CONTRIBUTION TO FAMILIAL BREAST CANCER OF INHERITED MUTATIONS IN THE BRCA2-INTERACTING PROTEIN PALB2 (original) (raw)
. Author manuscript; available in PMC: 2012 Mar 15.
Abstract
Inherited mutations in the BRCA2-interacting protein PALB2 are known to be associated with increased risks of breast cancer. In order to evaluate the contribution of PALB2 to familial breast cancer in the United States, we sequenced the coding sequences and flanking regulatory regions of the gene from constitutional genomic DNA of 1144 familial breast cancer patients with wildtype sequences at BRCA1 and BRCA2. Overall, 3.4% (33/972) of patients not selected by ancestry and 0% (0/172) of patients specifically of Ashkenazi Jewish ancestry were heterozygous for a nonsense, frameshift, or frameshift-associated splice mutation in PALB2. Mutations were detected in both male and female breast cancer patients. All mutations were individually rare: the 33 heterozygotes harbored 13 different mutations, 5 previously reported and 8 novel. PALB2 heterozygotes were 4-fold more likely to have a male relative with breast cancer (P=0.0003) and 6-fold more likely to have a relative with pancreatic cancer (P=0.002), and 1.3-fold more likely to have a relative with ovarian cancer (P=0.18). Compared to their female relatives without mutations, increased risk of breast cancer for female PALB2 heterozygotes was 2.3-fold (95%CI [1.5–4.2]) by age 55 and 3.4-fold (95%CI [2.4–5.9]) by age 85. Loss of the wildtype PALB2 allele was observed in laser dissected tumor specimens from heterozygous patients. Given this mutation prevalence and risk, consideration might be given to clinical testing of PALB2 by complete genomic sequencing for familial breast cancer patients with wildtype sequences at BRCA1 and BRCA2.
INTRODUCTION
Discovery and characterization of the breast cancer susceptibility genes BRCA1 and BRCA2 have led to major changes in both prevention and treatment. Genetic testing for inherited mutations offers the opportunity for risk reducing intervention (1). Therapeutic approaches that exploit the biological function of BRCA1 and BRCA2 have been proposed (2) and are now showing promise in the clinic (3, 4). Given this experience, there has been a great deal of interest in the identification and characterization of other genes responsible for inherited breast cancer (5). Among these genes is PALB2_,_ partner and localizer of BRCA2 (6), first characterized clinically in patients with Fanconi anemia, complementation group N (7, 8). It was soon discovered that heterozygosity for loss of function mutations at PALB2 increases risk of breast cancer 2- to 6-fold (5, 9). Inherited PALB2 mutations associated with increased risks of breast cancer have been identified in families from many parts of the world (10–18), but thus far, a heterogeneous American population has not been screened.
The purpose of the present project was to estimate the contribution of inherited mutations in PALB2 to familial breast cancer in a large series of patients from the United States and to characterize the spectrum of inherited breast-cancer-associated mutations in PALB2 in this heterogeneous population. For this purpose, we sequenced the complete coding and flanking regulatory regions of PALB2 from constitutional DNA of 1144 familial breast cancer patients, all previously determined to have wildtype sequences at BRCA1 and BRCA2.
Materials and Methods
Subjects
Participants were patients with a primary diagnosis of invasive breast cancer at any age and of any histologic subtype and with at least two first or second degree relatives with invasive breast cancer (19). Two series of participants were identified: one series identifying their ancestry as Ashkenazi Jewish (AJ) and the other series not selected for any specific ancestry. The series of Ashkenazi Jewish patients was enrolled in order to evaluate the possibility of founder mutations in this population in either known or novel breast cancer genes. Enrollment of probands, ascertainment of families, and genomic analysis proceeded identically for the two series. Potential participants were referred by physicians, genetic counselors, or themselves. All potential participants were interviewed prior to enrollment by a certified genetic counselor (J.B.M.) and informed consent was obtained for those meeting the criteria above and interested in enrolling. The consenting participant became the proband for her or his family. For each proband, all genetically informative adult relatives, whether or not affected with breast cancer, were also requested to enroll. Relatives were contacted first by the proband, then interviewed by the genetic counselor and informed consent obtained for those interested. Enrollment began in 1988 and is ongoing. All families in this study have been followed from their time of enrollment to the present by the genetic counselor. For all probands, BRCA1 and BRCA2 had been determined to be wildtype, in almost all cases based on commercial sequencing and BART analysis by Myriad Genetics (20). The two series combined included 1144 familial breast cancer patients without mutations in BRCA1 or BRCA2, of whom 172 participants (all female) were of AJ ancestry and 972 participants (959 females and 13 males) were of other ancestries. The study was approved by the University of Washington Human Subjects Division (IRB protocol 34173).
