Exploring the emerging role of the microbiome in cancer immunotherapy (original) (raw)
- 27k Accesses
- 216 Citations
- 81 Altmetric
- 7 Mentions
- Explore all metrics
Abstract
The activity of the commensal microbiota significantly impacts human health and has been linked to the development of many diseases, including cancer. Gnotobiotic animal models have shown that the microbiota has many effects on host physiology, including on the development and regulation of immune responses. More recently, evidence has indicated that the microbiota can more specifically influence the outcome of cancer immunotherapy. Therapeutic interventions to optimize microbiota composition to improve immunotherapy outcomes have shown promise in mouse studies. Ongoing endeavors are translating these pre-clinical findings to early stage clinical testing. In this review we summarize 1) basic methodologies and considerations for studies of host-microbiota interactions; 2) experimental evidence towards a causal link between gut microbiota composition and immunotherapeutic efficacy; 3) possible mechanisms governing the microbiota-mediated impact on immunotherapy efficacy. Moving forward, there is need for a deeper understanding of the underlying biological mechanisms that link specific bacterial strains to host immunity. Integrating microbiome effects with other tumor and host factors regulating immunotherapy responsiveness versus resistance could facilitate optimization of therapeutic outcomes.
Similar content being viewed by others
Background
The human body is a complex ecosystem inhabited and influenced by an abundance of microorganisms including bacteria, yeast, fungi, protozoa, archaea, and viruses, all of which collectively constitute the commensal microbiota. The commensal microbiota and the human host have co-evolved in a mutualistic relationship, in which each benefits the fitness of the other and the two can be collectively viewed as a superorganism. Much recent research has focused on the bacterial component of the microbiota. On average, a healthy human body is comprised of approximately 30 trillion cells and is inhabited by approximately 39 trillion bacterial cells [1]. The collection of genes within the commensal microbiota is defined as the commensal microbiome and vastly outnumbers human genes. The microbiota is capable of synthesizing or transforming a wide variety of metabolites, including hormones, essential vitamins, and other bioactive compounds, which cannot be otherwise acquired by the host [2]. These metabolites can modulate various biological functions, most notably the immune and nervous systems [3]. Alterations in the normal microbiota have been reported to contribute to the development of many diseases [4,5,6,7,8,9,10,11,12,13,14,15]. In the cancer context, some specific bacteria have been demonstrated to be involved in the process of carcinogenesis [15]. In addition, the microbiota has also been implicated in modulating the efficacy and toxicity of cancer therapy, including chemotherapy and immunotherapy [16]. Preclinical data suggest that modulation of the microbiota could become a novel strategy for improving the efficacy of immune-based therapies for cancer, in particular checkpoint blockade approaches targeting the CTLA-4 and PD-1 pathways [17, 18].
Establishment of commensal bacterial colonization in the human host
In adults the microbiota consists of about a dozen phyla, primarily Firmicutes and Bacteroidetes, followed by Actinobacteria, Proteobacteria, Fusobacteria, and others [19]. The relative proportions of these phyla vary between individuals and between anatomical sites. The GI tract is considered the most impactful site of host-microbe interactions. Various factors can influence the composition of the gut microbiota in a given individual, such as the composition of the maternal microbiota, mode of infant delivery (vaginal vs. C-section), diet, exposure to antibiotics and other medications, germline genetics of the host, and environmental factors [20]. Initial microbial exposure may occur as early as in utero, where the GI tract of the fetus may first be colonized by maternal bacteria through ingestion of amniotic fluid [21, 22]. After vaginal delivery, the neonatal microbiota resembles the mother’s vaginal microbiota and is undifferentiated across anatomical sites [23], but subsequently becomes shaped by the selective pressure of site-specific factors and by 3 years of age, an adult-like intestinal microbiota dominated by Firmicutes or Bacteroidetes is established. After this age, the microbiome composition in a healthy individual reaches a relatively stable state with minor fluctuations in physiological conditions, but strong and prolonged perturbations can occur in disease conditions or with antibiotics. At the species level there is enormous inter-individual heterogeneity in gut microbiomes, which has hindered efforts for clearly defining a core microbiome shared between healthy individuals. It has been suggested that the functional capacity of the microbiota, as depicted by abundances of genes involved in metabolic pathways, could constitute a metric better suited to define a core healthy microbiota [19, 24]. Indeed, the basic categories of metabolic pathways were more evenly represented across individuals as compared to bacterial taxonomy [19]. It remains to be determined whether this shared set of metabolic pathways is the major characteristic of a healthy microbiota.
Next generation sequencing methods in microbiome studies
Culturing of bacterial strains has been central to classical microbiology and has enabled the study of individual pathogens and some commensal bacteria. For most commensal bacteria, culture methods had not been optimized for their in vitro isolation and study. With recent improvements in methodology, a large proportion of commensal bacteria is now considered culturable [25, 26]. Culturomics is a strategy which incorporates multiple culture conditions, coupled with MALDI-TOF mass spectrometry and/or 16S ribosomal RNA (rRNA) or total genome sequencing for bacterial identification [27, 28]. This high-throughput approach can enable isolation and identification of commensals for further manipulation and mechanistic studies.
The most common method for taxonomic characterization of complex bacterial communities is based on selective amplification and sequencing of part of the gene encoding the 16S rRNA, part of the small ribosomal subunit in prokaryotes. This is a ubiquitous 1.5 kb gene, containing conserved sequences and hypervariable regions (nine regions: V1-V9), the latter being useful for bacterial taxonomic classification, as originally described by Woese and colleagues [29]. In the first step of this technique, a pair of universal primers targeting conserved sequences flanking a hypervariable region are used to generate an amplicon library, which is then sequenced. To account for sequencing errors, amplicons which share sequence similarity above a certain threshold are grouped into operational taxonomic units (OTUs). A representative amplicon is selected from each OTU bin and assigned a taxonomic identity based on cross-referencing to pre-existing databases [30,31,32]. All other amplicons in the OTU are also assigned the same identity. Thus, OTU binning can artificially decrease the observed diversity of a microbial community [33] and alternative methods for analysis have been proposed [34,35,36]. Because bacterial identification is based on a portion of the 16S rRNA gene, species level resolution is usually not feasible with this method and identification is typically limited to family or genus level [37]. Another consideration in 16S analyses is that most bacteria contain multiple copies of the 16S rRNA gene, which can lead to inaccurate quantitation of bacterial cells [38]. Additional bias can be introduced in the amplification step, depending on the choice of primers. Despite these limitations, the low cost and high-throughput potential of this technique make it the most commonly used for initial descriptive data.
Metagenomic shotgun sequencing generates short reads representing the whole genomic content within an environmental sample and is considered less biased than 16S rRNA gene amplicon sequencing, because it does not contain a PCR amplification step. However, this can result in contamination with human genomic DNA and requires higher sequence coverage to detect bacterial species of low abundance. This necessitates additional data storage, computing power, and more sophisticated analysis pipelines. Errors can also be introduced in the downstream analysis at the step of genome assembly or gene prediction [39]. Various bioinformatic tools have been developed for metagenome assembly, and databases have been established for gene prediction, but there is no consensus on the best strategy [40]. Compared to 16S rRNA gene amplicon sequencing, superior resolution down to species and strain level identity is feasible with shotgun sequencing because multiple marker gene sequences are used for taxonomic annotation [37]. This approach can also be used to characterize non-bacterial compartments of the commensal microbiota, including archaea, fungi or viruses. Another advantage of shotgun sequencing is that it can be used for characterization of the functional capacity encoded by the microbiome using gene prediction tools and databases [40]. By contrast, functional capacity can only be inferred indirectly from 16S rRNA amplicon sequencing data [41,42,43]. Each of these sequencing methods has its limitations, but the two can be integrated to improve the accuracy of bacterial identification and quantitation [44].
Impact of the commensal microbiota on immunity: insights from gnotobiotic mouse models
The role of the commensal microbiota in modulating host physiology becomes particularly evident when conventionally raised specific pathogen-free (SPF) mice are compared to germ-free (GF, axenic) mice. GF mice are defined as devoid of detectable microbiota during their life. The term gnotobiotic pertains to animals with known (defined) microbiota composition and encompasses GF, as well as ex-GF animals colonized with defined microbial communities. The commensal microbiota broadly impacts host physiology, and this has been mainly shown in studies with GF mice, which have inefficient energy extraction from the diet, abnormal fluid balance and electrolyte status, and disturbances in liver, lung, cardiovascular system, endocrine organ, nervous system, and immune system functions [45, 46].
Impact on local immunity
The gut microbiota is intimately involved in the development and regulation of the immune system, especially with respect to local mucosal immunity. This has been demonstrated in GF mice, which show deficiencies in the gastrointestinal immune compartment rendering them more susceptible to infections. However, such deficiencies can be corrected by colonization with commensal bacteria. For instance, in GF mice, the mucus-producing goblet cells are fewer and smaller. As a result, the mucus layer, the first line of defense against pathogens in the intestine, is thinner and has a different mucin composition [47, 48]. Additional examples of GI immune defects in GF mice include: 1) smaller mesenteric lymph nodes (MLN) and abnormal high endothelial venules with poor lymphocyte binding [49]; 2) fewer and smaller Peyer’s patches which lack germinal centers [50, 51]; and 3) lack of lymphoid follicles in the intestinal lamina propria (LP), but presence of nascent cryptopatches which can develop into functional isolated lymphoid follicles upon microbial colonization [52,53,54]. These local immune deficiencies are accompanied by a decreased number of LP CD4+ T cells, plasma cells, and decreased IgA production leading to further impaired intestinal barrier function [55, 56]. The presence of commensal bacteria is required not only for normalizing the LP CD4+ T cell numbers, but also for proper programming of the local Treg/Th17 balance. GF mice are almost completely devoid of Th17 cells, but have increased frequency of FoxP3+ T cells [57].
