ONCOGENE MUTATION PROFILING OF PEDIATRIC SOLID TUMORS REVEALS SIGNIFICANT SUBSETS OF EMBRYONAL RHABDOMYOSARCOMA AND NEUROBLASTOMA WITH MUTATED GENES IN GROWTH SIGNALING PATHWAYS (original) (raw)
. Author manuscript; available in PMC: 2013 Feb 1.
Abstract
Compared to the numerous broad screens for oncogene mutations in adult cancers, very few have been performed in pediatric solid tumors. To identify novel mutations and potential therapeutic targets in pediatric cancers, we performed a high-throughput Sequenom-based analysis in large sets of several major pediatric solid cancers, including neuroblastoma (NB), Ewing sarcoma (ES), rhabdomyosarcoma (RMS), and desmoplastic small round cell tumor (DSRCT).
Experimental Design
We designed a highly multiplexed Sequenom-based assay to interrogate 275 recurrent mutations across 29 genes. Genomic DNA was extracted from 192 NB, 75 ES, 89 RMS, and 24 DSRCT samples. All mutations were verified by Sanger sequencing.
Results
Mutations were identified in 13% of NB samples, 4% of ES samples, 21.1% of RMS samples, and no DSRCT samples. ALK mutations were present in 10.4% of NB samples. The remainder of NB mutations involved the BRAF, RAS, and MAP2K1 genes and were absent in samples harboring ALK mutations. Mutations were more common in embryonal RMS (ERMS) samples (28.3%) than alveolar RMS (ARMS) (3.5%). In addition to previously identified RAS and FGFR4 mutations, we report for the first time PIK3CA and CTNNB1 (Beta-Catenin) mutations in 4.9% and 3.3% of ERMS, respectively.
Conclusions
In ERMS, ES, and NB, we identified novel occurrences of several oncogene mutations recognized as drivers in other cancers. Overall, NB and ERMS contain significant subsets of cases with non-overlapping mutated genes in growth signaling pathways. Tumor profiling can identify a subset of pediatric solid tumor patients as candidates for kinase inhibitors or RAS-targeted therapies.
Keywords: mutation, rhabdomyosarcoma, neuroblastoma, Ewing sarcoma, desmoplastic small round cell tumor
Introduction
Many different tumor types harbor somatic gene mutations which contribute to tumor development and can also serve as promising therapeutic targets. Examples of therapeutically relevant mutations include KIT and PDGFRA mutations in gastrointestinal stromal tumors, EGFR mutations in lung adenocarcinomas, and BRAF mutations in melanoma.(1-4) More recently, activating ALK mutations have been identified in a subset of neuroblastomas, demonstrating the presence of therapeutically susceptible mutations in pediatric tumors.(5, 6)
Numerous large scale gene profiling studies have been performed to identify novel mutations in various tumor types.(7-9) However, broad mutational profiling studies have not been performed on a large cohort of NB and pediatric sarcoma samples. Furthermore, although ALK harbors recurrent mutations in neuroblastoma, ALK mutated tumors account for less than 15% of neuroblastoma cases (5), raising the possibility of alternative signaling gene mutations in additional subsets of this cancer.
The Sequenom MassARRAY technology utilizes a mass spectrometry-based genotyping approach which allows for more sensitive mutational analysis compared to traditional Sanger sequencing.(8) This platform also performs well using DNA extracted from archived paraffin embedded material. Furthermore, highly multiplexed PCR assays allow for efficient high-throughput screening of large tumor sample sets. (9)
In order to identify novel mutations in pediatric tumors, we utilized the Sequenom MassARRAY platform to perform a high-throughput sequencing analysis interrogating 275 point mutations across 29 oncogenes in large sample sets of four tumor types: NB, ES, RMS, and DSRCT. In addition to known recurrent somatically mutated genes, we included an extended panel of BRAF and PTPN11 mutation assays, based on the notion that known germline point mutations in these genes, defined in a constellation of conditions grouped together as RAS/MAPK syndromes (10, 11), might also occur as sporadic somatic mutations. Patients with these genetic disorders have an increased risk of pediatric neoplasia including rhabdomyosarcoma and neuroblastoma.(12, 13) The results of our mutation screen show that tumor mutation profiling can identify a subset of patients with NB and pediatric sarcomas who may be candidates for novel RAS, MEK, and PIK3CA inhibitors.
Methods
Tumor Samples
All frozen tumor samples and formalin-fixed paraffin embedded (FFPE) samples were procured at Memorial Sloan-Kettering Cancer Center (MSKCC) under IRB-approved protocols. Genomic DNA was extracted using the Qiagen DNeasy kit according to the manufacturer’s directions. DNA from 192 NB samples, 75 ES, 89 RMS, and 24 DSRCT samples and cell lines was collected. Cell lines used in the study are listed in Supplemental Table S1. Testing for EWSR1 and PAX/FKHR rearrangements was performed by RT-PCR or FISH, as previously described (14).
Curation of Oncogene Point Mutations
We queried the Sanger Institute COSMIC online database for recurrent non-synonymous point mutations known to occur in different human cancer types. Mutations were selected based on their susceptibility as a therapeutic target and mutation frequency. Furthermore, an extended list of BRAF and PTPN11 mutations was generated based on known germline mutations in RAS/MAPK syndromes (10, 11). Insertion and deletion type mutations were not included. A total of 275 point mutations (Supplemental Table S2) across 29 genes were selected for interrogation (Table 1). Input sequence files for assay design were generated by the MSKCC Bioinformatics Core Facility.
Table 1. Oncogenes included in the Sequenom panel.
The panel included 29 genes with known recurrent oncogenic mutations. 275 nucleotide changes were interrogated.
| AKT1 | FGFR3 | MET |
|---|---|---|
| AKT2 | FLT3 | NOTCH1 |
| AKT3 | GNAQ | NRAS |
| ALK | HRAS | PDGFRA |
| BRAF | IDH1 | PIK3CA |
| CDK4 | IDH2 | PIK3R1 |
| CTNNB1 | JAK2 | PTPN11 |
| EGFR | KIT | RET |
| ERBB2 | KRAS | SMO |
| FGFR2 | MAP2K1 |
Sequenom Based DNA Sequencing
For these assays we used the MassARRAY system (Sequenom), which is based on matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS). Genotyping by this method relies on the principle that mutant and wild-type alleles for a given point mutation produce single-allele base extension reaction products of a mass that is specific to the sequence of the product. Mutation calls are based on the mass differences between the wild-type product and the mutant products as resolved by MALDI-TOF MS. Amplification and extension primers were designed using Sequenom Assay Designer v3.1 software to target the curated list of mutations. Amplification primers were designed with a 10mer tag sequence to increase their mass so that they fall outside the range of detection of the MALDI-TOF MS. The sequences of primers are shown in Supplemental Table S3.
