Safely targeting cancer stem cells via selective catenin coactivator antagonism (original) (raw)
Abstract
Throughout our life, long-lived somatic stem cells (SSC) regenerate adult tissues both during homeostatic processes and repair after injury. The role of aberrant regulation of SSC has also recently gained prominence in the field of cancer research. Following malignant transformation, so termed cancer stem cells (CSC), endowed with the same properties as SSC (i.e. the ability to both self-renew and generate differentiated progenitors), play a major part in tumor initiation, therapy resistance and ultimately relapse. The same signaling pathways involved in regulating SSC maintenance are involved in the regulation of CSC. CSC exist in a wide array of tumor types, including leukemias, and brain, breast, prostate and colon tumors. Consequently, one of the key goals in cancer research over the past decade has been to develop therapeutic strategies to safely eliminate the CSC population without damaging the endogenous SSC population. A major hurdle to this goal lies in the identification of the key mechanisms that distinguish CSC from the normal endogenous tissue stem cells. This review will discuss the discovery of the specific CBP/catenin antagonist ICG-001 and the ongoing clinical development of the second generation CBP/catenin antagonist PRI-724. Importantly, specific CBP/catenin antagonists appear to have the ability to safely eliminate CSC by taking advantage of an intrinsic differential preference in the way SSC and CSC divide.
Keywords: Asymmetric, CREB-binding protein, p300, symmetric, Wnt/catenin
Metastasis, multi-drug resistance and disease relapse constitute the central challenge for the successful treatment of advanced malignancies. Tumor initiation, metastasis and disease relapse have all recently been attributed to subpopulations of self-renewing, highly tumorigenic, drug-resistant tumor cells termed cancer stem cells (CSC) or, alternatively, tumor initiating cells (TIC).1 CSC have many of the same attributes that their normal somatic stem cell (SSC) counterparts are endowed with, in that they have the ability to both self-renew and go on to more differentiated cell types. SSC, alternatively termed tissue stem cells, reside in specialized niches within tissues or organs (e.g. hematopoietic stem cells, neuronal stem cells and intestinal stem cells) and are critical during development as well as in the adult for both normal tissue homeostasis and regeneration after injury.2 Therefore, a recent major focus in cancer research has been to develop therapeutic strategies to safely eliminate the CSC population without deleterious effects on the normal SSC population.
To safely accomplish this goal, we need to identify the key mechanisms that regulate stem cell self-renewal versus differentiation and, even more critically, the features that distinguish the control of self-renewal of CSC from their normal endogenous SSC counterparts. However, essentially the same evolutionarily conserved signaling pathways that govern embryonic development (i.e. Wnt/β-catenin,3,4 Hedgehog5 and Notch6) appear to control the behavior of both normal SSC as well as CSC. CSC express similar markers and exhibit cellular behaviors highly reminiscent of SSC. One conclusion that can be drawn from the literature is that there are multiple points of intersection and crosstalk, including feedback and feed-forward loops, connecting the various signaling cascades that modulate “stemness.” The focus of this review will be on the role of nuclear catenin (both β-catenin and γ-catenin / plakoglobin) in the transcriptional regulation of CSC and the prospects for safely and effectively targeting catenin coactivator interactions to eliminate the CSC population in cancer.
Cancer Stem Cells and Their Role in Tumorigenesis
Cohnheim et al.7 first proposed the concept that cancer might arise from a rare population of cells with stem cell-like properties almost 150 years ago. More recently, increasing evidence has confirmed the existence of a small subset of cells termed cancer stem cells (CSC) or, alternatively, tumor initiating cells (TIC), which are believed to be responsible for tumor initiation, drug resistance and metastasis. Acknowledgement of the presence of CSC has forced a paradigm shift from the earlier models of tumor homogeneity towards one of tumor hierarchy, where CSC play the critical role.8 The CSC concept postulates that a small population of CSC provide for the long-term maintenance of the tumor, whereas the bulk of the tumor consists of rapidly proliferating and differentiated (albeit aberrantly or only partially differentiated) cells. CSC are able to self-renew,9 actively express telomerase10 and express multidrug resistance pathways.11,12 CSC are generally quiescent, but can give rise to rapidly dividing transient amplifying cells, which form the bulk of tumor cells (Fig.1). Despite some still existing controversy regarding the CSC hypothesis,13 it is clear that distinct cancer cell populations have enhanced tumorigenic capacity compared to bulk tumor cells. John Dick and colleagues first isolated CSC (known as leukemic stem cells [LSC]) from bulk acute myeloid leukemia cells in 1997.14 LSC maintained or reacquired the ability to proliferate indefinitely without proper differentiation.15 Over the past decade, a large number of studies have also identified CSC in solid tumors, including brain,16 melanoma,17 breast,18 liver,19 pancreatic20 and colon cancer.21
Fig 1.