Impact on systemic immunity
Systemic innate immune modulation is also influenced by the commensal microbiota, with multiple lines of evidence indicating stimulatory effects on myelopoiesis at the level of granulocyte-macrophage progenitors in the bone marrow and in the periphery, as well as on the function of DCs, macrophages, and neutrophils (reviewed in [58]). In many cases, these systemic effects have been attributed to circulating bacteria-derived molecules (microbe- or pathogen-associated molecular patterns, MAMPs and PAMPs, respectively), such as lipopolysaccharide (LPS), peptidoglycan, or flagellin, which when recognized by pattern-recognition receptors (PRRs) on innate immune cells, can signal via a MyD88-dependent pathway to enhance systemic innate immune cell responsiveness [58]. Bacterial metabolites, such as short-chain fatty acids (SCFA), the products of dietary fiber fermentation by the microbiota, have been implicated in stimulating DC generation in the bone marrow and their phagocytic capacity [59]. Systemic adaptive immunity is also stimulated by the presence of commensal bacteria, particularly the proper development of distant (non-mucosal) lymphoid tissues, such as the spleen and peripheral lymph nodes. This is evidenced by the poorly developed B cell follicles and T cells zones in these organs in GF mice, leading to decreased IgG levels in the serum [60, 61]. Commensal bacteria are also required for proper programming of the Th1/Th2 balance and in GF mice there is a bias towards Th2-type allergic responses, which can be corrected by colonization with commensal bacteria [62].
Specificity of microbiota-mediated immune programming
Different members of the commensal microbiota are not equivalent in their capacity to polarize T cell responses. For instance, in SPF mice the group of segmented filamentous bacteria (SFB), which colonize the mouse terminal ileum and adhere to the epithelial cells, are particularly potent inducers of Th17 cell differentiation [63]. SFB are not found within the human microbiota, but further studies have shown that other bacteria derived from human fecal samples are also capable of adhering to the epithelial layer and inducing Th17 cells when transferred to mice [64,65,66]. By contrast, Treg differentiation and function are strongly induced by Bacteroides fragilis [67] and Clostridium clusters XIVa, IV, and XVIII [68, 69]. Polysaccharide A (PSA) from the capsule of B. fragilis can polarize towards Th1-type responses [62]. Higher Bacteroidetes/Firmicutes ratio resulting from high-fiber diet increased the levels of circulating SCFAs and alleviated Th2 cell-mediated allergic airway inflammation by reducing the capacity of lung-resident DCs to drive Th2-type responses [59]. Monocolonization of GF mice with 52 different human commensal bacteria demonstrated that most of the species were capable of inducing alterations in the frequency and function of immune subsets within the intestinal LP, Peyer’s patches, MLN, and spleen. Some more notable effects were alterations in cytokine production in the LP and in frequencies of Treg, pDC, CD103+ dendritic cells (DCs), macrophages and mononuclear phagocytes [66]. Notably, many species were able to translocate to the MLN and spleen [66]. This is likely an artifact of the model, due to the poor intestinal barrier function in GF mice. Therefore, the mechanisms leading to the observed alterations in immune cell subset composition, especially those seen systemically, may not in all cases reflect the physiological state.
Practical considerations in the use of germ-free mouse models
SPF mice have been used to gain valuable insight in the impact of microbiota-host interactions on host physiology in health and disease. When it comes to clinical translatability, a question that arises regarding the degree of similarity between the microbiomes of humans and laboratory mice. Although a direct comparison between datasets from different studies can be blurred by differences in analysis platforms and protocols, a general consensus exists that on a phylum through family level, the microbiomes of SPF mice and humans are similar with both species being predominantly colonized by Bacteroidetes and Firmicutes [70, 71]. Comparison between datasets on a deeper taxonomic level is challenging because of limited representation of microbial genes in the current databases causing difficulties with genus, species and strain level annotation. A study comparing microbial metagenomes of humans and SPF mice of different genetic backgrounds and housed in different facilities showed that only 4% of microbial gene sequences were shared between humans and mice. Despite that discordance, functional annotation of the mouse and human microbiomes using the KEGG database revealed that 85% of the annotated gene orthologs were shared between mouse and human microbiomes [72]. Therefore, the murine organism as a host appears to have similar functional requirements for the commensal microbiota, which makes it an appropriate recipient of human microbiota for studying its effects on host physiology. A high value of GF mice in microbiome research is their utility in generating purely human microbiota-associated mouse models for studying microbe-host interactions and demonstrating causal effects of the microbiota on the health/disease states of the host. Indeed, successful transfer of microbiota from humans to GF mice often imprints the human health phenotype onto the murine recipient.
There are some differences between mice and humans which might affect the efficiency of human gut microbiota engraftment into mice or their spatial establishment throughout the GI tract. A potentially relevant difference in GI tract anatomy is the presence of a non-glandular fore-stomach in mice, which takes up two thirds of the stomach, has no secretory activity, and serves for temporary food storage. This allows for food to be ingested in bulk, but to be released for downstream digestion more gradually according to energy demands. The lack of gastric secretions in the fore-stomach results in higher pH of its contents (pH 4.8) [73] and the overall pH in the mouse stomach is 2.7–4.1, while in humans it can be as low as pH 1 [71]. The milder pH and the abundance of oligosaccharides in the mouse fore-stomach provide conditions for the bloom of Lactobacillae, whereas in humans, the stomach contains mainly Streptococcus, Prevotella spp. and Helicobacter pylori [71, 73]. Another difference is the presence of circular folds (plicae circularis) in the human small intestinal mucosa, which are absent in mice [71, [74](/article/10.1186/s40425-019-0574-4#ref-CR74 "Treuting PM, Arends MJ, Dintzis SM. Lower Gastrointestinal Tract. In: Comparative Anatomy and Histology [Internet]. Elsevier; 2018 [cited 2019 Jan 21]. p. 213–28. Available from: https://linkinghub.elsevier.com/retrieve/pii/B9780128029008000129
")\]. These structures could provide additional niche for mucus-associated bacteria \[[71](/article/10.1186/s40425-019-0574-4#ref-CR71 "Hugenholtz F, de Vos WM. Mouse models for human intestinal microbiota research: a critical evaluation. Cell Mol Life Sci. 2018;75(1):149–60.")\]. Mice also have a relatively large cecum, where microbial fermentation of indigestible fiber takes place, while in humans the cecum is small and of uncertain importance \[[74](/article/10.1186/s40425-019-0574-4#ref-CR74 "Treuting PM, Arends MJ, Dintzis SM. Lower Gastrointestinal Tract. In: Comparative Anatomy and Histology [Internet]. Elsevier; 2018 [cited 2019 Jan 21]. p. 213–28. Available from:
https://linkinghub.elsevier.com/retrieve/pii/B9780128029008000129
")\]. In humans, fermentation and production of vitamins K and B and SCFA occur in the colon, which is segmented into pouches (haustra). The cecal appendix in humans is enriched in gut-associated lymphoid tissue and in microbial burden and has been hypothesized to serve as a reservoir of beneficial bacteria which may replenish the microbiota after diarrhea or other disturbances \[[75](/article/10.1186/s40425-019-0574-4#ref-CR75 "Smith HF, Parker W, Kotzé SH, Laurin M. Multiple independent appearances of the cecal appendix in mammalian evolution and an investigation of related ecological and anatomical factors. Comptes Rendus Palevol. 2013 Aug 1;12(6):339–54.")\]. In mice, the appendix does not exist as a separated structure. Additional differences in GI tracts of humans and mice that might affect the fidelity of human microbiota transfer to mice include overall lower pH and oxygen tension in the mouse intestine, as well as differences in the glycan profile of the mucus, which might affect the growth of mucus-utilizing bacteria. Apart from differences in GI tract, the inability of some bacterial species to survive the conditions of the transfer, including storage outside the host, oxygen exposure, and longer time spent in the stomach, may also limit the fidelity of reconstitution in mice. Differences in diet between human donor and mouse recipient could additionally result in skewed engraftment profiles. The sex of the recipient mouse has also been shown to affect colonization fidelity \[[76](/article/10.1186/s40425-019-0574-4#ref-CR76 "Wang J, Wang J, Pang X, Zhao L, Tian L, Wang X. Sex differences in colonization of gut microbiota from a man with short-term vegetarian and inulin-supplemented diet in germ-free mice. Sci Rep. 2016 Oct 31;6:36137.")\].GF mice have many physiological defects, which can become a confounding factor in microbiome studies. Notably, due to compromised intestinal barrier function and immature immune system in GF mice microbial colonization could result in systemic translocation and abnormal magnitudes and sites of microbe-host interactions [66]. A more physiologically relevant mouse colonization would be the acquisition of experimental microbiota from the mother at birth. Thus, the offspring from artificially colonized by gavage ex-GF mice, can be used for experimentation. It has been shown that the microbiota from artificially colonized ex-GF mice bred in an isolator can be vertically transmitted to generations F1 and F2 without significant drift between generations [77]. The use of such offspring mice could also capture effects of microbiota-mediated epigenetic immune programming occurring in utero. In addition to proper guiding of immune system maturation, such natural colonization of offspring mice with a functionally complex microbiota could eliminate other confounding factors such as the metabolic and endocrine abnormalities characteristic of GF mice. Therefore, an important experimental tool is to generate gnotobiotic mouse colonies maintaining a stable and defined microbiota derived from individual human subjects, functionally recapitulating the complex SPF microbiota and normalizing mouse physiology [78]. Towards this goal, it has been shown that a small number of culturable bacterial strains can cover most of the functional potential of the gut microbiome [79, 80]. Individual strains of interest can then be introduced and their immunomodulatory roles can be studied in the context of more physiologically relevant conditions [80].
An alternative to using GF mice as human microbiota recipient is the use of antibiotic-treated SPF mice. Although SPF mice with intact microbiota are generally not receptive to human microbiota, engraftment can be substantially improved with certain antibiotic regimens, which deplete the bulk of the pre-existing commensals, thus opening up an niche for subsequent colonization [81, 82]. Such models can be a useful alternative in mechanistic studies with some mouse strains of genetically engineered mouse models unavailable in GF status. However, the potential contribution of residual non-depleted mouse microbiota should be considered in such experimental settings, including its influence not only on the host but also on the acquired human microbes.