The initial PCR amplification was performed in a 5-μL reaction mixture containing 10 to 20 ng of DNA/1.25x buffer; 1.625 mmol/L MgCl2; 500 μmol/L deoxynucleotide triphosphate (dNTP); 100 nmol/L from each primer; and 0.5 U HotStar TaqDNA Polymerase (Qiagen) under the following conditions: 95°C (15 minutes); 95°C (20 seconds): 56°C (30 seconds); and 72°C (60 seconds) for 45 cycles, and a final extension phase at 72°C (3 minutes). Remaining unincorporated dNTPs were dephosphorylated by adding 2 μL of a shrimp alkaline phosphatase cocktail containing 1.53 μL of water, 0.17 μL of reaction buffer (Sequenom), and 0.3 μL of shrimp alkaline phosphatase (Sequenom). The unincorporated dNTPs were then placed in a thermal cycler under the following conditions: 37°C for 40 minutes, 85°C for 5 minutes, and then held at 4°C indefinitely. A single-base extension reaction was then performed in a 2-μL TypePLEX reaction mix (Sequenom) consisting of 0.72 μL water; 0.20 μL TypePLEX 10x buffer (Sequenom); 0.10 μL TypePLEX terminator mix (Sequenom); 0.94 μL extension primer mixture; and 0.04 μL TypePLEX enzyme (Sequenom). Thermal cycling was performed under the following conditions: 94°C for 30 seconds followed by 40 cycles of (94°C for 5 seconds, 5 cycles of 52°C for 5 seconds and 80°C for 5 seconds), then at 72°C for 3 minutes, and was finally held at 4°C indefinitely. The reaction mixture was then desalted by adding 16 μL of water and 6 mg of cationic resin mixture, SpectroCLEAN (Sequenom), and placed in a rotating shaker for 30 minutes. Completed genotyping reactions were spotted in nanoliter volumes onto a matrix-arrayed silicon SpectroCHIP with 384 elements using the MassARRAY Nanodispenser (Sequenom). SpectroCHIPs were analyzed using the Autoflex MALDI-TOF MS (Bruker AXS, Madison, WI), and the spectra were processed using SpectroACQUIRE software (Sequenom) in real time. Genotype calls are automatically generated using complex mathematical algorithms according to the peak heights, noise-to-peak-height ratio, area under the curve, and anchoring of the peaks. Results are then linked to plate information created in MassARRAY Typer 4.0 software (Sequenom). Individual calls are manually reviewed and finalized.
Whole Genome Amplification (WGA)
WGA was performed on 62 RMS samples and 74 EWS samples for sequencing of FGFR4 and CTNNB1. For frozen tissue, the Repli-G Midi (Qiagen, Valencia, CA) was used to amplify 100 ng of genomic DNA. WGA DNA was assessed by Picogreen quantification followed by PCR amplification with two control amplicons. For FFPE samples, The Sigma GenomePlex (Sigma Aldrich) was used to amplify 100 ng of genomic DNA. WGA DNA was assessed by Picogreen quantification followed by PCR amplification with two control amplicons.
PCR Amplification and DNA Sequencing of WGA Samples
All exons of FGFR4 and exon 3 of CTNNB1 were amplified as amplicons of 500 bp or less, covering the exonic regions plus at least 50 bp of intronic sequences (Supplemental Table S4). M13 tails were added to the primers to facilitate Sanger sequencing. RMS samples were evaluated for FGFR4 and CTNNB1 mutations. ES samples were evaluated for CTNNB1 mutations. PCR reactions were carried out in 384 well plates, in a Duncan DT-24 water bath thermal cycler, with 10 ng of whole genome amplified DNA as template, using a touchdown PCR protocol with PCR reactions were carried out in 384 well plates, in a Duncan DT-24 water bath thermal cycler, with 10 ng of whole genome amplified DNA (Repli-G Midi, Qiagen) as template, using a touchdown PCR protocol with HotStart Kapa Fast Taq (Kapa Biosystems). The touchdown PCR method consisted of : 1 cycle of 95°C for 5 min; 3 cycles of 95°C for 30 sec, 64°C for 15 sec, 72°C for 30 sec; 3 cycles of 95°C for 30 sec, 62°C for 15 sec, 72°C for 30 sec; 3 cycles of 95°C for 30 sec, 60°C for 15 sec, 72°C for 30 sec; 37 cycles of 95°C for 30 sec, 58°C for 15 sec, 72°C for 30 sec; 1 cycle of 70°C for 5 min. Templates were purified using AMPure (Agencourt Biosciences, Beverly, MA). The purified PCR reactions were split into two, and sequenced bidirectionally with M13 forward and reverse primer and Big Dye Terminator Kit v.3.1 (Applied Biosystems, Foster City, CA), at Agencourt Biosciences. Dye terminators were removed using the CleanSEQ kit (Agencourt Biosciences), and sequence reactions were run on ABI PRISM 3730xl sequencing apparatus (Applied Biosystems, Foster City, CA). Mutations were detected using an automated detection pipeline at the MSKCC Bioinformatics Core. Bi-directional reads and mapping tables (to link read names to sample identifiers, gene names, read direction, and amplicon) were subjected to a QC filter which excludes reads that have an average phred score of < 10 for bases 100-200. Passing reads were assembled against the reference sequences for each gene, containing all coding and UTR exons including 5Kb upstream and downstream of the gene, using command line Consed 16.0 (PMID: 9521923). All traces for mutation calls were manually reviewed and validated with original non-WGA DNA.