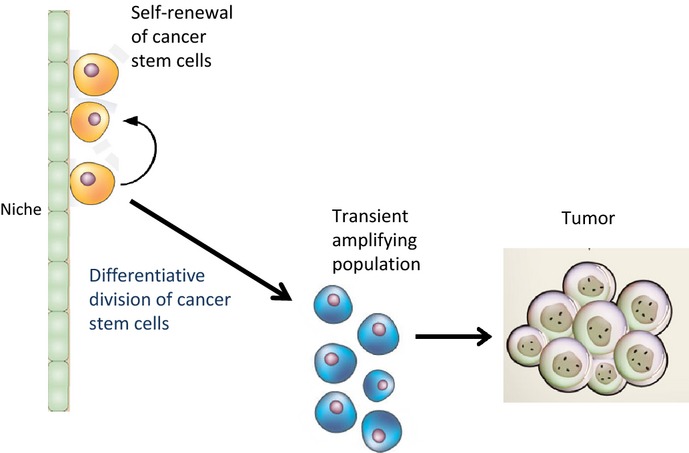
Cancer stem cells both self-renew and undergo differentiative divisions to maintain or expand the cancer stem cell population or generate transient amplifying cells that go on to rapidly divide to form the bulk of the tumor.
Catenin Dependent Transcription and “Stemness”
The entry of catenin (classically β-catenin, although other catenins [.g. γ-catenin/plakoglobin] may also play a critical role)22 into the nucleus and the subsequent transcriptional processes affected by β-catenin are classically controlled by the so termed “canonical Wnt” signaling cascade. However, there are a number of alternative signaling pathways that can induce the nuclear translocation of β-catenin and its subsequent participation in transcription. For example, the process of epithelial to mesenchymal transition (EMT) involves downregulation of E-cadherin, which normally binds cytoplasmic β-catenin in a complex with α-catenin that stabilizes epithelial architecture,23 leading to the subsequent nuclear translocation of β-catenin.24 EMT is a hallmark of metastasis25 and has also been implicated in the generation of CSC.26 A variety of receptor tyrosine kinases27 and non-receptor tyrosine kinases including Src28 and Abl29 can also disrupt the E-Cadherin/β-catenin interaction, thereby enhancing β-catenin mediated transcription. In addition, hypoxia30,31 and high glucose levels32 can also activate β-catenin mediated signaling. It is clear that a wide range of signaling molecules and cascades also influence β-catenin dynamics and β-catenin transcription.33–35
In collaboration with signals from a number of other key pathways (e.g. Notch, Hedgehog, JAK/Stat, BMP, Hippo and FGF/MAPK), Wnt glycoproteins and, in particular, nuclear β-catenin, play essential roles in balancing self-renewal versus differentiation of adult stem cells (Fig.2). However, there has been enormous controversy regarding whether Wnt signaling is important for proliferation and maintenance of potency (pluripotency or multipotency)1,36,37 or differentiation of stem/progenitor cells.38,39 Wnt/β-catenin signaling has been shown to maintain pluripotency in ES cells37 and expand neural stem/progenitors, thereby increasing brain size.40 However, Wnt/β-catenin signaling is also required for the differentiation of ES cells,41 as well as fate determination in neural crest stem cells.42 Clearly, Wnt/β-catenin signaling plays dichotomous roles in stem cell biology.
Fig 2.

The ultimate decision for a cell to retain potency or initiate differentiation is dependent upon numerous inputs from various signaling pathways (e.g. JAK/STAT, Wnt, Growth Factors, Hippo, Notch and Hedgehog) that also play critical roles during development. In the end, those multiple pathways must be integrated and funneled down into a simple decision point. β-catenin plays a central role in integrating these signals.
Wnt/Catenin, Cancer and Cancer Stem Cells
Wnt signaling is an enormously complex and ancient pathway that dates back to the first anaerobic metazoans. The Wnt/catenin pathway is critical in both normal embryonic development and throughout the life of the organism in virtually every tissue and organ system.
The pathway has emerged as a pivotal player in the specification and maintenance of stem cell lineages in multiple stem cell compartments in a wide array of tissues and organs, including intestines, the heart, and hematopoietic, neuronal and mammary glands.43 Therefore, not surprisingly, a recurrent theme in cancer biology involves the aberrant regulation of Wnt signaling.44,45 The discovery in 1991 that mutations in the tumor suppressor adenomatous polyposis coli (APC)46,47 were associated with >80% of sporadic colorectal cancers via aberrant activation of Wnt signaling provided significant rationale to therapeutically target this pathway. APC is the most frequently mutated gene in human cancers.48,49 However, mutations affecting the Wnt pathway are not restricted to colon cancer. Loss-of-function mutations in Axin have been found in hepatocellular carcinomas, and oncogenic β-catenin mutations, first described in colon cancer and melanoma,50 have also been found in a wide variety of solid tumors,51 including hepatocellular carcinomas,52 thyroid tumors53 and ovarian endometrioid adenocarcinomas.54
Safely Targeting Cancer Stem Cells
The significant role of aberrant Wnt signaling in cancer and CSC has engendered substantial efforts into the development of therapeutic approaches to target this pathway. However, a number of factors have thwarted progress in this field. First, the Wnt signaling cascade is bewilderingly complex, in that in mammals there are 19 Wnt ligands and more than 15 receptors and co-receptors distributed over seven protein families,55 and this represents only the tip of the iceberg in regards to the difficulty in attempting to develop safe and effective specific Wnt pathway therapeutics. Further adding to the complexity of targeting transcriptionally competent β-catenin is the fact that β-catenin can bind to a broad spectrum of transcription factors other than TCF/LEF. Transcriptionally active β-catenin therefore modulates a plethora of downstream biological processes, including pluripotency, EMT, oxidative stress and lineage commitment.56
Successful therapeutic manipulation of endogenous “stemness” (normal or cancerous) will require significant precision to affect the desired transformations without deleterious effects (depletion, in particular) to the normal stem cell populations.4 Thus, the ability to target aberrant catenin transcriptional signaling offers enormous promise. However, just like the Sword of Damocles, significant risks and concerns regarding targeting such a critical pathway in stem cell maintenance and tissue homeostasis are ever present.