Genomic DNA sequencing
PALB2 exons, flanking intronic regions (50–100 bp in length), and 5′- and 3′-UTRs were evaluated by Sanger sequencing of constitutional genomic DNA from all subjects. Genomic DNA isolated from peripheral blood lymphocytes was amplified by PCR using flanking intronic primers (Supplementary Table 1). Nested PCR and four overlapping amplicons were developed to fully cover the 1473 bp of exon 4. Multiple internal primers were used to sequence exon 5 in both directions. Amplicons were sequenced in both forward and reverse directions except as follows. For exon 9, to overcome the chances of non-specific products due to Alu sequences upstream of the splice site, the exon was amplified using nested primers from an outer product, then cycle-sequenced from the reverse direction. Similarly, exon 13 was only sequenced in the reverse direction. PCR products were purified and cycle-sequenced using the BigDye Terminator Cycle Sequencing chemistry (Applied Biosystems, Life Technologies Corp, Carlsbad, CA) and analyzed on a ABI PRISM® 3700 Genetic Analyzer (Applied Biosystems, Life Technologies Corp, Carlsbad, CA). All sequence variants were confirmed by replicate Sanger sequence, then evaluated for co-segregation with breast cancer in the family of the proband.
Analysis of transcripts
RNA and cDNA were isolated and transcript lengths evaluated as previously described (19). The possibility of nonsense mediated decay was evaluated by comparing electropherogram peak heights of mutant and wildtype alleles from transcript sequences. Multiple splice variants resulting from single genomic mutations were quantified by cloning PCR-amplified cDNA products into pCR2.1-TOPO cloning vectors (Invitrogen, Carlsbad, CA), then by transforming competent E. coli strains, then by sequencing individual clones. Cloning experiments were carried out in triplicate.
Genotyping family members and controls
In order to genotype all informative relatives of each proband with a mutation, TaqMan assays were designed for PALB2 mutations c.168delTTGT, c.509delGA, c.757delCT, c.1240C>T, c.2386G>T, c.2559C>T, c.2686insT, c.2919delAA, c.3022delC and restriction digests were designed for mutations c.196C>T (HinP1I), c.1653T>A (HpyCH4III), c.2718G>A (BsmAI), c.3113G>A (BstEII). In addition to family members, 960 unrelated anonymous controls were evaluated for each mutation. For TaqMan assays, DNA was diluted to 20ng, with all wells in the same assay containing the same amount of sample. Two controls without template and positive controls with the proband’s PALB2 mutation were run with every plate. After each real-time PCR amplification, an allelic discrimination plate was analyzed using Sequence Detection System (SDS) software (Applied Biosystems, CA), with the autocaller function enabled and 95% quality interval for allele calling. Fluorescence signals were plotted for test samples, for controls without template, and for positive controls, with X and Y axes representing each allele. Genotypes were called based on location on the scatterplot.
Genotypes of deceased family members and others not available for genotyping were called only if they could be reconstructed with certainty from surviving children, spouses, and siblings. No genotypes were imputed probabilistically.
In order to determine whether the mutation PALB2 c.3113 shared a common origin in the five patients in which it appears, all probands and their relatives carrying PALB2 c.3113 were genotyped with microsatellite markers on chromosome 16 developed for this purpose. The markers were selected to flank PALB2 at chr16:23,614,483–23,652,678 (hg19). The microsatellites were AC23 at chr16:21,732,554–21,732,600 (8 alleles in our series), 23GT at chr16:23,037,671–23,037,716 (9 alleles in our series), and TATG14 at chr16:23,749,512–23,749,567 (4 alleles in our series). Microsatellite sequences, PCR primers, and genotyping conditions are reported in Supplementary Table 2.
