L1 Retrotransposition in Human Neural Progenitor Cells (original) (raw)
. Author manuscript; available in PMC: 2010 Jul 23.
Published in final edited form as: Nature. 2009 Aug 5;460(7259):1127–1131. doi: 10.1038/nature08248
Abstract
Long Interspersed Element-1 (LINE-1 or L1) retrotransposons have dramatically impacted the human genome. L1s must retrotranspose in the germ-line or during early development to ensure their evolutionary success; yet the extent to which this process impacts somatic cells is poorly understood. We previously demonstrated that engineered human L1s can retrotranspose in adult rat hippocampus progenitor cells (NPCs) in vitro and in the mouse brain in vivo1. Here, we demonstrate that NPCs isolated from human fetal brain and NPCs derived from human embryonic stem cells (hESCs) support the retrotransposition of engineered human L1s in vitro. Furthermore, we developed a quantitative multiplex polymerase chain reaction that detected an increase in the copy number of endogenous L1s in the hippocampus and in several regions of adult human brains when compared to the copy number of endogenous L1s in heart or liver genomic DNAs from the same donor. These data suggest that de novo L1 retrotransposition events may occur in the human brain and, in principle, have the potential to contribute to individual somatic mosaicism.
The human nervous system is complex, containing approximately 1015 synapses with a vast diversity of neuronal cell types and connections that are influenced by complex and incompletely understood environmental and genetic factors2. Neural progenitor cells (NPCs) give rise to the three main lineages of the nervous system: neurons, astrocytes, and oligodendrocytes. To determine if human NPCs can support L1 retrotransposition, we transfected human fetal brain stem cells (hCNS-SCns) (Fig. 1A)3 with an expression construct containing a retrotransposition-competent human L1 driven from its native promoter (RC-L1; L1RP). The RC-L1 also contains a retrotransposition indicator cassette in its 3′ UTR, consisting of a reversed copy of the enhanced green fluorescent protein (EGFP) expression cassette, which is interrupted by an intron in the same transcriptional orientation as the RC-L14-7. The orientation of the cassette ensures that EGFP-positive cells will arise only if the RC-L1 undergoes retrotransposition (Fig. S1A).
Figure 1. L1 retrotransposition in hCNS-SCns.
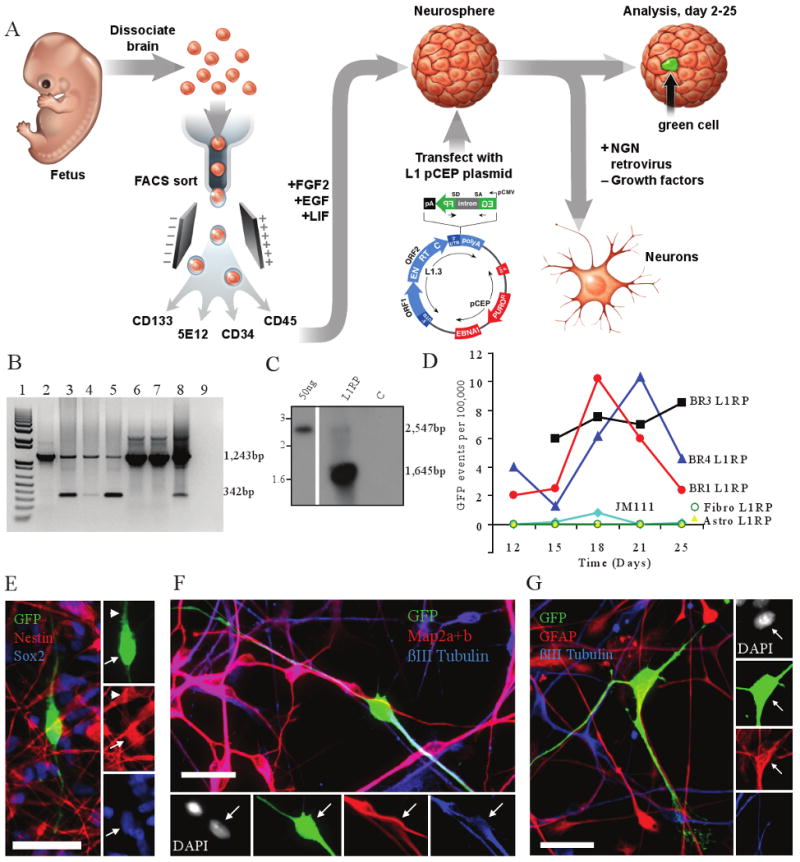
a, Experimental rationale. b, PCR of genomic DNA; 1,243bp product contains the intron; 342bp product indicates intron loss and retrotransposition. Lane 1, weight standards; Lane 2, hCNS-SCns transfected with JM111/L1RP; Lanes 3-5, three hCNS-SCns lines transfected with L1RP, Lane 6-7, primary astrocytes and fibroblasts transfected with L1RP; Lane 8, positive control; Lane 9, water. c, Southern blot of hCNS-SCns (line FBR-BR3); 2,547bp band represents plasmid; 1,645bp band is diagnostic for genomic insertion. d, Time course of L1 retrotransposition. e, EGFP-positive cells express Nestin and Sox2. f, EGFP-positive cells can differentiate to neurons (βIII tubulin and Map2a+2b positive). g, EGFP-positive cells can differentiate into glia (GFAP positive, βIII tubulin negative). Scale bar = 25 μm, arrows indicate co-labeled cell body; arrowheads indicate co-labeled processes.
