Natural resolution of inflammation (original) (raw)
. Author manuscript; available in PMC: 2014 May 15.
Published in final edited form as: Periodontol 2000. 2013 Oct;63(1):149–164. doi: 10.1111/prd.12034
Inflammation is an essential mechanism in human health and disease. The first description of inflammation by the Roman, Cornelius Celsus, in the 1st century defined the clinical symptoms of inflammatory diseases. Four cardinal signs of inflammation were identified: rubor et tumor cum calore et dolore (redness and swelling with heat and pain). Disease development was defined as an imbalance of the four cardinal signs (60, 61). In 1858, Rudolph Virchow's investigations of the cellular basis of pathology led to the inclusion of an additional cardinal sign, fucntio laesa (loss of function). Subsequent studies included the germ theory of disease in the late 19th century, proposed by Robert Koch and Louis Pasteur, which identified microorganisms as major inducers of the acute inflammatory response. Metchnikoff found that when neutrophils are ingested by tissue macrophages acute inflammation resolves. More recently, advanced cellular and molecular mechanisms governing the fate of inflammation have been identified. Acute inflammation is accepted to be a physiological response that occurs in vascularized tissues to defend the host and to maintain homeostasis.
Inflammation is initiated as a protective response to challenges with pathogens or foreign bodies, or injury, experienced by host tissues. This process is characterized by vascular dilation, enhanced permeability of capillaries, increased blood flow and leukocyte recruitment. Polymorphonuclear neutrophils are among the first leukocyte responders to accumulate in the inflamed site. These cells are crucial as the first line of defense of the innate immune system because of their phagocytotic and microbicidal functions. Next, mononuclear cells, monocytes and macrophages enter the inflammatory site and clear cellular debris and apoptotic polymorphonuclear neutrophils by phagocytosis without prolonging inflammation; a nonphlogistic (nonheat or fever producing) process (79).
Although the inflammatory response is protective, failure to remove noxious materials produced by neutrophils via phagocytosis, failure to clear apoptotic inflammatory cells and a delay of apoptosis characterize the chronic and pathological lesion. The incomplete elimination of leukocytes from a lesion is observed in susceptible individuals; acute inflammation fails to resolve and chronic disease and fibrosis develop (101). Accordingly, loss of resolution and failure to return tissue to homeostasis results in neutrophil-mediated destruction and chronic inflammation (103), which is a major cause of human inflammatory pathologies, including arthritis, asthma, cancers, cardiovascular diseases and periodontal diseases. Simply stated, since the time of Celsus, we have thought of inflammation in terms of induction. In recent years, we have identified the molecular mediators of inflammation induction (cytokines and chemokines) as our understanding of the process at the molecular level has evolved. We never paid any attention to attenuation; how is inflammation turned off? We always assumed that it was a passive process caused by the decay or cessation of inducers.
In order to maintain a healthy status, both the initiation of acute inflammation and its resolution must be efficient. It was widely accepted that the loss of pro-inflammatory mediators was the ‘turn off’ signal of inflammation, ending subsequent responses passively. Perhaps the most important discovery of the late 20th century regarding inflammation was the finding of Charles N. Serhan, which uncovered the molecules that mediate the resolution of inflammation (13, 54, 83). Resolution of inflammation, and the return to homeostasis, is not simply passive, but an active and highly regulated biochemical process that is now considered ‘programmed resolution’ at the tissue level. In this context, specialized immunoresolvents comprise a genus of endogenous molecules, including resolvins, lipoxins, protectins and maresins, which actively drive the termination of inflammation. These mediators trigger pathways that signal the physiologic end of the acute inflammatory phase (54, 91). Systematic studies have revealed the role of pro-resolution lipid mediators in the resolution of inflammation, which are the subject of this review.
Inflammatory response
The primary goal of the inflammatory response is to detect and eliminate factors that interfere with homeostasis. A typical inflammatory response consists of four components: inflammatory inducers; the detecting sensors; downstream mediators; and the target tissues that are affected. The type and the degree of inflammatory response activated is dependent on the nature of the inflammatory trigger (bacterial, viral or parasitic) and its persistence (64). Bacterial pathogens are recognized by pattern recognition receptors (1, 24), such as Toll-like receptors, which are expressed by tissue-resident macrophages. Binding of Toll-like receptors induces the production of inflammatory cytokines, chemokines and pro-inflammatory lipid mediators such as prostaglandins. These inflammatory mediators are the major players in establishing an effective inflammatory response and in clearance of the bacteria. Pro-inflammatory mediators, such as interleukin-1 beta, interleukin-6, tumor necrosis factor alpha and prostaglandin E2 are produced locally in the inflamed gingival tissues. Viral infections induce the production of interferon alpha and interferon beta and the activation of cytotoxic lymphocytes, whereas parasites lead to the production of interleukin-4, interleukin-5 and interleukin-13 by mast cells and basophils. Pro-inflammatory cyto-kines in the circulation induce leukocytosis and acute-phase proteins. With continued exposure, soluble antigens react with circulating specific antibody to form immune complexes that further amplify inflammation at sites of deposition.
Sensing of the inflammatory response by innate immune cells and resident cells triggers the production of mediators that modulate the fate of inflammation. Neutrophils, macrophages, dendritic cells and mast cells produce low-molecular-weight proteins – cytokines – that control the initiation of inflammation and its maintenance and regulate its amplitude and the duration of the response. The genetic regulation leading to the secretion of pro-inflammatory cytokines from a variety of cells is generally dependent on transcriptional activation by the nuclear protein, nuclear factor-kappaB (11, 37). The nuclear factor-kappaB-regulated pathways are activated by pattern recognition receptors, such as lipopolysaccharide, through a Toll-like receptor pathway (37). Chemokines are cytokines with chemoattractant functions that induce migration of cells to the site of infection. Once blood leukocytes exit a blood vessel, they are attracted by functional gradients of chemotactic factors to the site of infection (76, 107). Chemokines are synthesized by a variety of cells, including endothelial, epithelial and stromal cells, as well as by leukocytes.
In a classical acute inflammatory response, cellular events are temporally activated. Upon initial challenge, protein exudation increases and polymorphonuclear leukocytes (neutrophils) accumulate in inflamed tissue. Neutrophil infiltration follows a rapid response from sentinel cells prestationed in the tissues at the time of injury, including macrophages and mast cells (61). Polymorphonuclear neutrophil extravasation into injured or infected tissues requires adhesion to endothelial cells in the capillaries, a process mediated by surface molecules (selectins and integrins) on polymorphonuclear neutrophils and by intracellular adhesion molecules and vascular cell adhesion molecules on endothelial cells (23, 24). Selectins cause rolling of polymorphonuclear neutrophils along the capillary wall and integrins mediate firm attachment, allowing transcellular diapedesis of neutrophils to the infected tissue (23). As primary defenders, neutrophils transmigrate into tissues in large numbers to neutralize pathogens and promote the clearance of cellular and other debris by phagocytosis. As the lesion matures, neutrophils accumulate in the local tissue and die via apoptosis (programmed cell death). The initial accumulation of neutrophils is followed by a second wave of cellular infiltration, of mononuclear phagocytes (monocytes). Differentiation of monocytes into macrophages promotes the removal of apoptotic neutrophils and debris by nonphlogistic phagocytosis. Macrophages that have completed the elimination of apoptotic neutrophils are cleared from the inflamed tissue either by egression to the lymphatic system or by apoptosis, by a process termed efferocytosis. This temporal regulation of inflammation requires that this conserved cellular phenotype is highly regulated to clear the original insult (79). Failure to resolve the inflammatory response, or continuous activation of the responses, become harmful to the tissue and consequently develop into the chronic lesion that we call inflammatory diseases.
For many years, our understanding of inflammation was limited to an understanding of pro-inflammatory mediators. Currently, we realize that there are pro-inflammatory signals which are temporally followed by specific signals that drive resolution of inflammation. Many of these molecules are lipids. We now realize that, in addition to pro-inflammatory mediators that turn on inflammation, there is a separate set of lipid mediators that act as endogenous agonists to activate termination of inflammation by stimulating resolution. The following sections provide a more detailed look at both pro-inflammatory lipid mediators and resolution of inflammation lipid mediators in the initiation and resolution of acute inflammation.
Pro-inflammatory lipid mediators of acute inflammation
A large family of bioactive lipid mediators, the eicosanoids (20-lipid mediators), are involved in pathophysiologic processes, including those associated with host defense and inflammation (31, 36, 77). Arachidonic acid is a common endogenous precursor for the biosynthesis of eicosanoids and is derived from the _sn_-2 position of cell-membrane phospholipids through the action of the enzyme phospholipase A2, which releases arachidonic acid from the phospholipid membrane. The release of arachidonic acid constitutes the rate-determining step in the generation of eicosanoids produced by most phagocytic and immune cells. Arachidonic acid is rapidly converted to various potent lipid mediators in a cell-specific manner by cyclooxygenases, lipoxygenases or epoxygenases to yield prostaglandins, leukotrienes and endoperoxides, respectively.
Locally produced lipid mediators, such as prostaglandins, prostacyclin and thromboxanes, act as autacoids (short-lived molecules that signal immediately adjacent to, or at the site of, synthesis). These lipid mediators are synthesized from membrane-released arachidonic acid when cells are activated by stimuli, such as trauma and cytokines. Arachidonic acid is metabolized by two major enzyme pathways: cyclooxygenases and lipoxygenases. Cyclooxygenase-1 (constitutively expressed cyclooxygenase) is responsible for basal levels of prostaglandin synthesis, whereas cyclooxygenase-2 (inducible cyclooxygenase) catalyzes the conversion of arachidonic acid to lipid mediators during inflammation (96). Prostaglandins have 10 subclasses, of which D, E, F, G, H and I are the most important in inflammation (33, 49). Specifically, prostaglandin E2 is generated via prostaglandin E synthase in leukocytes, whereas prostaglandin I2 is generated via prostacyclin synthase in endothelial cells and thromboxanes are generated via thromboxane synthase in platelets (31).