Laser capture microdissection and genotyping breast tumor specimens
Tumor sections of 5–8 um from paraffin-embedded blocks were microdissected with the automated LCM Veritas System (Arcturus Molecular Devices, Life Technologies Corp, Carlsbad, CA) as previously described (21). Briefly, for each initial cap placement, laser was focused with the 7.5um spot size setting and the 10× objective. The pulse of the capture laser was adjusted to a 1000–1500 usec duration and 70mW power. Microdissection was performed with a medium laser spot size 12.43um and a 10x objective. Loss of heterozygosity was analyzed by direct sequencing of reconstituted tumor DNA at the site of the mutation. For all the PALB2 mutations for which tumor specimens were available, sequencing primers were designed to span maximum amplicon sizes of 250bp (Supplementary Table 3). Sequencing was performed as described above. Basecalls with PHRED scores >30 were included.
Statistical analysis
Frequencies of mutations in subgroups were compared by chi-square tests or Fisher’s exact tests, as appropriate. Odds ratios (OR), risk ratios (RR) and 95% confidence limits (CI) were calculated. Cumulative risks were estimated by Kaplan-Meier methods.
Results
Spectrum of PALB2 mutations
From genomic DNA of all 1144 participants, complete PALB2 coding sequences and flanking regulatory regions were evaluated for nonsense mutations, frameshift mutations, and splice mutations leading to out-of-frame message deletions. Of the 972 breast cancer probands of unselected ancestries, 33 (3.4%) carried a truncating mutation in PALB2 (Table 1). Of the 13 male probands, 2 (16%) carried a truncating mutation; of the 959 female probands, 31 (3.2%) carried a truncating mutation. Of the 172 breast cancer probands of AJ ancestry, zero carried such a mutation. Given the mutation frequency among patients of unselected ancestries, 5.5 carriers would be expected among the AJ patients (χ2=5.9, P=0.015). Thirteen different truncating mutations in PALB2 were observed, leading to stops in exons 3–11 (Figure 1).
Table 1.
Inherited truncating mutations in PALB2
| Genomic locale (hg19) | Exon | DNA mutation | Type | RNA mutation | Protein mutation | Stop codon | Freq cases (N=972*) | Freq controls (N=960) | Ancestry |
|---|---|---|---|---|---|---|---|---|---|
| chr16:23,649,207–210 | 3 | c.172-175 del TTGT | frameshift | - | S58fsX8** | 66 | 1 | 0 | Irish |
| chr16:23,649,186 | 3 | c.196 C>T | nonsense | - | Q66X** | 66 | 2 | 0 | Scots |
| chr16:23,647,357–358 | 4 | c.509-510 del GA | frameshift | - | R170fsX13 | 183 | 7 | 0 | German |
| chr16:23,647,109–110 | 4 | c.757-758 del CT | frameshift | - | L253fsX2 | 255 | 4 | 0 | German |
| chr16:23,646,627 | 4 | c.1240 C>T | nonsense | - | R414X** | 414 | 3 | 0 | German |
| chr16:23,646,214 | 4 | c.1653 T>A | nonsense | - | Y551X | 551 | 2 | 0 | French |
| chr16:23,641,089 | 5 | c.2386 G>T | nonsense | - | G796X | 796 | 1 | 0 | English |
| chr16:23,640,552 | 6 | c.2559 C>T | splice | r.2558-2586 del 29 | G853fsX20** | 873 | 1 | 0 | German |
| chr16:23,637,618 | 7 | c.2686 ins T | frameshift | - | S896fsX31** | 927 | 3 | 0 | German |
| chr16:23,637,587 | 7 | c.2718 G>A | nonsense | - | W906X** | 906 | 1 | 0 | English |
| chr16:23,634,364–365 | 9 | c.2920-2921 del AA | frameshift | - | K974fsX4** | 978 | 1 | 0 | English |
| chr16:23,632,770 | 10 | c.3026 del C | frameshift | - | P1009fsX5** | 1014 | 2 | 0 | German |
| chr16:23,632,683 | 10 | c.3113 G>A | splice | (a) r.2997-3113 del 117 | p.999-1038del (56%) | - | 5 | 0 | African |
| (b) r.3083-3113 del 31 | G1028fsX2 (40%) | 1030 | American (2), | ||||||
| .(c) - | W1038X (4%) | 1038 | English (3) |
Figure 1.

