapy. The leukemia cases in the X-SCID trial were largely responsive to treatment.
Those results have led to the use of lentiviral vectors. Lentivirus vectors are genetically engineered based on the noninfectious components of the HIV design and can carry and integrate a therapeutic gene into a cell. Lentiviruses also have the advantage of penetrating the nucleus and integrating genes into nondividing cells. Clinical trials of this approach are under way. [22] Additionally, adrenoleukodystrophy and beta-thalassemia have been treated with gene therapy of hematopoietic progenitors using a lentivirus vector.
Use of gene editing has also been reported. In one technique, the mutation site is specifically targeted, using a tool such as the clustered regularly interspaced short palindromic repeats (CRISPR) with Cas9 nuclease system, and the mutation is replaced with the normal sequence. A major challenge in using gene editing strategies to cure β-thalassemia is that large numbers of gene-corrected stem cells are needed to produce therapeutically significant levels of hemoglobin, . [22]
Key barriers to gene therapy include the following:
- Efficient gene transfer
- Optimal expression and regulation of transferred therapeutic genes
- Efficient targeting of gene transfer to safe areas of the genome
- The development of scalable and cost-effective methods of gene therapy
![]()