Differential Coactivator Modulation
To generate a transcriptionally active complex, β-catenin must recruit one of the two Kat3 transcriptional coactivators, cAMP response element binding protein (CREB)-binding protein (CBP) or its closely related homolog p300 (E1A-binding protein, 300 KDa), as well as other components of the basal transcriptional apparatus.57–59 Recent studies have documented that CBP and p300 interact with hundreds of proteins in their roles as master orchestrators of transcription. Due to their high degree of homology, these two coactivators have long been considered as largely redundant. However, accumulating evidence indicates that CBP and p300 are not redundant but have definitive and unique roles both in vitro and in vivo.60–62
Using the TopFlash reporter gene system in SW480 colon carcinoma cells, we identified ICG-001 from a library of 5000 secondary structure mimetics. ICG-001 (Fig.3) had an IC50 value of 3 μM in this assay. Using an affinity chromatography approach, we identified and subsequently validated that ICG-001 binds specifically and with high affinity (approximately 1 nM) to the coactivator CBP, but, importantly, not to its closely related homolog p300, despite the fact that these two coactivators are up to 93% identical, with even higher homology, at the amino acid level.63,64 We demonstrated that selectively blocking the interaction between CBP and β-catenin with ICG-001 led to the initiation of a differentiation program in a wide variety of stem/progenitor cells.65,66 This led us to develop our model of differential coactivator usage, which highlights the distinct roles of the coactivators CBP and p300 in catenin-mediated transcription.58 The critical decision by β-catenin to utilize either CBP or p300 is the first decision that guides the cell to either proliferate/maintain potency or initiate a differentiation transcriptional program, respectively (Fig.4).
Fig 3.
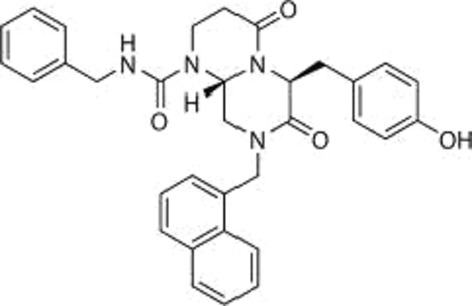
Chemical structure of the CBP/catenin antagonist ICG-001.
Fig 4.
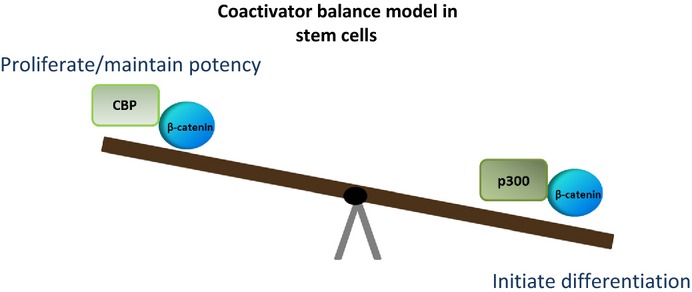
Wnt signaling is a complex pathway, believed to be involved in the regulation of divergent processes, including the maintenance of pluripotency and commitment to differentiation. We developed a model in which β-catenin/CBP-mediated transcription is critical for the maintenance of potency, whereas β-catenin/p300-mediated transcription is the first critical step to initiate differentiation. Hence, the balance between CBP and p300-mediated β-catenin transcription regulates the balance between maintenance of potency, and the initiation of commitment to differentiate in stem and progenitor cells.