When interpreting results from experiments with GF mice, it should also be considered that even though GF mice are devoid of detectable viable microbiota, they are exposed to microbial residues (MAMPs, PAMPs, or antigens) derived from dead bacteria in sterile diet and bedding [83]. If present in sufficient quantities, these molecules could theoretically affect immune functions in a similar manner as do intact viable bacteria. For instance, MAMPs/PAMPs can be recognized by PRRs on intestinal epithelium or mucosal immune cell subsets leading to downstream signaling. Bacterial antigens can be sampled directly from the intestinal lumen by DCs or can be transported to LP antigen-presenting cells (APCs) via passage through goblet cells. APCs, in turn, can migrate to MLN and activate adaptive immunity. Bacterial antigens may also be taken up by M cells to stimulate plasma cell development and IgA secretion in Peyer’s patches. Because GF mice have poor barrier function, MAMPs/PAMPs and antigens might also translocate into the circulation and affect systemic immunity. Commonly used sterile diets can have a range of levels of microbial residues. For instance, LPS content, as a measure of overall bacterial contamination in diets, shows a range of 1–100 EU/μg [84]. A sterile diet rich in microbial residue can induce maturation of the immune system in a similar manner (albeit less prominently), as does colonization with commensal bacteria, with particularly strong impact on CD4+ T cells and Treg cells in the MLN and IL-4 and IL-12 cytokine responses in spleen cells [84]. Indeed, a sterile chow that contained high level of microbial residues resulted in decreased Th2-type response to allergic sensitization of GF mice compared to a sterile diet that was poor in microbial residues [83]. Use of chemically defined ultra-filtered diet, rather than conventional sterile chow could uncouple the effects of microbial colonization from those of dietary microbial residue exposure.
Evidence linking the gut microbiome to cancer immunotherapy
Multiple studies support that gut microbes can profoundly influence the potency of immunotherapy and some chemotherapies with immunostimulatory functions (summarized in Table 1). Pioneering work in this field found that intestinal microbiota was essential for optimal responses to CpG-oligonucleotide immunotherapy which activates innate immune cells through TLR9 [85]. Similarly, the gut microbiota was found to shape the anti-cancer immune response by stimulating generation of a specific subset of “pathogenic” Th17 (pTh17) cells and memory Th1 immune response after treatment with immune-stimulatory chemotherapy cyclophosphamide [86]. Certain bacterial taxa in patients with hematologic malignancies are associated with efficacy of allogeneic hematopoietic stem cell transplantation (allo-HSCT) and decreased risk for graft-versus-host disease (GVHD) following therapy [87, 88]. Initial evidence for the contribution of specific microbes to immune checkpoint blockade (ICB) immunotherapy, including CTLA-4 and PD-1/PD-L1 blockade, was demonstrated in mouse models [17, 18]. B. fragilis was reported to enhance anti-CTLA-4 efficacy via a proposed mechanism involving the activation of Th1 cells with cross-reactivity to bacterial antigens and tumor neoantigens [18]. Oral administration of Bifidobacterium increased tumor infiltration and IFN-γ production by CD8+ tumor-specific T cells and improved both basal tumor control and anti-PD-L1 efficacy via a proposed mechanism involving increased activation of splenic and intratumoral DCs [17]. These mouse studies established the importance of the microbiome in cancer ICB therapy and inspired clinical pursuits to assess the microbiome’s impact on anti-CTLA-4 and anti-PD-1/PD-L1-based therapies in patients.
Results from multiple institutions have contributed to the growing consensus that the gut microbiome is linked to immunotherapy efficacy in cancer patients [44, 89,90,91,92]. DNA sequencing of stool samples collected prior to checkpoint blockade therapy identified an association between gut microbiome composition and subsequent therapeutic response. Distinct bacterial taxa were overrepresented in responder (R) patients, whereas other bacterial sequences were over-represented in non-responder (NR) patients. Importantly, only some of these identified bacteria were consistent across multiple studies. This discrepancy may reflect discordant biology—the patient populations were from geographically distinct locations, with potentially dissimilar environmental and genetic factors—but also may be explained by technical differences, such as fecal collection, storage and DNA extraction and sequencing methods, as well as downstream bioinformatic analysis. Moving beyond correlative studies, human microbiota “avatars” (GF mice colonized with patient stool-derived commensals) have been used to show the mechanistic contribution of the microbiota to treatment response. Mirroring patient data, mice reconstituted with R patient fecal material showed greater benefit from checkpoint blockade than mice colonized with NR fecal samples [44, 89, 90]. Beyond clinical efficacy rate, immune-related toxicity of ICB has also been linked to the composition of the gut microbiome. Based on stool samples collected from patients treated with an anti-CTLA-4 antibody, bacteria in the Bacteroidetes phylum were associated with lower incidence of treatment-induced colitis [93].
Table 1 Studies linking the gut microbiome composition to efficacy of cancer therapy. The table summarizes major findings from clinical and preclinical studies pointing to a link between gut bacteria and therapeutic outcomes in the context of various cancers and therapeutic regimens
Deciphering the biological mechanism of microbiome mediated immune modulation
These findings linking the gut microbiome to immunotherapy efficacy only scratch the surface of this complex interaction. Determining the biological mechanisms is critical for moving towards therapeutic manipulation of the microbiota to optimize patient response. Tractable mouse models are being utilized to explore the causal role gut bacteria play in treatment efficacy.
When it comes to exploring the possible mechanisms of microbiota-mediated modulation of anti-tumor immunity, two general questions arise. First, what is the nature of the messenger, which delivers a signal from the GI tract to the tumor and/or tumor-draining lymph node (TdLN)? Such a messenger would be able to enter the circulation in order to access the distant tumor site and can be classified as microbiota- or host-derived cell (live microbes or host immune cells) or molecule (MAMP/PAMP, microbial metabolite, or host cytokine). The second question is what is the nature of the immune effect that the messenger confers within the tumor? An immunosuppressive effect could be achieved by augmenting regulatory functions (Tregs, MDSCs, tumor-associated macrophages) or directly inhibiting anti-tumor immunity; an immunostimulatory effect could be achieved by alleviating regulatory functions or stimulating anti-tumor T cell responses (via antigenicity, adjuvanticity or bystander activation). The exact mechanisms of microbiota-mediated effects on tumor growth and efficacy of immunotherapy are only beginning to be understood. Figure 1 summarizes these hypothetical scenarios and early evidence is discussed below.
Fig. 1
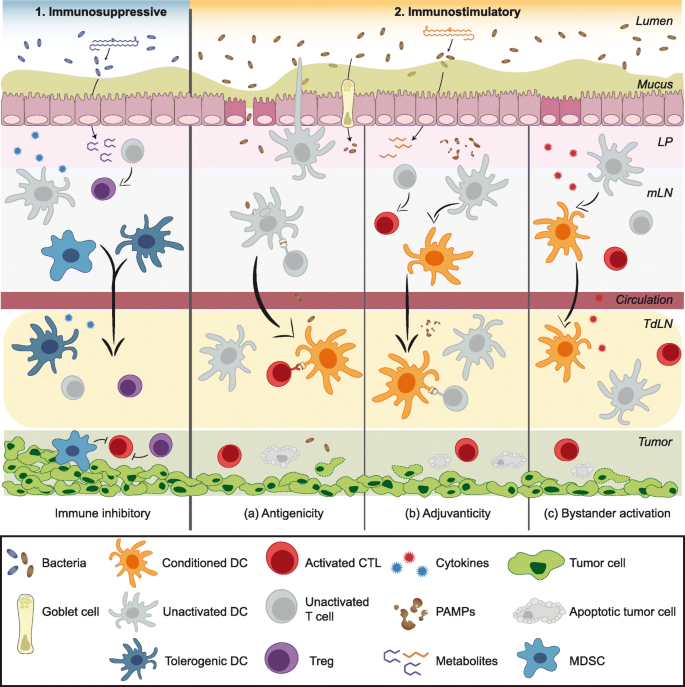
Possible mechanisms linking the gut microbiota to anti-tumor immunity. The composition of the gut microbiome may impact immunotherapy efficacy by either acting as (1) an immunosuppressive or (2) an immunostimulatory factor via various non-mutually exclusive mechanisms. (1) Certain commensal bacteria may suppress anti-tumor immunity by skewing immune subset balances towards suppressive phenotypes such as Tregs and MDSCs. Locally in mucosal sites, induction of immunosuppressive cells could be mediated by cytokines released by host cells (such as gut epithelium or immune cells) in response to microbial sensing. Immunosuppressive effects in distant sites, such as active immunosuppression in the TME, could be mediated by trafficking of locally induced suppressor cells. Additionally, bacterial metabolites with immunosuppressive properties might be released into the circulation and promote immunosuppressive cell functions in the TdLN and TME. Chronic inflammation caused by continuous stimulation by PAMPs/MAMPs or epithelial injury could also ultimately contribute to immunosuppression over time. (2) The immunostimulatory effects of the gut microbiota could be mediated by augmented antigenicity, adjuvanticity, or bystander T cell activation. (a) Antigenicity: Cross-reactive T cells driven by bacterial antigens that additionally recognize tumor-associated antigens is one conceivable mechanism. Luminal bacteria or bacterial antigens can be internalized by DCs in the LP via trans-endothelial dendrites extending through the epithelium into the lumen. Goblet cells and M cells can also serve as portals to deliver bacterial antigens to mucosal APCs. Alternatively, disruption of barrier function may allow for the translocation of luminal bacteria and bacterial antigens. Antigen-loaded DCs can migrate from the LP to the MLN and possibly to distant sites such as the TdLN, where they may prime cross-reactive anti-tumor CD8+ or CD4+ T cells, enhancing cytotoxic T lymphocyte (CTL) function in the TME. (b) Adjuvanticity: PAMPs/MAMPs may condition DCs to be more potent T cell activators, for instance by upregulating costimulatory molecule expression, enhancing antigen presentation, or boosting type I IFN production. Some microbial metabolites could alter immune cell function epigenetically or otherwise to poise innate and adaptive cells in a heightened activation state. (c) Bystander activation: A heightened inflammatory state in the TME driven by pro-inflammatory cytokines released in response to bacterial stimuli may contribute to tumor cell killing by T cell help provided by bacteria-specific T cells to tumor antigen-specific T cells
Live bacteria or MAMPs/PAMPs as messengers
Commensal bacteria have been identified in extra-gastrointestinal tissues typically considered to be sterile. Notably, Geller et al. identified bacteria within the TME in human pancreatic ductal adenocarcinoma [94]. In this study, viable bacteria were hypothesized to gain access to the cancerous lesions via a retrograde migration from the duodenum towards the pancreatic duct and were shown to decrease gemcitabine chemotherapy efficacy by metabolizing the active form of the drug. In terms of impact on immune function, it was experimentally shown that bacterial translocation into the MLN and spleen generated a Th1 memory response specific to the translocated species [86]. In the scenario of bacterial translocation, live bacteria gaining access to spleen, lymph nodes, or tumor may initiate a strong immune response by providing both foreign antigens and adjuvants (MAMPs/PAMPs). Consequently, tumor cell killing may ensue due to T cell cross-reactivity or bystander activation within the tumor microenvironment (TME). Thus, commensals might bolster anti-tumor immunity through both augmented antigenicity as well as adjuvanticity, as described below.