Lipoxygenases catalyze the formation of hydroxyeicosatetraenoic acids from arachidonic acid, leading to the formation of leukotrienes and other biologically active compounds (77). Leukotrienes are predominantly produced by inflammatory cells, including polymorphonuclear leukocytes, macrophages and mast cells. There are three distinct lipoxygenases that are cell specific: 5-lipoxygenase in myeloid cells; 12-lipoxygenase in platelets; and 15-lipoxygenase in epithelial/endothelial cells. Cellular activation by pathogens and immune complexes results in the activation of a sequential enzymatic reaction which includes cytosolic phospholipase A2 and 5-lipoxygenase. 5-Lipoxygenase converts released arachidonic acid to the epoxide leukotriene A4, which undergoes transformation by distinct pathways, one of which generates leukotriene B4, a potent regulator of neutrophil chemotaxis and leukocyte adhesion to endothelial cells (31). The end products of 12-lipoxygenase and 15-lipoxygenase are 12-hydroxyeicosatetraenoic acid and 15-hydroxyeicosatetraenoic acid, respectively, which are further metabolized (Fig. 1).
Fig. 1.
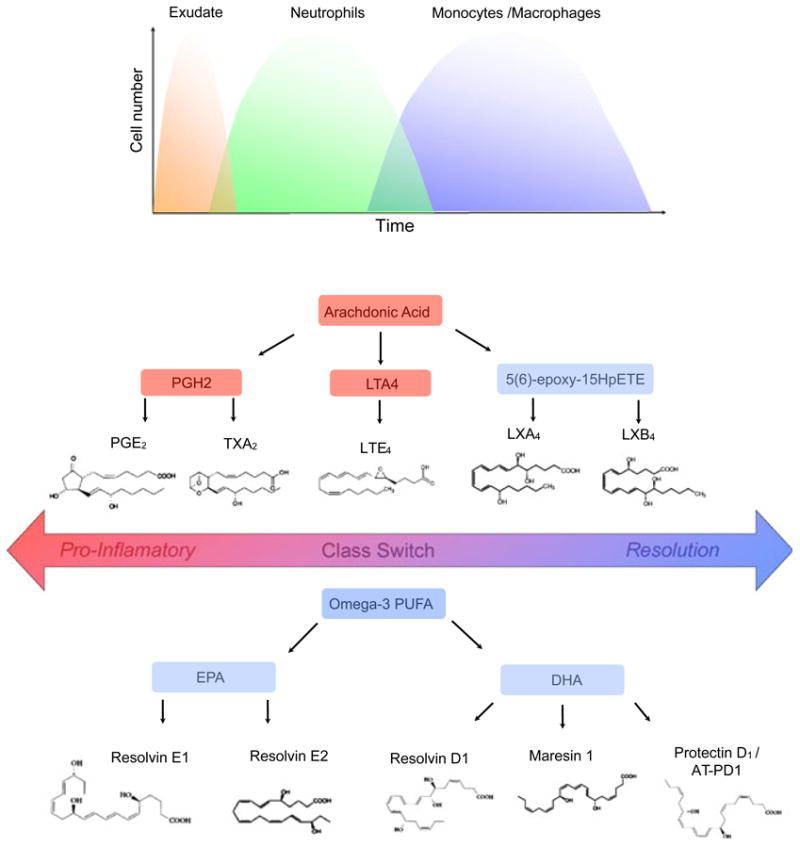
Acute inflammation and lipid mediators. The local acute inflammatory response is characterized by exudate formation and the initial recruitment of neutrophils followed by the recruitment of monocytes that differentiate into macrophages. Lipid mediators such as prostaglandins and leukotrienes, and cytokines and chemokines, regulate the well-coordinated initial events of acute inflammation. Arachidonic acid released from membrane phospholipids by phospholipase A2 can be further metabolized by (i) cyclooxygenase-1 and cyclooxygenase-2 to generate prosta-noids, including prostaglandins and thromboxanes; (ii) lipoxygenases to generate leukotrienes and hydroxy acids; and (iii) lipoxygenase interaction products (biosynthetic pathways to generate lipoxins). Lipoxins are generated during transcellular biosynthesis, which requires two cell types involving distinct lipoxygenases. Lipid mediator class-switches yield pro-resolution lipid mediators such as lipoxin A4 and the eicosapentaenoic acid-derived resolvins (i.e. resolvin E1 and resolvin E2) and docosahexaenoic acid-derived lipid mediators, including D-series resolvins, protectins and maresins. AT-PD1, aspirin triggered protectin D1; DHA, docosahexaenoic acid; EPA, eicosapentaenoic acid; HpETE, hydroperoxyeicosatetraenoic acid; LT, leukotriene; LX, lipoxin; PG, prostaglandin; PGH2, prostaglandin H2; PLA2, phospholipase A2; PUFA, polyunsaturated fatty acid; Rv, resolvin; TX, thromboxane.
Excessive production of inflammatory mediators, such as prostaglandins and leukotrienes, with an exacerbated sensing response to inflammatory triggers is correlated with progression from acute inflammation to chronic inflammation in many diseases. Favorable inflammatory processes are self-limiting, which implies the existence of endogenous anti-inflammation and pro-resolution pathways (82).
Lipid mediators of resolution of inflammation
The primary goal of the inflammatory response is clearance of the insult with resolution and restoration of tissue homeostasis (102). A rapid and complete elimination of leukocytes from a lesion is the ideal outcome. Restoration of tissue homeostasis through resolution pathways follows an acute inflammatory response that generates lipid mediators of inflammation (leukotrienes). Resolution of inflammation is initiated by an active class switch in the mediators, such as classic prostaglandins and leukotrienes, to the production of immunoresolvents. Endogenous lipid mediators, including resolvins, protectins, lipoxins and maresins, are biosynthesized during the resolution phase of acute inflammation (15). The pathways that generate these molecules are complex and will be described in the next section. Lipoxins are derived from endogenous fatty acids (arachidonic acid), while resolvins, protectins and maresins are derived from dietary fatty acids, specifically the ω-3 fatty acids found in fish oil. Functionally, these specialized lipid mediators stimulate and accelerate resolution via multifactorial mechanisms at the tissue level (Fig. 2).
Fig. 2.
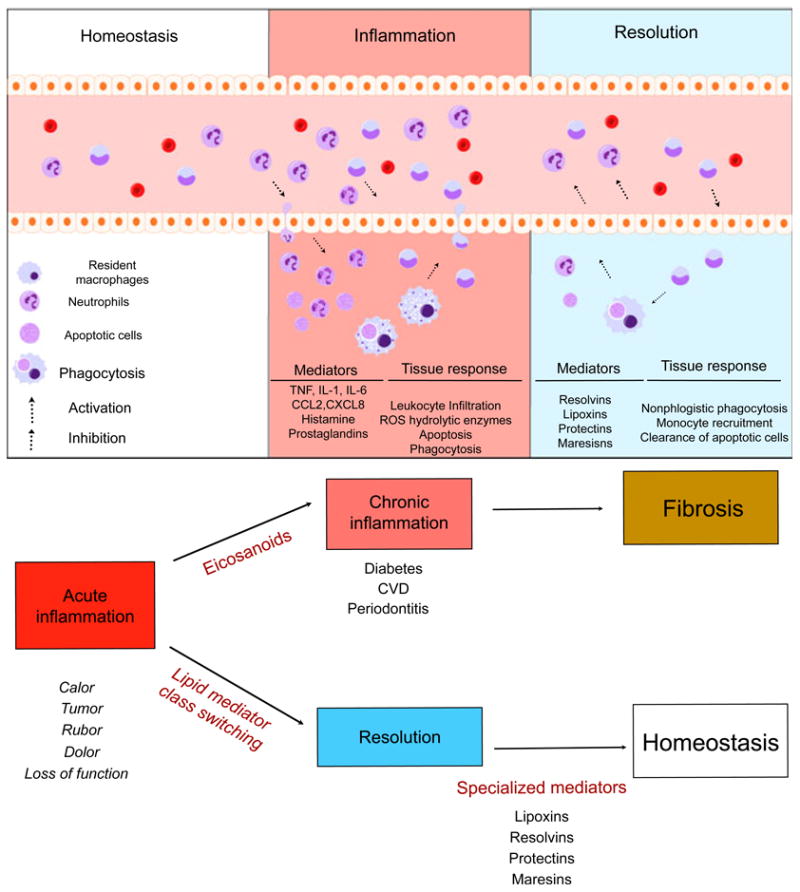
Natural inflammation resolution response. Acute inflammation is a self-limiting response. In conditions of homeostasis, resident cells maintain normal conditions and remove apoptotic cells and debris. Upon challenge to tissue, the inflammatory response begins. Cell communication mediators activate chemoattraction, vascular permeability and infiltration of leukocytes to the periphery. Microorganisms and dead cells are taken up by phagocytes. The outcome of acute inflammation (chronicity, fibrosis or resolution) is influenced by the type of factors and the degree to which they are involved in the response. Resolution is the re-establishment of normal (homeostasis) and is an actively regulated, well-coordinated process. In resolution, inhibition of leukocyte infiltration, nonphlogistic phagocytosis and vascular and tissue function return to normal. CCL2, chemokine (C-C motif) ligand 2; CVD, cardiovascular disease; CXCL8, chemokine (C-X-C motif) ligand 8; IL, interleukin; ROS, reactive oxygen species; TNF, tumor necrosis factor.
