History of myeloid derived suppressor cells (MDSCs) in the macro- and micro-environment of tumour-bearing hosts (original) (raw)
. Author manuscript; available in PMC: 2015 Mar 13.
Published in final edited form as: Nat Rev Cancer. 2013 Oct;13(10):739–752. doi: 10.1038/nrc3581
Abstract
Tumour-induced granulocytic hyperplasia is associated with tumour vasculogenesis and escape from immunity via T-cell suppression. Initially, these myeloid cells were identified as granulocytes or monocytes; however, recent studies revealed that this hyperplasia was associated with populations of multi-potent progenitor cells identified as myeloid-derived suppressor cells (MDSCs). The discovery and study of MDSCs have provided a wealth of information regarding tumour pathobiology, extended our understanding of neoplastic progression, and modified our approaches to immune adjuvant therapy. In this perspective, we discuss the history of MDSCs, their influence on tumour progression and metastasis, and the crosstalk between tumour cells, MDSCs, and the host macroenvironment.
INTRODUCTION
One characteristic of tumour progression, originally described in the early 1900s1, is an increase in extramedullary haematopoiesis (EMH) and neutrophilia that was later shown to result in immune evasion and tumour vascularization. Classically, this occurs within the host macroenvironment and is associated with increased serum haematopoietic, colony-stimulating activity2 and abnormal myeloid cell differentiation resulting in a bidirectional molecular crosstalk between tumour cells and myeloid progenitor cells. Originally, these abnormal myeloid cells were described as veto cells, null cells, or natural-suppressor (NS) cells and were later shown to inhibit lymphocyte numbers, cytotoxic T-lymphocyte (CTL) induction, and activity3. These cells lacked membrane markers for mature T-cells, B-cells, and natural killer (NK) cells, as well as, macrophages4, 5, resulting in the nomenclature of null cells. Initially, their phenotypic characterization was contentious, and it continues to be partially unresolved due to investigator-dependent phenotypic marker profiles and cellular heterogeneity6. Consistent with tumor heterogeneity7, there is a tumour-dependent variability in myeloid-cell expansion that may be associated with the secretion of differing cytokines and chemokines. In recent years, the concept of myeloid-derived suppressor cells (MDSCs) was introduced to reflect the abnormal nature of myelopoiesis in cancer, which is the focus of this review. These studies have revealed that circulating MDSC numbers correlate with a poor prognosis, tumour vasculogenesis, osteoporosis, and tumour evasion of host immunity8–10.
A direct relationship between tumour burden and MDSC frequency has been demonstrated in several mouse tumour models11, 12 and clinical studies8–10, as well as, an inverse correlation between MDSC and T-cell frequency in the peripheral blood (PB)12. While this may be model and, potentially, tumour dependent, a direct relationship between tumour burden and MDSC frequency and numbers is generally accepted. In support of this observation, the resection of solid tumours has been shown to decrease MDSC frequency in the PB and to reverse T-cell suppression13, 14. The increase in MDSCs depends both on tumour burden12, 15, 16 and the tumor-secreted factors17–19 regulating myeloid progenitor cell survival and expansion. Antibody-mediated depletion of MDSCs also restores T-cell frequency and function20, 21. Confirmation of these observations using transplantable tumours has been provided with a mouse mammary tumour virus (MMTV) c-erB_-2_ transgenic mouse model of breast cancer22. In this model, there was a direct association between the spontaneous development of metastatic mammary tumours and MDSC expansion. Similar observations have been observed clinically with solid tumours, including a direct relationship with tumour state and indirectly with T-cell dysfunction10.
HISTORY OF SUPPRESSIVE MYELOID CELLS; PHENOTYPES AND SUBSETS
In the mid-1960s, NS cells within tumour-bearing mice were reported to induce a leukemoid reaction that was related to the duration of tumour growth and myeloid-cell infiltration23, 24. These cells were not only associated with tumour growth, but were also a major component of inflammatory and haematopoietic processes3, including a presence in neonatal/newborn spleens, adult bone marrow (BM)3, and adult spleens following total body irradiation (TBI)4. Subsequent studies revealed an increase in NS cells in lymphoid and some parenchymal organs during tumour growth24, 25 and following Bacillus Chalmette-Guerin (BCG)26, 27 injection. The tumour-induced granulocytosis, associated lymphopenia24, and loss of T-cell function25 suggested a potential impact on cancer outcome, as well as, therapeutic potential if NS cells were down-regulated. This potential has been supported by studies demonstrating that decreasing myeloid cells in tumour-bearing mice is therapeutic13, 28. In the late 1970s, it was documented that leukemoid reaction(s) included cellular population(s), which could inhibit CTL induction29 and activity3. These cells, due to a lack of conventional membrane markers for T-cells, B-cells, NK cells, and macrophages, were also described as NS or “null” cells5, 30. Functionally, they inhibited T-cell proliferative responses, antibody production, and CTL induction. They also suppressed antitumour immune responses and promoted immune evasion.
Mouse myeloid suppressor cells
Initially, the phenotypic characterization of null or NS cells in mice was contentious due to a lack of phenotypic markers, and they were defined based on a suppressive function3. Subsequently, mouse studies identified their phenotype (BOX 1) using the expression of single membrane markers, including CD3431, Gr132, 33, or CD11b34. NS cells in tumour-bearing mice were also characterized as committed myeloid progenitor cells and quantified as cycling progenitor cells, or immature cells of monocyte-macrophage lineage using soft agar colony-forming assay35. NS activity was also reported to be mediated by multiple cell populations6, 36 including cells from the spleen or BM37. Early studies suggested that the most potent cyclophosphamide (CTX)-induced suppressor-cell population was derived from cells with a granulocyte-monocyte lineage that expressed CD11b6. This was supported by an observed 600-fold increase in granulocyte-macrophage colony-forming cells in the blood of mice following CTX injection as compared to untreated controls35. Cells with an NS phenotype and function were also identified at sites of intense haematopoiesis38 and found to be increased by tumour secretion38–40, or exogenous administration of haematopoietic growth factors including granulocyte-macrophage colony-stimulating factor (GM-CSF)41 and granulocyte-CSF (G-CSF)42. Thus, not only does tumour growth increase myeloid suppressor cells, but their numbers are also expanded by haematopoietic growth factor administration with potential impact on therapeutic intervention. It should be noted that cells with this phenotype are observed in the BM of naïve mice, but in the absence of T-cell suppressive activity. It has been suggested that in addition to their expansion, these immature cells need to be activated in order to be immunosuppressive43, such that in naïve mice these cells are not MDSCs.
Phenotypic markers on murine and human MDSCs.
The table below identifies phenotypic markers of murine and human MDSCs. Their phenotypes have been controversial and numerous marker subsets have been used to define MDSCs and their subsets. In some instances, the expression levels are identified as low (lo) or bright (Br) a difference that may be relevant to other markers as well. In this table, the cellular phenotypes have been subset into monocyte (Mo), granulocyte (G) and endothelial-committed MDSCs. These circulating endothelial progenitors (EPCs) have not been as well studied, and numerous markers have not been assessed (ND). Indeed, the relationship between EPCs and MDSCs remains obscure and are presented here to suggest a potential relationship. Review papers that discuss MDSC phenotypic markers are referenced for murine60, 81 and human76, 84, 174, 190, 191 studies.