Subsequently, we have identified several small molecules (IQ-1, ID-8 and, most recently, YH249/250) that selectively block the p300/β-catenin interaction, thereby increasing the CBP/β-catenin interaction, which maintains potency (pluripotency or multipotency) in a variety of stem cell populations, both in mouse and human.65,67–69 The therapeutic potential of the selective CBP/β-catenin antagonist ICG-001 has been examined in a variety of preclinical tumor models, where it has demonstrated the ability to safely eliminate drug-resistant tumor-initiating cells.70–72
Interestingly, CBP/β-catenin antagonists have also demonstrated efficacy in a variety of injury models, including pulmonary and renal fibrosis73,74 and myocardial infarction.75 It appears that the differential effects of CBP/β-catenin antagonists on CSC versus normal SSC (i.e. forced differentiation and elimination versus differentiation and enhanced repair without apparent depletion) are apparently cell intrinsic and not due to the selective targeting by CBP/β-catenin antagonists of CSC versus normal SSC. We proposed that CBP/β-catenin antagonists take advantage of the intrinsic propensity of CSC to increase their number of symmetric divisions at the expense of asymmetric divisions due to various mutations (e.g. p53 and PTEN).76,77 Normal endogenous long-term repopulating stem cells preferentially divide asymmetrically with one daughter cell remaining in the niche and the other going on to a transient amplifying cell required for generating the new tissue involved in repair processes.78 However, if CSC undergo more symmetric differentiative divisions when treated with CBP/catenin antagonists, the CSC in the niche will eventually be cleared out, whereas normal SSC that divide asymmetrically will always maintain one of the dividing daughter cells in the stem cell niche (Fig.5). This fundamental and cell intrinsic difference between SSC and CSC provides a unique opportunity to therapeutically target CSC without damaging the normal endogenous stem cell populations utilizing specific CBP/catenin antagonists.78
Fig 5.
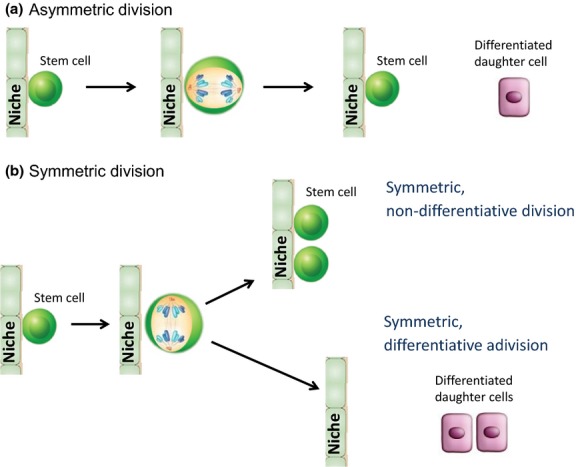
Model depicting symmetric and asymmetric modes of division. The intrinsic difference between normal somatic stem cells (SSC) and cancer stem cells (CSC) is that normal SSC favor asymmetric division whereas CSC favor symmetric divisions. Treatment of CSC with CBP/catenin antagonists causes CSC to undergo symmetric differentiative divisions, thereby eventually clearing CSC from the niche. In sharp contrast, SSC undergo asymmetric divisions when treated with CBP/catenin antagonists.
To The Clinic
Although the Wnt signaling pathway was discovered over 30 years ago, only recently have therapeutic agents that specifically target the Wnt pathway been introduced into clinical trials, although a few US Food and Drug Administration (FDA)-approved drugs do affect Wnt signaling, albeit non-specifically.4 Despite intensive investigation of the pathway and the unveiling of a multitude of potential therapeutic points of intervention in the pathway, as well as the identification of reagents that interfere with some of these targets, it is still unclear whether most approaches will provide both clinical efficacy and safety. To date, pre-clinical and clinical experience with both small molecules and biologics that target different points of intervention (porcupine, tankyrase, Fzd receptors and extracellular Wnt ligands) suggest that a therapeutic window does exist for the use of Wnt inhibitors in cancer patients. However, the full anti-tumor potential of these agents may not be realized due to side effects involving on target inhibition of Wnt/β-catenin signaling including intestinal toxicity and bone breakage.79
PRI-724, a specific CBP/catenin clinical compound
In principle, significant concerns about specificity could be raised about the use of small molecule inhibitors that target the coactivator protein CBP, which has perhaps as many as 500 molecular partners, including a wide array of transcription factors. However, to date, these concerns have not been borne out either pre-clinically or clinically. This is perhaps at first surprising and a full discussion of why a small molecule therapeutic that selectively targets the N-terminus of CBP has many therapeutic advantages is beyond the scope of this review.78 However, a few salient features are worth mentioning: (i) the extremely high biochemical selectivity of ICG-001/PRI-724 for its molecular target; (ii) the disruption of only a small subset of CBP interactions; and (iii) the unique properties of the two Kat3 coactivators, CBP and p300.
PRI-724 is a second generation specific CBP/catenin antagonist (IC50–150 nM) developed by Prism Pharma and partnered with Eisai Pharmaceuticals for oncology. PRI-724 proved to be extremely safe in pre-clinical investigational new drug enabling toxicology studies. The No Adverse Event Level for PRI-724 was 120 mg/kg/day in dogs given 28-day continuous infusion. An open label Phase Ia safety study in subjects with solid tumors, where the expression of the biomarker survivin/BIRC5 was measured by immuno-magnetic RT-PCR in circulating tumor cells for PRI-724 was initiated at USC in March 2011. The results of this trial were reported at ASCO in June 2013. In all, 18 patients were treated (dose escalation from 40 to 1280 mg/m2/day) via continuous infusion for 7 days. Just as had been observed in preclinical studies, PRI-724 had a very acceptable toxicity profile, with only one DLT of grade 3 of reversible hyperbilirubinemia. Downregulation of the biomarker survivin/BIRC5 with upregulation of the differentiation antigen CK20 in circulating tumor cells strongly correlated with increasing plasma concentrations of the drug.80 Additional trials with PRI-724 are underway, including combination trials with a modified Folfox6 regimen for refractory colorectal cancer patients, a Phase Ib trial for refractory pancreatic cancer patients in combination with gemcitabine and a Phase 1/2 trial for heme malignancies.