Lipoxins
Lipoxins are the natural proresolving molecules produced from endogenous fatty acids. Derived from arachidonic acid, lipoxins have potent anti-inflammatory and resolution actions (78, 86, 92). Lipoxins A4 and B4 were first isolated and identified as inhibitors of polymorphonuclear neutrophil infiltration and as stimulators of nonphlogistic recruitment of macrophages (14, 58). Three main pathways of lipoxin synthesis have been identified. In the first pathway, in human mucosal tissues such as the gastrointestinal tract, the airways and the oral cavity, sequential oxygenation of arachidonic acid by 15-lipoxygenase, and 5-lipoxygenase, followed by enzymatic hydrolysis, leads to the production of lipoxin A4 and lipoxin B4(54). In the second pathway, in blood vessels, 5-lip-oxygenase biosynthesizes lipoxin A4, and 12-lipoxygenase in platelets produces lipoxin B4. Lipoxin A4 regulates cellular functions through the activation of specific receptors (lipoxin A4 receptor/formyl peptide receptor 2 and G protein-coupled receptor 32); these receptors are expressed by neutrophils and monocytes. A third synthetic pathway is triggered by aspirin. Aspirin promotes the acetylation of cyclooxygenase-2, leading to a change in cyclooxygenase-2 activity and in the chirality of the products, which are termed aspirin-triggered lipoxins. A complete review of these pathways has been provided by Serhan & Chiang (84). Cells that express cyclooxygenase-2 include vascular endothelial cells, epithelial cells, macrophages and neutrophils. In addition to the synthesis of lipoxin, aspirin also blocks prostaglandin synthesis by acetylation of cyclooxygenase-2, inhibiting inflammation (85).
Resolvins
Resolvins are lipid mediators that are induced endogenously during the resolution phase of inflammation. These lipid mediators are biosynthesized from the precursor essential ω-3 polyunsaturated fatty acids eicosapentaenoic acid and docosahexaenoic acid derived from the diet (13, 106). The two major groups of the resolvin family have distinct chemical structures: E-series, derived from eicosapentaenoic acid; and D-series, derived from docosahexaenoic acid. E-series resolvins are produced by vascular endothelium via aspirin-modified cyclooxygenase-2 that converts eicosapentaenoic acid to 18R-hydroperoxyeicoapentaenoic acid and 18S-hydroperoxyeicoapentaenoic acid. These intermediates are rapidly taken up by human neutrophils and are metabolized to resolvin E1 and resolvin E2 by 5-lipoxygenase. Production of resolvin E1 is increased in the plasma of individuals taking aspirin or eicosapentaenoic acid, resulting in the amelioration of the clinical signs of inflammation (42, 62, 69). Similarly, docosahexaenoic acid-derived resolvins, D-series, have been shown to reduce inflammation by decreasing platelet-leukocyte adhesion, and aspirin-triggered docosahexaenoic acid conversion produces molecules with dual anti-inflammatory and pro-resolution functions (82).
Interaction between resolvins and specific receptors modulates the fate of innate immune cells and counter-regulates active inflammation. Selective target sites for resolvins are G protein-coupled receptors. Chemokine-like receptor 1 (also known as ChemR23) is a G protein-coupled receptor that is expressed on monocytes and dendritic cells. BLT1, a leukotriene receptor, is the resolvin E1 receptor on neutrophils (7, 8). Upon selective binding to the receptors, resolvin E1 attenuates nuclear factor-kappaB signaling and the production of pro-inflammatory cytokines, including tumor necrosis factor alpha. D-series resolvins target G protein-coupled receptor 32 and lipoxin A4 receptor/formyl peptide receptor 2 receptors, and, as shown recently, CB2 receptor is expressed on platelets and polymorphonuclear neutrophils (Fig. 3). Activation of CB2 leads to the inhibition of P-selectin expression, thus decreasing polymorphonuclear neutrophil chemotaxis. Resolvins induce the hallmark functions of resolution of inflammation, including prevention of neutrophil penetration, phagocytosis of apoptotic neutrophils to clear the lesion and enhancing clearance of inflammation within the lesion to promote tissue regeneration (13, 38, 80).
Fig. 3.
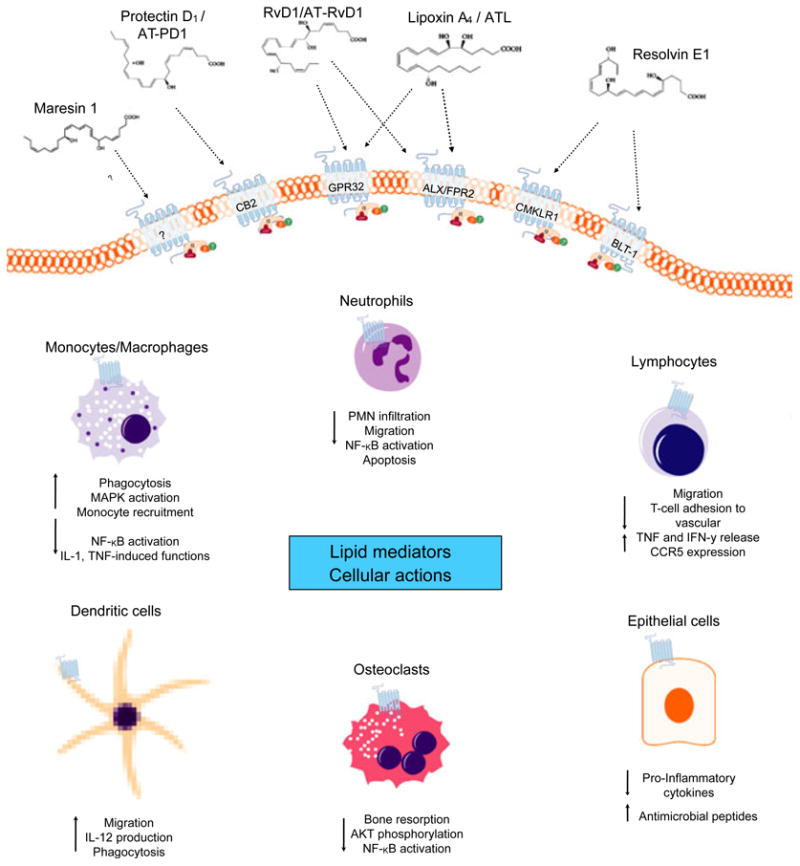
Interaction of cellular G protein-coupled receptor and lipid mediators. Biosynthesized pro-resolution lipid mediators interact selectively with G protein-coupled receptors. Receptor interactions with lipid mediators have been described in part. Lipoxin A4 stereoselective binding to the receptors lipoxin A4 receptor formyl peptide 2 and G protein-coupled receptor 32 activates anti-inflammation and resolution. Resolvin E1 (agonist) and the receptors chemokine-like receptor 1 and leukotriene B4 receptor type 1 have been shown to have positive influences on cell fate in inflammation. Resolvin D1 specifically interacts with both lipoxin A4 receptor and G protein-coupled receptor 32 on phagocytes, suggesting that each plays a role in resolving acute inflammation. The specic receptor interactions for protectin D1/aspirin-triggered protectin D1 and maresin 1 are yet to be defined; however, protectin D1 lipid mediators have been suggested to target cannabinoid receptor 2, CB2. Interactions between lipid mediators and cognitive receptors were found to influence in-vitro and in-vivo cellular functions with cell-type specificity. AKT, protein kinase B; ALX/FPR2, lipoxin A4 receptor; AT-PD1, aspirin-triggered protectin D1; ATL, aspirin-triggered lipoxin; BLT-1, leukotriene B4 receptor type 1; CB2, cannabinoid receptor 2; CCR5, chemokine (C-C motif) receptor 5; chemokine-like receptor 1; GPR32, G protein-coupled receptor 32; IFN-γ, interferon gamma; IL, interleukin; MAPK, mitogen-activated protein kinase; NF-κB, nuclear factor kappaB; PNM, polymorphonuclear neutrophil; TNF, tumor necrosis factor.
Protectins
Protectins are also biosynthesized via a lipoxygenasemediated pathway. This pathway converts docosahexaenoic acid into a 17S-hydroxyperoxide-containing intermediate that is rapidly taken up by leukocytes and converted into 10,17-Dihydroxydocosahexaenoic acid, known as protectin D1 (88) or neuroprotectin (17). The name accounts for the protective actions observed in neural tissues (57, 66) and within the immune system. Protectin D1 is also produced by human peripheral blood lymphocytes with a T-helper 2 phenotype; it reduces tumor necrosis factor alpha and interferon gamma secretion, blocks T-cell migration and promotes T-cell apoptosis (4). Recently, a novel protectin-synthesis pathway, called aspirin-triggered protectin D1, was found that utilizes aspirin-triggered cyclooxygenase-2 to synthesize epimeric 17R-hydroxyperoxide from docosahexaenoic acid and has shown a positive interaction with CB2 and peroxisome proliferator-activated receptor family receptors (17, 95). Both protectins reduce polymorphonuclear neutrophil transmigration through endothelial cells and enhance the clearance (efferocytosis) of apoptotic polymorphonuclear neutrophils by human macrophages (95).
Maresins
Macrophage mediators in resolving inflammation (maresins) were recently identified as primordial molecules produced by macrophages with homeostatic functions. Metabolopidomic approaches in peritonitis models led to the identification of a novel pathway of docosahexaenoic acid metabolism (82, 87, 93). Macrophage phagocytosis of apoptotic cells triggers the biosynthesis of resolvin E1, protectin D1, lipoxin A4 and maresin-1 (68). Conversion of docosahexaenoic acid into 14-hydroxy diHA was identified to occur via the 14-lipoxygenase pathway. Freshly prepared 14-H(p)docosahexaenoic acid is rapidly converted by macrophages into bioactive products (93). Maresin-1 effectively stimulates efferocytosis with human cells and also has regenerative functions.