A low level of L1RP retrotransposition, averaging 8-12 events per 100,000 cells, was observed in three different hCNS-SCns lines (BR1, BR3 and BR4; Fig. 1D). By comparison, an L1 containing two missense mutations in the ORF1-encoded protein (JM111/L1RP)5,8 did not retrotranspose (Figs. 1B & D). Controls demonstrated precise splicing of the intron from the retrotransposed EGFP gene (Fig. 1B, Figs. S1 & S4) and indicated that L1 retrotransposition events were detectable by both PCR and Southern blotting 3 months post-transfection (Fig. 1C). Moreover, RT-PCR revealed that hCNS-SCns express endogenous L1 transcripts and that some transcripts are derived from the human-specific (L1Hs) subfamily 4,9,10 (Fig. S6A-B, Table S4-5).
To determine if L1 retrotransposition occurred in undifferentiated cells, we conducted immunocytochemical localization of cell type restricted markers in EGFP-positive hCNS-SCns. These cells expressed neural stem cell markers, including Sox2, Nestin, Musashi-1 and Sox1 (Figs. 1E, S2A-B), and some co-labeled with Ki-67, indicating that they continued to proliferate (Fig. S2C). EGFP-positive hCNS-SCns also could be differentiated to cells of both the neuronal and glial lineages (Figs. 1F & 1G). Notably, L1RP did not retrotranspose using our experimental conditions in primary human astrocytes or fibroblasts, although a low level of endogenous L1 expression was detected in both cell types (Figs. 1D, S2D-E, S6A-B).
We next used two different protocols to derive NPCs from five human embryonic stem cell lines (hESCs; Fig 2A). As in our previous study1, NPC differentiation led to a ∼25-fold increase in L1 promoter activity over a 2-day period and then declined (Fig. 2C); there also was a ∼250-fold increase in synapsin promoter activity during differentiation (Fig. S4B). H13B-derived NPCs expressed both endogenous L1 RNA and ORF1p, although the level of ORF1p expression was less than in the H13B hESC line (Fig. 2D). HUES6-derived NPCs also expressed endogenous L1 RNA (Fig. S6A-B) and sequencing indicated some transcripts are derived from the L1Hs subfamily (Table S4-5). Similar studies performed with fetal brain, liver, and skin samples showed evidence of endogenous L1 transcription (Fig. S6C-D, Table S4-5).
Figure 2. L1 retrotransposition in hESC-derived NPCs.