Augmented antigenicity due to cross-reactivity to bacteria and tumor antigens
Some data suggest a mechanistic role for T cell epitopes shared between bacteria and tumor cells [18, 89, 95]. Under this proposed model, cross-reactive T cells primed against bacterial antigens might exert anti-tumor effects either by providing help (CD4+ T cells) or through direct killing (CD8+ T cells). In a preclinical study, adoptive transfer of _B. fragilis-_reactive CD4+ T cells conferred enhanced tumor control and restored anti-CTLA-4 efficacy in GF mice [18]. Peripheral immune cells isolated from patients receiving immune checkpoint blockade (ICB) treatment and assayed for in vitro T cell IFN-γ production following stimulation with certain bacteria showed an association with progression-free survival (PFS), whereas non-specific T cell activation with polyclonal activators demonstrated no relation to ICB response [89]. Balachandran et al. found intra-tumoral and circulating T cell clones with specificity to both neoantigens and predicted cross-reactivity with microbial epitopes [95].
Adjuvanticity of MAMPs/PAMPs
Microbiota-derived MAMPs or PAMPs can traverse the mucosal barrier and enter the circulation. For instance, serum from healthy individuals was demonstrated to contain stimuli capable of activating a range of TLR and NOD receptors [96]. In the cancer context, bacterial LPS aberrantly entering the circulation following total body irradiation augmented the activity of adoptive T cell therapy in mouse models [97]. Additionally, nucleic acids from bacteria have also been shown to act as natural adjuvants [98]. In particular, the unmethylated CpG dinucleotides enriched in prokaryotes are potent activators via TLR9. These pro-inflammatory microbial products can trigger at least partial activation of innate immune cells such as DCs. Such conditioned APCs might possess enhanced capacity to prime anti-tumor T cells. Evidence for heightened DC activation stemming from distinct microbiome compositions was illustrated in Sivan et al. who showed that splenic DCs isolated from mice colonized with Bifidobacterium sp. showed superior priming of naïve CD8+ T cells ex vivo [17]. Enrichment in Faecalibacterium genus in patients with metastatic melanoma associated with responsiveness to ICB therapy was also associated with increase in antigen processing and presentation markers in the tumor [90].
Microbial metabolites as messengers
Gut bacteria produce various bioactive molecules as byproducts of their metabolism. These metabolites can exhibit diverse effects on the host, including modulating the immune system [99]. SCFAs are one of the most extensively characterized classes of microbial metabolites known to shape host immunity [100]. Through anaerobic fermentation, bacteria break down complex carbohydrates into SCFAs such as acetate, butyrate, and propionate. These metabolites are the primary energy source consumed by intestinal epithelial cells [101] and can also affect cytokine production [102], macrophage and DC function [59, 103], and B cell class switching [104]. SCFAs can additionally act to inhibit histone deacetylases, facilitating Treg differentiation [105]. By mimicking human signaling molecules, SCFAs can also act as ligands for G-protein coupled receptors [106]. Other bacterial metabolites relevant to host immunity include retinoic acid and co-metabolites, such as polyamines and aryl hydrocarbon receptor ligands [107]. These small molecules can impact immunity by acting as signaling molecules, epigenetic regulators, and metabolic switches and may ultimately shape anti-tumor immunity.
Given the predicted importance of bacterial metabolic contribution to host immunity and immunotherapy efficacy, there is significant interest in identifying both the specific bacteria exerting immune modulatory effects, as well as the functional and metabolic characteristics of these bacteria_._ To address this question, metagenomic and metatranscriptomic sequencing approaches coupled with metabolomic analysis of patient serum and stool samples will be critical for a more complete characterization of the biosynthetic pathways present within a given microbiome. Insights into metabolic contributions of the microbiome in the context of immunotherapy also may lead to new candidate therapeutic strategies, either through provision of desired metabolites as drugs, or via genetic manipulation of selected commensals for clinical administration.
Host cytokines as messengers
Another potential mechanism by which gut bacteria could modulate systemic immune responses is through local induction of soluble immunomodulatory factors that then disseminate systemically. Circulating cytokines may shift the activation threshold of key immune subsets within the TME or TdLN, thus leading to augmented adaptive immune responses in the context of immunotherapy. Candidate mechanisms include increased production of type I interferons, IL-12 and TNFα, or decreased production of immune suppressive cytokines such as IL-10 and TGF-β. As an example, segmented filamentous bacteria can induce secretion of IL-22 from type 3 innate lymphoid cells in mice, causing production of serum amyloid A in the terminal ileum which, in turn, acts on the LP DCs to drive Th17 polarization [63, 108]. In cancer models, oral administration of Akkermansia muciniphila improved the efficacy of PD-1 blockade in an IL-12–dependent manner in mice [89].
Immune cells as messengers
A recurring theme in many of the described mechanistic studies is that innate immune cells, often DCs, represent the central cell type affected by perturbations within the commensal community [17, 18, 85, 86, 109, 110]. DCs are key microbial sensors that bridge innate to adaptive immunity and are also critical for molding T cell responses within the TME. Microbial signals might only need to function locally in the LP and MLN to drive DC function and the subsequent delivery of the immunomodulatory effect to the TME might be carried out by the DCs themselves or downstream by T cells. Various innate immune cells have been shown capable of exiting the intestinal LP and translocating to the spleen and peripheral lymph nodes under steady state [111].
Different mechanisms of microbial sensing by DCs might be in play in the context of a damaged versus intact intestinal barrier. Compromised barrier integrity could allow for translocation of live bacteria or microbial products into the circulation. These could then be recognized by PRRs on innate immune cells, such as DCs, and affect downstream innate and adaptive immunity. Such potential mechanisms may contribute to microbiota-mediated modulation of anti-tumor immunity in situations of gut inflammation, such as with total body irradiation, chemotherapy agents that cause mucositis, or with anti-CTLA-4 treatment where 11% of patients experience colitis and 34% develop diarrhea [112]. However, anti-PD-1 therapy shows only 2% incidence of colitis [112], suggesting that additional mechanisms likely exist, by which commensals shape host immunity. On the other hand, in the context of an intact barrier, mucosal DCs constantly sample bacterial-derived antigens via various mechanisms. For instance, a subset of DCs in the LP are reported to be capable of extending dendrites between epithelial cells to sample the lumen [113]. DCs may also acquire proteins via goblet cell channels [114] or microfold cells (M cells) [115]. Bacterial antigen-loaded DCs could induce immune tolerance to commensal bacteria, or they could prime bacterial antigen-reactive T cells, which in some instances might be capable of cross-reacting with tumor antigens [18, 89, 95] or in other cases might provide bystander help during anti-tumor responses. In this respect, understanding the mechanisms driving tolerogenicity vs. immunogenicity might provide insight into the mechanisms of microbiota impact on antitumor immunity.
Given the complexity of the commensal-host interaction, the diversity of the microbiome, and inter-individual variability, it is likely that multiple modalities contribute to the impact of the microbiota on immunotherapy efficacy. Furthermore, the relative contribution of the microbiome will need to be integrated along with other dimensions affecting the potency of immunotherapy, including germline genetic determinants and tumor cell-intrinsic oncogenic alterations [116,117,118]. Determining the relative contribution of all these factors and the most translatable aspects to human health will require careful experimental design in cancer patients to test hypotheses stemming from murine experiments.
Potential future clinical applications
Use of antibiotics in conjunction with immunotherapy
The collective evidence linking the gut microbiome to immunotherapy efficacy creates exciting opportunities to improve clinical treatment strategies. A straightforward implication is that antibiotics administration to patients receiving cancer immunotherapies should be pursued with caution. Routy et al. found that antibiotics administration to patients in conjunction with immunotherapy was associated with shorter PFS and shorter overall survival (OS) [89] and these results have recently been supported by an additional retrospective analysis [119]. Additionally, greater bacterial diversity was associated with higher response rates to anti-PD-1 therapy [89, 90]. These data among others (reviewed in [120]) suggest that antibiotics may have detrimental effects on patient outcomes with checkpoint blockade immunotherapy, which should prompt discretion in their administration. However, one could also imagine that some patients may have an abundance of bacterial entities that dominantly promote immune suppression, such as through expansion of FoxP3+ Tregs. In those defined instances, appropriate antibiotics might decrease the abundance of such immune regulatory bacteria, perhaps allowing immune-potentiating bacteria to bloom and support improved tumor control. Studies are ongoing in reconstituted GFM to test these ideas.
Use of the microbiome as a prognostic biomarker
The modulatory effects of the microbiome could foreseeably offer multiple avenues of clinical intervention. Microbiome composition could be considered as a complementary prognostic or predictive biomarker for treatment outcomes. Higher bacterial diversity in the gut (but not the oral microbiome) was identified to be associated with better response rates to ICB [90]. More specifically, certain bacteria were found to be enriched in anti-PD-1 responders while other species were enriched in non-responders. These data suggest that fecal DNA sequencing prior to therapy, by quantifying the community richness and the relative proportion of putatively identified “beneficial” or “detrimental” bacteria, may be suggestive of outcome and ultimately help guide treatment decisions. Prospectively designed clinical studies to validate these associations will be key to define the utility of these approaches. In the future, the composition of the microbiome may be one parameter incorporated with other known correlates of outcome such as T cell infiltration and tumor mutational burden to 1) predict potential efficacy with a given immunotherapy and 2) inform additional interventions via the microbiota to improve immunotherapy potency or alternatively decrease treatment related toxicity.
Therapeutic interventions to modulate microbiome composition and function
Preclinical evidence extends the correlative relationship between the microbiome and response observed in patients to support a causal role. This scenario opens the exciting possibility to improve efficacy by manipulating the gut flora. Intervention strategies range from less precise or “blunt” approaches to more targeted therapeutic approaches (described in Fig. 2).