As mentioned above, CBP/β-catenin antagonists have also demonstrated efficacy in a wide variety of injury models, including pulmonary and renal fibrosis73,74 and myocardial infarction.75 Given the apparent safety of these agents both pre-clinically and clinically, additional clinical trials targeting these indications are anticipated in the future.
Acknowledgments
We would like to thank Dr Jia-Ling Teo for a critical review of and assistance with the preparation of this manuscript.
Disclosure Statement
The corresponding author is a consultant and equity holder in Prism Pharma Co. Ltd.
References
- Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–11. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- Singh SR. Stem cell niche in tissue homeostasis, aging and cancer. Curr Med Chem. 2012;19:5965–74. doi: 10.2174/092986712804485917. [DOI] [PubMed] [Google Scholar]
- Miki T, Yasuda SY, Kahn M. “Wnt/Β-catenin signaling in embryonic stem cell self-renewal and somatic cell reprogramming. Stem Cell Rev. 2011;7:836–46. doi: 10.1007/s12015-011-9275-1. [DOI] [PubMed] [Google Scholar]
- Takahashi-Yanaga F, Kahn M. Targeting Wnt signaling: can we safely eradicate cancer stem cells? Clin Cancer Res. 2010;16:3153–62. doi: 10.1158/1078-0432.CCR-09-2943. [DOI] [PubMed] [Google Scholar]
- Merchant AA, Matsui W. Targeting hedgehog – a cancer stem cell pathway. Clin Cancer Res. 2010;16:3130–40. doi: 10.1158/1078-0432.CCR-09-2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Sato C, Cerletti M, Wagers A. Notch signaling in the regulation of stem cell self-renewal and differentiation. Curr Top Dev Biol. 2010;92:367–409. doi: 10.1016/S0070-2153(10)92012-7. [DOI] [PubMed] [Google Scholar]
- Cohnheim J. Ueber Entzuendung und Eiterung [in German] Path Anat Physiol Klin Med. 1867;40:1–79. [Google Scholar]
- Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8:755–68. doi: 10.1038/nrc2499. [DOI] [PubMed] [Google Scholar]
- O'Brien CA, Kreso A, Jamieson CH. Cancer stem cells and self-renewal. Clin Cancer Res. 2010;16:3113–20. doi: 10.1158/1078-0432.CCR-09-2824. [DOI] [PubMed] [Google Scholar]
- Armanios M, Greider CW. Telomerase and cancer stem cells. Cold Spring Harb Symp Quant Biol. 2005;70:205–8. doi: 10.1101/sqb.2005.70.030. [DOI] [PubMed] [Google Scholar]
- Clevers H. The cancer stem cell: premises, promises and challenges. Nat Med. 2011;17:313–9. doi: 10.1038/nm.2304. [DOI] [PubMed] [Google Scholar]
- Visvader JE, Lindeman GJ. Stem cells and cancer – the promise and puzzles. Mol Oncol. 2010;4:369–72. doi: 10.1016/j.molonc.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly PN, Dakic A, Adams JM, Nutt SL, Strasser A. Tumor growth need not be driven by rare cancer stem cells. Science. 2007;317:337. doi: 10.1126/science.1142596. [DOI] [PubMed] [Google Scholar]
- Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730–7. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- Jamieson CH, Weissman IL, Passegué E. Chronic versus acute myelogenous leukemia: a question of self-renewal. Cancer Cell. 2004;6:531–3. doi: 10.1016/j.ccr.2004.12.005. [DOI] [PubMed] [Google Scholar]
- Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- Fang D, Nguyen TK, Leishear K, et al. A tumorigenic subpopulation with stem cell properties in melanomas. Cancer Res. 2005;65:9328–37. doi: 10.1158/0008-5472.CAN-05-1343. [DOI] [PubMed] [Google Scholar]
- Dontu G, El-Ashry D, Wicha MS. Breast cancer, stem/progenitor cells and the estrogen receptor. Trends Endocrinol Metab. 2004;15:193–7. doi: 10.1016/j.tem.2004.05.011. [DOI] [PubMed] [Google Scholar]
- Ma S, Chan KW, Hu L, et al. Identification and characterization of tumorigenic liver cancer stem/progenitor cells. Gastroenterology. 2007;132:2542–56. doi: 10.1053/j.gastro.2007.04.025. [DOI] [PubMed] [Google Scholar]
- Li C, Heidt DG, Dalerba P, et al. Identification of Pancreatic Cancer Stem Cells. Cancer Res. 2007;67:1030–7. doi: 10.1158/0008-5472.CAN-06-2030. [DOI] [PubMed] [Google Scholar]
- O'Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106–10. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- Kim YM, Ma H, Oehler V, et al. The gamma catenin/CBP complex maintains survivin transcription in β-catenin deficient/depleted cancer cells. Curr Cancer Drug Targets. 2011;11:213–25. doi: 10.2174/156800911794328420. [DOI] [PubMed] [Google Scholar]