Truncating mutations in PALB2. Thirteen different truncating mutations in PALB2 were detected in familial breast cancer patients. On the PALB2 gene sequence, frameshift mutations are indicated in red, nonsense mutations in purple, and splice mutations in blue. On the PALB2 protein, yellow symbols indicate low complexity regions (LCR), the red symbol the coiled coil domain, and green symbols the WD40-like repeats that comprise beta propeller structures.
Analysis of alternate transcripts
Identification of aberrant splice mutations was based on analysis of transcripts. For each sample with a single basepair genomic variant in coding sequence or in flanking intronic regions, cDNA was generated from lymphoblast RNA by RT-PCR of PALB2 message. Of the 19 such variants observed in the cohort, two substitutions, c.2559C>T and c.3113G>A, led to altered splicing, out-of-frame deletions in the PALB2 message, and hence premature stops (Figure 2). At PALB2 c.3113G>A in exon 10, three different messages were produced. Cloning and sequencing multiple messages indicated that 56% of mutant messages included an in-frame 117 bp deletion, 40% of messages included an out-of-frame 31 bp deletion, and 4% of messages were an immediate stop at residue 1038 due to the creation of a nonsense codon at this site. Other rare PALB2 variants in the 1144 probands included silent and missense substitutions in coding sequence, variants in the 5′ UTR and flanking intronic sequences, and variants at splice sites leading to in-frame deletions in transcripts (Supplementary Table 4, Supplementary Figure 1).
Figure 2.
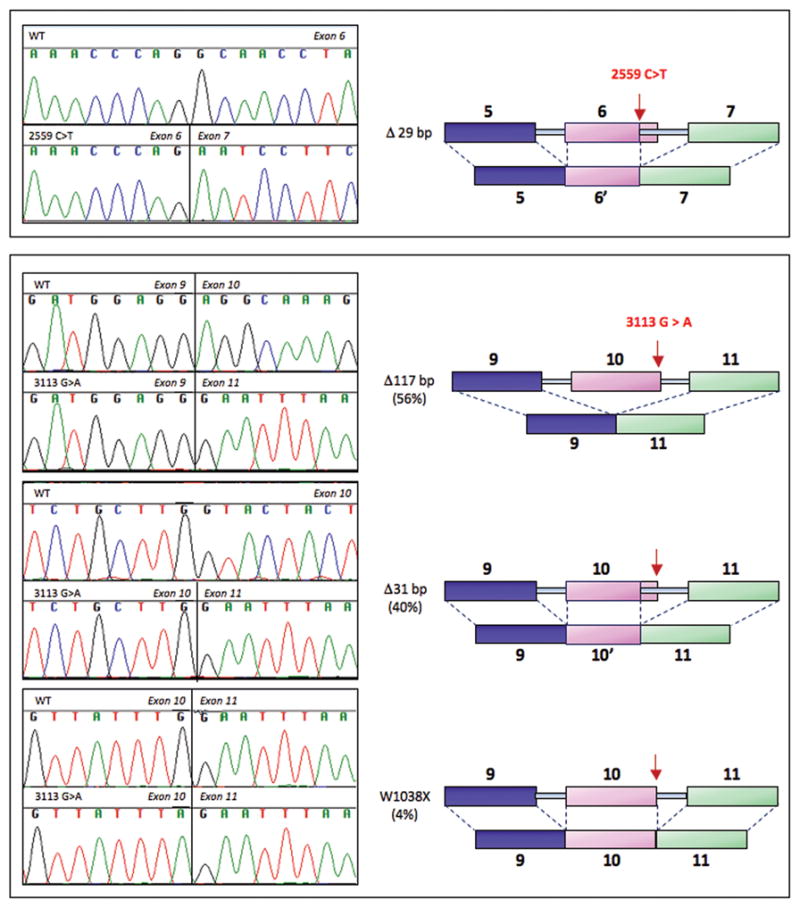
Splice mutations in PALB2. Effects on splicing of single bp genomic substitutions in PALB2 were evaluated by sequencing transcripts. PALB2 c.2559C>T leads to altered splicing and deletion of 29 bp in the PALB2 message. PALB2 c.3113G>A produces three different PALB2 messages: complete deletion of exon 10 (117 bp), use of an alternate splice site within exon 10 and deletion of 31 bp, and an immediate stop at codon 1038.