| Murine | Mo-MDSC | G-MDSC | EPC |
|---|---|---|---|
| CCR2 | +++ | +++ | +++ |
| CXCR4 | +++ | +++ | +++ |
| CXCR2 | +++ | +++ | +++ |
| CD11b | +++ | +++ | +++ |
| CD11c | subset | subset | ND |
| CD16low | +++ | +++ | ND |
| CD31 | --- | --- | +++ |
| CD45 | +++ | +++ | +++ |
| CD62L | lo | lo | ND |
| CD80 | +++ | +++ | ND |
| CD115 (M-CSF R) | ++ | subset | ND |
| CD117 (cKIT) | --- | --- | ND |
| CD124 (IL-4Ralpha) | +++ | +++ | ND |
| F4/80 | +++ | --- | +++ |
| Gr1 | lo | Br | +++ |
| Ly6C | +++ | lo | +++ |
| Ly6G | --- | +++ | ND |
| MHC I | +++ | +++ | ND |
| MHC II | some models | Some models | --- |
| Sca-1 | subset | subset | +++ |
| TIE-2 | --- | --- | +++ |
| VEGFR1 (Flt-1) | +++ | +++ | |
| VEGFR2 (KDR/Flk-1) | +++ | +++ | +++ |
| VLA-4 (CD49d) | subset | --- | ND |
| Human | |||
| CCR2 | ++ | --- | === |
| CXCR4 | +++ | +++ | +++ |
| CD11b+ | +++ | +++ | +++ |
| CD11c+ | subset | subset | ND |
| CD13 | subset | subset | ND |
| CD14 | +++ | lo | +++ |
| CD15 | --- | +++ | ND |
| CD16low | subset | subset | ND |
| CD31 | --- | --- | +++ |
| CD33 | subset | subset | subset |
| CD34 | subset | subset | +++ |
| CD38 | subset | subset | +++ |
| CD39 | +++ | +++ | ND |
| CD45 | +++ | +++ | +++ |
| CD62L | lo | lo | ND |
| CD66b (CEACAM8) | --- | +++ | ND |
| CD115 (M-CSF R) | ++ | --- | ND |
| CD117 (cKIT) | lo | ++ | +++ |
| CD124 (IL-4Ralpha) | +++ | +++ | ND |
| HLA-DR negative | +++ | +++ | +++ |
| Lin negative | +++ | +++ | +++ |
| Tie-2 | +++ | --- | +++ |
| VEGFR1 (Flt-1) | +++ | +++ | +++ |
| VEGFR2 (KDR/Flk-1) | +++ | +++ | +++ |
| VLA-4 (CD49d) | subset | subset | ND |
Phenotypic heterogeneity44, 45 without consensus regarding the cellular phenotype(s) that induces T-cell dysfunction broadened the already diverse nomenclature to include immature myeloid cells (iMCs), myeloid suppressor cells (MSCs), and Gr1+ myeloid cells46, 47. Not until the Gr1+CD11b+ cellular phenotype was suggested in the late 1990s, was there general agreement that a cell population existed, classified as NS cells, that was distinct from monocytes and granulocytes48, 49. In 2007, the nomenclature controversy was largely resolved when a letter was published50 and a consensus51 reached identifying MDSC as the term for this cellular phenotype and function. However, MDSC does not testify to the immaturity of these cells, and some investigators continue to describe them as iMCs. Because, iMC does not speak to T-cell suppression, MDSC has become the preferred terminology52, however, although the nomenclature differs, the cellular phenotypes for MDSCs and iMCs remain consistent.
Cytology (macrophage and granulocytic) and gene expression (differential expression of inducible nitric oxide synthase (iNOS) and arginase) studies support MDSC heterogeneity, and recently, markers that subdivide mouse MDSCs have been identified based on expression of Ly-6C and Ly-6G53. CD11b+Ly-6GlowLy-6Chigh cells have a monocytic-like morphology, preferentially express nitric oxide synthase 2 (NOS2), have increased T-cell suppressive activity, and are identified as monocytic-MDSCs (Mo-MDSCs)53. This contrasts with CD11b+Ly-6G+Ly-6Clow cells that have a granulocyte-like morphology, express high levels of arginase type 1 (ARG1), and are identified as granulocytic-MDSCs (G-MDSCs). These cells have a polymorphonuclear morphology and, therefore, the term polymorphonuclear (PMN)-MDSC is used interchangeably with G-MDSC. In the majority of tumour-bearing mice, the G-MDSC population consistently increases, whereas the Mo-MDSC frequency is significantly increased in a limited number of tumour models53. Further, even in mice whose tumours have substantial increases in Mo-MDSCs, the G-MDSC population is also increased. Thus, the G-MDSC subset is the predominant MDSC population in tumour-bearing mice53. This subset distinction can also be clarified by CD11b and Gr-1 staining54. Two cellular fractions are generally recognized, a Gr-1Br subset, mainly composed of G-MDSCs, and a Gr-1int subset encompassing Mo-MDSCs17.
Additional MDSC subsets have been based on the intensity of Gr1 expression (Gr1lo, Gr1int, Gr1Br)17 or the use of alternative phenotypic markers including F4/80 or MHCII expression markers, which are also used to identify tumour-associated macrophages (TAMs). TAMs are a cellular population that histologically can be confused with MDSCs, but are defined as mature, differentiated macrophages55. Distinguishing MDSCs from macrophages and, to a lesser extent, granulocytes can be technically challenging. Mouse Mo-MDSCs have been classified as CD11b+Ly6Glo/intLy6Chi cells that express low levels of F4/80 (a marker for macrophages) and higher levels of Gr1 than TAMs56. Further, both MDSCs and TAMs express interleukin 4 receptor α (IL4Rα; also known as CD124) and M-CSF receptor (also known as CD115)56. Mo-MDSCs are a mixture of myeloid progenitor cells in varying stages of differentiation55, 57, which can differentiate into macrophages, dendritic cells (DCs), or granulocytes. Studies to distinguish TAMs from Mo-MDSCs have linked inducible NOS2 expression to the overall differentiation and maturation stage. NOS2+ cells do not generally express granulocyte markers, including chemokine (C-X-C motif) receptor 2 (CXCR2) and lymphocyte antigen 6G (Ly6G), which are expressed by NOS2− MDSCs58. However, intracellular NOS2 staining of tumour-infiltrating MDSCs has also identified a NOS2+ cellular subset with a ring-shaped nuclei58. This subset has T-suppressive activity, and the ring-shaped nuclei are characteristic of immature neutrophils. Thus, tumours affect haematopoietic progenitor cell differentiation, resulting in the accumulation of cells with neutrophil-like ring nuclei, but no macrophage differentiation markers, such as F4/80. This is consistent with prior studies that demonstrated cellular phenotypes in the BM with the potential to differentiate into macrophages in response to M-CSF59. Further, this cellular subset, as well as, Mo-MDSCs can differentiate into F4/80+ TAMs in the tumour microenvironment60. In one study, MDSCs were subset based on CD49d expression with CD49d+ MDSCs demonstrating a monocytic phenotype and immunosuppressive activity. In this study CD49d− MDSCs were granulocytic with little T-cell inhibitory activity61. However, subsequent studies were unable to distinguish between the granulocytic and iNOS+ MDSCs based on CD49d expression58. Studies using rodent MDSC subsets have been paralleled by human studies that, in general, revealed similar phenotypes including differences in enzyme expression by MDSC subsets and differentiation into granulocytic and monocytic subsets62.
Human MDSCs
In the mid-1990s, G-CSF or GM-CSF stem-cell mobilization for autologous PB stem-cell transplantation, predominantly for the treatment of patients with breast cancer or lymphoma, was found to result in myeloid cells with T-cell suppressive activity that were defined as suppressive CD14+ monocytes63–65. Indeed, studies of monocyte associated suppressive activity revealed a direct relationship between the number of CD14+ cells and suppressor cell function in mobilized stem cell products64, as well as, characteristics of cells with a monocyte lineage66. Subsequent studies of breast cancer patients following stem cell transplantation revealed that T-cell number and function inversely correlated with T-cell suppressor activity and CD14 cell frequency in the PB post transplantation38, 64 and from patients with advanced disease67. This inhibitory cell was isolated from PB stem cell (PBSC) products and shown to be a CD14+ myeloid cell68 comprising up to 40% of the cells in the apheresis product. Further, 10% or more of PB leukocytes following peripheral stem cell transplant (PSCT)69 were shown to be CD14+ with T-cell suppressive activity65.