- Onder TT, Gupta PB, Mani SA, et al. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 2008;68:3645–54. doi: 10.1158/0008-5472.CAN-07-2938. [DOI] [PubMed] [Google Scholar]
- Kim K, Daniels KJ, Ha ED. Tissue-specific expression of beta-catenin in normal mesenchyme and uveal melanomas and its effect on invasiveness. Exp Cell Res. 1998;245:79–90. doi: 10.1006/excr.1998.4238. [DOI] [PubMed] [Google Scholar]
- Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. 2011;147:275–92. doi: 10.1016/j.cell.2011.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani SA, Guo W, Liao MJ, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–15. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagh PK, Gary JK, Zinser GM, et al. β-Catenin is required for Ron receptor-induced mammary tumorigenesis. Oncogene. 2011;30:3694–704. doi: 10.1038/onc.2011.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coluccia AM, Benati D, Dekhil H, et al. SKI-606 decreases growth and motility of colorectal cancer cells by preventing pp60(c-Src)-dependent tyrosine phosphorylation of beta-catenin and its nuclear signaling. Cancer Res. 2006;66:2279–86. doi: 10.1158/0008-5472.CAN-05-2057. [DOI] [PubMed] [Google Scholar]
- Ress A, Moelling K. Bcr interferes with beta-catenin-Tcf1 interaction. FEBS Lett. 2006;580:1227–30. doi: 10.1016/j.febslet.2006.01.034. [DOI] [PubMed] [Google Scholar]
- Mazumdar J, Obrien WT, Johnson RS, et al. O2 regulates stem cells through Wnt/β-catenin signalling. Nat Cell Biol. 2010;12:1007–13. doi: 10.1038/ncb2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kida A, Kahn M. Hypoxia selects for a quiescent, CML stem/leukemia initiating-like population dependent on CBP/catenin transcription. Curr Mol Pharmacol. 2013;6:204–10. doi: 10.2174/1874467207666140219121219. [DOI] [PubMed] [Google Scholar]
- Chocarro-Calvo A, García-Martínez JM, Ardila-González S, De la Vieja A, García-Jiménez C. Glucose-induced β-catenin acetylation enhances Wnt signaling in cancer. Mol Cell. 2013;49:474–86. doi: 10.1016/j.molcel.2012.11.022. [DOI] [PubMed] [Google Scholar]
- Brembeck FH, Rosário M, Birchmeier W. Balancing cell adhesion and Wnt signaling, the key role of beta-catenin. Curr Opin Genet Dev. 2006;16:51–9. doi: 10.1016/j.gde.2005.12.007. [DOI] [PubMed] [Google Scholar]
- van Veelen W, Le NH, Helvensteijn W, et al. β-catenin tyrosine 654 phosphorylation increases Wnt signalling and intestinal tumorigenesis. Gut. 2011;60:1204–12. doi: 10.1136/gut.2010.233460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabata A. Prostaglandin E2 and pain – an update. Biol Pharm Bull. 2011;34:1170–3. doi: 10.1248/bpb.34.1170. [DOI] [PubMed] [Google Scholar]
- Ginis , Luo Y, Miura T, et al. Differences between human and mouse embryonic stem cells. Dev Biol. 2004;269:360–80. doi: 10.1016/j.ydbio.2003.12.034. [DOI] [PubMed] [Google Scholar]
- Sato N, Meijer L, Skaltsounis L, Greengard P, Brivanlou AH. Maintenance of pluripotency in human and mouse embryonic stem cells through activation of Wnt signaling by a pharmacological GSK-3-specific inhibitor. Nat Med. 2004;10:55–63. doi: 10.1038/nm979. [DOI] [PubMed] [Google Scholar]
- Muroyama Y, Kondoh H, Takada S. Wnt proteins promote neuronal differentiation in neural stem cell culture. Biochem Biophys Res Commun. 2004;313:915–21. doi: 10.1016/j.bbrc.2003.12.023. [DOI] [PubMed] [Google Scholar]
- Dravid G, Ye Z, Hammond H, et al. Defining the role of Wnt/beta-catenin signaling in the survival, proliferation, and self-renewal of human embryonic stem cells. Stem Cells. 2005;23:1489–501. doi: 10.1634/stemcells.2005-0034. [DOI] [PubMed] [Google Scholar]
- Chenn A, Walsh CA. Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science. 2002;297:365–9. doi: 10.1126/science.1074192. [DOI] [PubMed] [Google Scholar]
- Zehner D, Fujita Y, Hulsken J, et al. beta-Catenin signals regulate cell growth and the balance between progenitor cell expansion and differentiation in the nervous system. Dev Biol. 2003;258:406–18. doi: 10.1016/s0012-1606(03)00123-4. [DOI] [PubMed] [Google Scholar]