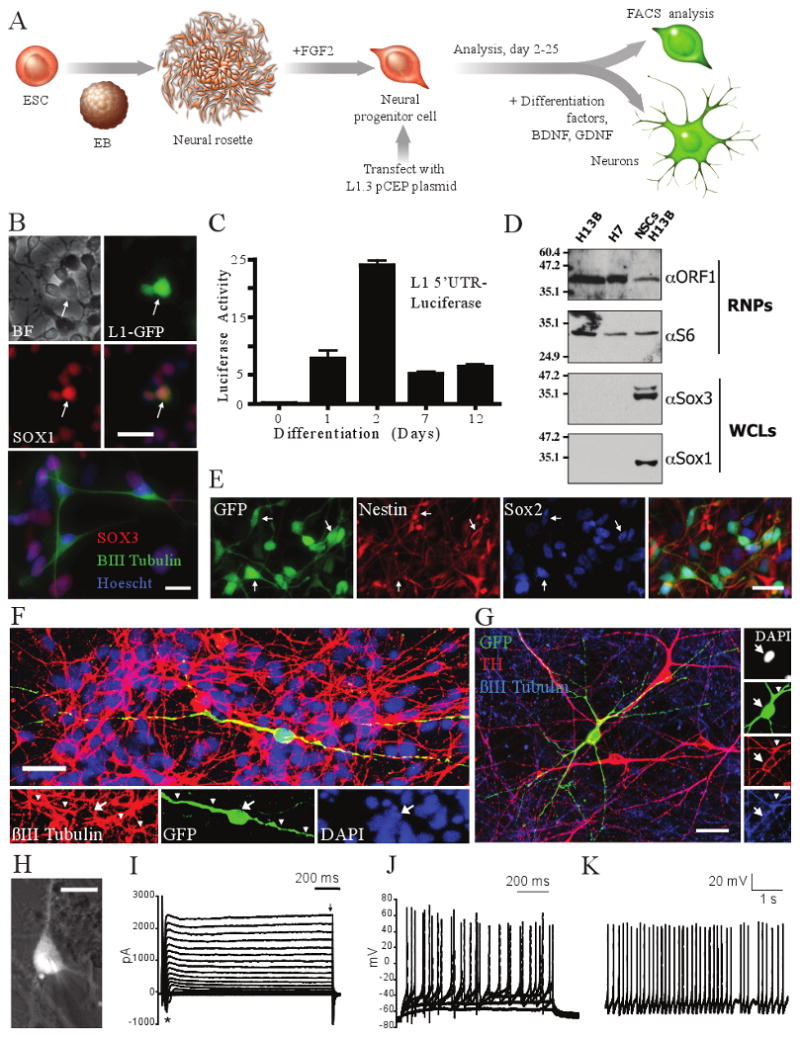
a, Experimental rationale. b, L1 retrotransposition in H13B (top, LRE3-GFP) and H7 (bottom, LRE3-mneo1)-derived NPCs (BF = brightfield). G418-resistant foci can express progenitor (SOX3) and neuronal (βIII tubulin) markers. c, L1 5′ UTR is induced upon differentiation. d, H13B-derived NPCs express endogenous ORF1p. RNP= ribonucleoprotein particle samples; WCL= whole cell lysate. e-g, EGFP-positive, HUES6-derived NPCs express SOX2 and Nestin and can differentiate to be tyrosine hydroxylase (TH) positive. Scale bar = 25 μm. h, LRE-EGFP positive neuron from which (I)-(K) were obtained. Scale bar = 10 μm. i, Transient Na+ (asterisk) and sustained K+ currents (arrow) in response to voltage step depolarizations j, Suprathreshold responses to somatic current injections. k, Spontaneous action potentials (Vm = −50 mV). Arrows indicate cell soma co-localization; arrowheads indicate co-labeled processes.
RC-L1 retrotransposition was readily detected at varying efficiencies in hESC-derived NPC lines (Table S1, lab G and M; Fig. S1, S4F-G). Again, we determined that JM111/L1RP could not retrotranspose (Table S1), that EGFP-positive NPCs expressed canonical neural stem cell markers (Fig. 2B, 2E, S3C, S3D), and that EGFP-positive HUES6-derived NPCs could be differentiated to cells of both the neuronal and glial lineages (Figs. 2F, S3E-F). The variability in retrotransposition efficiencies in hES-derived NPCs likely depended on multiple factors (see Table S1 for specific details).
Characterization of EGFP-positive neurons revealed that some expressed subtype-specific markers (tyrosine hydroxylase (Fig. 2G) and GABA (data not shown)) and whole-cell perforated patch clamp recording demonstrated that some HUES6-derived NPCs are functional (Fig. 2H-K; n=4 cells). Finally, we demonstrated that an RC-L1 tagged with neomycin or blasticidin retrotransposition indicator cassettes could retrotranspose in NPCs (Fig. S1 & S4C-E)5,11. Some G418-resistant foci also expressed SOX3 and could be differentiated to a neuronal lineage (Fig. 2B).
We next characterized 19 retrotransposition events from EGFP-positive NPCs (Fig. S7B; Table S2). Comparison of the pre- and post-integration sites demonstrated that retrotransposition occurred into an actual or inferred L1 endonuclease consensus cleavage site (5′-TTTT/A and derivatives). Five events were flanked by target site duplications, and no large deletions were detected at the insertion site5,9,12 (Fig. S7B; Table 2). Interestingly, 16 of 19 retrotransposition events were fewer than 100 kB from a gene and some occurred in the vicinity of a neuronally expressed gene1,12,13.
Notably, we consistently observed higher L1 retrotransposition efficiencies in hESC-derived NPCs when compared to fetal NPCs. A Euclidian distance map based on exon-array expression analysis14 indicated that hCNS-SCns cluster closer to HUES6 cells, whereas HUES6-derived NPCs cluster closer to fetal brain (Fig. S11A). Thus, hESC-derived NPCs and hCNS-SCns may represent different developmental stages in progenitor differentiation. That being stated, we conclude that engineered human L1s can retrotranspose in human NPCs.