In the late 1990s, human MDSCs were also described in patients with head and neck cancer based on CD34 expression and T-cell suppression in patients with GM-CSF-secreting cancers38, 64, 70. However, these MDSCs were lineage-negative (Lin−) CD34+ cells, a phenotype that also defines haematopoietic progenitor cells. Thereafter, the MDSC phenotype was extended to include a phenotype that included negativity for human leukocyte antigen D-related (HLA-DR) expression and a myeloid marker71. In cancer patients, MDSCs express either or both of the common myeloid markers CD33 or CD11b, are Lin−, and or are HLA-DR−71. Thus, human MDSCs were initially defined as HLA-DR−CD33+71 or CD14−CD11b+ cells72 with both phenotypes identifying cell populations with T-cell suppressive activity. Recently, more rigorous marker subsets have defined MDSCs, including Mo-MDSC expression of CD14+/dull 73, 74 and G-MDSC expression of CD15+, as observed primarily in patients with advanced renal-cell cancer (RCC)67, 75. The expression of CD14 on MDSCs as a Mo-MDSC, initially controversial, has become accepted. Nonetheless, CD14 expression is difficult to use as a lineage marker as PMNs also express lower levels of this marker. Thus, differentiation between CD14 bright versus dim in the assessment of MDSCs is critical. Human MDSCs have been further subdivided based on maturity76, although, definition of these subsets has not yet reached consensus. The identification of a precursor subset supports MDSC plasticity and their potential to differentiate into not only granulocytes, monocytes, and DCs, but also osteoclasts (OCs) and endothelial cells (ECs). Now, there are additional phenotypic parameters under investigation including, but not limited to, high CD66b levels, low levels of CD62L, low CD16 expression76, vascular endothelial growth-factor receptor 1 (VEGFR1) expression75, and S100A977, although the latter phenotype is not membrane expressed and cannot be used to isolate cells.
Myeloid cell populations are the predominant population in the PB, both in number and function, tumour infiltration, and supporting tumour growth and progression78. Although granulocytes are the first line of defense against tumour challenge, they also have a pro-tumourigenic role providing an immune paradox. The basis for this functional MDSC “plasticity,” observed in both mice and humans, is based on their capability to express different functional profiles in response to varying environmental signals including cytokines, growth factors and acidosis, hypoxia, interstitial pressure, and glucose levels. Hypoxia and hypoxia-inducible factor-1 alpha (HIF-1α) in the tumor microenvironment are partially responsible for the regulation of ARG-1 and NOS expression by murine MDSC and their differentiation. Similarly, the tumor-dependent profile of secreted cytokines also regulates MDSC differentiation supporting the conversion between MDSC phenotypes, differentiation, and activation. However, the exact combination and sequence of tumour-derived and microenvironmental factors that regulate MDSC mobilization, proliferation, abnormal differentiation, and activation remain poorly understood. Consequently, the current focus is directed towards mechanisms and molecules driving the protumoural skewing and the phenotypic heterogeneity of circulating and tumour infiltrating MDSCs. Similar to mouse MDSCs, human MDSCs are haematopoietic progenitors that can differentiate into not only granulocytes and monocytes, but also ECs and OCs. While specific growth factors drive MDSCs to differentiate, whether differing MDSC profiles between tumours are due to tumour heterogeneity, or if a profile consistency exists based on tumour histiotype, is unstudied.
Mechanisms of myeloid cell and tumour cross-regulation
Haematopoietic abnormalities in tumour-bearing hosts, originally described as neutrophilia characterized by abundant less mature cells79, have long been observed1. Mechanistically, this is associated with tumour secretion of cytokines and chemokines that induce myeloid cell proliferation, accumulation, and tumour infiltration18, 22, 80. Recently, it was reported that many of these accumulating cells were not mature cells as originally described, but rather progenitor cells with immunosuppressive activity and the ability to differentiate not only into granulocytes, histiocytes, and DCs, but also EC precursors and OCs. Myeloid cells respond to a wide range of stimuli most notability their molecular microenvironment and the heterogeneous milieu of tumor-secreted cytokines. The plasma cytokines and chemokines differ between tumour-bearing hosts (both in humans and mice), resulting in tumour-dependent proliferation, site(s) of accumulation, and infiltration, which have made it difficult to determine the crucial factors that result in MDSC activity in the tumour microenvironment. Further, not only do tumours secrete growth factors that induce myelopoiesis and chemokines that mobilize and marginate MDSCs, they may also limit their maturation and differentiation thereby contributing to their accumulation81. Thus, tumour necrosis factor-alpha (TNF-α) impairs MDSC maturation82 by regulating the receptor for advanced glycation end products (RAGE) and its ligands (S100A8 and S100A9 proteins)83. Additional, proinflammatory mediators, such as IL-1 beta (IL-1β), IL-6, leukotriene D4 (LTD4), and prostaglandin E2 (PGE2) that regulate MDSC accumulation and activation, may also affect MDSC differentiation, but remain to be assessed for these functions. The heterogeneity in the processes (BOX 2) that regulate MDSC proliferation, accumulation, and trafficking have been documented in both rodent models53 and clinically84. These processes include mobilization and proliferation (most notably as EMH), maturation, arrest, trafficking, and margination all of which contribute to organ and tumour accumulation. Indeed, MDSCs within tumours have been shown in the 4T1 tumour model to be derived from splenic and hepatic EMH prior to tumour infiltration11. Therefore, macroenvironmental changes, such as types and extent of circulating cytokines and chemokines, have a critical role in regulating infiltrating cells in the tumour microenvironment such that MDSC pathobiology is regulated by the macroenvironment via altered myelopoiesis and differentiation56, 85, 86 in association with tumour-derived haematopoietic growth factors and chemokines.
Since the first characterization of MDSCs, previously known as NS or null cells, they have been controversial. They are similar to monocytes and granulocytes with common morphologic and phenotypic features. However, MDSCs result from the expansion of myeloid precursor cells and are defined as iMCs with potent immune suppressive activity. They have an impaired ability to differentiate into mature myeloid cells, and accumulate in peripheral lymphoid and parenchymal organs. Despite, phenotypic homogeneity, MDSCs do not exist in healthy hosts, at least in the absence of activation, and are only observed during chronic pathological conditions, such as infections, inflammation, or cancer. Further, the mechanisms responsible for MDSC expansion, trafficking, abnormal differentiation, and function remain to be clarified. The table below identifies the mechanisms and some of the mediators that regulate MDSCs and is patterned on the mobilization of hematopoietic progenitors, which are the "normal" cellular population most similar to MDSCs.