Novel concepts of inflammation resolution
The local lipid mediators constitute a new genus of anti-inflammatory and pro-resolving endogenous compounds that have proven to be very potent in treating a number of inflammation-associated models of human disease. Lipoxin A4/aspirin-triggered lipoxins and resolvin E1 have been shown to inhibit neutrophil recruitment, attenuate pro-inflammatory gene expression and reduce the severity of colitis in a murine model. Infiltration of polymorphonuclear neutrophils and lymphatic removal of phagocytes were observed when resolvin E1, resolvin D2, protectin D1, lipoxin and maresin were used to ameliorate colitis (9, 45, 44). Most recently in a similar model, resolvin D1 has been associated with the regulation of microRNAs and target genes and with the reduction of leukotriene B4, prostaglandin D2, thromboxane A2, prostaglandin F2α and thromboxane A2 in peritoneal exudates (29, 51). Treatment with maresin-1 has been demonstrated to stimulate tissue regeneration in planaria. Consistently in periodontal disease models, lipoxin A4/aspirin-triggered lipoxins prevented connective tissue and bone loss (104). Treatment of experimental periodontitis with lipid mediators resulted in complete resolution of inflammation and remarkable regeneration of both soft and bone tissues; a restoration of homeostasis. It has been suggested that defective resolution of inflammation mechanisms underlies the inflammatory phenotype presented in chronic diseases and that lipid mediators can rescue this phenotype. That pro-resolution lipid mediators play a role as natural molecules in the maintenance of homeostasis is evident with promising potential as therapeutic agents for human diseases (Table 1).
Table 1. Resolution of lipid mediator cellular actions.
| Lipid mediator | Cellular function | References |
|---|---|---|
| Lipoxin A4 / aspirin-triggered lipoxin | Decreases adherence of leukocytes | Papayianni et al. (71) |
| Reduces vascular leakage | Takano et al. (98) | |
| Lowers prostaglandin E2 levels in exudates | Pouliot et al. (73) | |
| Reduces the number of apoptotic neutrophils | Perretti et al. (72) | |
| Inhibits neutrophil recruitment | Serhan et al. (90) | |
| Attenuates expression of the nuclear factor-kappaB gene | Chiang et al. (20) | |
| Blocks leukocyte adhesion protein-1 and chemotaxis | Ohira et al. (70) | |
| Promotes lymphatic removal of phagocytes | Bannenberg et al. (13) | |
| Activates FLRP2/Lipoxin A4 receptor receptor internalization and phagocytosis | Maderna et al. (59) | |
| Inhibits T-cell adhesion to vascular and salivary epithelium | Chintamani et al. (21) | |
| Enhances microbial phagocytic function | Tobin et al. (100) | |
| Resolvin E1 | Inhibits neutrophil infiltration | Serhan et al. (86) |
| Modulates chemokine/cytokine synthesis | Bannenberg et al. (13) | |
| Promotes healing of inflamed tissues and bone regeneration | Hasturk et al. (39) | |
| Enhances phagocytosis | Schwab et al. (80) | |
| Activates lymphatic removal of phagocytes | Van Dyke et al. (101) | |
| Attenuates systemic production of C-reactive protein and interleukin-1 | Hasturk et al. (38) | |
| Reduces eosinophil and lymphocyte recruitment | Aoki et al. (5) | |
| Regulates adipokines | Gonzalez-Periz et al. (35) | |
| Decreases inflammatory actions of cyclooxygenase-2 | Li et al. (56) | |
| Attenuate expression of the nuclear factor-kappaB gene | Ishida et al. (46) | |
| Lowers the number of monocytes | Li et al. (56) | |
| Increases CD55 expression on epithelial cells and polymorphonuclear cell clearance | Campbell et al. (18) | |
| Rescues impaired phagocytosis in LAP patient macrophages | Freedman et al. (30) | |
| Prevents rejection of allograft | Levy et al. (55) | |
| Activates anti-apoptotic signals | El Kebir et al. (27) | |
| Resolvin D1 | Inhibits neutrophil recruitment | Serhan et al. (89) |
| Anti-hyperalgesic properties | Albert et al. (2) | |
| Shortens resolution interval | Hong et al. (43) | |
| Reduces oxidative stress-mediated inflammation | Spite et al. (97) | |
| Attenuates agonist pain molecules | Bang et al. (12) | |
| Induces macrophage phagocytosis | Krishnamoorthy et al. (52) | |
| Stimulates the M2 macrophage phenotype | Titos et al. (99) | |
| Temporally regulates microRNAs | Recchiuti et al. (75) | |
| Reduces cytokines in bronchoalveolar lavage fluid | Wang et al. (105) | |
| Ameliorates insulin sensitivity | Hellman et al. (40) | |
| Enhances microbial clearance | Chiang et al. (19) | |
| Reduces levels of prostaglandins and leukotrienes | Norling et al. (67) | |
| Protectin D1/AT-PD1 | Decreases inflammatory actions of cyclooxygenase-2 | Marcheselli et al. (63) |
| Inhibits neutrophil infiltration | Bannenberg et al. (13) | |
| Modulates chemokine/cytokine synthesis | Serhan et al. (88) | |
| Regulates T-cell migration | Ariel et al. (6) | |
| Regulates macrophage function | Duffield et al. (25) | |
| Up-regulates CCR5 expression on apoptotic leukocytes | Schwab et al. (80) | |
| Prevents hepatocyte steatosis | Gonzalez-Periz et al. (35) | |
| Inhibits pain signals | Bang et al. (12) | |
| Suppresses eosinophil chemotaxis and adhesion | Miyata et al. (65) | |
| Maresin -1 | Reduces neutrophil numbers in exudate | Serhan et al. (93) |
| Enhances macrophage phagocytic functions | Serhan et al. (87) | |
| Decreases transendothelial polymorphonuclear cell migration | Shinohara et al. (95) |
Potential implications for periodontal diseases
Periodontal diseases, including gingivitis and periodontitis, are inflammatory diseases initiated by microbial biofilms. Uncontrolled inflammation causes loss of function and progression of disease. The host response to the biofilm destroys the periodontium in the pathogenesis of the disease. While an acute inflammatory response that is resolved in a timely manner prevents tissue injury, inadequate resolution and failure to return tissue to homeostasis results in neutrophil-mediated destruction and chronic inflammation. Classic studies performed in the 1980s showed that pharmacological anti-inflammatory products prevent the progression of periodontal disease (33, 41, 44, 94). It is now clear that an essential goal for intervention in inflammatory disease is the return to tissue homeostasis. Restoration of tissue health through resolution pathways is initiated following an acute inflammatory response that generates lipid mediators of inflammation, including the classic eicosanoids, prostanoids, prostacyclins and leukotrienes. The class switch that initiates resolution gives rise to the synthesis of immunoresolvents through pathways that are partially known (54, 83). Lipid mediator class switching is driven, in part, by cyclooxygenase-derived prostaglandins E2 and D2, via transcriptional regulation of enzymes involved in lipoxin biosynthesis. The immunoresolvents generated include ω-6 polyunsaturated fatty acids, arachidonic acid-derived lipoxins, aspirin-triggered lipoxins, ω-3 polyunsaturated fatty acids, eicosapentaenoic acid-derived resolvins, docosahexaenoic acid-derived resolvins D, protectins and maresins.
In an attempt to understand the role of resolution mediators in disease prevention, animal models for periodontitis were developed. Over-expression of 15-lipoxygenase type I in rabbits increased the endogenous levels of lipoxin A4, protecting the host from developing periodontal disease (103). In addition, resolvin E1, when topically applied to the tissues, also ameliorated signs of disease activity, decreasing bone loss by 95% and reducing the number of tissue neutrophils. Leukocyte infiltration was also decreased when resolvin-1 was used in a mouse dorsal air pouch model (47). Similarly, in human cells obtained from subjects with localized aggressive periodontitis, resolvin E1 and lipoxin A4 decreased neutrophil superoxide production in response to tumor necrosis factor alpha and the bacterial surrogate peptide, N-formylmethionyl-leucyl-phenylalanine, by 80% (30). Recently, a clinical trial of patients with moderate to severe periodontitis evaluated dietary supplementation of ω-3 polyunsaturated acids and aspirin in addition to scaling and root planing; pocket depths were reduced and clinical attachment levels were increased, with lower levels of inflammatory mediators in saliva compared with scaling and root planing alone (28). The potential for treatment of periodontal diseases with one or a combination of specialized lipid mediators is clear and requires further investigation.
Systemic impact of uncontrolled periodontal inflammation
Uncontrolled (chronic) inflammation is a hallmark of various human diseases, including diabetes, cardiovascular diseases and periodontitis. Despite advances in our knowledge of the causes and risk factors associated with chronic periodontitis, there are no signs of decline in prevalence. According to the Centers for Disease Control (2012), 64.7 million adults have periodontal disease (26). Periodontal diseases, including gingivitis and periodontitis, are leukocyte-driven inflammatory diseases characterized by soft-tissue loss and osteoclast-mediated bone loss (101).The etiology of periodontitis is recognized to be multifactorial, and a number of behavioral, environmental, microbial, systemic and genetic risk factors influence host susceptibility and disease progression (50). The impact of a dysregulated inflammatory response has potential harm to the systemic health of individuals.
Inflammatory diseases, such as type 2 diabetes and chronic periodontal diseases, have a reciprocal relationship. Among subjects diagnosed with diabetes, one-third present with severe periodontal disease, and adult patients with severe periodontitis have increased risk of poor glycemic control, increasing the risk of oral and systemic complications. Hyperglycemia resulting from type 2 diabetes can impact polymorphonuclear neutrophils and monocytes through various mechanisms, including the accumulation of advanced-glycation end products, which leads to an increase of extracellular production of superoxide by leukocytes and increased secretion of pro-inflammatory cytokines, such as interleukin-1 beta, insulin-like growth factor, tumor necrosis factor alpha and matrix metalloproteinases (53). Owing to the failure of resolution pathways, increased inflammation in periodontitis leads to the prolonged activation of polymorphonuclear neutrophils and monocytes with ineffective clearance of bacteria, consequently aggravating systemic diabetes regulation.