Breast cancer risks to mutation carriers
As each PALB2 mutation was identified in a breast cancer proband, all available affected and unaffected relatives were genotyped. For deceased relatives, genotypes were reconstructed as possible from surviving adult children, spouses, and siblings. Genotypes were assigned only if they could be reconstructed with certainty. For 83 female relatives, affected and unaffected, with unambiguous PALB2 genotypes, cumulative risks of breast cancer were compared for those carrying a PALB2 mutation versus those with wildtype PALB2 genotypes. Ratios of these risks provided estimates of relative risks, cumulative by age, associated with PALB2 truncating mutations. Relative risks were 2.3-fold (95%CI [1.5–4.2]) by age 55 and 3.4-fold (95%CI [2.4–5.9]) at age 85, consistent with those previously reported (4).
Population origins of PALB2 mutations
Of the 13 PALB2 mutations identified in this series, five have been previously reported and eight were novel (Table 1). Ancestries of patients carrying PALB2 mutations, as proportions of all patients of each ancestry in our series, are indicated in Table 2. In some of the patients of German, French, and English origin, we detected mutations originally discovered in patients of the same ancestries (7–9). A mutation previously discovered in a French-Canadian family (12) was not detected in patients of that ancestry in our series. Three mutations offered particularly interesting histories. PALB2 c.3113G>A appeared in three patients of English ancestry and two patients of African-American ancestry. These five patients shared a haplotype of length >2MB defined by microsatellite markers on chromosome 16 flanking PALB2. Among the patients’ relatives who were wildtype at c.3113G>A, none shared this complete haplotype. The allele-specific haplotype suggests a single origin for this mutation, which has previously been reported in British families (9). A second historically interesting mutation was c.509-510delGA, carried by seven patients, all of whom reported German ancestry on their familial lineages with histories of breast cancer. Further exploration of these patients’ pedigrees revealed a common ancestor who immigrated to the U.S. from Germany in the 19th century. PALB2 c.509-510delGA has also been reported in families from Poland (17), suggesting this mutation may have a central European origin. Finally, the two patients carrying PALB2 c.196C>T shared a distant ancestor who immigrated from Scotland.
Table 2.
Ancestries of participants
| Origin of lineage with breast cancer, as reported by the participant | Cases genotyped | Cases with PALB2 mutations |
|---|---|---|
| Series 1: Ashkenazi Jewish ancestry | ||
| Ashkenazi Jewish | 172 | |
| Series 2: Unselected ancestry | ||
| African American | 18 | 2 |
| American Indian or Inuit | 33 | |
| Austrian | 4 | |
| Basque | 3 | |
| Belgian | 2 | |
| Chinese | 4 | |
| Czech | 7 | |
| Danish | 13 | |
| Dutch | 20 | |
| English | 225 | 6 |
| European not otherwise known | 3 | |
| Filipino | 1 | |
| French | 25 | 2 |
| French Canadian | 21 | |
| German | 257 | 20 |
| Greek | 6 | |
| Hungarian | 3 | |
| Icelandic | 2 | |
| Irish | 116 | 1 |
| Italian | 65 | |
| Japanese | 1 | |
| Korean | 2 | |
| Lebanese | 2 | |
| Mexican | 13 | |
| Norwegian | 12 | |
| Polish | 11 | |
| Portuguese | 4 | |
| Puerto Rican | 2 | |
| Romanian | 3 | |
| Russian | 3 | |
| Scots | 9 | 2 |
| Scots Irish | 31 | |
| Slovenian | 4 | |
| Swedish | 22 | |
| Swiss | 17 | |
| Turkish | 2 | |
| Welsh | 6 | |
| Series 2 total | 972 | 33 |
Male breast cancer, pancreatic cancer, and ovarian cancer in PALB2 families
The cancer profiles of PALB2 families (Table 3) were similar to cancer profiles of BRCA2 families. Male or female patients with PALB2 mutations were significantly more likely than patients without PALB2 mutations to have a relative with male breast cancer or with pancreatic cancer. In contrast, PALB2 genotype was not significantly associated with the likelihood of having a relative with ovarian cancer or with age at onset of female breast cancer (50.0±11.9 years among PALB2 heterozygotes vs 50.2±6.8 years among patients not carrying PALB2 mutations). Patients with PALB2 mutations were more likely to be probands of families with at least six cases of female breast cancer, perhaps reflecting the possibility that some families in our series with relatively few cases of breast cancer do not carry mutations in any susceptibility genes. It should be noted that most of the patients with PALB2 mutations (17 of 33) were not probands of these extremely high-incidence families.