Several studies have reported an inverse correlation between L1 expression and the methylation status of the CpG island in their 5′ UTRs15-16. Thus, we performed bisulfite conversion analyses on genomic DNAs derived from matched brain and skin tissue samples from two 80- to 82-day-old fetuses (Fig 3A; one male/one female sample). We then amplified a portion of the L1 5′UTR containing 20 CpG sites and sequenced the resultant amplicons. Interestingly, the L1 5′ UTR exhibited significantly less methylation in both brain samples when compared to the matched skin sample (Two-sample Kolmogorov-Smirnov test P≤0.0079 day 80 female, P ≤ 0.0034 day 82 male; Fig. 3B). The analysis of individual L1 5′ UTR sequences, demonstrated the greatest variation between the brain and skin at CpG residues located near the 3′ end of the amplicon, and six amplicons from the brain samples were unmethylated (Fig. 3E; S8A-B). Restricting this analysis to 10 L1s from both brain and skin with highest sequence homology to an RC-L1 revealed 19/20 sequences were derived from the L1Hs subfamily (data not shown), and one L1Hs element from the brain was completely unmethylated (Fig. 3C). In all cases, control experiments showed that the bisulfite conversion efficiency was >90% (Fig. S8C).
Figure 3. Methylation analysis and chromatin immunoprecipitation (ChIP) for the endogenous human L1 5′ UTR.
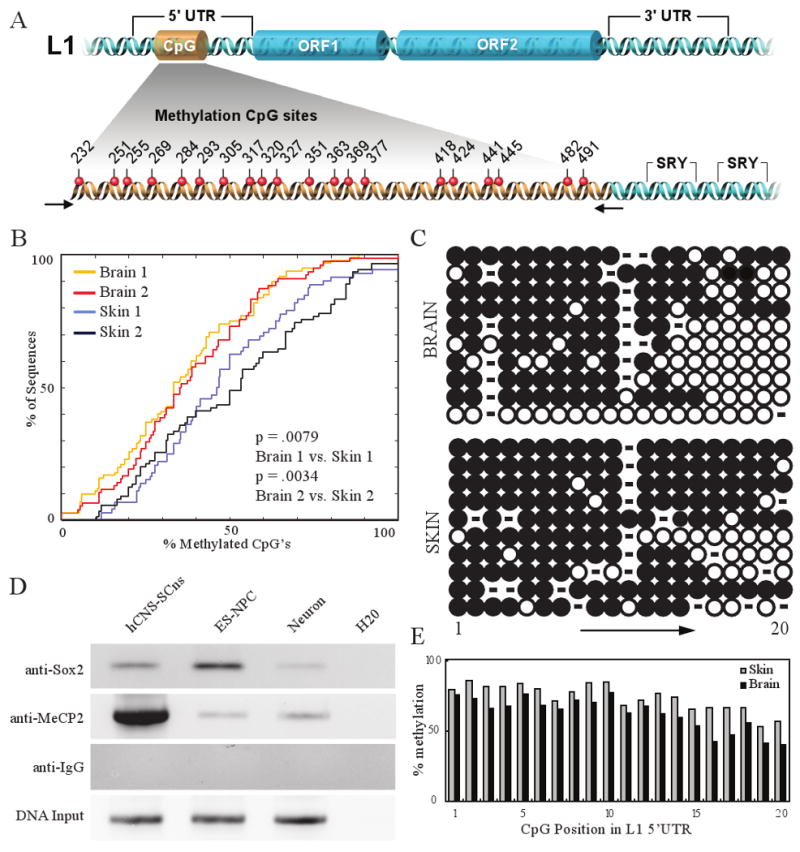
a, Schematic illustrating the L1 CpG island, and SRY/SOX2 binding sites. b, Cumulative distribution function (CDF) plot, comparing overall methylation and collapsing CpG sites into a single data point, (two-sample Kolmogorov-Smirnov test). c, Individual methylation of sequences exhibiting highest sequence similarity to consensus RC-L1s. Open circles = unmethylated, closed circles = methylated CpG dinucleotides. d, ChIP identifying MeCP2 and Sox2 occupying the endogenous human L1 promoter, extracts were analyzed by PCR towards the L1 5′ UTR SRY binding region (Sox2 immunoprecipitation) or CpG island region (MeCP2 immunoprecipitation). e, CpG dinucleotides exhibited higher methylation at the 5′ end of the CpG island; higher methylation overall was observed in skin samples.