| Factors and mechanisms regulating MDSCs | |
|---|---|
| Process mechanism | Examples of mediators |
| MDSC proliferation in the BM and EMH12, 20–23, 48, 91, 95 | G-CSF, GM-CSF, SCF |
| MDSCs have an impaired differentiation45, 69, 78–80 | TNF, VEGFA |
| Mobilization of MDSCs can occur from the marrow and sites of EMH23, 175, 182 | CXCL8, G-CSF, GM-CSF |
| Chemotaxis of MDSCs to an inflammatory or tumor site80, 87, 192 | CCL2, CXCL12 |
| Organ or tumor arrest of circulating MDSCs12, 126, 129 | VLA4, C5a |
| Activation of MDSCs resulting in increased enzyme secretion and T-cell suppression183, 184 | PGE2, LTD4, TLR agonists |
| MDSCs can differentiate into OCs, monocytes, granulocytes, OCs, ECs or DCs68, 103, 111 | ATRA, Vitamin D |
MACROENVIRONMENTAL REGULATION OF MDSCS
Regulation of MDSC proliferation
Building on our understanding of tumour progression and metastasis, tumour-secreted chemokines, growth factors, and enzymes that contribute to myeloid-cell expansion, differentiation, and recruitment also have been suggested to regulate tumour progression and metastasis. The effect of MDSCs on tumour progression and metastasis is observed in vivo by down-regulation of neutrophilia following tumour resection13, 14, 28, 87, intervention using antibodies to myeloid cells18, 88, or molecular growth-factor inhibitors20. The increase in circulating myeloid cells in tumor-bearing hosts, originally termed reactive neutrophilia or emergency granulopoiesis, was associated with an increased frequency of iMCs. Improvements in biomarkers identified neutrophilia with an increase in circulating colony forming unit in culture (CFU-c) and, recently, MDSCs. Mechanistic studies using tumour-conditioned media (TCM) to cultures of spleen15, BM56, or isolated myeloid/progenitor cell subsets53, 89 have identified mediators and effected MDSC survival, proliferation, differentiation, and function, as well as, T-cell proliferation and DC function90 and differentiation33. Tumours that stimulate MDSC proliferation have been shown to secrete VEGF32, 33, 81, 91, GM-CSF40, stem cell factor (SCF)92, FMS-like tyrosine kinase 3 ligand (FLT3-L)93, G-CSF17, 18, 41, and/or macrophage-colony stimulating factor (M-CSF)57. Indeed, studies using transplantable tumours linked tumour GM-CSF secretion to MDSC expansion17, 40, 41, 94 by the knock down of GM-CSF in tumor cell lines resulting in an altered MDSC number and subset distribution following tumour growth17. Consistent with such studies, and depending on the tumour studied, antibody or molecular inhibitor neutralization of GM-CSF, or its receptor15, 94, 95, M-CSF96, 97, VEGF91, 98, S100A8/A981, G-CSF18, or SCF92, have also been shown to inhibit MDSC accumulation, MDSC recruitment to the tumour microenvironment, and delay tumour development (Table 1). In a mouse model of mammary cancer (BALB-neuT mice, which develop c-erb-2-driven mammary tumours), investigators reported a direct correlation between tumour progression and numbers of immature Gr1+CD11b+CD131+cells with immunosuppressive activity91. G-CSF, similar to GM-CSF, can also facilitate G-MDSC accumulation, as well as, regulate granulopoiesis and neutrophil mobilization99. Increased G-CSF levels have been shown to inhibit innate and adaptive immunity100 and are aberrantly expressed by diverse human tumours101. G-CSF production also correlates with atypical MDSC-like responses in mouse tumour models22, 80; however, the correlation between G-CSF and MDSC accumulation has focused largely on systemic MDSC responses102 with limited studies assessing the impact of G-CSF on Mo- versus G-MDSC subsets11. While it remains unclear whether a specific growth factor is unique to a defined tumour histiotype, tumour secretion of growth factors regulates MDSC proliferation.
Table 1.
| Therapeutic Agent | Target | Type of Cancer Tested | Effect on MDSCs |
|---|---|---|---|
| Neutralizing Ab to receptor tyrosine kinase (cKit)92 | Growth factor | Colon carcinoma (mice) | Inhibition of proliferation |
| Prokineticin 2 (PROK2)-specific Ab183 | Growth factor | Tumours of human & mouse origin in nude mice | Inhibition of PMN/MDSC expansion & tumour recruitment |
| Neutralizing Ab to granulocyte-macrophage colony-stimulating factor (GM-CSF)15 | Growth factor | Pancreatic Cancer (mice) | Inhibition of proliferation |
| Neutralizing Ab to granulocyte colony-stimulating factor (G-CSF)42 | Growth factor | Colon carcinoma (mice) | Inhibition of proliferation |
| Colony stimulating factor 1 receptor (CSF1R) & protein kinase (KIT) tyrosine kinase inhibitor (TKI) (PLX3397)97 | Growth factor | Breast cancer (human) | Inhibition of TAM recruitment |
| CSF1R antagonist (GW2580)96 | Growth factor | Lung carcinoma & prostate cancer (mice) | Inhibition of MDSC & macrophage proliferation |
| Vascular endothelial growth factor (VEGF)-trap98 | Growth factor | Solid tumours (human) | No activity |
| Ab to VEGF (Avastin)185 | Growth factor | Metastatic renal cell carcinoma (RCC) (human) | Weak inhibition of proliferation |
| Ab to interleukin 6 receptor (IL-6R) & gemcitabine141 | Interleukin & Cytotoxic | Carcinoma (mice) | Inhibits accumulation of Mo- & G-MDSCs |
| CXC chemokine receptor 2 (CXCR2) (S-265610) & CXCR4 (AMD3100) antagonists151 | Chemokine | mammary cancer (mice) | Altered recruitment of iMCs to the tumour |
| Neutralizing Ab to chemokine (C-C motif) ligand 2 (CCL2)192 | Chemokine | Mammary carcinoma (mouse & human xenografts) | Targeting inflammatory monocytes & macrophages |
| Gemcitabine145 | Cytotoxic | Lung & mammary cancer (mice) | Inhibition of proliferation |
| Doxorubicin-cyclophosphamide10 | Cytotoxic | Breast cancer (human) | Weak inhibition of proliferation |
| 5-fluorouracil142 | Cytotoxic | Thymoma (mice) | Inhibition of MDSC expansion |
| Docetaxel143 | Cytotoxic | Mammary carcinoma (mice) | Inhibition of MDSC expansion & macrophage polarization to M1 phenotype |
| Cyclooxygenase 2 (COX2) inhibitor (Celebrex)181 | Enzyme inhibitor (Inhib.) | Mammary carcinoma (mice) | Inhibition of proliferation/activation |
| Amino-biphosphonate180, 193 | Enzyme inhib. | Mammary tumours (mice) | Inhibition of proliferation |
| Phosphodiesterase-5 (PDE5) inhibitors144 | Enzyme inhib. | Mammary and colon carcinoma and fibrosarcoma (mice) | Inhibition of proliferation & T-cell suppression |
| TKI (sunitinib)67, 95, 182, 184 | Signal inhib. | RCC (human) | Inhibition of proliferation |
| Peroxisome proliferator–activated receptor-g (PPARg) inhibitor179 | Signal inhib. | Carcinoma & myeloid sarcoma (mice) | Induces proliferation of MDSCs |
In vitro mechanistic studies to assess MDSC regulation have used TCM and demonstrated that TCM, but not control media, can induce the proliferation of c-kit+ MDSC precursors15; resulting in a cell population that is predominantly Gr-1+CD11b+103. In one study, proliferation and differentiation of c-kit+ MDSC splenocytes was only observed with GM-CSF and not with recombinant VEGF or SCF, and only minimally with IL-615. However, an intermediate level of proliferation was observed with high concentrations of M-CSF. More critically, addition of a neutralizing GM-CSF antibody to the TCM culture abrogated MDSC generation from c-kit+ precursors. Neutralizing antibodies to other cytokines, such as SCF, IL-6, or IL-1β, had no effect suggesting that GM-CSF is both necessary and sufficient to generate functional, immunosuppressive MDSCs in vitro. Thus, GM-CSF was suggested to be the tumour-associated proliferative factor for MDSC differentiation and proliferation (i.e., secreted by tumours, not by normal epithelial cells, resulting in suppressive MDSCs from c-kit+ splenocytes)15.