Table 3.
Cancers in relatives of participants
| Cancer in relatives | PALB2 genotype of participant | |||
|---|---|---|---|---|
| Mutation N=33 | Wildtype N=939 | RR [95% CI] | P value | |
| Male breast cancer | 9 | 62 | 4.13 [2.25, 7.58] | 0.0003 |
| Pancreatic cancer | 5 | 24 | 5.93 [2.41, 14.56] | 0.002 |
| Ovarian cancer | 18 | 388 | 1.32 [0.96, 1.82] | 0.18 |
| ≥ female breast cancers | 16 | 280 | 1.63 [1.13, 2.34] | 0.036 |
Loss of heterozygosity in tumors from patients with inherited PALB2 mutations
For seven female breast cancer patients with PALB2 mutations, paraffin embedded breast tumor specimens were available, enabling dissection of tumor cells by laser capture microscopy and evaluation of loss of heterozygosity at PALB2. In each of these specimens, loss of the wildtype PALB2 allele was observed (Figure 3). At least for these patients, loss of heterozygosity of the wildtype PALB2 allele suggests that PALB2 acts as a conventional inherited tumor suppressor gene.
Figure 3.

Loss of heterozygosity of wildtype PALB2 alleles in tumor tissue. Tumor tissue was available from seven patients with five different mutations. In all cases, cancer cells isolated by laser dissection carried only the mutant PALB2 allele. Each panel indicates the sequence at the mutant allele (arrow) for a wildtype control (WT), for constitutional genomic DNA from the carrier (MUT), and for laser-dissected tumor cells (Tumor).
DISCUSSION
In a series of familial breast cancer patients of unselected ancestry, with wildtype sequences of BRCA1 and BRCA2, the prevalence of inherited PALB2 mutations was 3.4%. Thirteen different protein truncating mutations were identified, all of which were individually rare. Five mutations had been previously reported; eight were encountered for the first time in this series. Both the occurrence of PALB2 mutations among male and female breast cancer patients and the 4-fold higher frequency of male breast cancer among relatives of PALB2 heterozygotes suggest that PALB2 predisposes to both male and female breast cancer. Cancer profiles of these families are also consistent with the suggestion that PALB2 is a susceptibility gene for pancreatic cancer (22). Ovarian cancer was more common among relatives of PALB2 heterozygotes (55% of PALB2 families included a relative with ovarian cancer) than among relatives of other cases (41% of non-PALB2 families included a relative with ovarian cancer), but this difference was not significant given this sample size. Direct evaluation of large numbers of ovarian cancer patients will be useful in determining the role of inherited mutations in PALB2 in this malignancy. The distributions of ages at diagnosis of breast cancer patients with PALB2 mutations were very similar to those of the other familial breast cancer patients in the series, suggesting that in the context of familial breast cancer, PALB2 does not lead to significantly younger diagnosis. It is very unlikely that there is a founder allele of PALB2 among patients of Ashkenazi Jewish ancestry, but any family, of any ancestry, could harbor a private cancer-predisposing allele of PALB2.
The prevalence of inherited mutations in PALB2 in non-BRCA1, non-BRCA2 familial breast cancer patients is approximately the same as the prevalence of inherited mutations in CHEK2 in a similar cohort (19). It has been suggested that genetic testing for CHEK2 c.1100delC in the clinical setting is now timely, with targeted surveillance and medical follow up for mutation carriers (23). We agree and join others (24) in suggesting that similar consideration be extended to PALB2. The breast cancer risk associated with protein truncating mutations in PALB2 appears greater than the risk associated with CHEK2 c.1100delC. The challenge is that in the U.S., no one mutation of PALB2 is sufficiently frequent to represent a substantial fraction of PALB2 mutations, so that the entire gene must be sequenced to capture the responsible alleles. Simultaneous detection of all mutations in all known breast cancer genes will make such expanded screening more feasible (25).