Previous data suggested that Sox2 and MeCP2 could associate with the L1 promoter and repress L1 transcription under some experimental conditions1,17. Two putative SRY/Sox2 binding sites are located in the L1 5′ UTR immediately 3′ to the CpG island (Fig. 3A, S11B)18. Thus, we performed chromatin immunoprecipitation (ChIP) for Sox2 and MeCP2 in hCNS-SCns, HUES6-derived NPCs, and HUES6-derived neurons. Sox2 associated with the L1 5′ UTR in a pattern that correlates with the decrease in SOX2 expression observed during neural differentiation (Fig. 3D, S4H). MeCP2 expression was lower in both hCNS-SCns and HUES6-derived NPCs than in neurons (Fig. S4H), and both hCNS-SCns and HUES6-derived NPCs expressed similar levels and types of L1 transcripts (Fig. S6A-B). However, higher levels of MeCP2 were detected in association with the L1 promoter in hCNS-SCns than in HUES6-derived NPCs. We hypothesize that less L1 promoter methylation in the developing brain may correlate with increased L1 transcription and perhaps L1 retrotransposition, and the differential interaction of Sox2 and MeCP2 with L1 regulatory sequences may modulate L1 activity in different neuronal cell types.
Although NPCs are useful to monitor L1 activity, they only allow monitoring a single L1 expressed from a privileged context. By comparison, the average human genome contains ∼80-100 active L1s whose expression may be affected by chromatin structure4. Therefore, we developed a quantitative multiplexing PCR strategy to investigate endogenous L1 activity in the human brain, hypothesizing that active retrotransposition would result in increased L1 content in the brain as compared to other tissues (Fig. 4A).
Figure 4. Multiplex quantitative PCR analyses of L1 copy number in human tissues.
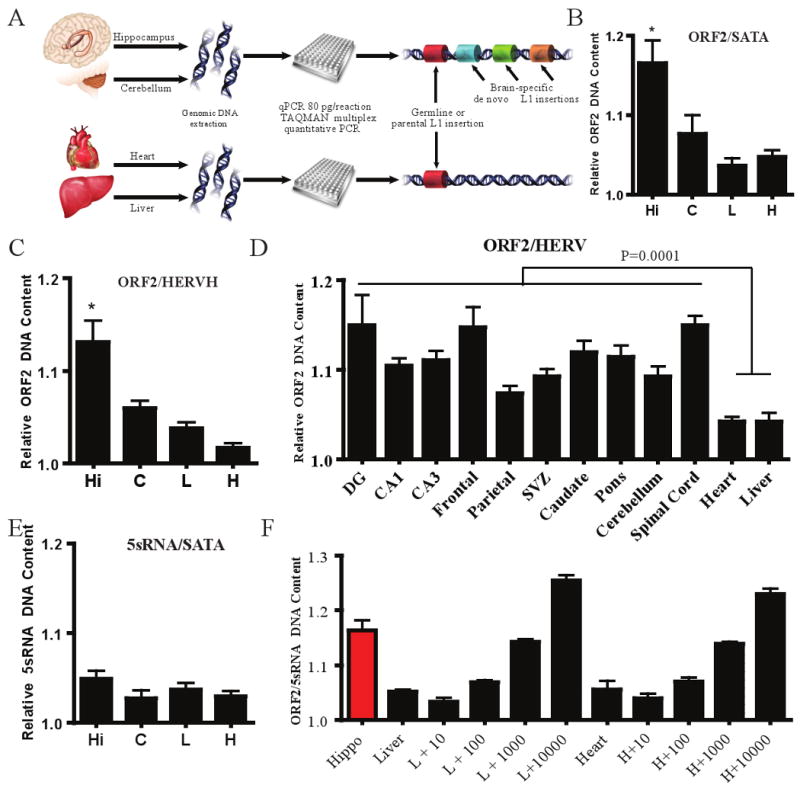
a, Experimental schematic. b-c, Relative quantity of L1, standardized such that the lowest liver value was normalized to 1.0. Hi = hippocampus, C = cerebellum, H = heart, and L = liver. Additional L1 ORF2 assays with other internals controls, Fig. S9-10. Error bars all s.e.m. *, p<0.05 (repeated measures one-way ANOVA with Bonferroni correction, n=3 individuals, with 3 repeat samples from each tissue). d, Ten samples from various brain regions (n=3 individuals) compared to somatic liver and heart. One-way t-test, p ≤ 0.0001 with 34 degrees of freedom. e, Multiplexing of 5S rDNA with α -satellite indicated no significant change, p≤0.5054. f, Hippocampal tissue compared to liver and heart spiked with estimated plasmid copy numbers of L1 (10, 100, 1,000, and 10,000 copies).