PCR Amplification and DNA Sequencing for Mutation Validation
All identified mutations were verified by standard Sanger sequencing. Forward and reverse primers are listed in Supplemental Table S5. Each PCR reaction was performed in a 50-μL volume mixture containing 100 ng of genomic DNA; forward and reverse primers (20 pmol each); 200 μM of each dNTP; 1.5 mmol/L MgCl2; 1X Qiagen PCR buffer containing 1.5 mmol/L MgCl2 and 2.5 units of HotStarTaq DNA polymerase (Qiagen). The PCR amplification was carried out under the following conditions: 1 cycle at 95°C (15 minutes); 40 cycles at 94°C (30 seconds), at 60°C (30 seconds), and at 72°C (60 seconds); and a final extension step at 72°C (10 minutes). Amplified products were purified using Spin Columns (Qiagen) and sequenced in both directions using the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems), according to the manufacturer’s protocol, on an ABI3730xl running ABI Prism DNA Sequence Analysis Software (Applied Biosystems).
Results
Mutations were identified by Sequenom analysis in 13% (25/192) of NB samples, 4% (3/75) of ES samples, 20.2% of RMS samples (18/89), and 0% of DSRCT samples (Table 2). Clinical information on the patients with mutated samples is provided in Supplemental Table S6. Mutations were found in 28.3% (17/60) of embryonal RMS (ERMS) tumors, with 13/60 (21.7%) identified by Sequenom analysis and the remainder identified by direct sequencing of FGFR4 (described below). Mutations were observed in 3.5% (1/29) of alveolar RMS (ARMS) samples. The majority of NB mutations were activating ALK mutations (Table 3), accounting for 10.4% of the tumor samples, a proportion consistent with previous reports (15). The remainder of NB mutations involved BRAF, RAS, and MAP2K1 genes and were not seen in samples harboring ALK mutations.
Table 2. Summary of identified mutations.
Mutations were identified in three of the four tumor types: A) Rhabdomyosarcoma. FGFR4 mutations were identified by direct sequencing. B) Neuroblastoma (ALK mutations are not included) C) Ewing Sarcoma. No mutations were identified in any Desmoplastic Small Round Cell tumors.
| A. Rhabdomyosarcoma (n=89) | ||
|---|---|---|
| Sample ID | Histology | Gene Mutation |
| R60 | ERMS | BRAF_1799T>A (V600E) |
| R11 | ERMS | CTNNB1_121A>G (T41A), NRAS_181C>A (Q61K) |
| R7 | ERMS | CTNNB1_134C>A (S45Y) |
| R13 | ERMS | FGFR4_1648G>C (V550L) |
| R31 (RMS559 cell line) | ERMS | FGFR4_1648G>C (V550L) |
| R36 | ERMS | FGFR4_1648G>C (V550L) |
| R6 | ERMS | FGFR4_1648G>C (V550L) |
| R51 (SMS-CTR cell line) | ERMS | HRAS_181C>A (Q61K) |
| R61 | ERMS | KRAS_34G>T (G12C) |
| R50 | ERMS | KRAS_35G>A (G12D) |
| R71 | ARMS | KRAS_38G>A (G13D) |
| R44 | ERMS | NRAS 181C>A (Q61K) |
| R29 | ERMS | NRAS_181C>A (Q61K) |
| R35 (RD cell line) | ERMS | NRAS_183A>T (Q61H) |
| R1 | ERMS | PIK3CA_1624G>A (E542K) |
| R43 | ERMS | PIK3CA_1633G>A (E545K) |
| R48 | ERMS | PIK3CA_3140A>G (H1047R) |
| R33 | ERMS | PTPN11_226G>C (E76Q) |
| B. Neuroblastomas (n=192) | |||
|---|---|---|---|
| Sample ID | MYCN Amplification | ALK Mutation | Gene Mutation |
| NB32 | no | no | BRAF_1783T>C (F595L) |
| NB158 | no | no | BRAF_1799T>A (V600E) |
| NB23 | yes | no | KRAS_35G>T (G12V) |
| NB139 | no | no | MAP2K1_171G>T (K57N) |
| NB151 | no | no | NRAS_181C>A (Q61K) |
| C. Ewing sarcomas (n=75) | ||
|---|---|---|
| Sample ID | Translocation | Gene Mutation |
| ES2 (A673 cell line) | EWS-FLI1 | BRAF_1799T>A (V600E) |
| ES9 | EWS-FLI1 | CTNNB1 133_135delTCT (S45del) |
| ES17 | EWS-ERG | NRAS_181C>A (Q61K) |
Table 3. ALK mutations detected by the Sequenom panel.
ALK mutations were identified in 20/192 (10.4%) NB samples. No additional mutations were identified in this cohort of tumors. ALK mutations were not identified in RMS, ES, or DSRCT samples.
| Neuroblastoma Sample | ALK Mutation |
|---|---|
| NB58 | F1174C |
| NB10 | F1174L |
| NB174 | F1174L |
| NB191 | F1174L |
| NB3 | F1174L |
| NB30 | F1174L |
| NB39 | F1174L |
| NB54 | F1174L |
| NB81 | F1174L |
| NB83 | F1174L |
| NB188 | F1174L |
| NB62 | F1245C |
| NB12 | F1245V |
| NB109 | R1275Q |
| NB129 | R1275Q |
| NB134 | R1275Q |
| NB135 | R1275Q |
| NB171 | R1275Q |
| NB2 | R1275Q |
| NB44 | R1275Q |
RAS family mutations were found in a subset of NB, RMS, and ES samples. 11.7% (7/60) of ERMS samples, 3.5% (1/29) of ARMS samples, 1% (2/192) of NB samples, and 1.3% (1/75) of ES samples harbored RAS mutations on our panel.
BRAF mutations were identified in 1% (2/192) of NB samples, 1.7% (1/60) of ERMS samples, and 1.3% (1/75) of ES samples. The ERMS and ES samples harbored the activating BRAFV600E mutation. One NB sample carried the BRAFV600E mutation, while the other carried the less common BRAFD594V mutation. A MAP2K1K57N mutation was identified in one NB sample.
PIK3CA mutations were identified in 5% (3/60) of ERMS cases, but were not seen in any other tumor type in our screen (Figure 1). Two of the 3 mutations were in the helical domain of the gene (E542K and E545K). The other mutation was located in the kinase domain (H1047R).
Figure 1. PIK3CA mutations in rhabdomyosarcoma.