Fig. 2

Microbiota-oriented interventions to improve immunotherapy treatment. While stable on a global scale, the gut microbiota regularly undergoes small fluctuations and is amenable to strategies which could shape the commensal community to either help improve patient response rates to immunotherapy or prevent treatment-related toxicity such as colitis. These approaches range from complex community transfers in the form of (a) fecal microbiota transplantation (FMT) which may have many effects on the recipient, to delivery of (g) a single microbial metabolite with a specific immune-modulatory effect. Additional approaches include (b) modulating macronutrient or prebiotic intake to shift bacterial communities, (c) targeting broad classes of bacteria with antibiotics, (d) administration of a select number of known beneficial bacterial species, or (e) a single defined bacterial isolate. Bacteriophages (f) or viruses that infect and kill selected bacteria, could also be used as a means of selectively depleting a detrimental bacterial population
One such approach is fecal microbiota transplantation (FMT). For example, fecal samples could be prepared from anti-PD-1 responders that show a favorable composition of commensal bacteria, then transplanted endoscopically or prepared for oral delivery into patients who are anti-PD-1-resistant and show an unfavorable composition of gut microbes. This approach would parallel the strategies being used to treat refractory Clostridium difficile infection in patients [121]. This approach delivers a complex community and the promise to transfer its beneficial effect. However, FMT is clouded by uncertainties related to the imprecise definition of a favorable microbiota, the possibility of delivering immune-regulatory bacteria, and the potential to transfer disease-promoting bacteria such as those contributing to obesity or even carcinogenesis.
A subtler means of intervention may include modulating the existing commensal community via prebiotics or dietary changes to favor the expansion of beneficial bacteria that require specific substrates, or conversely, “starving” detrimental bacteria of their required nutrients. For example, short-term changes in human macronutrient consumption towards a high fat, low fiber animal-based diet increased bile-tolerant microorganisms (Alistipes, Bilophila and Bacteroides) and decreased levels of Firmicutes that metabolize dietary plant polysaccharides (Roseburia, Eubacterium rectale and Ruminococcus bromii) [122]. Similarly, antibiotics could be considered a means of targeting immune-regulatory bacteria. Both of these approaches lack the precision to modulate very specific bacterial populations, however, and may have variable effects depending on the starting state of the commensal community.
Alternatively, beneficial or immune-potentiating bacteria could be prepared as a probiotic and provided as an immunotherapy adjuvant. Once molecular mechanisms are determined, genetic manipulation of the selected bacteria could be utilized to maximize beneficial effects. Historically, certain bacterial species have been some of the most amenable organisms to genetic manipulation, and the breadth of tools available to study and modify bacteria continues to expand. This technology allows the modification of a bacterium’s existing function or the introduction of completely novel genes [123]. For example, a Bacteroides strain modified to carry a gene cluster to utilize porphyran stabilized its engraftment into mice fed a porphyran-supplemented diet [124]. This strategy effectively creates a unique metabolic niche for the exogenous microbe and presents a potential means to facilitate probiotic efficacy. Bacteria may also be genetically modified to drive expression of a metabolite of interest [125]. For well-characterized bacteria such as Escherichia coli, genetic manipulation is routine, but for many human commensals, incomplete genomic information leaves fewer tools available for these strategies currently. To circumvent this limitation, it is possible to express bacterial genes of interest heterologously in common laboratory hosts such as E. coli or Bacillus subtilis [125]. An alternative approach to adding beneficial bacteria to the microbiota is selective depletion of harmful species from the community. Bacteriophages are viruses that can infect and kill bacteria and are naturally present in the microbiome where they play a key role in preserving community equilibrium. Some phages have been used preclinically to decrease pathogenic bacteria while leaving the commensal community intact, and could be further engineered to target certain bacterial species or strains [123].
Finally, if a bacterial metabolic pathway is identified along with defined metabolic products that mediate improved anti-tumor immunity and immunotherapy, then small molecule entities could be tested as candidate immune-potentiating drugs. In all cases, appropriately controlled clinical trials will be required to validate any potential microbiome-based therapy and to assess benefits and risks. Clinical trials to evaluate the impact of fecal microbiome transplant and probiotic administration with checkpoint inhibitors are already underway [126].
Conclusion – the future for the microbiome and immunotherapy
Given the intricacy of the microbiome, it will be challenging to tease out the essential mechanistic elements in such a complex system. Even if two individuals harbor the same species of bacteria, there can be variation of each bacterium at the strain level, which could yield divergent functions upon interaction with the host. Moreover, two identical strains in two disparate communities may contribute differently to their collective consortium and thus function differently with respect to the host. As such, tremendous care will need to be taken when assigning specific functional attributes to given commensal bacteria. Furthermore, a large majority of focus on cancer immunotherapy and the microbiome has investigated the contribution of bacteria but has yet to thoroughly investigate non-bacterial components including viruses, fungi and protozoa. Evidence in non-cancer disease models has indicated that the mycobiome (fungi) and the virome (viruses) can regulate systemic immunity. For example, manipulation of the mycobiome by oral antifungal drugs increased the severity of allergic airway disease in mice and was dependent on gut-resident CX3CR1+ mononuclear phagocytes [127, 128]. The virome, encompassing bacteriophages, mammalian viruses, and the endogenous retroviruses, is estimated to contain ten-fold more particles than bacterial microbes [129]. Supporting the link between the intestinal virome and host immunity, alterations in viral communities have been observed in the context of human immunodeficiency virus [130], and inflammatory bowel disease [131] and have been associated with autoimmune disorders including Type 1 diabetes [132, 133]. Incorporating a pan-kingdom view of the microbiome will likely lead to a more holistic understanding its impact on cancer treatment.
Looking forward, it is important to recognize that the microbiome contributes only one dimension to the many facets that govern the interface between cancer and the host immune response. Cancer cells grow and evolve under the selective pressure of therapy, and molecular evolution of the tumor could still occur when the microbiome is manipulated to maximize immunotherapy efficacy. In addition, it is conceivable that the composition of the microbiome similarly may evolve over the course of cancer progression and therapy administration. This variation offers additional research challenges, but with this pliability also comes exciting promise for intervention and exploiting the host-microbiome interdependency to deliver more potent therapy. In the future, it will be important to consider the microbiota as one of several parameters to be incorporated into considerations of personalized cancer therapy.
Abbreviations
Allo-HSCT:
Allogeneic hematopoietic stem cell transplantation
APCs:
Antigen-presenting cells
CTL:
Cytotoxic T lymphocyte
CTLA-4:
Cytotoxic T-lymphocyte-associated protein 4
DC:
Dendritic cell
GF:
Germ-free
GVHD:
Graft-versus-host disease
ICB:
Immune checkpoint blockade
LP:
Lamina propria
MALDI-TOF:
Matrix-assisted laser desorption ionization time-of-flight
MAMP:
Microbe-associated molecular pattern
MDSC:
Myeloid-derived suppressor cell
MLN:
Mesenteric lymph nodes
OS:
Overall survival
OTU:
Operational taxonomic unit
PD-1:
Programmed cell death protein 1
PD-L1:
Programmed death-ligand 1
PAMP:
Pathogen-associated molecular pattern
PFS:
Progression-free survival
PRR:
Pattern recognition receptor
PSA:
Polysaccharide A
SCFA:
Short-chain fatty acids
SFB:
Segmented filamentous bacteria
SPF:
Specific pathogen-free
TdLN:
Tumor-draining lymph node
TME:
Tumor microenvironment
References
- Sender R, Fuchs S, Milo R. Revised estimates for the number of human and Bacteria cells in the body. PLoS Biol. 2016;14(8):e1002533.
Article CAS PubMed PubMed Central Google Scholar - Lepage P, Leclerc MC, Joossens M, Mondot S, Blottière HM, Raes J, et al. A metagenomic insight into our gut’s microbiome. Gut. 2013;62(1):146–58.
Article PubMed Google Scholar - Vernocchi P, Del Chierico F, Putignani L. Gut microbiota profiling: metabolomics based approach to unravel compounds affecting human health. Front Microbiol. 2016;7:1144.
Article PubMed PubMed Central Google Scholar - Tai N, Wong FS, Wen L. The role of gut microbiota in the development of type 1, obesity and type 2 diabetes mellitus. Rev Endocr Metab Disord. 2015;16(1):55–65.
Article CAS PubMed PubMed Central Google Scholar - Geuking MB, Köller Y, Rupp S, McCoy KD. The interplay between the gut microbiota and the immune system. Gut Microbes. 2014;5(3):411–8.
Article PubMed PubMed Central Google Scholar - Matsuoka K, Kanai T. The gut microbiota and inflammatory bowel disease. Semin Immunopathol. 2015;37:47–55.
Article CAS PubMed Google Scholar - Fujimura KE, Lynch SV. Microbiota in allergy and asthma and the emerging relationship with the gut microbiome. Cell Host Microbe. 2015 May 13;17(5):592–602.
Article CAS PubMed PubMed Central Google Scholar - Collins SM. A role for the gut microbiota in IBS. Nat Rev Gastroenterol Hepatol. 2014 Aug;11(8):497–505.
Article CAS PubMed Google Scholar - Zhang X, Zhang D, Jia H, Feng Q, Wang D, Liang D, et al. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat Med. 2015 Aug;21(8):895–905.
Article CAS PubMed Google Scholar - Vieira SM, Pagovich OE, Kriegel MA. Diet, microbiota and autoimmune diseases. Lupus. 2014 May;23(6):518–26.
Article CAS PubMed PubMed Central Google Scholar - Miele L, Giorgio V, Alberelli MA, De Candia E, Gasbarrini A, Grieco A. Impact of gut microbiota on obesity, diabetes, and cardiovascular disease risk. Curr Cardiol Rep. 2015;17(12):120.
Article PubMed Google Scholar - Torres-Fuentes C, Schellekens H, Dinan TG, Cryan JF. The microbiota–gut–brain axis in obesity. Lancet Gastroenterol Hepatol. 2017;2(10):747–56.
Article PubMed Google Scholar - Mulak A, Bonaz B. Brain-gut-microbiota axis in Parkinson’s disease. World J Gastroenterol. 2015 Oct 7;21(37):10609–20.
Article CAS PubMed PubMed Central Google Scholar - Blanton LV, Barratt MJ, Charbonneau MR, Ahmed T, Gordon JI. Childhood undernutrition, the gut microbiota, and microbiota-directed therapeutics. Science. 2016 Jun 24;352(6293):1533.
Article CAS PubMed Google Scholar - Zhang H, Sun L. When human cells meet bacteria: precision medicine for cancers using the microbiota. Am J Cancer Res. 2018;8(7):1157–75.
Article PubMed PubMed Central Google Scholar - Panebianco C, Andriulli A, Pazienza V. Pharmacomicrobiomics: exploiting the drug-microbiota interactions in anticancer therapies. Microbiome. 2018 May 22;6(1):92.