Briefly, we designed Taqman probes against a conserved 3′ region of ORF2 (conjugated with the VIC fluorophore), in addition to a number of control probes (conjugated with the 6FAM fluorophore). Controls were designed against the L1 5′UTR and other non-mobile DNA sequences in the genome that are higher (e.g., α satellite19) or lower in copy numbers (e.g., HERVH and 5S rDNA gene) than ORF2. In addition since the majority of L1 retrotransposition events are 5′ truncated9,20,21, we reasoned that the L1 5′ UTR probes should detect a smaller copy number increase than the L1 ORF2 probes. Each probe set amplified a single product of the predicted size (Fig. S10B). Moreover, sequencing PCR products derived from both ORF2 probe sets revealed enrichment for members of the L1Hs subfamily (Table S3).
We next isolated genomic DNA from the hippocampus, cerebellum, liver and heart from three adult humans. We consistently observed a statistically significant increase in L1 ORF2 content in the hippocampus when compared to heart and liver samples from the same individual (Fig. 4B-C, S9A, Fig. S10A). Notably, two individuals (1079 & 1846) showed more dramatic copy number differences than a third (4590) (Fig. S10A). Controls demonstrated that the ratio of the 5S rDNA gene to α satellite DNA between each tissue remained relatively constant (Fig. 4E).
We extended this analysis to 10 brain regions from three additional individuals (Fig. 4D, S9B). The samples were derived from the frontal and parietal cortex, spinal cord, caudate, CA1 and CA3 areas of the hippocampus, and pons, as well as from the hippocampal dentate gyrus (DG) and the subventricular zone (SVZ)22. As above, there was marked variation between different brain areas and between individuals (Fig. S9C). However, an unpaired t-test comparing all the grouped brain samples to the heart and liver DNA again revealed a small, but statistically significant increase in ORF2 content in the brain (Fig 4D).
To independently corroborate the observed increase in L1 copy number in the hippocampus and cerebellum samples, we spiked 80 pg of liver and heart genomic DNA (approximately 12 genomes) from individual 1846 with a calculated quantity of L1 plasmid, then we repeated the multiplexing approach to assay ORF2 quantity relative to 5S rDNA internal control (Fig. 4F). Three replications of this experiment indicated that the hippocampus samples contained approximately 1,000 more L1 copies than the heart or liver genomic DNAs, suggesting a theoretical increase in ORF2 of approximately 80 copies/cell. The spiked L1 copies were in the form of a plasmid, which likely affects the copy number estimates, providing an estimate of relative change and not precise quantification of the absolute number of L1s per cell. Ultimately, proof that endogenous L1s are retrotransposing in the brain requires identification of new retrotransposition events in individual somatic cells.
The large degree of variability in L1-ORF2 copy numbers between brain regions and individuals may represent unsystematic rates of L1 retrotransposition or an additional level of regulation that requires further elucidation. That being stated, our in vitro findings in NPCs coupled with the observed L1-ORF2 copy number changes in the brain make it tempting to speculate that somatic retrotransposition events occur during early stages of human nervous system development. This study contributes to a body of evidence indicating that engineered L1s can retrotranspose during early development, and in select somatic cells1,6,23-25. Future experiments will determine whether endogenous L1s truly retrotranspose in the brain and whether these events are simply ‘genomic noise’ or have the potential to impact neurogenesis and/or neuronal function.
Methods Summary
Cell culture, transfection, and analysis
Fetal hCNS-SCns lines3 and hESCs24,26 were cultured as previously described. Neural progenitors were derived from hESCs as previously described14,27. NPCs were transfected by nucleofection (Amaxa Biosystems), and either maintained as progenitors in the presence of FGF-2 or differentiated as previously described14. Cells were transfected with L1s containing an EGFP retrotransposition cassette in pCEP4 (Invitrogen) that lacks the CMV promoter and contains a puromycin resistance gene7.
Cell lysates
Ribonucleoprotein particles were isolated and analyzed as previously described8.
Luciferase assays were performed as previously described1.
Chromatin immunoprecipitation was performed utilizing primers towards the L1 5′UTR and a ChIP assay kit (Upstate/Millipore) as per manufacturer's protocol.
Bisulfite Analysis
Fetal tissues were obtained from the Birth Defects Research Lab at the Univ. of Washington. Bisulfite conversions were performed by manufacturer's instructions utilizing the Epitect kit (Quiagen). BLASTN (http://blast.ncbi.nlm.nih.gov/Blast.cgi) was used to align sequences to a database of full-length L1s.