- Hari L, Brault V, Kleber M, et al. Lineage-specific requirements of beta-catenin in neural crest development. J Cell Biol. 2002;159:867–80. doi: 10.1083/jcb.200209039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kühl SJ, Kühl M. On the role of Wnt/β-catenin signaling in stem cells. Biochim Biophys Acta. 2013;1830:2297–306. doi: 10.1016/j.bbagen.2012.08.010. [DOI] [PubMed] [Google Scholar]
- Polakis P. Drugging Wnt signalling in cancer. EMBO J. 2012;31:2737–46. doi: 10.1038/emboj.2012.126. . Erratum in:. 2012; : 3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastas JN, Moon RT. WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer. 2013;13:11–26. doi: 10.1038/nrc3419. [DOI] [PubMed] [Google Scholar]
- Groden J, Thliveris A, Samowitz W, et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell. 1991;66:589–600. doi: 10.1016/0092-8674(81)90021-0. [DOI] [PubMed] [Google Scholar]
- Kinzler KW, Nilbert MC, Su LK, et al. Identification of FAP locus genes from chromosome 5q21. Science. 1991;253:661–5. doi: 10.1126/science.1651562. [DOI] [PubMed] [Google Scholar]
- Clements WM, Lowy AM, Grode J. Adenomatous polyposis coli/beta-catenin interaction and downstream targets: altered gene expression in gastrointestinal tumors. Clin Colorectal Cancer. 2003;3:113–20. doi: 10.3816/ccc.2003.n.018. [DOI] [PubMed] [Google Scholar]
- Drier Y, Lawrence MS, Carter SL, et al. Somatic rearrangements across cancer reveal classes of samples with distinct patterns of DNA breakage and rearrangement-induced hypermutability. Genome Res. 2013;23:228–35. doi: 10.1101/gr.141382.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinfeld B, Robbins O, El-Gamil M, Albert I, Porfiri E, Polakis P. Stabilization of beta-catenin by genetic defects in melanoma cell lines. Science. 1997;275:1790–2. doi: 10.1126/science.275.5307.1790. [DOI] [PubMed] [Google Scholar]
- Herr P, Hausmann G, Basler K. WNT secretion and signalling in human disease. Trends Mol Med. 2012;18:483–93. doi: 10.1016/j.molmed.2012.06.008. [DOI] [PubMed] [Google Scholar]
- Han ZG. Functional genomic studies: insights into the pathogenesis of liver cancer. Annu Rev Genomics Hum Genet. 2012;13:171–205. doi: 10.1146/annurev-genom-090711-163752. [DOI] [PubMed] [Google Scholar]
- Sastre-Perona A, Santisteban P. Role of the wnt pathway in thyroid cancer. Front Endocrinol (Lausanne) 2012;3:31. doi: 10.3389/fendo.2012.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratcliffe TA, Monk BJ, Planutis K, Holcombe RF. Wnt signaling in ovarian tumorigenesis. Int J Gynecol Cancer. 2008;18:954–62. doi: 10.1111/j.1525-1438.2007.01127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niehrs C. The complex world of WNT receptor signalling. Nat Rev Mol Cell Biol. 2012;13:767–79. doi: 10.1038/nrm3470. [DOI] [PubMed] [Google Scholar]
- Le NH, Franken P, Fodde R. Tumour-stroma interactions in colorectal cancer: converging on beta-catenin activation and cancer stemness. Br J Cancer. 2008;98:1886–93. doi: 10.1038/sj.bjc.6604401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon RT. Wnt/beta-catenin pathway. Sci STKE. 2005;271:cm1. doi: 10.1126/stke.2712005cm1. [DOI] [PubMed] [Google Scholar]
- Teo J-L, Kahn M. The Wnt signaling pathway in cellular proliferation and differentiation: a tale of two coactivators. Adv Drug Deliv Rev. 2010;62:1149–55. doi: 10.1016/j.addr.2010.09.012. [DOI] [PubMed] [Google Scholar]
- Angers S, Moon RT. Proximal events in Wnt signal transduction. Nat Rev Mol Cell Biol. 2009;10:468–77. doi: 10.1038/nrm2717. [DOI] [PubMed] [Google Scholar]
- Kung AL, Rebel VI, Bronson RT, et al. Gene dose-dependent control of hematopoiesis and hematologic tumor suppression by CBP. Genes Dev. 2000;14:272–7. [PMC free article] [PubMed] [Google Scholar]
- Yamauchi T, Oike Y, Kamon J, et al. Increased insulin sensitivity despite lipodystrophy in Crebbp heterozygous mice. Nat Genet. 2002;30:221–6. doi: 10.1038/ng829. [DOI] [PubMed] [Google Scholar]
- Roth FJ, Shikama N, Henzen C, et al. Differential role of p300 and CBP acetyltransferase during myogenesis: p300 acts upstream of MyoD and Myf5. EMBO J. 2003;2003:5186–96. doi: 10.1093/emboj/cdg473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan M, Kahn M. Investigating Wnt signaling: a chemogenomic safari. Drug Discov Today. 2005;10:1467–74. doi: 10.1016/S1359-6446(05)03613-5. [DOI] [PubMed] [Google Scholar]