Article PubMed PubMed Central Google Scholar - Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science. 2015 Nov 27;350(6264):1084–9.
Article CAS PubMed PubMed Central Google Scholar - Vétizou M, Pitt JM, Daillère R, Lepage P, Waldschmitt N, Flament C, et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science. 2015 Nov 27;350(6264):1079–84.
Article CAS PubMed PubMed Central Google Scholar - Structure, Function and Diversity of the healthy human microbiome. Nature. 2012;486(7402):207–214.
- Tanaka M, Nakayama J. Development of the gut microbiota in infancy and its impact on health in later life. Allergol Int. 2017 Oct 1;66(4):515–22.
Article CAS PubMed Google Scholar - Indrio F, Martini S, Francavilla R, Corvaglia L, Cristofori F, Mastrolia SA, et al. Epigenetic matters: the link between early nutrition, microbiome, and long-term health development. Front Pediatr. 2017;5:178.
Article PubMed PubMed Central Google Scholar - Perez-Muñoz ME, Arrieta M-C, Ramer-Tait AE, Walter J. A critical assessment of the “sterile womb” and “in utero colonization” hypotheses: implications for research on the pioneer infant microbiome. Microbiome. 2017;5(1):48.
Article PubMed PubMed Central Google Scholar - Dominguez-Bello MG, Costello EK, Contreras M, Magris M, Hidalgo G, Fierer N, et al. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci U S A. 2010 Jun 29;107(26):11971–5.
Article PubMed PubMed Central Google Scholar - The Integrative Human Microbiome Project. Dynamic analysis of microbiome-host omics profiles during periods of human health and disease. Cell Host Microbe. 2014;16(3):276–89.
Article CAS Google Scholar - Browne HP, Forster SC, Anonye BO, Kumar N, Neville BA, Stares MD, et al. Culturing of ‘unculturable’ human microbiota reveals novel taxa and extensive sporulation. Nature. 2016 May 4;533(7604):543–6.
Article CAS PubMed PubMed Central Google Scholar - Goodman AL, Kallstrom G, Faith JJ, Reyes A, Moore A, Dantas G, et al. Extensive personal human gut microbiota culture collections characterized and manipulated in gnotobiotic mice. PNAS. 2011 Apr 12;108(15):6252–7.
Article PubMed PubMed Central Google Scholar - Lagier J-C, Hugon P, Khelaifia S, Fournier P-E, La Scola B, Raoult D. The rebirth of culture in microbiology through the example of Culturomics to study human gut microbiota. Clin Microbiol Rev. 2015 Jan;28(1):237–64.
Article CAS PubMed PubMed Central Google Scholar - Bilen M, Dufour J-C, Lagier J-C, Cadoret F, Daoud Z, Dubourg G, et al. The contribution of culturomics to the repertoire of isolated human bacterial and archaeal species. Microbiome. 2018 May 24;6(1):94.
Article PubMed PubMed Central Google Scholar - Woese CR, Fox GE. Phylogenetic structure of the prokaryotic domain: the primary kingdoms. Proc Natl Acad Sci U S A. 1977 Nov;74(11):5088–90.
Article CAS PubMed PubMed Central Google Scholar - Cole JR, Wang Q, Fish JA, Chai B, McGarrell DM, Sun Y, et al. Ribosomal database project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014;42(Database issue):D633–42.
Article CAS PubMed Google Scholar - DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006;72(7):5069–72.
Article CAS PubMed PubMed Central Google Scholar - Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013 Jan;41(Database issue):D590–6.
CAS PubMed Google Scholar - Nguyen N-P, Warnow T, Pop M. White B. A perspective on 16S rRNA operational taxonomic unit clustering using sequence similarity. NPJ Biofilms Microbiomes. 2016;2:16004.
Article PubMed PubMed Central Google Scholar - Edgar RC. UNOISE2: improved error-correction for Illumina 16S and ITS amplicon sequencing. bioRxiv. 2016:081257.
- Amir A, McDonald D, Navas-Molina JA, Kopylova E, Morton JT, Xu ZZ, et al. Deblur rapidly resolves single-nucleotide community sequence patterns. mSystems. 2017;2(2):e00191–16.
- Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: high resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13(7):581–3.
Article CAS PubMed PubMed Central Google Scholar - Ma J, Prince A, Aagaard KM. Use of whole genome shotgun metagenomics: a practical guide for the microbiome-minded physician scientist. Semin Reprod Med. 2014 Jan;32(01):005–13.
Article CAS Google Scholar - Kembel SW, Wu M, Eisen JA, Green JL. Incorporating 16S gene copy number information improves estimates of microbial diversity and abundance. PLoS Comput Biol. 2012 Oct 25;8(10):e1002743.
Article CAS PubMed PubMed Central Google Scholar - Prakash T, Taylor TD. Functional assignment of metagenomic data: challenges and applications. Brief Bioinform. 2012 Nov;13(6):711–27.
Article PubMed PubMed Central Google Scholar - D’Argenio V. Human microbiome acquisition and Bioinformatic challenges in metagenomic studies. Int J Mol Sci. 2018;19(2).
- Langille MGI, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 2013 Sep;31(9):814–21.
Article CAS PubMed PubMed Central Google Scholar - Aßhauer KP, Wemheuer B, Daniel R, Meinicke P. Tax4Fun: predicting functional profiles from metagenomic 16S rRNA data. Bioinformatics. 2015 Sep 1;31(17):2882–4.
Article CAS PubMed PubMed Central Google Scholar - Iwai S, Weinmaier T, Schmidt BL, Albertson DG, Poloso NJ, Dabbagh K, et al. Piphillin: improved prediction of metagenomic content by direct inference from human microbiomes. PLoS One. 2016 Nov 7;11(11):e0166104.
Article CAS PubMed PubMed Central Google Scholar - Matson V, Fessler J, Bao R, Chongsuwat T, Zha Y, Alegre M-L, et al. The commensal microbiome is associated with anti–PD-1 efficacy in metastatic melanoma patients. Science. 2018 Jan 5;359(6371):104–8.
Article CAS PubMed PubMed Central Google Scholar - Al-Asmakh M, Zadjali F. Use of germ-free animal models in microbiota-related research. J Microbiol Biotechnol. 2015 Oct 28;25(10):1583–8.
Article PubMed Google Scholar - Luczynski P, McVey Neufeld K-A, Oriach CS, Clarke G, Dinan TG, Cryan JF. Growing up in a bubble: using germ-free animals to assess the influence of the gut microbiota on brain and behavior. Int J Neuropsychopharmacol. 2016;19(8).
- Deplancke B, Gaskins HR. Microbial modulation of innate defense: goblet cells and the intestinal mucus layer. Am J Clin Nutr. 2001 Jun 1;73(6):1131S–41S.
Article CAS PubMed Google Scholar - Smith K, McCoy KD, Macpherson AJ. Use of axenic animals in studying the adaptation of mammals to their commensal intestinal microbiota. Semin Immunol. 2007;19(2):59–69.
Article CAS PubMed Google Scholar - Manolios N, Geczy CL, Schrieber L. High endothelial venule morphology and function are inducible in germ-free mice: a possible role for interferon-gamma. Cell Immunol. 1988 Nov;117(1):136–51.
Article CAS PubMed Google Scholar - Weinstein PD, Cebra JJ. The preference for switching to IgA expression by Peyer’s patch germinal center B cells is likely due to the intrinsic influence of their microenvironment. J Immunol. 1991;147(12):4126–35.
CAS PubMed Google Scholar - Lécuyer E, Rakotobe S, Lengliné-Garnier H, Lebreton C, Picard M, Juste C, et al. Segmented filamentous bacterium uses secondary and tertiary lymphoid tissues to induce gut IgA and specific T helper 17 cell responses. Immunity. 2014 Apr 17;40(4):608–20.
Article CAS PubMed Google Scholar - Pabst O, Herbrand H, Friedrichsen M, Velaga S, Dorsch M, Berhardt G, et al. Adaptation of solitary intestinal lymphoid tissue in response to microbiota and chemokine receptor CCR7 signaling. J Immunol. 2006 Nov 15;177(10):6824–32.
Article CAS PubMed Google Scholar - Bouskra D, Brézillon C, Bérard M, Werts C, Varona R, Boneca IG, et al. Lymphoid tissue genesis induced by commensals through NOD1 regulates intestinal homeostasis. Nature. 2008 Nov;456(7221):507–10.
Article CAS PubMed Google Scholar - Pabst O, Herbrand H, Worbs T, Friedrichsen M, Yan S, Hoffmann MW, et al. Cryptopatches and isolated lymphoid follicles: dynamic lymphoid tissues dispensable for the generation of intraepithelial lymphocytes. Eur J Immunol. 2005;35(1):98–107.
Article CAS PubMed Google Scholar - Macpherson AJ, Martinic MM, Harris N. The functions of mucosal T cells in containing the indigenous commensal flora of the intestine. Cell Mol Life Sci. 2002 Dec;59(12):2088–96.
Article CAS PubMed Google Scholar - Macpherson AJ, Hunziker L, McCoy K, Lamarre A. IgA responses in the intestinal mucosa against pathogenic and non-pathogenic microorganisms. Microbes Infect. 2001 Oct;3(12):1021–35.
Article CAS PubMed Google Scholar - Ivanov II, de Llanos Frutos R, Manel N, Yoshinaga K, Rifkin DB, Sartor RB, et al. Specific microbiota direct the differentiation of Th17 cells in the mucosa of the small intestine. Cell Host Microbe. 2008;4(4):337–49.
Article CAS PubMed PubMed Central Google Scholar - Gorjifard S, Goldszmid RS. Microbiota—myeloid cell crosstalk beyond the gut. J Leukoc Biol. 2016;100(5):865–79.
Article CAS PubMed PubMed Central Google Scholar - Trompette A, Gollwitzer ES, Yadava K, Sichelstiel AK, Sprenger N, Ngom-Bru C, et al. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat Med. 2014 Feb;20(2):159–66.
Article CAS PubMed Google Scholar - Bauer H, Horowitz RE, Levenson SM, Popper H. The response of the lymphatic tissue to the microbial flora. Studies on germfree mice. Am J Pathol. 1963;42:471–83.
CAS PubMed PubMed Central Google Scholar - Benveniste J, Lespinats G, Adam C, Salomon JC. Immunoglobulins in intact, immunized, and contaminated axenic mice: study of serum IgA. J Immunol. 1971;107(6):1647–55.