In cardiovascular disease studies, periodontal disease was shown to be a contributing factor. Severe periodontal disease affects 10–15% of the general population and has been linked to cardiovascular disease in cross-sectional and cohort studies (48, 74). Studies reported that elevated cell- and cytokine-mediated markers of inflammation, including C-reactive protein, fibrinogen and various cytokines, are associated with periodontal diseases. By reducing the progression of periodontal disease, the levels of inflammatory markers common to both diseases (i.e. interleukin-6, tumor necrosis factor alpha and C-reactive protein) are decreased, which might, in turn, decrease the severity of vascular disease. After adjusting for risk factors, such as smoking, diabetes, alcohol intake, obesity and blood pressure, subjects with periodontal disease had a 1.14- to 1.59-fold greater risk of developing cardiovascular heart disease compared with those without periodontal disease (10).
Diseases associated with uncontrolled acute inflammation are characterized by a lack of activation of resolution programs and by the inappropriate release and maintenance of high levels of toxic substances and pro-inflammatory mediators, which may result in damage to host tissues and prolong the inflammatory response (13). In experimental animal models, compelling evidence demonstrated the actions of pro-resolution mediators in the regulation of local and systemic mediators of inflammation, elucidating the role of endogenous mediators in resolving the systemic acute inflammatory processes and in disease regulation (Table 2).
Table 2. Agonists of inflammation resolution in disease models.
| Disease models/species | Actions | References |
|---|---|---|
| Periodontitis/rabbit | Lipoxin A4/aspirin-triggered lipoxin | |
| Prevents connective tissue and bone loss | Serhan et al. (90) | |
| Accelerates healing of inflamed tissues | Serhan et al. (90) | |
| Ceases infiltration of neutrophils | Serhan et al. (90) | |
| Resolvin E1 | ||
| Reduces bone loss | Hasturk et al. (39) | |
| Regenerates lost soft tissue and bone tissue | Hasturk et al. (38) | |
| Peritonitis/mouse | Lipoxin A4/aspirin-triggered lipoxin | |
| Stops neutrophil recruitment | Bannenberg et al. (13) | |
| Promotes lymphatic removal of phagocytes | Schwab et al. (80) | |
| Resolvin E1 | ||
| Decreases polymorphonuclear cell infiltration | Arita et al. (7) | |
| Regulates chemokine and cytokine production | Schwab et al. (80) | |
| Promotes lymphatic removal of phagocytes | Bannenberg et al. (13) | |
| Resolvin D1 | ||
| Shortens resolution interval | Spite et al. (97) | |
| Regulates microRNAs | Recchiuti et al. (75) | |
| Reduces concentrations of leukotriene B4, prostaglandin D2, prostaglandin F2 and thromboxane A2 in exudates | Krishnamoorthy et al. (51) Norling et al. (67) | |
| Lowers antibiotic requirement | Chiang et al. (19) | |
| Increases animal survival | Chiang et al. (19) | |
| Reduces bacterial titers | Chiang et al. (19) | |
| Protectin D1 | ||
| Promotes local clearance of apoptotic cells | Bannenberg et al. (13) | |
| Regulates lymphatic removal of phagocytes | Ariel et al. (6) | |
| Modulates T-cell migration | Schwab et al. (80) | |
| Maresin-1 | ||
| Blocks infiltration of polymorphonuclear cells into the peritonium | Serhan et al. (93) | |
| Colitis/mouse | Lipoxin A4/aspirin-triggered lipoxin | |
| Reduces severe colitis | Aliberti et al. (3) | |
| Attenuates pro-inflammatory mediators of gene expression | Gewirtz et al. (34) | |
| Inhibits weight loss | Gewirtz et al. (34) | |
| Reduces immune dysfunction | Wallace et al. (104) | |
| Resolvin E1 | ||
| Improves animal survival rate | Arita et al. (7) | |
| Reduces weight loss | Arita et al. (7) | |
| Promotes lipopolysaccharide detoxification | Campbell et al. (18) | |
| Stops neutrophil recruitment | Ishida et al. (46) | |
| AT-Resolvin D1 | ||
| Reduces disease activity index | Bento et al. (17) | |
| Attenuates pro-inflammatory mediators of gene expression | Bento et al. (17) | |
| Attenuates neutrophil recruitment | Bento et al. (17) | |
| Resolvin D2 | ||
| Improves disease activity index | Bento et al. (17) | |
| Reduces colonic polymorphonuclear cell infiltration | Bento et al. (17) | |
| Retinopathy/mouse | Resolvin E1/Resolvin D1/Protectin D1 | |
| Protects against neovascularization | Connor et al. (22) | |
| Tissue regeneration/mouse | Resolvin E1 | |
| Promotes bone regeneration in calvaria defects | Gao et al. (32) | |
| Tissue regeneration/planaria | Maresin-1 | |
| Stimulates tissue regeneration after surgical damage | Serhan et al. (87) |
Conclusion
The primary role of acute inflammation is protection of the host. It is initiated by neutrophils in response to challenge. The fate of this process is determined by the balance between the presence of mediators and sensors that either amplify the inflammatory process or control the return to normal health. It is now evident that resolution of inflammation is modulated by protective mediators, such as arachidonic acid-derived lipoxins, aspirin-triggered lipoxins, ω3-eicosapentaenoic acid-derived resolvins of the E-series, docosahexaenoic acid-derived resolvins of the D series, protectins and maresins. The selective interaction of lipid mediators with G protein-coupled receptors of innate immune cells induces cessation of leukocyte infiltration; the return to normal of vascular permeability/edema; polymorphonuclear neutrophil death (mostly via apoptosis); nonphlogistic infiltration of monocyte/macrophages; and removal (by macrophages) of apoptotic polymorphonuclear neutrophils, foreign agents (bacteria) and necrotic debris from the site. These cellular events achieve the ideal outcome of inflammation, namely resolution, with return to pre-disease homeostasis.
Acknowledgments
Supported in part by USPHS grants DE15566, DE19938 and T32-DE007327.
References
- 1.Aderem A, Ulevitch RJ. Toll-like receptors in the induction of the innate immune response. Nature. 2000;406:782–787. doi: 10.1038/35021228. [DOI] [PubMed] [Google Scholar]
- 2.Albert CM, Campos H, Stampfer MJ, Ridker PM, Manson JE, Willett WC, Ma J. Blood levels of long-chain n-3 fatty acids and the risk of sudden death. N Engl J Med. 2002;346:1113–1118. doi: 10.1056/NEJMoa012918. [DOI] [PubMed] [Google Scholar]
- 3.Aliberti J, Hieny S, Reis e Sousa C, Serhan CN, Sher A. Lipoxin-mediated inhibition of IL-12 production by DCs: a mechanism for regulation of microbial immunity. Nat Immunol. 2002;3:76–82. doi: 10.1038/ni745. [DOI] [PubMed] [Google Scholar]
- 4.Anderson P, Delgado M. Endogenous anti-inflammatory neuropeptides and pro-resolving lipid mediators: a new therapeutic approach for immune disorders. J Cell Mol Med. 2008;12:1830–1847. doi: 10.1111/j.1582-4934.2008.00387.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aoki H, Hisada T, Ishizuka T, Utsugi M, Kawata T, Shimizu Y, Okajima F, Dobashi K, Mori M. Resolvin E1 dampens airway inflammation and hyperresponsiveness in a murine model of asthma. Biochem Biophys Res Commun. 2008;367:509–515. doi: 10.1016/j.bbrc.2008.01.012. [DOI] [PubMed] [Google Scholar]
- 6.Ariel A, Fredman G, Sun YP, Kantarci A, Van Dyke TE, Luster AD, Serhan CN. Apoptotic neutrophils and T cells sequester chemokines during immune response resolution through modulation of CCR5 expression. Nat Immunol. 2006;7:1209–1216. doi: 10.1038/ni1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arita M, Bianchini F, Aliberti J, Sher A, Chiang N, Hong S, Yang R, Petasis NA, Serhan CN. Stereochemical assignment, antiinflammatory properties, and receptor for the omega-3 lipid mediator resolvin E1. J Exp Med. 2005;201:713–722. doi: 10.1084/jem.20042031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arita M, Ohira T, Sun YP, Elangovan S, Chiang N, Serhan CN. Resolvin E1 selectively interacts with leukotriene B4 receptor BLT1 and ChemR23 to regulate inflammation. J Immunol. 2007;178:3912–3917. doi: 10.4049/jimmunol.178.6.3912. [DOI] [PubMed] [Google Scholar]
- 9.Arita M, Yoshida M, Hong S, Tjonahen E, Glickman JN, Pe-tasis NA, Blumberg RS, Serhan CN. Resolvin E1, an endogenous lipid mediator derived from omega-3 eicosapentaenoic acid, protects against 2,4,6-trinitrobenzene sulfonic acid-induced colitis. Proc Natl Acad Sci USA. 2005;102:7671–7676. doi: 10.1073/pnas.0409271102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bahekar AA, Singh S, Saha S, Molnar J, Arora R. The prevalence and incidence of coronary heart disease is significantly increased in periodontitis: a meta-analysis. Am Heart J. 2007;154:830–837. doi: 10.1016/j.ahj.2007.06.037. [DOI] [PubMed] [Google Scholar]