Recently, organ-16 and tumour-dependent104 MDSC proliferation was shown to rely, in part, on tumour-derived microenvironmental and macroenvironmental growth factor levels, which vary in a tumour and anatomical manner3, 12, 16, 95. In one study, MDSC numbers in the spleen directly correlated with tumour G-CSF transcript levels, whereas MDSC numbers in the tumour directly correlated with splenic GM-CSF transcript levels, tumour volume, and tumour cell numbers14. The observation of EMH in reticuloendothelial6 and parenchymal tissues3, 24 is well established; however, its role in MDSC accumulation has been little studied32, 105. Indeed, adult, non-tumour-bearing mice have few splenic progenitor cells33; however, when stressed, haematopoietic progenitor cells mobilize from the marrow, arrest, and accumulate in lymphoid and parenchymal organs106. Studies using 4T1 tumour-bearing mice revealed a splenic myeloid-cell reservoir that consists primarily of CD11b+Ly6GBr cells11. Circulating MDSCs arrest and accumulate in the splenic marginal zones and periarteriolar lymphatic sheaths, migrate to the red pulp, and proliferate within the subcapsular red pulp. The Gr1dull subset of MDSCs was reported to have a higher proliferation rate in the BM107; although, both MDSC subsets proliferate in the spleen11, 108. Similarly, clinical and rodent therapeutic strategies (Table 1) to induce MDSC differentiation, such as the administration of all trans-retinoic acid (ATRA)109, 110 or vitamin D70, 111, have also been shown to reduce MDSC number and/or function.
The proinflammatory proteins, S100A8 and S100A9, can also support MDSC expansion and block myeloid precursor differentiation. S100A8 and S100A9 are highly expressed in the tumour microenvironment, potentially supporting MDSC recruitment112. Tumour secretion of VEGF, G-CSF, and GM-CSF, as discussed above, is also at least partially responsible for MDSC expansion91. However, other mechanisms are involved including matrix metallopeptidase-9 (MMP9) secretion and the associated remodeling of the ECM, promotion of angiogenesis, and stimulation of VEGF production113.
Regulation of MDSC mobilization
The haematopoietic system cycles continually from a small population of pluripotent haematopoietic stem cells (HSCs) as part of a delicate balance between quiescence and proliferation in addition to self-renewal and differentiation. Quiescence and self-renewal are tightly regulated by both cell-intrinsic signals and environmental factors114. However, tumours subvert this niche-signal-mediated maintenance of HSC dormancy leading to rapid HSC proliferation and differentiation resulting in a selective loss of lymphocytes with maintenance of myeloid lineage cells. In adults under steady-state conditions, HSCs and progenitor cells are primarily found in the BM, although low numbers circulate in the blood and traffic to haematopoietic and non-haematopoietic organs115 where they proliferate and differentiate as EMH116. Mobilization is the first step in myeloid progenitor trafficking to inflammatory sites. However, there has been little study into the mechanisms governing MDSC mobilization, their subsequent trafficking to sites of EMH and tumours, and the suppression of these processes (Table 1). These processes include, but are not limited to, alterations in the adhesive interactions between myeloid progenitors and stromal cells during transmigration through the subendothelial basal lamina and the EC layer117. During steady-state haematopoiesis, GM-CSF and G-CSF not only regulate proliferation, but also (directly or indirectly) mobilization from the BM and spleen118. In addition, growth factor administration or secretion from tumours results in myeloid hyperplasia thereby increasing prosteroid and neutral protease secretion that also regulates mobilization, myeloid progenitor survival, and their distribution between marginal and circulating pools119. Because MDSCs include myeloid-committed haematopoietic progenitors and are likely fixed at an immature differentiation stage120, the mechanisms associated with their mobilization may be similar to those of haematopoietic progenitor cells.
Treatment of humans or mice with selective antagonists of CXCR4, CXCR4-blocking antibodies, or CXCL12 expression121, results in a rapid (1–4 hours) mobilization of neutrophils, myeloid progenitors, and MDSCs57, 121 into the blood. This suggests that disrupting CXCR4 signaling can contribute to G-CSF-induced mobilization and granulopoiesis89, 121. G-CSF administration also induces protease secretion in the BM microenvironment contributing to cellular mobilization122. The rapid mobilization by chemokines (minutes to hours) compared with G-CSF (days) supports multiple mechanisms. Accumulating evidence suggests that CXCL12 has a key role in controlling myeloid cell mobilization and homeostasis123. Thus, mice deficient in CXCL12 or CXCR4 die perinatally with neutrophilia and fail to establish BM myelopoiesis124. CXCL12 also acts as a retention signal, constraining CXCR4-expressing haematopoietic progenitor cells to the BM. However, CXCL12 is subject to NH2- and COOH-terminal proteolytic modification, reducing its chemotactic activity125.
Regulation of MDSC trafficking and arrest
Chemokines have a role in leukocyte recruitment and arrest in the tumour microenvironment as do members of the integrin and selectin adhesion molecule families126. A number of chemokines89, 112 attract myeloid cells to the tumour microenvironment127, as well as, stimulate the secretion of matrix-degrading enzymes127. However, myeloid cells also express integrins128, 129 providing another mechanisms to regulate cellular trafficking within vascular microenvironments130. One of these integrins is α4β1, a receptor for vascular cell adhesion molecule (VCAM) and fibronectin, that selectively promotes the arrest of endothelial progenitor cells131 and monocytes130, 132, to neovascular tissue. Human CD34+ and mouse Lin−Sca1+ progenitor cells, as well as, BM-derived myeloid cells (CD14+CD11b+) adhere to tumour endothelium in vivo via integrin α4β1 (VLA-4). The blockade of α4β1 in tumour-bearing mice suppresses myeloid progenitor tumour infiltration and reduces blood vessel density. Integrins also mediate progenitor cell arrest at sites of ischemia133 such that circulating endothelial progenitor cell (EPC) adhesion to endothelial monolayers requires β2 integrin expression.
Adoptive transfer studies16, 57 with carboxyfluorescein succinimidyl ester (CFSE)-labeled MDSCs from tumour-bearing mice demonstrated that MDSCs rapidly transverse the pulmonary capillary networks and accumulate in the spleens16. In contrast, injection into naïve mice results in MDSC arrest primarily in the lungs. Consistent with these findings, an analysis of MDSC proliferation by BrdU labeling revealed proliferation predominantly within the spleens of both naïve and tumour-bearing mice16. MDSC accumulation is also a result of prolonged MDSC survival, decreased apoptosis, and significantly increased proliferation and accumulation within the spleens, but not the BM, of tumour-bearing mice. Support for this observation was provided by Ki-67 staining, which revealed that MDSCs proliferated in the cords of the sub-capsular red pulp, with Gr1+ cells infiltrating throughout the red pulp and marginal zones38. In these studies using 4T1 tumour-bearing mice, the spleen, but not the BM, was shown to be the primary site of MDSC proliferation, as well as, providing a reservoir of Ly6G+ MDSCs that can rapidly demarginate and recirculate16. Further, this organ-specific proliferation is tumour unique134, and the frequency of Mo- versus G-MDSCs subsets differs between tumours and organs16 such that in 4T1 tumour-bearing mice, G-MDSCs have a significantly higher cellular frequency than Mo-MDSCs in the PB and spleen53, 60, 95. This higher frequency is also present in the liver, but not the lungs. Most MDSC studies have focused on MDSCs within the PB, spleen, or tumours; however, MDSCs also accumulate in the liver12, 135, including following adoptive transfer of splenic MDSCs11. Further, MDSC tumour infiltration is tumour dependent and associated with a high frequency and absolute number of MDSCs infiltrating the spleen, liver, lungs, and PB16.