CAS PubMed Google Scholar - Mazmanian SK, Liu CH, Tzianabos AO, Kasper DL. An immunomodulatory molecule of symbiotic Bacteria directs maturation of the host immune system. Cell. 2005 Jul 15;122(1):107–18.
Article CAS PubMed Google Scholar - Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009 Oct 30;139(3):485–98.
Article CAS PubMed PubMed Central Google Scholar - Atarashi K, Tanoue T, Ando M, Kamada N, Nagano Y, Narushima S, et al. Th17 cell induction by adhesion of microbes to intestinal epithelial cells. Cell. 2015 Oct 8;163(2):367–80.
Article CAS PubMed PubMed Central Google Scholar - Tan TG, Sefik E, Geva-Zatorsky N, Kua L, Naskar D, Teng F, et al. Identifying species of symbiont bacteria from the human gut that, alone, can induce intestinal Th17 cells in mice. Proc Natl Acad Sci U S A. 2016 Dec 13;113(50):E8141–50.
Article CAS PubMed PubMed Central Google Scholar - Geva-Zatorsky N, Sefik E, Kua L, Pasman L, Tan TG, Ortiz-Lopez A, et al. Mining the human gut microbiota for immunomodulatory organisms. Cell. 2017 Feb 23;168(5):928–943.e11.
Article CAS PubMed PubMed Central Google Scholar - Round JL, Mazmanian SK. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc Natl Acad Sci U S A. 2010 Jul 6;107(27):12204–9.
Article PubMed PubMed Central Google Scholar - Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science. 2011 Jan 21;331(6015):337–41.
Article CAS PubMed Google Scholar - Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, et al. Treg induction by a rationally selected mixture of clostridia strains from the human microbiota. Nature. 2013 Aug;500(7461):232–6.
Article CAS PubMed Google Scholar - Nguyen TLA, Vieira-Silva S, Liston A, Raes J. How informative is the mouse for human gut microbiota research? Dis Model Mech. 2015 Jan;8(1):1–16.
Article CAS PubMed PubMed Central Google Scholar - Hugenholtz F, de Vos WM. Mouse models for human intestinal microbiota research: a critical evaluation. Cell Mol Life Sci. 2018;75(1):149–60.
Article CAS PubMed Google Scholar - Xiao L, Feng Q, Liang S, Sonne SB, Xia Z, Qiu X, et al. A catalog of the mouse gut metagenome. Nat Biotechnol. 2015 Oct;33(10):1103–8.
Article CAS PubMed Google Scholar - Gärtner K. The forestomach of rats and mice, an effective device supporting digestive metabolism in muridae (review). J Exp Anim Sci. 2002 Jan 1;42(1):1–20.
Article Google Scholar - Treuting PM, Arends MJ, Dintzis SM. Lower Gastrointestinal Tract. In: Comparative Anatomy and Histology [Internet]. Elsevier; 2018 [cited 2019 Jan 21]. p. 213–28. Available from: https://linkinghub.elsevier.com/retrieve/pii/B9780128029008000129
- Smith HF, Parker W, Kotzé SH, Laurin M. Multiple independent appearances of the cecal appendix in mammalian evolution and an investigation of related ecological and anatomical factors. Comptes Rendus Palevol. 2013 Aug 1;12(6):339–54.
Article Google Scholar - Wang J, Wang J, Pang X, Zhao L, Tian L, Wang X. Sex differences in colonization of gut microbiota from a man with short-term vegetarian and inulin-supplemented diet in germ-free mice. Sci Rep. 2016 Oct 31;6:36137.
Article CAS PubMed PubMed Central Google Scholar - Lundberg R, Bahl MI, Licht TR, Toft MF, Hansen AK. Microbiota composition of simultaneously colonized mice housed under either a gnotobiotic isolator or individually ventilated cage regime. Sci Rep. 2017 Feb 7;7:42245.
Article CAS PubMed PubMed Central Google Scholar - Macpherson AJ, McCoy KD. Standardised animal models of host microbial mutualism. Mucosal Immunol. 2015;8(3):476–86.
Article CAS PubMed Google Scholar - Lagkouvardos I, Pukall R, Abt B, Foesel BU, Meier-Kolthoff JP, Kumar N, et al. The mouse intestinal bacterial collection (miBC) provides host-specific insight into cultured diversity and functional potential of the gut microbiota. Nat Microbiol. 2016 Oct;1(10):16131.
Article CAS PubMed Google Scholar - Desai MS, Seekatz AM, Koropatkin NM, Kamada N, Hickey CA, Wolter M, et al. A dietary Fiber-deprived gut microbiota degrades the colonic mucus barrier and enhances pathogen susceptibility. Cell. 2016 Nov 17;167(5):1339–1353.e21.
Article CAS PubMed PubMed Central Google Scholar - Staley C, Kaiser T, Beura LK, Hamilton MJ, Weingarden AR, Bobr A, et al. Stable engraftment of human microbiota into mice with a single oral gavage following antibiotic conditioning. Microbiome. 2017;5(1):87.
Article PubMed PubMed Central Google Scholar - Hintze KJ, Cox JE, Rompato G, Benninghoff AD, Ward RE, Broadbent J, et al. Broad scope method for creating humanized animal models for animal health and disease research through antibiotic treatment and human fecal transfer. Gut Microbes. 2014 Mar 1;5(2):183–91.
Article PubMed PubMed Central Google Scholar - Schwarzer M, Srutkova D, Hermanova P, Leulier F, Kozakova H, Schabussova I. Diet matters: endotoxin in the diet impacts the level of allergic sensitization in germ-free mice. PLoS One. 2017 Jan 4;12(1):e0167786.
Article PubMed PubMed Central Google Scholar - Hrncir T, Stepankova R, Kozakova H, Hudcovic T, Tlaskalova-Hogenova H. Gut microbiota and lipopolysaccharide content of the diet influence development of regulatory T cells: studies in germ-free mice. BMC Immunol. 2008 Nov 6;9(1):65.
Article CAS PubMed PubMed Central Google Scholar - Iida N, Dzutsev A, Stewart CA, Smith L, Bouladoux N, Weingarten RA, et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science. 2013 Nov 22;342(6161):967–70.
Article CAS PubMed PubMed Central Google Scholar - Viaud S, Saccheri F, Mignot G, Yamazaki T, Daillère R, Hannani D, et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science. 2013 Nov 22;342(6161):971–6.
Article CAS PubMed PubMed Central Google Scholar - Jenq RR, Taur Y, Devlin SM, Ponce DM, Goldberg JD, Ahr KF, et al. Intestinal Blautia is associated with reduced death from graft-versus-host disease. Biol Blood and Marrow Transplant. 2015 Aug 1;21(8):1373–83.
Article Google Scholar - Peled JU, Devlin SM, Staffas A, Lumish M, Khanin R, Littmann ER, et al. Intestinal microbiota and relapse after hematopoietic-cell transplantation. J Clin Oncol. 2017 May 20;35(15):1650–9.
Article PubMed PubMed Central Google Scholar - Routy B, Chatelier EL, Derosa L, Duong CPM, Alou MT, Daillère R, et al. Gut microbiome influences efficacy of PD-1–based immunotherapy against epithelial tumors. Science. 2018;359(6371):91–7.
Article CAS PubMed Google Scholar - Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti–PD-1 immunotherapy in melanoma patients. Science. 2017:eaan4236.
- Frankel AE, Coughlin LA, Kim J, Froehlich TW, Xie Y, Frenkel EP, et al. Metagenomic shotgun sequencing and unbiased Metabolomic profiling identify specific human gut microbiota and metabolites associated with immune checkpoint therapy efficacy in melanoma patients. Neoplasia. 2017 Sep 15;19(10):848–55.
Article CAS PubMed PubMed Central Google Scholar - Chaput N, Lepage P, Coutzac C, Soularue E, Le Roux K, Monot C, et al. Baseline gut microbiota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab. Ann Oncol. 2017 Jun 1;28(6):1368–79.
Article CAS PubMed Google Scholar - Dubin K, Callahan MK, Ren B, Khanin R, Viale A, Ling L, et al. Intestinal microbiome analyses identify melanoma patients at risk for checkpoint-blockade-induced colitis. Nat Commun. 2016 Feb 2;7:10391.
Article CAS PubMed PubMed Central Google Scholar - Geller LT, Barzily-Rokni M, Danino T, Jonas OH, Shental N, Nejman D, et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science. 2017 Sep 15;357(6356):1156–60.
Article CAS PubMed PubMed Central Google Scholar - Balachandran VP, Łuksza M, Zhao JN, Makarov V, Moral JA, Remark R, et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature. 2017 Nov;551(7681):512.
Article CAS PubMed PubMed Central Google Scholar - Thaiss CA, Levy M, Grosheva I, Zheng D, Soffer E, Blacher E, et al. Hyperglycemia drives intestinal barrier dysfunction and risk for enteric infection. Science. 2018 Mar 23;359(6382):1376–83.
Article CAS PubMed Google Scholar - Paulos CM, Wrzesinski C, Kaiser A, Hinrichs CS, Chieppa M, Cassard L, et al. Microbial translocation augments the function of adoptively transferred self/tumor-specific CD8+ T cells via TLR4 signaling. J Clin Invest. 2007;117(8):2197–204.
Article CAS PubMed PubMed Central Google Scholar - Hall JA, Bouladoux N, Sun CM, Wohlfert EA, Blank RB, Zhu Q, et al. Commensal DNA limits regulatory T cell conversion and is a natural adjuvant of intestinal immune responses. Immunity. 2008 Oct 17;29(4):637–49.
Article CAS PubMed PubMed Central Google Scholar - Rooks MG, Garrett WS. Gut microbiota, metabolites and host immunity. Nat Rev Immunol. 2016 Jun;16(6):341–52.
Article CAS PubMed PubMed Central Google Scholar - Morrison DJ, Preston T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes. 2016;7(3):189–200.
Article PubMed PubMed Central Google Scholar - den BG, van EK, Groen AK, Venema K, Reijngoud D-J, Bakker BM. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J Lipid Res. 2013;54(9):2325–40.
Article CAS Google Scholar - Iraporda C, Errea A, Romanin DE, Cayet D, Pereyra E, Pignataro O, et al. Lactate and short chain fatty acids produced by microbial fermentation downregulate proinflammatory responses in intestinal epithelial cells and myeloid cells. Immunobiology. 2015 Oct;220(10):1161–9.