- 11.Baldwin AS., Jr The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 12.Bang S, Yoo S, Yang TJ, Cho H, Hwang SW. 17(R)-resolvin D1 specifically inhibits transient receptor potential ion channel vanilloid 3 leading to peripheral antinociception. Br J Pharmacol. 2012;165:683–692. doi: 10.1111/j.1476-5381.2011.01568.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bannenberg GL, Chiang N, Ariel A, Arita M, Tjonahen E, Gotlinger KH, Hong S, Serhan CN. Molecular circuits of resolution: formation and actions of resolvins and protectins. J Immunol. 2005;174:4345–4355. doi: 10.4049/jimmunol.174.7.4345. [DOI] [PubMed] [Google Scholar]
- 14.Bannenberg G, Moussignac RL, Gronert K, Devchand PR, Schmidt BA, Guilford WJ, Bauman JG, Subramanyam B, Perez HD, Parkinson JF, Serhan CN. Lipoxins and novel 15-epi-lipoxin analogs display potent anti-inflammatory actions after oral administration. Br J Pharmacol. 2004;143:43–52. doi: 10.1038/sj.bjp.0705912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bannenberg G, Serhan CN. Specialized pro-resolving lipid mediators in the inflammatory response: An update. Biochim Biophys Acta. 2010;1801:1260–1273. doi: 10.1016/j.bbalip.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bazan NG, Eady TN, Khoutorova L, Atkins KD, Hong S, Lu Y, Zhang C, Jun B, Obenaus A, Fredman G, Zhu M, Winkler JW, Petasis NA, Serhan CN, Belayev L. Novel aspirin-triggered neuroprotectin D1 attenuates cerebral ischemic injury after experimental stroke. Exp Neurol. 2012;236:122–130. doi: 10.1016/j.expneurol.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bento AF, Claudino RF, Dutra RC, Marcon R, Calixto JB. Omega-3 fatty acid-derived mediators 17(R)-hydroxy docosahexaenoic acid, aspirin-triggered resolvin D1 and resolvin D2 prevent experimental colitis in mice. J Immunol. 2011;187:1957–1969. doi: 10.4049/jimmunol.1101305. [DOI] [PubMed] [Google Scholar]
- 18.Campbell EL, MacManus CF, Kominsky DJ, Keely S, Glover LE, Bowers BE, Scully M, Bruyninckx WJ, Colgan SP. Resolvin E1-induced intestinal alkaline phosphatase promotes resolution of inflammation through LPS detoxification. Proc Natl Acad Sci USA. 2010;107:14298–14303. doi: 10.1073/pnas.0914730107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chiang N, Fredman G, Backhed F, Oh SF, Vickery T, Schmidt BA, Serhan CN. Infection regulates pro-resolving mediators that lower antibiotic requirements. Nature. 2012;484:524–528. doi: 10.1038/nature11042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chiang N, Takano T, Arita M, Watanabe S, Serhan CN. A novel rat lipoxin A4 receptor that is conserved in structure and function. Br J Pharmacol. 2003;139:89–98. doi: 10.1038/sj.bjp.0705220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chinthamani S, Odusanwo O, Mondal N, Nelson J, Neela-megham S, Baker OJ. Lipoxin A4 inhibits immune cell binding to salivary epithelium and vascular endothelium. Am J Physiol Cell Physiol. 2012;302:C968–C978. doi: 10.1152/ajpcell.00259.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Connor KM, SanGiovanni JP, Lofqvist C, Aderman CM, Chen J, Higuchi A, Hong S, Pravda EA, Majchrzak S, Carper D, Hellstrom A, Kang JX, Chew EY, Salem N, Jr, Serhan CN, Smith LE. Increased dietary intake of omega-3-polyunsaturated fatty acids reduces pathological retinal angiogenesis. Nat Med. 2007;13:868–873. doi: 10.1038/nm1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cotran RS, Majno G. The delayed and prolonged vascular leakage in inflammation. I. Topography of the leaking vessels after thermal injury. Am J Pathol. 1964;45:261–281. [PMC free article] [PubMed] [Google Scholar]
- 24.Darveau RP. Periodontitis: a polymicrobial disruption of host homeostasis. Nat Rev Microbiol. 2010;8:481–490. doi: 10.1038/nrmicro2337. [DOI] [PubMed] [Google Scholar]
- 25.Duffield JS, Hong S, Vaidya VS, Lu Y, Fredman G, Serhan CN, Bonventre JV. Resolvin D series and protectin D1 mitigate acute kidney injury. J Immunol. 2006;177:5902–5911. doi: 10.4049/jimmunol.177.9.5902. [DOI] [PubMed] [Google Scholar]
- 26.Eke PI, Dye BA, Wei L, Thornton-Evans GO, Genco RJ. Cdc Periodontal Disease Surveillance workgroup: James Beck GDRP. Prevalence of periodontitis in adults in the United States: 2009 and 2010. J Dent Res. 2012;91:914–920. doi: 10.1177/0022034512457373. [DOI] [PubMed] [Google Scholar]
- 27.El Kebir D, Gjorstrup P, Filep JG. Resolvin E1 promotes phagocytosis-induced neutrophil apoptosis and accelerates resolution of pulmonary inflammation. Proc Natl Acad Sci USA. 2012;109:14983–14988. doi: 10.1073/pnas.1206641109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.El-Sharkawy H, Aboelsaad N, Eliwa M, Darweesh M, Alshahat M, Kantarci A, Hasturk H, Van Dyke TE. Adjunctive treatment of chronic periodontitis with daily dietary supplementation with omega-3 Fatty acids and low-dose aspirin. J Periodontol. 2010;81:1635–1643. doi: 10.1902/jop.2010.090628. [DOI] [PubMed] [Google Scholar]
- 29.Fredman G, Li Y, Dalli J, Chiang N, Serhan CN. Self-limited versus delayed resolution of acute inflammation: temporal regulation of pro-resolving mediators and microRNA. Sci Rep. 2012;2:639. doi: 10.1038/srep00639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fredman G, Oh SF, Ayilavarapu S, Hasturk H, Serhan CN, Van Dyke TE. Impaired phagocytosis in localized aggressive periodontitis: rescue by Resolvin E1. PLoS ONE. 2011;6:e24422. doi: 10.1371/journal.pone.0024422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Funk CD. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science. 2001;294:1871–1875. doi: 10.1126/science.294.5548.1871. [DOI] [PubMed] [Google Scholar]
- 32.Gao L, Faibish D, Fredman G, Herrera BS, Chiang N, Serhan CN, Van Dyke TE, Gyurko R. Resolvin E1 and chemokine-like receptor 1 mediate bone preservation. J Immunol. 2013;190:689–694. doi: 10.4049/jimmunol.1103688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gemmell E, Marshall RI, Seymour GJ. Cytokines and prostaglandins in immune homeostasis and tissue destruction in periodontal disease. Periodontol 2000. 1997;14:112–143. doi: 10.1111/j.1600-0757.1997.tb00194.x. [DOI] [PubMed] [Google Scholar]
- 34.Gewirtz AT, Collier-Hyams LS, Young AN, Kucharzik T, Guilford WJ, Parkinson JF, Williams IR, Neish AS, Madara JL. Lipoxin a4 analogs attenuate induction of intestinal epithelial proinflammatory gene expression and reduce the severity of dextran sodium sulfate-induced colitis. J Immunol. 2002;168:5260–5267. doi: 10.4049/jimmunol.168.10.5260. [DOI] [PubMed] [Google Scholar]
- 35.Gonzalez-Periz A, Horrillo R, Ferre N, Gronert K, Dong B, Moran-Salvador E, Titos E, Martinez-Clemente M, Lopez-Parra M, Arroyo V, Claria J. Obesity-induced insulin resistance and hepatic steatosis are alleviated by omega-3 fatty acids: a role for resolvins and protectins. FASEB J. 2009;23:1946–1957. doi: 10.1096/fj.08-125674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hamberg M, Svensson J, Samuelsson B. Thromboxanes: a new group of biologically active compounds derived from prostaglandin endoperoxides. Proc Natl Acad Sci USA. 1975;72:2994–2998. doi: 10.1073/pnas.72.8.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hanada T, Yoshimura A. Regulation of cytokine signaling and inflammation. Cytokine Growth Factor Rev. 2002;13:413–421. doi: 10.1016/s1359-6101(02)00026-6. [DOI] [PubMed] [Google Scholar]
- 38.Hasturk H, Kantarci A, Goguet-Surmenian E, Blackwood A, Andry C, Serhan CN, Van Dyke TE. Resolvin E1 regulates inflammation at the cellular and tissue level and restores tissue homeostasis in vivo. J Immunol. 2007;179:7021–7029. doi: 10.4049/jimmunol.179.10.7021. [DOI] [PubMed] [Google Scholar]
- 39.Hasturk H, Kantarci A, Ohira T, Arita M, Ebrahimi N, Chiang N, Petasis NA, Levy BD, Serhan CN, Van Dyke TE. RvE1 protects from local inflammation and osteoclast-mediated bone destruction in periodontitis. FASEB J. 2006;20:401–403. doi: 10.1096/fj.05-4724fje. [DOI] [PubMed] [Google Scholar]
- 40.Hellmann J, Tang Y, Kosuri M, Bhatnagar A, Spite M. Resolvin D1 decreases adipose tissue macrophage accumulation and improves insulin sensitivity in obese-diabetic mice. FASEB J. 2011;25:2399–2407. doi: 10.1096/fj.10-178657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hirata H. A role of prostaglandins in pathogenesis of periodontal disease. Studies of calcium releasing action of prostaglandins from bone and changes of its levels in gingiva (author's transl) Hiroshima Daigaku Shigaku Zasshi. 1980;12:252–264. [PubMed] [Google Scholar]