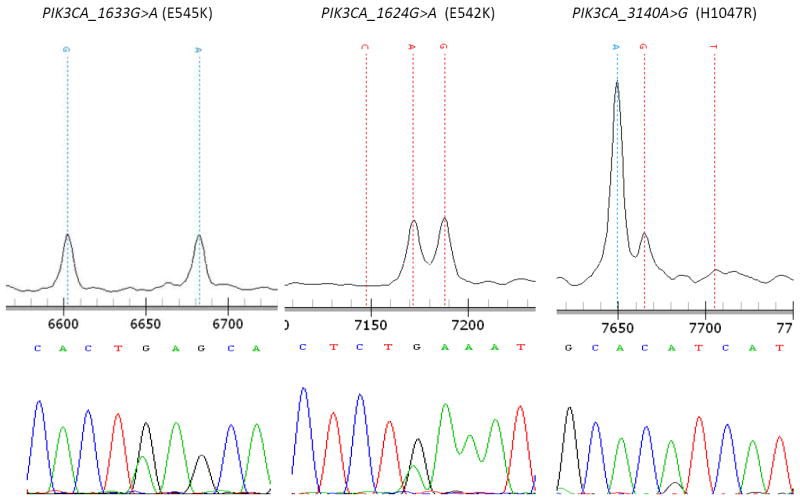
3 mutations were identified by Sequenom analysis and verified by direct sequencing. E542K and E545K mutations involve the helical domain of PIK3CA. The H1047R mutation involves the kinase domain of PIK3CA.
CTNNB1 mutations were identified in 1.3% (1/75) of ES samples, and 3.3% (2/60) of ERMS samples (Figure 2). One of the ERMS samples contained both an NRAS and a CTNNB1 gene mutation.
Figure 2. CTNNB1 mutations in rhabdomyosarcoma.
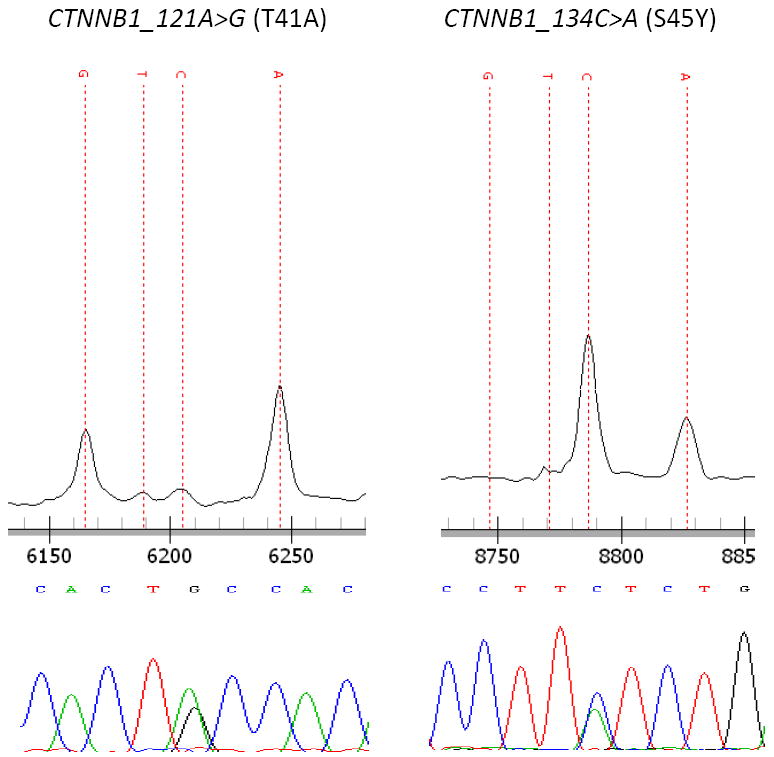
2 mutations were identified by Sequenom analysis and verified by direct sequencing.
A PTPN11 mutation was identified in 1.7% (1/60) ERMS samples. The tumor was from a patient diagnosed with Neurofibromatosis type 1. Sequencing of normal tissue from this patient confirmed the PTPN11 mutation to be somatic.
Full sequencing of the coding exons of FGFR4 and beta-catenin was performed in 43 ERMS and 19 ARMS samples. Similarly, full sequencing of beta-catenin was performed on 76 ES tumor samples. The V550L FGFR4 mutation was identified in 9.3% (4/43) ERMS tumors and none of the ARMS tumors. Sequencing of matched normal tissue, available for 2 of the 4 samples, confirmed the FGFR4 mutations to be somatic in both cases. Of the remaining 2 _FGFR4_-mutated samples, one lacked corresponding normal tissue and the other was a cell line (RMS 559). Additional CTNNB1 mutations, aside from those detected by the Sequenom assay, were not found in any of the RMS or ES samples.
Discussion
Multiple large scale cancer genomics efforts are underway to better characterize tumors and to help identify new therapeutic targets but few of these include pediatric solid tumors. Several genes have been examined for mutations in various pediatric tumor types (5, 16-27) but the present study represents the most expansive “hotspot” mutation profiling for these tumor types that has been reported to date. Here, we used a mass spectrometry-based platform to survey recurrent point mutations in 29 cancer genes in 380 cases of 4 major pediatric solid cancers.
The frequency of mutations found in our study was lower than in most adult onset carcinomas. Given the embryonal origin of these pediatric cancers, it is also possible that different genes are recurrently mutated in these tumor types. More comprehensive approaches such as whole exome sequencing would be needed to identify such mutations. However, a significant proportion of ERMS tumor samples were found to harbor various targetable mutations from our focused screen (Figure 3). RAS mutations are known to occur in a minority of ERMS patients.(17, 28) More recently, Paulson et al identified RAS mutations in 11/26 (42%) of ERMS samples. Furthermore, they identified NF1 gene deletions in 15% of ERMS samples, representing an alternative mechanism of RAS pathway activation.(29) In our study, 7 cases of ERMS harbored a RAS mutation. A codon 61 mutation of NRAS was found in 4 samples, including the RD cell line, as previously reported.(30) Two cases had KRAS codon 12 mutations. The ERMS cell line SMS-CTR was found to have an HRAS Q61K mutation. One of the 29 ARMS samples was found to have a RAS mutation. It was the sole mutation found in our cohort of ARMS samples. Our mutation data suggests that overall, RAS mutations are more typical of translocation-negative RMS but this difference may require a larger sample set to confirm statistically.