Article CAS PubMed Google Scholar - Gurav A, Sivaprakasam S, Bhutia YD, Boettger T, Singh N, Ganapathy V. Slc5a8, a Na+−coupled high-affinity transporter for short-chain fatty acids, is a conditional tumor suppressor in colon that protects against colitis and colon cancer under low-fiber dietary conditions. Biochem J. 2015;469(2):267–78.
Article CAS PubMed Google Scholar - White CA, Pone EJ, Lam T, Tat C, Hayama KL, Li G, et al. HDAC inhibitors upregulate B cell microRNAs that silence AID and Blimp-1 expression for epigenetic modulation of antibody and autoantibody responses. J Immunol. 2014;193(12):5933–50.
Article CAS PubMed Google Scholar - Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature. 2013 Dec 19;504(7480):451–5.
Article CAS PubMed PubMed Central Google Scholar - Cohen LJ, Esterhazy D, Kim S-H, Lemetre C, Aguilar RR, Gordon EA, et al. Commensal bacteria make GPCR ligands that mimic human signalling molecules. Nature. 2017 Sep;549(7670):48–53.
Article CAS PubMed PubMed Central Google Scholar - Levy M, Thaiss CA, Elinav E. Metabolites: messengers between the microbiota and the immune system. Genes Dev. 2016 Jul 15;30(14):1589–97.
Article CAS PubMed PubMed Central Google Scholar - Sano T, Huang W, Hall JA, Yang Y, Chen A, Gavzy SJ, et al. An IL-23R/IL-22 circuit regulates epithelial serum amyloid a to promote local effector Th17 responses. Cell. 2015 Oct 8;163(2):381–93.
Article CAS PubMed PubMed Central Google Scholar - Uribe-Herranz M, Bittinger K, Rafail S, Guedan S, Pierini S, Tanes C, et al. Gut microbiota modulates adoptive cell therapy via CD8α dendritic cells and IL-12. JCI Insight. 2018 Feb 22;3(4).
- Tanoue T, Morita S, Plichta DR, Skelly AN, Suda W, Sugiura Y, et al. A defined commensal consortium elicits CD8 T cells and anti-cancer immunity. Nature. 2019:1.
- Morton AM, Sefik E, Upadhyay R, Weissleder R, Benoist C, Mathis D. Endoscopic photoconversion reveals unexpectedly broad leukocyte trafficking to and from the gut. PNAS. 2014 May 6;111(18):6696–701.
Article CAS PubMed PubMed Central Google Scholar - Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob J-J, Cowey CL, et al. Overall survival with combined Nivolumab and Ipilimumab in advanced melanoma. N Engl J Med. 2017 Oct 5;377(14):1345–56.
Article CAS PubMed PubMed Central Google Scholar - Arques JL, Hautefort I, Ivory K, Bertelli E, Regoli M, Clare S, et al. Salmonella induces Flagellin- and MyD88-dependent migration of Bacteria-capturing dendritic cells into the gut lumen. Gastroenterology. 2009 Aug 1;137(2):579–587.e2.
Article PubMed Google Scholar - McDole JR, Wheeler LW, McDonald KG, Wang B, Konjufca V, Knoop KA, et al. Goblet cells deliver luminal antigen to CD103+ dendritic cells in the small intestine. Nature. 2012;483(7389):345–9.
Article CAS PubMed PubMed Central Google Scholar - Mowat AM, Agace WW. Regional specialization within the intestinal immune system. Nat Rev Immunol. 2014 Oct;14(10):667–85.
Article CAS PubMed Google Scholar - Queirolo P, Morabito A, Laurent S, Lastraioli S, Piccioli P, Ascierto PA, et al. Association of CTLA-4 polymorphisms with improved overall survival in melanoma patients treated with CTLA-4 blockade: a pilot study. Cancer Investig. 2013 Jun 1;31(5):336–45.
Article CAS Google Scholar - Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature. 2015 Jul;523(7559):231–5.
Article CAS PubMed Google Scholar - Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, et al. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov. 2016 Feb;6(2):202–16.
Article CAS PubMed Google Scholar - Derosa L, Hellmann MD, Spaziano M, Halpenny D, Fidelle M, Rizvi H, et al. Negative association of antibiotics on clinical activity of immune checkpoint inhibitors in patients with advanced renal cell and non-small-cell lung cancer. Ann Oncol. 2018 Jun 1;29(6):1437–44.
Article CAS PubMed PubMed Central Google Scholar - Galloway-Peña JR, Jenq RR, Shelburne SA. Can Consideration of the Microbiome Improve Antimicrobial Utilization and Treatment Outcomes in the Oncology Patient? Clin Cancer Res. 2017;23(13):3263–8.
Article PubMed PubMed Central Google Scholar - Rohlke F, Stollman N. Fecal microbiota transplantation in relapsing Clostridium difficile infection. Ther Adv Gastroenterol. 2012 Nov;5(6):403–20.
Article Google Scholar - David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014 Jan;505(7484):559–63.
Article CAS PubMed Google Scholar - Lim B, Zimmermann M, Barry NA, Goodman AL. Engineered regulatory systems modulate gene expression of human commensals in the gut. Cell. 2017 Apr 20;169(3):547–558.e15.
Article CAS PubMed PubMed Central Google Scholar - Shepherd ES, DeLoache WC, Pruss KM, Whitaker WR, Sonnenburg JL. An exclusive metabolic niche enables strain engraftment in the gut microbiota. Nature. 2018;557(7705):434–8.
Article CAS PubMed PubMed Central Google Scholar - Guo C-J, Chang F-Y, Wyche TP, Backus KM, Acker TM, Funabashi M, et al. Discovery of reactive microbiota-derived metabolites that inhibit host proteases. Cell. 2017;168(3):517–526.e18.
Article CAS PubMed PubMed Central Google Scholar - Mullard A. Oncologists tap the microbiome in bid to improve immunotherapy outcomes. Nat Rev Drug Discov. 2018 Feb 16;17:153–5.
Article CAS PubMed Google Scholar - Wheeler ML, Limon JJ, Bar AS, Leal CA, Gargus M, Tang J, et al. Immunological consequences of intestinal fungal Dysbiosis. Cell Host Microbe. 2016 Jun 8;19(6):865–73.
Article CAS PubMed PubMed Central Google Scholar - Li X, Leonardi I, Semon A, Doron I, Gao IH, Putzel GG, et al. Response to fungal Dysbiosis by gut-resident CX3CR1+ mononuclear phagocytes aggravates allergic airway disease. Cell Host Microbe. 2018 Dec 12;24(6):847–856.e4.
Article CAS PubMed PubMed Central Google Scholar - Filyk HA, Osborne LC. The multibiome: the intestinal Ecosystem’s influence on immune homeostasis, health, and disease. EBioMedicine. 2016 Nov 1;13:46–54.
Article PubMed PubMed Central Google Scholar - Monaco CL, Gootenberg DB, Zhao G, Handley SA, Ghebremichael MS, Lim ES, et al. Altered Virome and bacterial microbiome in human immunodeficiency virus-associated acquired immunodeficiency syndrome. Cell Host Microbe. 2016 Mar 9;19(3):311–22.
Article CAS PubMed PubMed Central Google Scholar - Norman JM, Handley SA, Baldridge MT, Droit L, Liu CY, Keller BC, et al. Disease-specific alterations in the enteric Virome in inflammatory bowel disease. Cell. 2015 Jan 29;160(3):447–60.
Article CAS PubMed PubMed Central Google Scholar - Richardson SJ, Horwitz MS. Is type 1 diabetes “going viral”? Diabetes. 2014 Jul 1;63(7):2203–5.
Article PubMed Google Scholar - Zhao G, Vatanen T, Droit L, Park A, Kostic AD, Poon TW, et al. Intestinal virome changes precede autoimmunity in type I diabetes-susceptible children. Proc Natl Acad Sci U S A. 2017 Jul 25;114(30):E6166–75.
Article CAS PubMed PubMed Central Google Scholar
Acknowledgements
Not applicable.
Funding
This work was supported by NIH grant R35 CA210098, a Team Science Award from the Melanoma Research Alliance, the American Cancer Society–Jules L. Plangere Jr. Family Foundation Professorship in Cancer Immunotherapy (T.F.G), funds from the University of Chicago Medicine Comprehensive Cancer Center, the University of Chicago Cancer Biology Training program (T32 CA009594) (J.F.), the NCI Predoctoral to Postdoctoral Fellow Transition Award (F99/K00) (J.F.), and the NCI Postdoctoral Fellowship Award (F32 CA236296) (V.M.).
Availability of data and materials
Not applicable.
Author information
Author notes
- Jessica Fessler and Vyara Matson contributed equally to this work.
Authors and Affiliations
- Department of Pathology, The University of Chicago, Chicago, IL, USA
Jessica Fessler, Vyara Matson & Thomas F. Gajewski - Department of Medicine, Section of Hematology/Oncology, The University of Chicago, 5841 S. Maryland Ave., MC2115, Chicago, IL, 60637, USA
Thomas F. Gajewski
Authors
- Jessica Fessler
You can also search for this author inPubMed Google Scholar - Vyara Matson
You can also search for this author inPubMed Google Scholar - Thomas F. Gajewski
You can also search for this author inPubMed Google Scholar
Contributions
JF and VM performed the literature search and produced the first draft of the manuscript. TFG edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Correspondence toThomas F. Gajewski.
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
T.F.G. is an advisory board member for Roche-Genentech, Merck, Abbvie, Bayer, Aduro, and Fog Pharma. T.F.G. receives research support from Roche-Genentech, BMS, Merck, Incyte, Seattle Genetics, and Ono. T.F.G. is a shareholder/cofounder of Jounce Therapeutics. The University of Chicago holds a licensing arrangement with Evelo. T.F.G., J.F., and V.M. are inventors on U.S. patent US20160354416 A1 submitted by the University of Chicago.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article
Cite this article
Fessler, J., Matson, V. & Gajewski, T.F. Exploring the emerging role of the microbiome in cancer immunotherapy.j. immunotherapy cancer 7, 108 (2019). https://doi.org/10.1186/s40425-019-0574-4
- Received: 03 November 2018
- Accepted: 22 March 2019
- Published: 17 April 2019
- DOI: https://doi.org/10.1186/s40425-019-0574-4