PCR
Adult human tissues were obtained from the NICDH Brain and Tissue Bank for Developmental Disorders (University of Maryland, Baltimore, MD). Taqman probes and primers were designed using L1 Base (http://l1base.molgen.mpg.de/) and copy number estimates were based on the UCSC genome browser (http://genome.ucsc.edu). Experiments were performed on an ABI Prism 7000 sequence detection system (Applied Biosystems). For each tissue, three separate tissue samples were extracted and considered as repeated measures. Whole genome size was estimated based on the equation, cell genomic DNA content = 3*109(#bps) * 2(diploid) *660 (MW 1bp) * 1.67*1012 (weight 1 dalton), resulting in the approximation that one cell contains 6.6 pg genomic DNA 28. Therefore, the 80pg of genomic DNA utilized per reaction is derived from approximately 12 cells.
Inverse PCR was performed as previously described1,24.
Other methods
More details can be found in the Supplementary Methods.
Supplementary Material
Supplementary Data
Acknowledgments
We thank J. Simon for excellent schematic drawings, M.L. Gage and Drs. J. Kim and H. Kopera for editorial comments, B. Miller and R. Keithley for cell culture assistance, C.T. Carson for hESC advice, D. Chambers and J. Barrie for flow cytometry assistance, L. Randolph-Moore for molecular advice, B. Aimone for statistics advice, T. Liang for microarray assistance, and Y. Lineu and J. Mosher for helpful comments. We also thank T. Fanning and M. Klymkowsky for the ORF1p and Sox3 antibodies, respectively. F.H.G. and N.G.C. are supported by the Picower Foundation, Lookout Fund, and the California Institute for Regenerative Medicine (CIRM). J.L.G.P. is supported by Plan Estabilizacion Grupos SNS ENCYT 2015 (EMER07/56, Instituto de Salud Carlos III, Spain) and through the IRG-FP7-PEOPLE-2007 Marie Curie program. K.S.O. was supported by grants GM069985 and NS048187 from the National Institutes of Health. J.V.M. was supported by grants GM082970 and GM069985 from the National Institutes of Health and by the Howard Hughes Medical Institute. Work in the laboratories of K.S.O. and J.V.M. only used NIH-approved stem cell lines.
Footnotes
Author contributions: N.G.C. and F.H.G. directed the project. J.V.M and J.L.G.P. directed aspects of the project conducted at Michigan. N.G.C., J.L.G-P, J.V.M., and F.H.G. designed experiments and drafted the manuscript. N.G.C., F.H.G, J.L.G-P., G.E.P. performed the experiments. G.W.Y. and M.T.L. carried out bioinformatics data analysis. Y.M. performed electrophysiology experiments. M.M. and K.S.O. provided hESC culture and NPC differentiation assistance. All authors commented on or contributed to the current manuscript.
References
- 1.Muotri AR, et al. Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition. Nature. 2005;435(7044):903–910. doi: 10.1038/nature03663. [DOI] [PubMed] [Google Scholar]
- 2.Tang Y, Nyengaard JR, De Groot DM, Gundersen HJ. Synapse. 3. Vol. 41. New York, N.Y: 2001. Total regional and global number of synapses in the human brain neocortex; pp. 258–273. [DOI] [PubMed] [Google Scholar]
- 3.Uchida N, et al. Direct isolation of human central nervous system stem cells. Proc Natl Acad Sci U S A. 2000;97(26):14720–14725. doi: 10.1073/pnas.97.26.14720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brouha B, et al. Hot L1s account for the bulk of retrotransposition in the human population. Proc Natl Acad Sci U S A. 2003;100(9):5280–5285. doi: 10.1073/pnas.0831042100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moran JV, et al. High frequency retrotransposition in cultured mammalian cells. Cell. 1996;87(5):917–927. doi: 10.1016/s0092-8674(00)81998-4. [DOI] [PubMed] [Google Scholar]
- 6.Ostertag EM, et al. A mouse model of human L1 retrotransposition. Nat Genet. 2002;32(4):655–660. doi: 10.1038/ng1022. [DOI] [PubMed] [Google Scholar]
- 7.Ostertag EM, Prak ET, DeBerardinis RJ, Moran JV, Kazazian HH., Jr Determination of L1 retrotransposition kinetics in cultured cells. Nucleic Acids Res. 2000;28(6):1418–1423. doi: 10.1093/nar/28.6.1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kulpa DA, Moran JV. Ribonucleoprotein particle formation is necessary but not sufficient for LINE-1 retrotransposition. Hum Mol Genet. 2005;14(21):3237–3248. doi: 10.1093/hmg/ddi354. [DOI] [PubMed] [Google Scholar]