MDSC REGULATION OF TUMOUR PROGRESSION AND METASTASIS
The tumour regulation of MDSCs is bidirectional, and activated MDSCs secrete chemokines, cytokines, and enzymes136 that contribute to tumour cell invasion, proliferation, survival, adhesion, and chemoattraction resulting in a crosstalk that can impact tumour progression, invasion, and metastasis137. Tumour progression and metastasis are multistage processes that are facilitated by MDSCs as documented by inverse correlations between outcome parameters and myeloid cell infiltration of tumours and circulating MDSC frequency10, 138. The stages of tumour progression and metastasis and their regulation by MDSCs are shown in Figure 2. Studies in the late 1970s, stimulated research into the pathobiology of metastasis building on the studies by Paget who developed the “seed and soil” hypothesis139. Since then, our understanding has exploded as to the tumour-promoting role of chronic inflammation. We have identified many of the key soluble and cellular mediators involved in this process, and promising studies in mice focused on the control of MDSC number and function have emerged. The adoptive transfer of MDSCs has been shown to significantly promote tumour growth in animal models113, 140, and Gr1+ cell depletion in tumour-bearing mice inhibits tumour growth, reduces metastasis, and prolongs survival20, 21, 141. Some chemotherapeutic agents preferentially suppress MDSCs including gemcitabine141, 5-fluorouracil142, and docetaxel143. Numerous studies of tumour-bearing mice, and a few clinical studies using therapeutics that target MDSCs or their function, have shown that in mice MDSC reduction delays tumour initiation, progression, and prolongs survival (Table 1)110, 144,145.
Figure 2. The role of MDSCs in the process of cancer progression and metastasis.

The process of cancer metastasis consists of sequential, interlinked, and selective steps with some stochastic elements. The outcome of each step is influenced by the interaction of metastatic cellular subpopulations with homeostatic factors and cells. Each step of the metastatic cascade is potentially rate limiting such that failure of a tumour cell to complete any step effectively impedes that portion of the process. Therefore, the formation of clinically relevant metastases represents the survival and growth of selected subpopulations of cells that preexist in primary tumours. The interactions between MDSCs and tumour cells during tumour progression and metastasis are highlighted. The role of tumour cells is indicated by green text, and those of MDSCs and other myeloid cells are indicated in red.
Definitive proof that a clinical reversal in MDSC number or function can significantly delay tumour progression or growth remains to be established. However, what has been demonstrated recently is that immune evasion limits cancer immunotherapies; and MDSC suppressive activity appears to be one of the more prevalent mechanisms present in cancer patients providing an obstacle to immune based intervention. It should be stressed that the mechanism of T-cell suppression and the responsible MDSC subset critical to immune subversion remains controversial. Thus, in contrast to results to date, adding interventional strategies that reverse MDSC immune suppressive activity supports adjuvant immunotherapy110, 146 and slowing of tumor growth147. MDSC diversity results in multiple roles during tumour progression, and future studies must define the mechanisms underlying their different functions with respect to tumour progression. Clearly, investigators should pursue other therapeutic mechanisms to regulate MDSCs based on their multiple pro-tumourigenic activities (Figure 2).
MDSC regulation of tumour invasion
Tumour and myeloid cell secretion of enzymes facilitate invasion, and positive correlations have been reported between proteases, such as MMP9, and tumour malignancy148. MicroRNA-49489 and HIF-1α149, 150 also have a role in regulating the non-immunological activity of MDSCs by enhancing their ability to infiltrate tumor tissue and, in the case of MicroRNA-494, to facilitate tumor invasion via the upregulation of MMPs89. Therefore, MDSCs, which secrete high levels of proteolytic enzymes, may facilitate invasion into the host stroma, but also intravasation through capillary basement membranes resulting in increased numbers of circulating tumour cells. MDSCs promote tumour invasion by the secretion of MMPs150, which have an essential role in extracellular matrix degradation113, 151. Support for an MDSC role in tumour invasion was provided by studies that used a mammary carcinoma lacking the type II transforming growth factor beta (TGFβ) receptor151 that resulted in an enhanced incidence of metastasis and an increased MDSC infiltrate152. The enhanced invasion was MMP-dependent; however, it was not clear if the MMPs were secreted by MDSCs or by the carcinoma cells in response to MDSC stimulation151. Both cell populations are likely involved as MDSCs derived from tumour-bearing hosts have increased MMP levels compared to “MDSCs” from non-tumour-bearing hosts151 suggesting that some of the increased invasive ability may be from MDSC-derived MMP secretion.
MDSC regulation of tumor cell arrest and niche formation
While metastasis is an inefficient process153, such that few tumour cells survive trafficking through the circulation, arrest, and interactions with host immunity, survival is facilitated by the formation of heterotypic emboli secondary to tumour cell interactions with myeloid cells and platelets154. These emboli physically protect tumour cells from the host immunity, as well as, larger emboli that more frequently arrest within the organ parenchyma. However, once tumour cells have extravasated, their growth is regulated by the cellular and growth factor milieu in the microenvironment155, which has been termed a metastatic niche156. These niches are controlled, in part, by macroenvironmental changes including the number of circulating MDSCs and EPCs. Such niches have been suggested to act as chemoattractants157; however, this may be irrelevant as tumour cells circulate and arrest based on physical constraints and adhesion factor expression. Further, increases in MDSC and EPC numbers, at least in tumour-bearing mice, occur in numerous solid organs, including the spleen, lung, and liver. Indeed, in rodent models MDSC frequency is highest in the spleen; however, in mice this site is abnormal for metastatic formation by carcinomas. In contrast, MDSCs also occur at a high frequency in lungs and livers, which are frequent metastatic sites. However, as reported by Hart, et al., even when organ fragments are implanted ectopically, specific organ milieu regulate tumour cell arrest and growth suggesting that mechanisms, in addition to myeloid cell expansion and angiogenesis, are critical, notably hormonal and organ-specific growth factors155. Thus, metastasis is both selective and inefficient158, and only a few cells survive to form a micrometastasis, a process partially regulated by MDSCs and other myeloid cells.
MDSC regulation of vasculogenesis and angiogenesis
Infiltrating and circulating MDSCs also have been suggested to have a role in tumour angiogenesis and vasculogenesis. Tumor growth can create an imbalance between inhibitory and stimulatory angiogenic molecules resulting in EC proliferation and recruitment. Haematopoietic and EPC proliferation results in an increase in their circulating number, which, in response to tumour-derived chemokine secretion, recruits leukocytes to nascent primary and secondary tumor foci contributing to the neovasculature. This increased vascularity facilitates tumour growth and access to the circulation and is associated with a positive correlation between vascular density and tumour grade159.
Angiogenesis, the formation of new vessels from preexisting ECs, is not the only mechanism of tumour neovascularization. Tumour vasculature can also arise by vasculogenesis, i.e., de novo vessel formation by the recruitment, migration, proliferation, and differentiation of BM-derived EPCs160. Mobilized from the BM by growth factors or chemotherapy161, EPCs become involved in tumour vasculogenesis162. Further, inhibiting EPC mobilization can slow or prevent tumour growth163, and several studies have shown that circulating EPC numbers correlate with clinical outcomes164. A major regulator, which can affect EPC proliferation and differentiation, is VEGFA, which can attract EPCs and is blocked by neutralizing VEGFA antibody91, 165. Taken together, these studies indicate that tumour-secreted factors can induce the proliferation, differentiation, and support the survival of EPCs and ECs166.
The extent of MDSC-derived tumour vasculature may depend on the tumour such that EPCs are found at high levels in mice bearing Lewis lung carcinomas, but are absent in mice bearing B16 melanomas, despite relatively similar levels of microvasculature167. In addition to directly contributing to the vasculature, myeloid progenitor cells also promote tumour growth. Admixing myeloid cells with tumour cells, under conditions that promote vascular leukocyte generation, results in larger and more highly vascularized tumours168. Both G-CSF and VEGFA mobilize EPCs into the peripheral circulation and promote angiogenesis169. Indeed, splenic MDSCs contribute to tumour vasculogenesis by directly differentiating into EPCs113; however, a subset of circulating progenitor cells is CD11b−170, and as such, Gr1+CD11b+ cells may be precursors that differentiate and participate in tumour vasculogenesis(BOX 1). Since EPCs can become part of the tumour vasculature170, their mobilization in response to tumour secretion of VEGF, G-CSF, or GM-CSF, contributes to tumour vascular formation. In contrast to tumour-bearing mice, non-tumour-bearing mice injected with G-CSF do not increase circulating EPCs, suggesting that additional tumour-associated cytokines are required, such as VEGFA, which can also mobilize EPCs from the BM169.