Figure 3. Oncogenic mutations identified in ERMS samples.
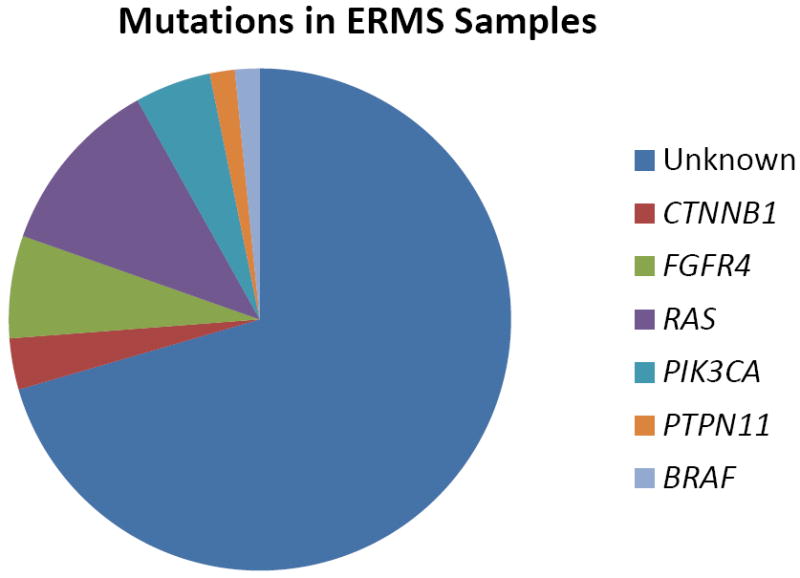
Mutations in one of 6 known cancer genes were identified in approximately 28% of all ERMS samples tested.
To our knowledge, RAS mutations have not been described in NB or ES. Whereas two studies have reported completely negative results(16, 19), another study described a rare codon 59 NRAS mutation in a NB cell line, which was not seen when the original tumor DNA was tested.(18) Our study shows that RAS mutations may play a role in rare cases of ES and NB.
BRAF point mutations have been studied in various pediatric solid tumors. For instance, mutations have been described in a significant proportion of low-grade pediatric astrocytomas.(31) A BRAFV600E mutation has been previously identified in the ES cell line A673 as well as in 2 RMS cell lines not used in our study.(1) However BRAF mutations have not been reported in NB cases.(21) Our study findings indicate that BRAF mutations are rare in these tumor types, but may play an oncogenic role in a small subset of sarcoma and NB patients. The BRAFV600E mutation was identified in a single ERMS tumor sample. Furthermore, 2/190 NB tumor samples harbored BRAF mutations, and another NB was found to have an activating mutation of MAP2K1 (MEK1), indicating a role of MAPK/ERK pathway activation in a subset of NB patients.
Unlike adult-onset tumors, mutations in the PIK3CA gene have not been commonly reported in pediatric tumor studies. One report of PIK3CA mutations in NB samples found 2 point mutations in 69 samples. However, neither of these substitutions were the frequent gain-of-function mutations (E542K, E545K, H1047R) often seen in other solid tumor types. Therefore, the effect on kinase activity of these mutations is unknown.(24) We did not identify any PIK3CA mutations in our larger set of NB cases.
Recently, 18% of myxoid/round-cell liposarcomas were found to have PIK3CA mutations localized to the helical and kinase domains, and these conferred a worse prognosis than wild-type PIK3CA within the same patient population.(32) PIK3CA mutations have not been previously reported in pediatric sarcomas. We identified PIK3CA mutations in 3/60 (5%) of ERMS samples, including two mutations in the helical domain and one in the kinase domain. Both mutations are known to be oncogenic.(33) Activation of AKT through the insulin-like growth factor pathway has been well established in RMS.(34-36) Our data suggest the possibility of an additional role of activating PIK3CA mutations in activating AKT in some ERMS patients. Furthermore, all 3 _PIK3CA_-mutant ERMS patients died of relapsed disease suggesting a more aggressive phenotype than is typical for ERMS. An expanded RMS sample set will be necessary to rigorously assess the impact of PIK3CA mutations on outcomes. In vitro evaluation of novel PIK3CA inhibitors in RMS cell lines along with efforts to establish RMS cell lines harboring PIK3CA mutations are currently in progress.
CTNNB1 (Beta-catenin) mutations have been identified in several embryonal pediatric tumor types. Activating CTNNB1 mutations occur in 10-15% of medulloblastoma cases and seem to define a distinct subgroup with improved overall outcomes.(27) CTNNB1 mutations have also been well characterized in hepatoblastomas and pediatric Wilms tumors.(20, 37) In a recent report, an evaluation of the CTNNB1 gene in 14 RMS samples did not identify any gene mutations.(38) In our screen, we identified a single mutated ES sample, and 2 RMS cases with mutations. Due to the heterogeneity of mutations in exon 3 of this gene, many of which were not included in our Sequenom panel, Sanger sequencing of exon 3 was performed in ES and RMS samples but this did not identify any additional mutations. However, these findings identify a small subset of RMS patients in which activation of the CTNNB1 pathway may play a role in tumor development. Intriguingly, both RMS patients with CTNNB1 mutations are alive and disease free for at least 6 years since diagnosis, including one patient diagnosed with stage 4 disease. This raises the possibility of improved outcomes in RMS patients with CTNNB1 mutations, similar to the patients with _CTNNB1_-mutated medulloblastoma.
There is an increased incidence of RMS and other childhood cancers in genetic syndromes with germline RAS/MAPK pathway mutations, including Cardio-facio-cutaneous Syndrome, Costello Syndrome, and Noonan Syndrome. Along with RAS genes, PTPN11 and BRAF are two of the most frequently mutated genes in this group of syndromes, with the germline mutations overlapping only partly with recurrent somatic mutations in these genes.(10, 11, 13) Therefore, we interrogated an extensive range of possible mutations in these genes to assess the possibility that mutations previously reported as germline may occasionally be acquired in sporadic RMS cases. However, we identified only one PTPN11 and one BRAF mutation in our survey of RMS samples.