- 42.Ho KJ, Spite M, Owens CD, Lancero H, Kroemer AH, Pande R, Creager MA, Serhan CN, Conte MS. Aspirin-triggered lipoxin and resolvin E1 modulate vascular smooth muscle phenotype and correlate with peripheral atherosclerosis. Am J Pathol. 2010;177:2116–2123. doi: 10.2353/ajpath.2010.091082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hong S, Gronert K, Devchand PR, Moussignac RL, Serhan CN. Novel docosatrienes and 17S-resolvins generated from docosahexaenoic acid in murine brain, human blood, and glial cells. Autacoids in anti-inflammation. J Biol Chem. 2003;278:14677–14687. doi: 10.1074/jbc.M300218200. [DOI] [PubMed] [Google Scholar]
- 44.Howell TH. Blocking periodontal disease progression with anti-inflammatory agents. J Periodontol. 1993;64:828–833. doi: 10.1902/jop.1993.64.8s.828. [DOI] [PubMed] [Google Scholar]
- 45.Hudert CA, Weylandt KH, Lu Y, Wang J, Hong S, Dignass A, Serhan CN, Kang JX. Transgenic mice rich in endogenous omega-3 fatty acids are protected from colitis. Proc Natl Acad Sci USA. 2006;103:11276–11281. doi: 10.1073/pnas.0601280103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ishida T, Yoshida M, Arita M, Nishitani Y, Nishiumi S, Masuda A, Mizuno S, Takagawa T, Morita Y, Kutsumi H, Inokuchi H, Serhan CN, Blumberg RS, Azuma T. Resolvin E1, an endogenous lipid mediator derived from eicosapentaenoic acid, prevents dextran sulfate sodium-induced colitis. Inflamm Bowel Dis. 2010;16:87–95. doi: 10.1002/ibd.21029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ishizuka T, Hisada T, Aoki H, Mori M. Resolvin E1: a novel lipid mediator in the resolution of allergic airway inflammation. Expert Rev Clin Immunol. 2008;4:669–672. doi: 10.1586/1744666X.4.6.669. [DOI] [PubMed] [Google Scholar]
- 48.Janket SJ, Baird AE, Chuang SK, Jones JA. Meta-analysis of periodontal disease and risk of coronary heart disease and stroke. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2003;95:559–569. doi: 10.1067/moe.2003.107. [DOI] [PubMed] [Google Scholar]
- 49.Kalinski P. Regulation of immune responses by prostaglandin E2. J Immunol. 2012;188:21–28. doi: 10.4049/jimmunol.1101029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kinane DF, Demuth DR, Gorr SU, Hajishengallis GN, Martin MH. Human variability in innate immunity. Periodontol 2000. 2007;45:14–34. doi: 10.1111/j.1600-0757.2007.00220.x. [DOI] [PubMed] [Google Scholar]
- 51.Krishnamoorthy S, Recchiuti A, Chiang N, Fredman G, Serhan CN. Resolvin D1 receptor stereoselectivity and regulation of inflammation and proresolving microRNAs. Am J Pathol. 2012;180:2018–2027. doi: 10.1016/j.ajpath.2012.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Krishnamoorthy S, Recchiuti A, Chiang N, Yacoubian S, Lee CH, Yang R, Petasis NA, Serhan CN. Resolvin D1 binds human phagocytes with evidence for proresolving receptors. Proc Natl Acad Sci USA. 2010;107:1660–1665. doi: 10.1073/pnas.0907342107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lalla E, Papapanou PN. Diabetes mellitus and periodontitis: a tale of two common interrelated diseases. Nat Rev Endocrinol. 2011;7:738–748. doi: 10.1038/nrendo.2011.106. [DOI] [PubMed] [Google Scholar]
- 54.Levy BD, Clish CB, Schmidt B, Gronert K, Serhan CN. Lipid mediator class switching during acute inflammation: signals in resolution. Nat Immunol. 2001;2:612–619. doi: 10.1038/89759. [DOI] [PubMed] [Google Scholar]
- 55.Levy BD, Zhang QY, Bonnans C, Primo V, Reilly JJ, Perkins DL, Liang Y, Amin Arnaout M, Nikolic B, Serhan CN. The endogenous pro-resolving mediators lipoxin A4 and resolvin E1 preserve organ function in allograft rejection. Prostaglandins Leukot Essent Fatty Acids. 2011;84:43–50. doi: 10.1016/j.plefa.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li N, He J, Schwartz CE, Gjorstrup P, Bazan HE. Resolvin E1 improves tear production and decreases inflammation in a dry eye mouse model. J Ocul Pharmacol Ther. 2010;26:431–439. doi: 10.1089/jop.2010.0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lukiw WJ, Cui JG, Marcheselli VL, Bodker M, Botkjaer A, Gotlinger K, Serhan CN, Bazan NG. A role for docosahexaenoic acid-derived neuroprotectin D1 in neural cell survival and Alzheimer disease. J Clin Investig. 2005;115:2774–2783. doi: 10.1172/JCI25420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Maddox JF, Colgan SP, Clish CB, Petasis NA, Fokin VV, Serhan CN. Lipoxin B4 regulates human monocyte/neutrophil adherence and motility: design of stable lipoxin B4 analogs with increased biologic activity. FASEB J. 1998;12:487–494. doi: 10.1096/fasebj.12.6.487. [DOI] [PubMed] [Google Scholar]
- 59.Maderna P, Cottell DC, Toivonen T, Dufton N, Dalli J, Perretti M, Godson C. FPR2/ALX receptor expression and internalization are critical for lipoxin A4 and annexin-derived peptide-stimulated phagocytosis. FASEB J. 2010;24:4240–4249. doi: 10.1096/fj.10-159913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Majno G. The healing hand Man and Wound in the Acient World. Cambridge: Harvard University Press; 1975. [Google Scholar]
- 61.Majno G, Joris I. Cells Tissues and Disease. Osxford: Oxford University; 2004. [Google Scholar]
- 62.Makriyannis A, Nikas SP. Aspirin-triggered metabolites of EFAs. Chem Biol. 2011;18:1208–1209. doi: 10.1016/j.chembiol.2011.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Marcheselli VL, Hong S, Lukiw WJ, Tian XH, Gronert K, Musto A, Hardy M, Gimenez JM, Chiang N, Serhan CN, Bazan NG. Novel docosanoids inhibit brain ischemia-reperfusion-mediated leukocyte infiltration and pro-inflammatory gene expression. J Biol Chem. 2003;278:43807–43817. doi: 10.1074/jbc.M305841200. [DOI] [PubMed] [Google Scholar]
- 64.Medzhitov R. Inflammation 2010: new adventures of an old flame. Cell. 2010;140:771–776. doi: 10.1016/j.cell.2010.03.006. [DOI] [PubMed] [Google Scholar]
- 65.Miyata J, Fukunaga K, Iwamoto R, Isobe Y, Niimi K, Takamiya R, Takihara T, Tomomatsu K, Suzuki Y, Oguma T, Sayama K, Arai H, Betsuyaku T, Arita M, Asano K. Dysregulated synthesis of protectin D1 in eosinophils from patients with severe asthma. J Allergy Clin Immunol. 2013;131:353–360. e351–352. doi: 10.1016/j.jaci.2012.07.048. [DOI] [PubMed] [Google Scholar]
- 66.Mukherjee PK, Marcheselli VL, Serhan CN, Bazan NG. Neuroprotectin D1: a docosahexaenoic acid-derived docosatriene protects human retinal pigment epithelial cells from oxidative stress. Proc Natl Acad Sci USA. 2004;101:8491–8496. doi: 10.1073/pnas.0402531101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Norling LV, Dalli J, Flower RJ, Serhan CN, Perretti M. Resolvin D1 limits polymorphonuclear leukocyte recruitment to inflammatory loci: receptor-dependent actions. Arterioscler Thromb Vasc Biol. 2012;32:1970–1978. doi: 10.1161/ATVBAHA.112.249508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Norling LV, Serhan CN. Profiling in resolving inflammatory exudates identifies novel anti-inflammatory and pro-resolving mediators and signals for termination. J Intern Med. 2010;268:15–24. doi: 10.1111/j.1365-2796.2010.02235.x. [DOI] [PubMed] [Google Scholar]
- 69.Oh SF, Pillai PS, Recchiuti A, Yang R, Serhan CN. Pro-resolving actions and stereoselective biosynthesis of 18S E-series resolvins in human leukocytes and murine inflammation. J Clin Investig. 2011;121:569–581. doi: 10.1172/JCI42545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ohira T, Bannenberg G, Arita M, Takahashi M, Ge Q, Van Dyke TE, Stahl GL, Serhan CN, Badwey JA. A stable aspirin-triggered lipoxin A4 analog blocks phosphorylation of leukocyte-specific protein 1 in human neutrophils. J Immunol. 2004;173:2091–2098. doi: 10.4049/jimmunol.173.3.2091. [DOI] [PubMed] [Google Scholar]
- 71.Papayianni A, Serhan CN, Phillips ML, Rennke HG, Brady HR. Transcellular biosynthesis of lipoxin A4 during adhesion of platelets and neutrophils in experimental immune complex glomerulonephritis. Kidney Int. 1995;47:1295–1302. doi: 10.1038/ki.1995.184. [DOI] [PubMed] [Google Scholar]
- 72.Perretti M, Chiang N, La M, Fierro IM, Marullo S, Getting SJ, Solito E, Serhan CN. Endogenous lipid- and peptide-derived anti-inflammatory pathways generated with glucocorticoid and aspirin treatment activate the lipoxin A4 receptor. Nat Med. 2002;8:1296–1302. doi: 10.1038/nm786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pouliot M, Clish CB, Petasis NA, Van Dyke TE, Serhan CN. Lipoxin A(4) analogues inhibit leukocyte recruitment to Porphyromonas gingivalis: a role for cyclooxygenase-2 and lipoxins in periodontal disease. Biochemistry. 2000;39:4761–4768. doi: 10.1021/bi992551b. [DOI] [PubMed] [Google Scholar]