MDSC regulation of host defense
The tumour microenvironment, once established, can inhibit host immunity. Tumours are not passive targets for host immunity; rather, they can down regulate anti-tumour immune responses through multiple mechanisms including tumour-secreted factors, as well as, mechanisms that result from changes in tissue homeostasis. During tumour progression and immunotherapy, tumor cell populations may be selected with increased resistance to immunosurveillance and immunotherapy. In addition, variants arise that can interfere with multiple stages of immune cell development, differentiation, migration, and cytotoxicity158. Thus, multiple aspects of antitumour immunity are subject to regulation by the tumour microenvironment171.
Although there are multiple mechanisms supporting tumour escape from host immunity172, in recent years, MDSCs have been a focus primarily because they are ubiquitous and associated with disease progression and poor prognosis173, 174. Many studies that examined hosts with advanced tumour burden also reported reduced T-cell frequency and function. The relationship between tumour burden and immunosuppression raises the question of whether tumour-induced immunosuppression is reversible by surgical removal of the primary tumour. Several studies have assessed immunosuppression after primary tumour resection in mice13, 14, 28, and varying effects on immunosuppression have been reported including partial recovery of T-cell function13, 28. VEGFA is an immunosuppressive cytokine that blocks normal myeloid-cell differentiation, resulting in increased MDSC-mediated inhibition of T-cell activity46, 71, 175. Thus, surgery may reverse immunosuppression in some cases, and this is associated with a reduction in immunosuppressive factors, thereby supporting immune recovery. However, immunosuppression may reoccur as metastatic lesions grow and inhibitory cytokine levels increase. Numerous reviews of the mechanisms associated with MDSC suppression of T-cell function have been written in recent years which focus is on NOS2176, reactive oxygen species (ROS), ARG1177, cyclooxygenase 2 (COX2), and potentially upregulation of IL-10 as primary mechanisms. The reader is directed to any of the recent reviews on this subject for additional information43, 73, 178.
MDSCS AS THERAPEUTIC TARGETS AND POTENTIAL REGULATORS OF CANCER THERAPY
Numerous enzymes, growth factors, and cytokines regulate the MDSC lifecycle, including their expansion, trafficking, differentiation, and function (BOX 2). Inhibitors of most of these regulatory mediators are identified in Table 1. These molecular inhibitors include a variety of drugs that target tyrosine kinases67, such as PDE5144, peroxisome proliferator-activated receptor gamma (PPARγ)179, amino-bisphosphonates180, and COX2181, in addition to the antibodies and molecular inhibitors of cytokines and chemokines discussed above. Indeed, several recent reviews have focused on this aspect of MDSC pathobiology173; therefore, we will briefly discuss the ability of MDSCs to inhibit responses to standard-of-care therapeutics. For example, VEGFR2 tyrosine kinase inhibitors (TKIs) that target angiogenesis also partially inhibit the tumour growth stimulatory effects of G-CSF via MDSC proliferation, an observation supporting a role for VEGFA in tumor progression and evasion of host immunity. Clinically, administration of the TKI inhibitor, sunitinib, to metastatic RCC patients significantly reduces MDSC numbers in the PB67, 182. MDSCs may also contribute to resistance to sunitinib therapy via secretion of the Bombina variagata peptide 8 homologue (BV8)183, potentially secondary to its upregulation by tumour-secreted G-CSF19. IL-8 also contributes to sunitinib resistance as shown in a RCC xenograft model in which sunitinib-induced resistance was reversed by co-treatment with anti-IL-8184. Since IL-8 has potent proangiogenic activity, its up-regulation may induce proangiogenic pathways that allow tumours to escape the anti-angiogenic activity of sunitinib. Additional growth factors may also block therapeutic responses VEGFA signaling inhibitors by recruiting myeloid cells that promote angiogenesis and immune tolerance185, 186. In addition to factors that directly drive angiogenesis187, mediators may also function by recruiting myeloid cells to release angiogenic factors188. The BM-derived myeloid cells that produce angiogenic factors and the accompanying escape from VEGFA inhibitors have also been observed in preclinical tumour models95, 186. Tumours resistant to inhibitors of VEGFA signaling can secrete cytokines that recruit myeloid cells that promote angiogenesis and immune tolerance185. In support of a role for chemokines in tumour progression, plasma levels of CXCL12 have been shown to correlate with metastasis in bevacizumab (monoclonal antibody to VEGF)-treated patients with advanced rectal cancer189.
CONCLUDING REMARKS
In recent years, there has been a heightened interest in MDSCs and their biological function in tumor pathobiology. As this timeline emphasizes, their contribution to tumor pathologic processes goes far beyond immune suppression. MDSCs are a heterogeneous population of immature haematopoietic cells that have a role in immune tolerance, tumour progression, and metastasis. Tumour progression is associated with increased numbers of these iMCs in the primary tumour microenvironment and circulation, which in turn is associated with a poor prognosis, supporting a role in metastasis. Recently, the controversy regarding the phenotype of human MDSCs has resolved with agreement concerning which subset(s) should be monitored, i.e., Lin−HLA-DR− and CD11b+ or CD33+ cells, with additional subsetting based on CD14dim or CD15+ expression. This consensus will greatly facilitate studies into the clinical relevance of tumour-infiltrating myeloid cells and their role in tumour progression and metastasis. The mechanisms responsible for the abnormal differentiation of hematopoietic progenitor cells into MDSCs remain to be clearly elucidated and provide a goal for near term studies.
Figure 1. Timeline: Historical Progression of MDSCs in the Literature.