- Emami KH, Nguyen C, Ma H, et al. A small molecule inhibitor of beta-catenin/CREB-binding protein transcription [corrected] Proc Natl Acad Sci USA. 2004;101:12682–7. doi: 10.1073/pnas.0404875101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa K, Yasuda SY, Teo JL, et al. Wnt signaling orchestration with a small molecule DYRK inhibitor provides long-term xeno-free human pluripotent cell expansion. Stem Cells Transl Med. 2012;1:18–28. doi: 10.5966/sctm.2011-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee ER, Laflamme MA, Papayannopoulou T, Kahn M, Murry CE, Henderson WR., Jr Human embryonic stem cells differentiated to lung lineage-specific cells ameliorate pulmonary fibrosis in a xenograft transplant mouse model. PLoS ONE. 2012;7:e33165. doi: 10.1371/journal.pone.0033165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyabayashi T, Teo JL, Yamamoto M, McMillan M, Nguyen C, Kahn M. Wnt/beta-catenin/CBP signaling maintains long-term murine embryonic stem cell pluripotency. Proc Natl Acad Sci USA. 2007;104:5668–73. doi: 10.1073/pnas.0701331104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marson A, Foreman R, Chevalier B, et al. Wnt signaling promotes reprogramming of somatic cells to pluripotency. Cell Stem Cell. 2008;3:132–5. doi: 10.1016/j.stem.2008.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenke-Layland K, Nsair A, Van Handel B, et al. Recapitulation of the embryonic cardiovascular progenitor cell niche. Biomaterials. 2011;2:2748–56. doi: 10.1016/j.biomaterials.2010.12.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wend P, Fang L, Zhu Q, et al. Wnt/β-catenin signalling induces MLL to create epigenetic changes in salivary gland tumours. EMBO J. 2013;32:1977–89. doi: 10.1038/emboj.2013.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gang EJ, Hsieh YT, Pham J, et al. Small-molecule inhibition of CBP/catenin interactions eliminates drug-resistant clones in acute lymphoblastic leukemia. Oncogene. 2014;33:2169–78. doi: 10.1038/onc.2013.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He K, Xu T, Xu Y, Ring A, Kahn M, Goldkorn A. Cancer cells acquire a drug resistant, highly tumorigenic, cancer stem-like phenotype through modulation of the PI3K/Akt/β-catenin/CBP pathway. Int J Cancer. 2014;134:43–54. doi: 10.1002/ijc.28341. [DOI] [PubMed] [Google Scholar]
- Henderson WR, Jr, Chi EY, Ye X, et al. Inhibition of Wnt/beta-catenin/CREB binding protein (CBP) signaling reverses pulmonary fibrosis. Proc Natl Acad Sci USA. 2010;107:14309–14. doi: 10.1073/pnas.1001520107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao S, He W, Li Y, et al. Targeted inhibition of β-catenin/CBP signaling ameliorates renal interstitial fibrosis. J Am Soc Nephrol. 2011;22:1642–53. doi: 10.1681/ASN.2010101079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki T, Hwang H, Nguyen C, Kloner RA, Kahn M. The small molecule Wnt signaling modulator ICG-001 improves contractile function in chronically infarcted rat myocardium. PLoS ONE. 2013;8:e75010. doi: 10.1371/journal.pone.0075010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicalese A, Bonizzi G, Pasi CE, et al. The tumor suppressor p53 regulates polarity of self-renewing divisions in mammary stem cells. Cell. 2009;138:1083–95. doi: 10.1016/j.cell.2009.06.048. [DOI] [PubMed] [Google Scholar]
- Lee JY, Nakada D, Yilmaz OH, et al. mTOR activation induces tumor suppressors that inhibit leukemogenesis and deplete hematopoietic stem cells after Pten deletion. Cell Stem Cell. 2010;7:593–905. doi: 10.1016/j.stem.2010.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn M. Symmetric division versus asymmetric division: a tale of two coactivators. Future Med Chem. 2011;3:1745–63. doi: 10.4155/fmc.11.126. [DOI] [PubMed] [Google Scholar]
- Sakata T, Chen JK. Chemical ‘Jekyll and Hyde's: small-molecule inhibitors of developmental signaling pathways. Chem Soc Rev. 2011;40:4318–31. doi: 10.1039/c1cs15019g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Khoueiry AB, Ning Y, Yang D, et al. A phase I first-in-human study of PRI-724 in patients (pts) with advanced solid tumors. J Clin Oncol. 2013;31(Suppl):2501. Abstract. [Google Scholar]