Testing for PALB2 and other breast cancer susceptibility genes will add complexity to the clinical interpretation of results. In particular, variants of uncertain significance will be identified. This study focused on PALB2 mutations leading to protein truncation. Individual missense mutations in PALB2 will ultimately need to be evaluated as they have been for BRCA1 and BRCA2 (26, 27).
The possibility of genetic testing for PALB2 provides another example of the complexity of the transition from research laboratory to clinic. The involvement of medical geneticists and genetic counselors in assessing risk is particularly critical when, as here, consequences of genetic testing can entail substantial medical and surgical decisions (28–30).
Supplementary Material
1
Acknowledgments
Supported by the Breast Cancer Research Foundation, Komen Foundation for the Cure, and NIH grants 5R01ES013160 (M.C.K, S.C., B.M.N., T.W., S.S., J.B.M., M.K.L.), and 5U01ES017156 (J.A.S.)
References
- 1.National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: breast cancer screening and diagnosis. J Natl Comp Cancer Network. 2009;7:1060–96. doi: 10.6004/jnccn.2009.0070. [DOI] [PubMed] [Google Scholar]
- 2.Ashworth A. A synthetic lethal therapeutic approach: poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J Clin Oncol. 2008;26:3785–90. doi: 10.1200/JCO.2008.16.0812. [DOI] [PubMed] [Google Scholar]
- 3.Fong PC, Boss DS, Yap TA, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361:123–34. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 4.Tutt A, Robson M, Garber JE, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. 2010;376:235–44. doi: 10.1016/S0140-6736(10)60892-6. [DOI] [PubMed] [Google Scholar]
- 5.Hollestelle A, Wasielewski M, Martens JW, Schutte M. Discovering moderate-risk breast cancer susceptibility genes. Curr Opin Genet Dev. 2010;20:268–76. doi: 10.1016/j.gde.2010.02.009. [DOI] [PubMed] [Google Scholar]
- 6.Xia B, Sheng Q, Nakanishi K, et al. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Molec Cell. 2006;22:719–29. doi: 10.1016/j.molcel.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 7.Xia B, Dorsman JC, Ameziane N, et al. Fanconi anemia is associated with a defect in the BRCA2 partner PALB2. Nature Genet. 2007;39:159–61. doi: 10.1038/ng1942. [DOI] [PubMed] [Google Scholar]
- 8.Reid S, Schindler D, Hanenberg H, et al. Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nature Genet. 2007;39:162–4. doi: 10.1038/ng1947. [DOI] [PubMed] [Google Scholar]
- 9.Rahman N, Seal S, Thompson D, et al. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nature Genet. 2007;39:165–7. doi: 10.1038/ng1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Erkko H, Xia B, Nikkilä J, et al. A recurrent mutation in PALB2 in Finnish cancer families. Nature. 2007;446:316–9. doi: 10.1038/nature05609. [DOI] [PubMed] [Google Scholar]
- 11.Tischkowitz M, Xia B, Sabbaghian N, et al. Analysis of PALB2/FANCN-associated breast cancer families. Proc Natl Acad Sci USA. 2007;104:6788–93. doi: 10.1073/pnas.0701724104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Foulkes WD, Ghadirian P, Akbari MR, et al. Identification of a novel truncating PALB2 mutation and analysis of its contribution to early-onset breast cancer in French-Canadian women. Breast Cancer Res. 2007;9:R83. doi: 10.1186/bcr1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cao AY, Huang J, Hu Z, et al. The prevalence of PALB2 germline mutations in BRCA1/BRCA2 negative Chinese women with early onset breast cancer or affected relatives. Breast Cancer Res Treat. 2009;114:457–62. doi: 10.1007/s10549-008-0036-z. [DOI] [PubMed] [Google Scholar]
- 14.García MJ, Fernández V, Osorio A, et al. Analysis of FANCB and FANCN/PALB2 Fanconi anemia genes in BRCA1/2-negative Spanish breast cancer families. Breast Cancer Res Treat. 2009;113:545–51. doi: 10.1007/s10549-008-9945-0. [DOI] [PubMed] [Google Scholar]