FGFR4 mutations were recently identified in 7.5% of primary RMS tumors with mutations involving amino acids 535 and 550 leading to increased receptor phosphorylation and tumor progression. Mutations involving these amino acids were also most susceptible to FGFR4 inhibition.(39) We identified FGFR4 mutations in 4 out of 63 tumors tested (6.3%). All 4 samples were ERMS, including the cell line RMS559. Interestingly, all 4 mutations led to a V550L amino acid change.
Our panel included several recently described oncogenes which have not been extensively evaluated in pediatric solid tumors, including the IDH and GNAQ genes. IDH1 and IDH2 mutations were initially identified as frequently mutated in adult onset gliomas(40), and soon after were identified in a significant percentage of cytogenetically normal AML samples.(41) Frequent mutations in the GNAQ gene have been recently identified in melanocytic tumors leading to activation of the MAPK pathway.(42) A large analysis of glial, epithelial, and stromal type adult onset tumors did not identify any mutations in non-melanocytic tumors.(43) Our assay performed a mutational survey of the primary hotspots of these genes; namely R132 and R172 of IDH1 and IDH2, and Q209 of GNAQ. We did not identify any mutations in either gene in our samples. Interestingly, IDH mutation studies of pediatric gliomas reveal that they occur in children 14 years of age or older, and not in younger children.(44) Similarly, IDH mutations are not characteristic of pediatric AML(45), indicating that these mutations may be much more likely to exist in adult-onset tumor types. Recently, a large mutational survey of the IDH gene in chondrosarcomas and chondromas revealed mutations in over 50% of cases, with the most frequent mutation involving the R132 codon.(46) Further mutational studies in other sarcoma types are warranted.
This analysis identified several novel occurrences of known oncogene mutations in pediatric tumors including PIK3CA and CTNNB1 mutations in RMS, CTNNB1 mutations in ES, and RAS, BRAF, and MAP2K1 mutations in NB. No mutations were identified in a relatively large sample size of DSRCT. The paucity of mutations identified in ES samples, and the absence of mutations identified in DSRCT samples may be a function of the characteristic aberrant transcription factors generated by the specific translocations found in the two tumor types. The broad transcriptional dysregulation caused by these chimeric proteins may affect multiple oncogenic pathways, reducing the selective pressure for additional oncogenic mutations for tumor formation and progression. However, whole genome mutational studies on ES and DSRCT need to be performed to confirm this hypothesis. A significant proportion of ERMS harbor oncogenic mutations. Including FGFR4 mutations, 28.3% (17/60) of ERMS samples had an identifiable oncogenic mutation (figure 3).
Several mutations identified are of present interest in terms of targeted agents in clinical development. MEK inhibition has been recognized as an effective modality against melanomas harboring BRAF mutations.(47) Various MEK inhibitors are currently in phase III trials for patients with _BRAF_-mutant melanoma. Similarly, many different PIK3CA inhibitors are under development with numerous early phase studies open for adult-onset tumor types with corresponding mutations.(48) ALK inhibition has been recognized as effective across various tumor cell lines with ALK mutations or rearrangements.(49) The ALK inhibitor crizotinib is currently in phase I/II studies for refractory pediatric solid tumor patients, and phase III studies for lung cancer patients harboring ALK rearrangements. Recent studies have also demonstrated the activity of MEK inhibitors against a subset of _RAS_-mutant tumors.(50) As more agents inhibiting mutated oncoproteins and their associated downstream signaling pathways become available, mutational genotyping of pediatric solid tumors such as RMS will serve as an important tool for identifying targeted therapy options for individual patients.
Supplementary Material
1
Statement of translational relevance.
To identify novel mutations and potential therapeutic targets in pediatric cancers, we performed a high-throughput screen to interrogate 275 recurrent mutation sites across 29 cancer genes in 192 neuroblastomas (NB), 75 Ewing sarcomas (ES), 89 rhabdomyosarcomas (RMS), and 24 desmoplastic small round cell tumors (DSRCT). In embryonal RMS (ERMS), ES, and NB, we identified novel occurrences of several oncogene mutations recognized as drivers in other cancers, including for the first time PIK3CA and CTNNB1 (beta-catenin) mutations in ERMS. Overall, NB and ERMS contain significant subsets of cases with non-overlapping mutated genes in growth signaling pathways. In particular, a substantial minority of ERMS show mutations in the RAS pathway. As more agents inhibiting mutated oncoproteins and their associated downstream signaling pathways become available, mutational genotyping of pediatric solid tumors such as RMS will serve as an important tool for identifying targeted therapy options for individual patients.
Acknowledgments
We thank Dr. Adriana Heguy and the personnel of the Beene Translational Oncology Core Facility (MSKCC), Alex Lash in the MSKCC Bioinformatics Core Facility, Drs. Leonard Wexler and Christine Pratilas from the MSKCC Department of Pediatrics for longtime support and helpful discussions, respectively, and Dr. Jonathan Fletcher from the Dana-Farber Cancer Institute, Dr. Jun Nishio from the Fukuoka University, Dr. Hajime Hosoi from the Kyoto Prefectural University of Medicine, and Dr. Michio Kaneko from the University of Tsukuba for providing us cell lines.
Ismail Yilmaz is currently at Gulhane Military Medical Academy in Istanbul, Turkey. Frederic Barr and Chyau-Yueh Lau are currently at the National Cancer Institute in Bethesda, Maryland.
Supported in part by NIH P01 grant CA106450 (ML), the Ewing Sarcoma Research Fund (ML), The Joanna Mcafee Childhood Cancer Foundation (FB), The Alveolar Rhabdomyosarcoma Research Fund (FB), and by a generous donation from M.B. Zuckerman (ML). The MSKCC Sequenom facility was supported by the Anbinder Fund.