- 9.Moran J, Gilbert N. Mammalian LINE-1 retrotransposons and related elements. ASM Press; Washington, DC: 2002. [Google Scholar]
- 10.Myers JS, et al. A comprehensive analysis of recently integrated human Ta L1 elements. Am J Hum Genet. 2002;71(2):312–326. doi: 10.1086/341718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morrish TA, et al. DNA repair mediated by endonuclease-independent LINE-1 retrotransposition. Nat Genet. 2002;31(2):159–165. doi: 10.1038/ng898. [DOI] [PubMed] [Google Scholar]
- 12.Gilbert N, Lutz S, Morrish TA, Moran JV. Multiple fates of L1 retrotransposition intermediates in cultured human cells. Mol Cell Biol. 2005;25(17):7780–7795. doi: 10.1128/MCB.25.17.7780-7795.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Symer DE, et al. Human l1 retrotransposition is associated with genetic instability in vivo. Cell. 2002;110(3):327–338. doi: 10.1016/s0092-8674(02)00839-5. [DOI] [PubMed] [Google Scholar]
- 14.Yeo GW, et al. Alternative splicing events identified in human embryonic stem cells and neural progenitors. PLoS computational biology. 2007;3(10):1951–1967. doi: 10.1371/journal.pcbi.0030196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bourc'his D, Bestor TH. Meiotic catastrophe and retrotransposon reactivation in male germ cells lacking Dnmt3L. Nature. 2004;431(7004):96–99. doi: 10.1038/nature02886. [DOI] [PubMed] [Google Scholar]
- 16.Takai D, Jones PA. The CpG island searcher: a new WWW resource. In Silico Biol. 2003;3(3):235–240. [PubMed] [Google Scholar]
- 17.Yu F, Zingler N, Schumann G, Stratling WH. Methyl-CpG-binding protein 2 represses LINE-1 expression and retrotransposition but not Alu transcription. Nucleic Acids Res. 2001;29(21):4493–4501. doi: 10.1093/nar/29.21.4493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tchenio T, Casella JF, Heidmann T. Members of the SRY family regulate the human LINE retrotransposons. Nucleic Acids Res. 2000;28(2):411–415. doi: 10.1093/nar/28.2.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee C, Wevrick R, Fisher RB, Ferguson-Smith MA, Lin CC. Human centromeric DNAs. Human genetics. 1997;100(3-4):291–304. doi: 10.1007/s004390050508. [DOI] [PubMed] [Google Scholar]
- 20.Pavlicek A, Paces J, Zika R, Hejnar J. Length distribution of long interspersed nucleotide elements (LINEs) and processed pseudogenes of human endogenous retroviruses: implications for retrotransposition and pseudogene detection. Gene. 2002;300(1-2):189–194. doi: 10.1016/s0378-1119(02)01047-8. [DOI] [PubMed] [Google Scholar]
- 21.Grimaldi G, Skowronski J, Singer MF. Defining the beginning and end of KpnI family segments. Embo J. 1984;3(8):1753–1759. doi: 10.1002/j.1460-2075.1984.tb02042.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gage FH. Mammalian neural stem cells. Science. 2000;287(5457):1433–1438. doi: 10.1126/science.287.5457.1433. [DOI] [PubMed] [Google Scholar]
- 23.Prak ET, Dodson AW, Farkash EA, Kazazian HH., Jr Tracking an embryonic L1 retrotransposition event. Proc Natl Acad Sci U S A. 2003;100(4):1832–1837. doi: 10.1073/pnas.0337627100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garcia-Perez JL, et al. LINE-1 retrotransposition in human embryonic stem cells. Hum Mol Genet. 2007;16(13):1569–1577. doi: 10.1093/hmg/ddm105. [DOI] [PubMed] [Google Scholar]
- 25.van den Hurk JA, et al. L1 retrotransposition can occur early in human embryonic development. Hum Mol Genet. 2007;16(13):1587–1592. doi: 10.1093/hmg/ddm108. [DOI] [PubMed] [Google Scholar]
- 26.Thomson JA, et al. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282(5391):1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- 27.Zhang SC, Wernig M, Duncan ID, Brustle O, Thomson JA. In vitro differentiation of transplantable neural precursors from human embryonic stem cells. Nat Biotechnol. 2001;19(12):1129–1133. doi: 10.1038/nbt1201-1129. [DOI] [PubMed] [Google Scholar]
- 28.Forslund O, et al. Nucleotide sequence and phylogenetic classification of candidate human papilloma virus type 92. Virology. 2003;312(2):255–260. doi: 10.1016/s0042-6822(03)00391-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data