- 15.Sluiter M, Mew S, van Rensburg EJ. PALB2 sequence variants in young South African breast cancer patients. Fam Cancer. 2009;8:347–53. doi: 10.1007/s10689-009-9241-0. [DOI] [PubMed] [Google Scholar]
- 16.Papi L, Putignano AL, Congregati C, et al. A PALB2 germline mutation associated with hereditary breast cancer in Italy. Fam Cancer. 2010;9:181–5. doi: 10.1007/s10689-009-9295-z. [DOI] [PubMed] [Google Scholar]
- 17.Dansonka-Mieszkowska A, Kluska A, Moes J, et al. A novel germline PALB2 deletion in Polish breast and ovarian cancer patients. BMC Med Genet. 2010;11:20. doi: 10.1186/1471-2350-11-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tischkowitz M, Xia B. PALB2/FANCN: Recombining cancer and Fanconi anemia. Cancer Res. 2010;70:7353–9. doi: 10.1158/0008-5472.CAN-10-1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walsh T, Casadei S, Coats KH, et al. Spectrum of mutations in BRCA1, BRCA2, CHEK2, and TP53 in families at high risk of breast cancer. JAMA. 2006;295:1379–88. doi: 10.1001/jama.295.12.1379. [DOI] [PubMed] [Google Scholar]
- 20.Frank TS, Deffenbaugh AM, Reid JE, et al. Clinical characteristics of individuals with germline mutations in BRCA1 and BRCA2: Analysis of 10,000 individuals. J Clin Oncol. 2002;20:1480–90. doi: 10.1200/JCO.2002.20.6.1480. [DOI] [PubMed] [Google Scholar]
- 21.Norquist BM, Garcia RL, Allison KH, et al. The molecular pathogenesis of hereditary ovarian carcinoma: Alterations in the tubal epithelium of women with BRCA1 and BRCA2 mutations. Cancer. 2010;116:5261–71. doi: 10.1002/cncr.25439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jones S, Hruban RH, Kamiyama M, et al. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science. 2009;324:217. doi: 10.1126/science.1171202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Narod SA. Testing for CHEK2 in the cancer genetics clinic: ready for prime time? Clin Genet. 2010;78:1–7. doi: 10.1111/j.1399-0004.2010.01402.x. [DOI] [PubMed] [Google Scholar]
- 24.Erkko H, Dowty JG, Nikkilä J, et al. Penetrance analysis of the PALB2 c. 1592delT founder mutation. Clin Cancer Res. 2008;14:4667–71. doi: 10.1158/1078-0432.CCR-08-0210. [DOI] [PubMed] [Google Scholar]
- 25.Walsh T, Lee MK, Casadei S, et al. Detection of inherited mutations for breast and ovarian cancer using genomic capture and massively parallel sequencing. Proc Natl Acad Sci USA. 2010;13:12629–33. doi: 10.1073/pnas.1007983107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Easton DF, Deffenbaugh AM, Pruss D, et al. A systematic genetic assessment of 1433 sequence variants of unknown clinical significance in the BRCA1 and BRCA2 breast cancer-predisposition genes. Am J Hum Genet. 2007;81:873–83. doi: 10.1086/521032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Borg A, Haile RW, Malone KE, et al. Characterization of BRCA1 and BRCA2 deleterious mutations and variants of unknown clinical significance in unilateral and bilateral breast cancer: the WECARE study. Hum Mutat. 2010;31:E1200–40. doi: 10.1002/humu.21202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hogarth S, Javitt G, Melzer D. The current landscape for direct-to-consumer genetic testing: legal, ethical, and policy issues. Annu Rev Genomics Hum Genet. 2008;9:161–82. doi: 10.1146/annurev.genom.9.081307.164319. [DOI] [PubMed] [Google Scholar]
- 29.European Society of Human Genetics. Statement of the ESHG on direct-to-consumer genetic testing for health-related purposes. Eur J Hum Genet. 2010;18:1271–3. doi: 10.1038/ejhg.2010.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.ASHG comments to NIH in response to Genetic Testing Registry request for information. http://www.ashg.org/pages/statement_7_19_10.shtml
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1