References
- 1.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 2.Heinrich MC, Corless CL, Demetri GD, Blanke CD, von Mehren M, Joensuu H, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21:4342–9. doi: 10.1200/JCO.2003.04.190. [DOI] [PubMed] [Google Scholar]
- 3.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–39. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 4.Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101:13306–11. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.George RE, Sanda T, Hanna M, Frohling S, Luther W, 2nd, Zhang J, et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature. 2008;455:975–8. doi: 10.1038/nature07397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Janoueix-Lerosey I, Lequin D, Brugieres L, Ribeiro A, de Pontual L, Combaret V, et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature. 2008;455:967–70. doi: 10.1038/nature07398. [DOI] [PubMed] [Google Scholar]
- 7.Ikediobi ON, Davies H, Bignell G, Edkins S, Stevens C, O’Meara S, et al. Mutation analysis of 24 known cancer genes in the NCI-60 cell line set. Mol Cancer Ther. 2006;5:2606–12. doi: 10.1158/1535-7163.MCT-06-0433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thomas RK, Baker AC, Debiasi RM, Winckler W, Laframboise T, Lin WM, et al. High-throughput oncogene mutation profiling in human cancer. Nat Genet. 2007;39:347–51. doi: 10.1038/ng1975. [DOI] [PubMed] [Google Scholar]
- 9.MacConaill LE, Campbell CD, Kehoe SM, Bass AJ, Hatton C, Niu L, et al. Profiling critical cancer gene mutations in clinical tumor samples. PLoS One. 2009;4:e7887. doi: 10.1371/journal.pone.0007887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nava C, Hanna N, Michot C, Pereira S, Pouvreau N, Niihori T, et al. Cardio-facio-cutaneous and Noonan syndromes due to mutations in the RAS/MAPK signalling pathway: genotype-phenotype relationships and overlap with Costello syndrome. J Med Genet. 2007;44:763–71. doi: 10.1136/jmg.2007.050450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rodriguez-Viciana P, Tetsu O, Tidyman WE, Estep AL, Conger BA, Cruz MS, et al. Germline mutations in genes within the MAPK pathway cause cardio-facio-cutaneous syndrome. Science. 2006;311:1287–90. doi: 10.1126/science.1124642. [DOI] [PubMed] [Google Scholar]
- 12.Moschovi M, Touliatou V, Papadopoulou A, Mayakou MA, Nikolaidou-Karpathiou P, Kitsiou-Tzeli S. Rhabdomyosarcoma in a patient with Noonan syndrome phenotype and review of the literature. J Pediatr Hematol Oncol. 2007;29:341–4. doi: 10.1097/MPH.0b013e31805d8f57. [DOI] [PubMed] [Google Scholar]
- 13.Schubbert S, Shannon K, Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer. 2007;7:295–308. doi: 10.1038/nrc2109. [DOI] [PubMed] [Google Scholar]
- 14.Huang HY, Illei PB, Zhao Z, Mazumdar M, Huvos AG, Healey JH, et al. Ewing sarcomas with p53 mutation or p16/p14ARF homozygous deletion: a highly lethal subset associated with poor chemoresponse. J Clin Oncol. 2005;23:548–58. doi: 10.1200/JCO.2005.02.081. [DOI] [PubMed] [Google Scholar]
- 15.Mosse YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF, et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature. 2008;455:930–5. doi: 10.1038/nature07261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ballas K, Lyons J, Janssen JW, Bartram CR. Incidence of ras gene mutations in neuroblastoma. Eur J Pediatr. 1988;147:313–4. doi: 10.1007/BF00442704. [DOI] [PubMed] [Google Scholar]
- 17.Stratton MR, Fisher C, Gusterson BA, Cooper CS. Detection of point mutations in N-ras and K-ras genes of human embryonal rhabdomyosarcomas using oligonucleotide probes and the polymerase chain reaction. Cancer Res. 1989;49:6324–7. [PubMed] [Google Scholar]
- 18.Moley JF, Brother MB, Wells SA, Spengler BA, Biedler JL, Brodeur GM. Low frequency of ras gene mutations in neuroblastomas, pheochromocytomas, and medullary thyroid cancers. Cancer Res. 1991;51:1596–9. [PubMed] [Google Scholar]
- 19.Radig K, Schneider-Stock R, Rose I, Mittler U, Oda Y, Roessner A. p53 and ras mutations in Ewing’s sarcoma. Pathol Res Pract. 1998;194:157–62. doi: 10.1016/S0344-0338(98)80016-2. [DOI] [PubMed] [Google Scholar]
- 20.Blaker H, Hofmann WJ, Rieker RJ, Penzel R, Graf M, Otto HF. Beta-catenin accumulation and mutation of the CTNNB1 gene in hepatoblastoma. Genes Chromosomes Cancer. 1999;25:399–402. [PubMed] [Google Scholar]
- 21.Miao J, Kusafuka T, Fukuzawa M. Hotspot mutations of BRAF gene are not associated with pediatric solid neoplasms. Oncol Rep. 2004;12:1269–72. [PubMed] [Google Scholar]
- 22.Gustafsson B, Angelini S, Sander B, Christensson B, Hemminki K, Kumar R. Mutations in the BRAF and N-ras genes in childhood acute lymphoblastic leukaemia. Leukemia. 2005;19:310–2. doi: 10.1038/sj.leu.2403589. [DOI] [PubMed] [Google Scholar]
- 23.Chen Y, Takita J, Hiwatari M, Igarashi T, Hanada R, Kikuchi A, et al. Mutations of the PTPN11 and RAS genes in rhabdomyosarcoma and pediatric hematological malignancies. Genes Chromosomes Cancer. 2006;45:583–91. doi: 10.1002/gcc.20322. [DOI] [PubMed] [Google Scholar]
- 24.Dam V, Morgan BT, Mazanek P, Hogarty MD. Mutations in PIK3CA are infrequent in neuroblastoma. BMC Cancer. 2006;6:177. doi: 10.1186/1471-2407-6-177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thompson MC, Fuller C, Hogg TL, Dalton J, Finkelstein D, Lau CC, et al. Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. J Clin Oncol. 2006;24:1924–31. doi: 10.1200/JCO.2005.04.4974. [DOI] [PubMed] [Google Scholar]
- 26.Curia MC, Zuckermann M, De Lellis L, Catalano T, Lattanzio R, Aceto G, et al. Sporadic childhood hepatoblastomas show activation of beta-catenin, mismatch repair defects and p53 mutations. Mod Pathol. 2008;21:7–14. doi: 10.1038/modpathol.3800977. [DOI] [PubMed] [Google Scholar]