- 74.Pussinen PJ, Nyyssonen K, Alfthan G, Salonen R, Laukkanen JA, Salonen JT. Serum antibody levels to Actinobacillus actinomycetemcomitans predict the risk for coronary heart disease. Arterioscler Thromb Vasc Biol. 2005;25:833–838. doi: 10.1161/01.ATV.0000157982.69663.59. [DOI] [PubMed] [Google Scholar]
- 75.Recchiuti A, Krishnamoorthy S, Fredman G, Chiang N, Serhan CN. MicroRNAs in resolution of acute inflammation: identification of novel resolvin D1-miRNA circuits. FASEB J. 2011;25:544–560. doi: 10.1096/fj.10-169599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rossi D, Zlotnik A. The biology of chemokines and their receptors. Annu Rev Immunol. 2000;18:217–242. doi: 10.1146/annurev.immunol.18.1.217. [DOI] [PubMed] [Google Scholar]
- 77.Samuelsson B, Borgeat P, Hammarstrom S, Murphy RC. Leukotrienes: a new group of biologically active compounds. Adv Prostaglandin Thromboxane Res. 1980;6:1–18. [PubMed] [Google Scholar]
- 78.Samuelsson B, Dahlen SE, Lindgren JA, Rouzer CA, Serhan CN. Leukotrienes and lipoxins: structures, biosynthesis, and biological effects. Science. 1987;237:1171–1176. doi: 10.1126/science.2820055. [DOI] [PubMed] [Google Scholar]
- 79.Savill J, Dransfield I, Gregory C, Haslett C. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat Rev Immunol. 2002;2:965–975. doi: 10.1038/nri957. [DOI] [PubMed] [Google Scholar]
- 80.Schwab JM, Chiang N, Arita M, Serhan CN. Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature. 2007;447:869–874. doi: 10.1038/nature05877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Serhan CN. A search for endogenous mechanisms of anti-inflammation uncovers novel chemical mediators: missing links to resolution. Histochem Cell Biol. 2004;122:305–321. doi: 10.1007/s00418-004-0695-8. [DOI] [PubMed] [Google Scholar]
- 82.Serhan CN. Systems approach to inflammation resolution: identification of novel anti-inflammatory and pro-resolving mediators. J Thromb Haemost. 2009;7(Suppl 1):44–48. doi: 10.1111/j.1538-7836.2009.03396.x. [DOI] [PubMed] [Google Scholar]
- 83.Serhan CN, Chiang N. Lipid-derived mediators in endogenous anti-inflammation and resolution: lipoxins and aspirin-triggered 15-epi-lipoxins. ScientificWorldJournal. 2002;2:169–204. doi: 10.1100/tsw.2002.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Serhan CN, Chiang N. Novel endogenous small molecules as the checkpoint controllers in inflammation and resolution: entree for resoleomics. Rheum Dis Clin North Am. 2004;30:69–95. doi: 10.1016/S0889-857X(03)00117-0. [DOI] [PubMed] [Google Scholar]
- 85.Serhan CN, Chiang N. Endogenous pro-resolving and anti-inflammatory lipid mediators: a new pharmacologic genus. Br J Pharmacol. 2008;153(Suppl 1):S200–S215. doi: 10.1038/sj.bjp.0707489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Serhan CN, Clish CB, Brannon J, Colgan SP, Chiang N, Gronert K. Novel functional sets of lipid-derived mediators with antiinflammatory actions generated from omega-3 fatty acids via cyclooxygenase 2-nonsteroidal antiinflammatory drugs and transcellular processing. J Exp Med. 2000;192:1197–1204. doi: 10.1084/jem.192.8.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Serhan CN, Dalli J, Karamnov S, Choi A, Park CK, Xu ZZ, Ji RR, Zhu M, Petasis NA. Macrophage proresolving mediator maresin 1 stimulates tissue regeneration and controls pain. FASEB J. 2012;26:1755–1765. doi: 10.1096/fj.11-201442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Serhan CN, Gotlinger K, Hong S, Lu Y, Siegelman J, Baer T, Yang R, Colgan SP, Petasis NA. Anti-inflammatory actions of neuroprotectin D1/protectin D1 and its natural stereoisomers: assignments of dihydroxy-containing docosatrienes. J Immunol. 2006;176:1848–1859. doi: 10.4049/jimmunol.176.3.1848. [DOI] [PubMed] [Google Scholar]
- 89.Serhan CN, Hong S, Gronert K, Colgan SP, Devchand PR, Mirick G, Moussignac RL. Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J Exp Med. 2002;196:1025–1037. doi: 10.1084/jem.20020760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Serhan CN, Jain A, Marleau S, Clish C, Kantarci A, Behbehani B, Colgan SP, Stahl GL, Merched A, Petasis NA, Chan L, Van Dyke TE. Reduced inflammation and tissue damage in transgenic rabbits overexpressing 15-lipoxygenase and endogenous anti-inflammatory lipid mediators. J Immunol. 2003;171:6856–6865. doi: 10.4049/jimmunol.171.12.6856. [DOI] [PubMed] [Google Scholar]
- 91.Serhan CN, Levy BD, Clish CB, Gronert K, Chiang N. Lipoxins, aspirin-triggered 15-epi-lipoxin stable analogs and their receptors in anti-inflammation: a window for therapeutic opportunity. Ernst Schering Res Found Workshop. 2000;31:143–185. doi: 10.1007/978-3-662-04047-8_8. [DOI] [PubMed] [Google Scholar]
- 92.Serhan CN, Maddox JF, Petasis NA, Akritopoulou-Zanze I, Papayianni A, Brady HR, Colgan SP, Madara JL. Design of lipoxin A4 stable analogs that block transmigration and adhesion of human neutrophils. Biochemistry. 1995;34:14609–14615. doi: 10.1021/bi00044a041. [DOI] [PubMed] [Google Scholar]
- 93.Serhan CN, Yang R, Martinod K, Kasuga K, Pillai PS, Porter TF, Oh SF, Spite M. Maresins: novel macrophage mediators with potent antiinflammatory and proresolving actions. J Exp Med. 2009;206:15–23. doi: 10.1084/jem.20081880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Seymour RA, Heasman PA. Drugs and the periodontium. J Clin Periodontol. 1988;15:1–16. doi: 10.1111/j.1600-051x.1988.tb01549.x. [DOI] [PubMed] [Google Scholar]
- 95.Shinohara M, Mirakaj V, Serhan CN. Functional metabolomics reveals novel active products in the DHA metabolome. Front Immunol. 2012;3:81. doi: 10.3389/fimmu.2012.00081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Smith WL, DeWitt DL, Garavito RM. Cyclooxygenases: structural, cellular, and molecular biology. Annu Rev Biochem. 2000;69:145–182. doi: 10.1146/annurev.biochem.69.1.145. [DOI] [PubMed] [Google Scholar]
- 97.Spite M, Summers L, Porter TF, Srivastava S, Bhatnagar A, Serhan CN. Resolvin D1 controls inflammation initiated by glutathione-lipid conjugates formed during oxidative stress. Br J Pharmacol. 2009;158:1062–1073. doi: 10.1111/j.1476-5381.2009.00234.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Takano T, Fiore S, Maddox JF, Brady HR, Petasis NA, Serhan CN. Aspirin-triggered 15-epi-lipoxin A4 (LXA4) and LXA4 stable analogues are potent inhibitors of acute inflammation: evidence for anti-inflammatory receptors. J Exp Med. 1997;185:1693–1704. doi: 10.1084/jem.185.9.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Titos E, Rius B, Gonzalez-Periz A, Lopez-Vicario C, Moran-Salvador E, Martinez-Clemente M, Arroyo V, Claria J. Resolvin D1 and its precursor docosahexaenoic acid promote resolution of adipose tissue inflammation by eliciting macrophage polarization toward an M2-like phenotype. J Immunol. 2011;187:5408–5418. doi: 10.4049/jimmunol.1100225. [DOI] [PubMed] [Google Scholar]
- 100.Tobin DM, Roca FJ, Oh SF, McFarland R, Vickery TW, Ray JP, Ko DC, Zou Y, Bang ND, Chau TT, Vary JC, Hawn TR, Dunstan SJ, Farrar JJ, Thwaites GE, King MC, Serhan CN, Ramakrishnan L. Host genotype-specific therapies can optimize the inflammatory response to mycobacterial infections. Cell. 2012;148:434–446. doi: 10.1016/j.cell.2011.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Van Dyke TE. Cellular and molecular susceptibility determinants for periodontitis. Periodontol 2000. 2007;45:10–13. doi: 10.1111/j.1600-0757.2007.00228.x. [DOI] [PubMed] [Google Scholar]
- 102.Van Dyke TE, Kornman KS. Inflammation and factors that may regulate inflammatory response. J Periodontol. 2008;79:1503–1507. doi: 10.1902/jop.2008.080239. [DOI] [PubMed] [Google Scholar]
- 103.Van Dyke TE, Serhan CN. Resolution of inflammation: a new paradigm for the pathogenesis of periodontal diseases. J Dent Res. 2003;82:82–90. doi: 10.1177/154405910308200202. [DOI] [PubMed] [Google Scholar]
- 104.Wallace JL, Fiorucci S. A magic bullet for mucosal protection and aspirin is the trigger! Trends Pharmacol Sci. 2003;24:323–326. doi: 10.1016/S0165-6147(03)00166-4. [DOI] [PubMed] [Google Scholar]
- 105.Wang B, Gong X, Wan JY, Zhang L, Zhang Z, Li HZ, Min S. Resolvin D1 protects mice from LPS-induced acute lung injury. Pulm Pharmacol Ther. 2011;24:434–441. doi: 10.1016/j.pupt.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 106.Yang R, Chiang N, Oh SF, Serhan CN. Metabolomics-lipidomics of eicosanoids and docosanoids generated by phagocytes. Curr Protoc Immunol. 2011 doi: 10.1002/0471142735.im1426s95. Chapter 14: Unit 14 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zlotnik A, Yoshie O. Chemokines: a new classification system and their role in immunity. Immunity. 2000;12:121–127. doi: 10.1016/s1074-7613(00)80165-x. [DOI] [PubMed] [Google Scholar]