- 27.Fattet S, Haberler C, Legoix P, Varlet P, Lellouch-Tubiana A, Lair S, et al. Beta-catenin status in paediatric medulloblastomas: correlation of immunohistochemical expression with mutational status, genetic profiles, and clinical characteristics. J Pathol. 2009;218:86–94. doi: 10.1002/path.2514. [DOI] [PubMed] [Google Scholar]
- 28.Martinelli S, McDowell HP, Vigne SD, Kokai G, Uccini S, Tartaglia M, et al. RAS signaling dysregulation in human embryonal Rhabdomyosarcoma. Genes Chromosomes Cancer. 2009;48:975–82. doi: 10.1002/gcc.20702. [DOI] [PubMed] [Google Scholar]
- 29.Paulson V, Chandler G, Rakheja D, Galindo RL, Wilson K, Amatruda JF, et al. High-resolution array CGH identifies common mechanisms that drive embryonal rhabdomyosarcoma pathogenesis. Genes Chromosomes Cancer. 2011;50:397–408. doi: 10.1002/gcc.20864. [DOI] [PubMed] [Google Scholar]
- 30.Chardin P, Yeramian P, Madaule P, Tavitian A. N-ras gene activation in the RD human rhabdomyosarcoma cell line. Int J Cancer. 1985;35:647–52. doi: 10.1002/ijc.2910350513. [DOI] [PubMed] [Google Scholar]
- 31.Schiffman JD, Hodgson JG, VandenBerg SR, Flaherty P, Polley MY, Yu M, et al. Oncogenic BRAF mutation with CDKN2A inactivation is characteristic of a subset of pediatric malignant astrocytomas. Cancer Res. 2010;70:512–9. doi: 10.1158/0008-5472.CAN-09-1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barretina J, Taylor BS, Banerji S, Ramos AH, Lagos-Quintana M, Decarolis PL, et al. Subtype-specific genomic alterations define new targets for soft-tissue sarcoma therapy. Nat Genet. 2010;42:715–21. doi: 10.1038/ng.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ligresti G, Militello L, Steelman LS, Cavallaro A, Basile F, Nicoletti F, et al. PIK3CA mutations in human solid tumors: role in sensitivity to various therapeutic approaches. Cell Cycle. 2009;8:1352–8. doi: 10.4161/cc.8.9.8255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cen L, Hsieh FC, Lin HJ, Chen CS, Qualman SJ, Lin J. PDK-1/AKT pathway as a novel therapeutic target in rhabdomyosarcoma cells using OSU-03012 compound. Br J Cancer. 2007;97:785–91. doi: 10.1038/sj.bjc.6603952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cao L, Yu Y, Darko I, Currier D, Mayeenuddin LH, Wan X, et al. Addiction to elevated insulin-like growth factor I receptor and initial modulation of the AKT pathway define the responsiveness of rhabdomyosarcoma to the targeting antibody. Cancer Res. 2008;68:8039–48. doi: 10.1158/0008-5472.CAN-08-1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wan X, Harkavy B, Shen N, Grohar P, Helman LJ. Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene. 2007;26:1932–40. doi: 10.1038/sj.onc.1209990. [DOI] [PubMed] [Google Scholar]
- 37.Su MC, Huang WC, Lien HC. Beta-catenin expression and mutation in adult and pediatric Wilms’ tumors. APMIS. 2008;116:771–8. doi: 10.1111/j.1600-0463.2008.00914.x. [DOI] [PubMed] [Google Scholar]
- 38.Bouron-Dal Soglio D, Rougemont AL, Absi R, Giroux LM, Sanchez R, Barrette S, et al. Beta-catenin mutation does not seem to have an effect on the tumorigenesis of pediatric rhabdomyosarcomas. Pediatr Dev Pathol. 2009;12:371–3. doi: 10.2350/08-11-0553.1. [DOI] [PubMed] [Google Scholar]
- 39.Taylor JGt, Cheuk AT, Tsang PS, Chung JY, Song YK, Desai K, et al. Identification of FGFR4-activating mutations in human rhabdomyosarcomas that promote metastasis in xenotransplanted models. J Clin Invest. 2009;119:3395–407. doi: 10.1172/JCI39703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–12. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361:1058–66. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Van Raamsdonk CD, Bezrookove V, Green G, Bauer J, Gaugler L, O’Brien JM, et al. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature. 2009;457:599–602. doi: 10.1038/nature07586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lamba S, Felicioni L, Buttitta F, Bleeker FE, Malatesta S, Corbo V, et al. Mutational profile of GNAQQ209 in human tumors. PLoS One. 2009;4:e6833. doi: 10.1371/journal.pone.0006833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pollack IF, Hamilton RL, Sobol RW, Nikiforova MN, Lyons-Weiler MA, Laframboise WA, et al. IDH1 mutations are common in malignant gliomas arising in adolescents: a report from the Children’s Oncology Group. Childs Nerv Syst. 2011;27:87–94. doi: 10.1007/s00381-010-1264-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ho PA, Alonzo TA, Kopecky KJ, Miller KL, Kuhn J, Zeng R, et al. Molecular alterations of the IDH1 gene in AML: a Children’s Oncology Group and Southwest Oncology Group study. Leukemia. 2010;24:909–13. doi: 10.1038/leu.2010.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Amary MF, Bacsi K, Maggiani F, Damato S, Halai D, Berisha F, et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J Pathol. 2011 doi: 10.1002/path.2913. [DOI] [PubMed] [Google Scholar]
- 47.Solit DB, Garraway LA, Pratilas CA, Sawai A, Getz G, Basso A, et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature. 2006;439:358–62. doi: 10.1038/nature04304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ogita S, Lorusso P. Targeting phosphatidylinositol 3 kinase (PI3K)-Akt beyond rapalogs. Target Oncol. 2011 doi: 10.1007/s11523-011-0176-7. [DOI] [PubMed] [Google Scholar]
- 49.McDermott U, Iafrate AJ, Gray NS, Shioda T, Classon M, Maheswaran S, et al. Genomic alterations of anaplastic lymphoma kinase may sensitize tumors to anaplastic lymphoma kinase inhibitors. Cancer Res. 2008;68:3389–95. doi: 10.1158/0008-5472.CAN-07-6186. [DOI] [PubMed] [Google Scholar]
- 50.Gilmartin AG, Bleam MR, Groy A, Moss KG, Minthorn EA, Kulkarni SG, et al. GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin Cancer Res. 2011;17:989–1000. doi: 10.1158/1078-0432.CCR-10-2200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1