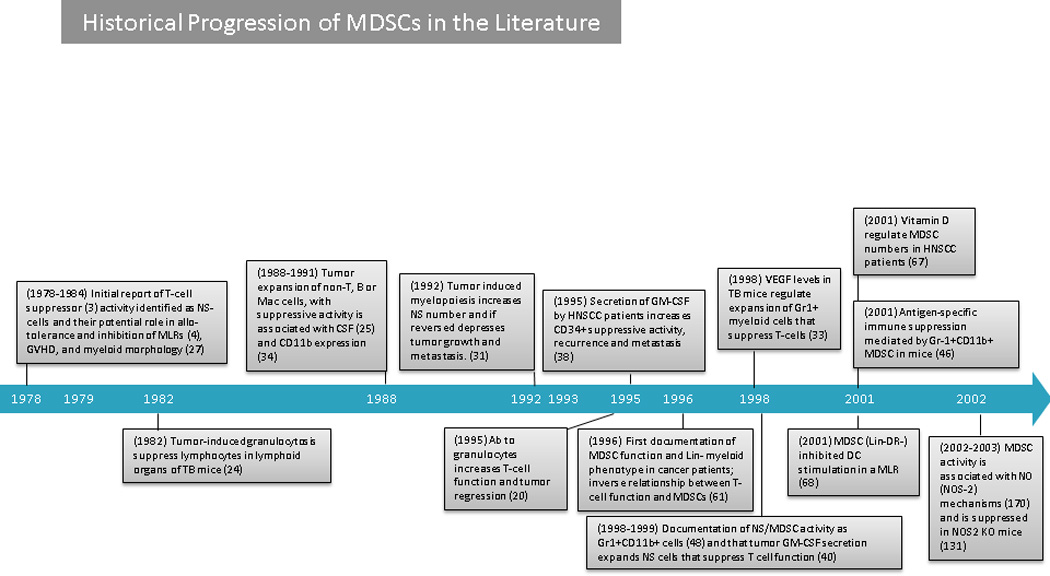
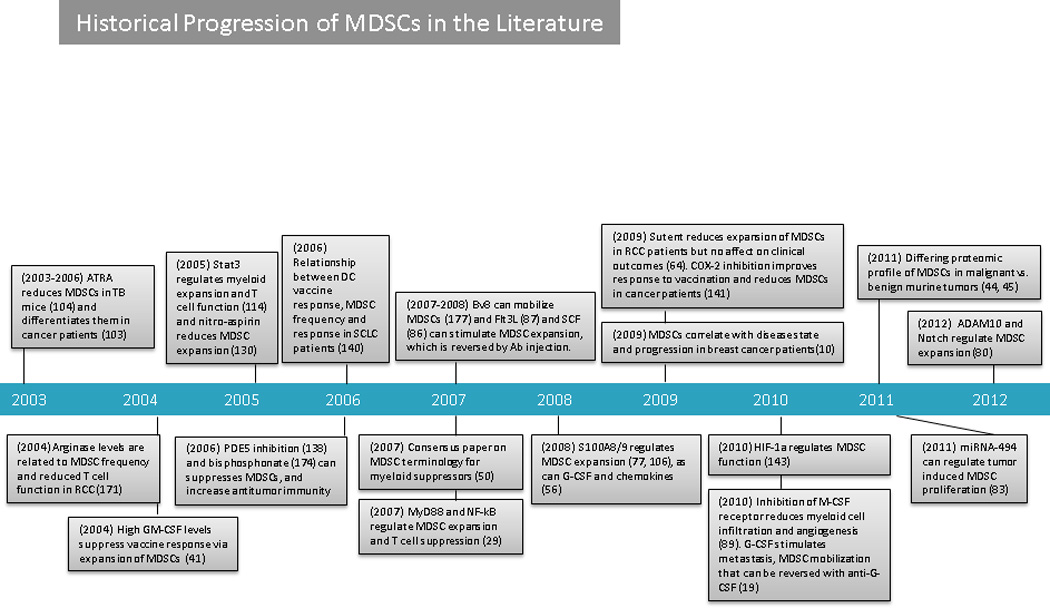
Antibody (Ab); a disintegrin and metallopeptidase domain-containing protein10 (ADAM10); all-trans retinoic acid (ATRA); bombina variagata peptide 8 (BV8); colony stimulating factor (CSF); FMS-like tyrosine kinase 3 (FLT3); granulocyte colony stimulating factor (G-CSF); granulocyte-macrophage colony stimulating factor (GM-CSF); graft versus host disease (GVHD); hypoxia inducible factor 1 alpha (HIF-1α); head and neck squamous cell carcinoma (HNSCC); knock out (KO); macrophage colony stimulating factor (M-CSF); myeloid-derived suppressor cell (MDSC); micro RNA 494 (miRNA-494); mixed lymphocyte reaction (MLR): myeloid differentiation primary response 88 (MYd88); nuclear factor kappa beta (NF-Kβ); nitric oxide (NO): nitric oxide synthase 2 (NOS2); natural suppressor (NS); phosphodiesterase type 5 (PDE5); renal cell carcinoma (RCC); signal transducer and activator of transcription 3 (STAT3); tumour bearing (TB); and, vascular endothelial growth factor (VEGF).
Online Summary.
- EMH and neutrophilia have been recognized as a cornerstone of neoplasia since the early 1900s. However, the specific role, phenotypes, and nomenclature for these myleopoietic cells have only been studied since the late 1970s.
- Currently, mouse MDSCs are defined as Gr1+CD11b+ cells, and human MDSCs are defined as Lin-HLA-DR-CD11b+. These cells are further subdivided into granulocytic and monocytic committed variants, as well as, being defined using additional and alternative phenotypic markers.
- MDSCs have a role in all steps of tumour progression and metastasis. However, important questions remain, as well as, the need for focused studies to fully understand the mechanisms regulating these processes.
- The crosstalk between tumour cells and myeloid cells is critical to these processes, although, the majority of studies have focused on the effect of tumours on myeloid cell proliferation, trafficking, biodistribution, and survival. Future studies need to assess the effect of myeloid cells on tumour cell signaling and molecular expression.
Glossary
Extramedullary hematopoiesis
(EMH) is the proliferation of hematopoietic cells outside of the bone marrow in response to pathologic processes, such as tumor growth, and can occur at different locations, such as the liver and spleen.
Host macroenvironment
Includes the systemic factors (growth factors, cells, tissues, and organs) that can regulate tumor growth, progression, and metastasis including factors, such as age, menopausal status, body mass index, as well as, overall immune status.
Tumor microenvironment
The milieu surrounding tumors including normal cells, blood vessels, soluble factors, and molecules that can influence and be influenced by the tumour’s growth.
Leukemoid reactions
A peripheral blood phenotype resembling that of leukemia, or indistinguishable from it, based on cellular morphologic appearance; observed in some infectious diseases, inflammatory conditions, and neoplasia.
Margination
A process that occurs during the early phases of inflammation; as a result of capillary dilation and slowing of the bloodstream, leukocytes occupy the periphery of the cross-sectional lumen and adhere to the ECs that line the vessels.
Mobilization
The release of hematopoietic progenitor cells from the bone marrow into the peripheral blood.
Plasticity
The capacity of cells with the same genotype to vary in differentiation, in phenotype, or in function in response to varying environmental conditions.
Neutrophilia
An absolute or relative increase in the normal number of neutrophils and their precursors in the circulating blood that may be associated with acute infections, malignancy, or following severe hemorrhage, or neutropenia (compensatory neutrophilia).
Tumor angiogenesis
The growth of blood vessels and capillary beds from existing vessels into a solid tumor.
Tumor vasculogenesis
The formation of new capillaries and blood vessels by EC progenitors.
Biographies
Dr. Talmadge has over 45 years’ experience in cancer research and tumour immunology and is currently head of the Transplantation Immunology Lab at the University of Nebraska Medical Center. His laboratory is focused on stem cell transplantation, immune response to transplantation and immunotherapy, and gene and cellular therapeutics.
Dmitry I. Gabrilovich is a Christopher M. Davis Professor in Cancer Research and Program Leader, Translational Tumor Immunology at the Wistar Institute. His lab is studying myeloid-derived suppressor cells in cancer and the molecular mechanisms of DC differentiation.
Footnotes
Competing Interest Statement
The authors have declared no competing financial interests.
References
- 1.Sonnenfeld A. Leukamische reaktiones bei carcinoma. Zeitschrift f Klin Med. 1929;111 [Google Scholar]
- 2.Robinson WA. Granulocytosis in Neoplasia. Ann NY Acad Sci. 1965;230:212–218. doi: 10.1111/j.1749-6632.1974.tb14451.x. [DOI] [PubMed] [Google Scholar]
- 3.Bennett JA, Rao VS, Mitchell MS. Systemic bacillus Calmette-Guerin (BCG) activates natural suppressor cells. Proc Natl Acad Sci U S A. 1978;75:5142–5144. doi: 10.1073/pnas.75.10.5142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Slavin S, Strober S. Induction of allograft tolerance after total lymphoid irradiation (TLI): development of suppressor cells of the mixed leukocyte reaction (MLR) J Immunol. 1979;123:942–946. [PubMed] [Google Scholar]
- 5.Duwe AK, Singhal SK. The immunoregulatory role of bone marrow. I. Suppression of the induction of antibody responses to T-dependent and T-independent antigens by cells in the bone marrow. Cell Immunol. 1979;43:362–371. doi: 10.1016/0008-8749(79)90180-1. [DOI] [PubMed] [Google Scholar]
- 6.Brooks-Kaiser JC, Bourque LA, Hoskin DW. Heterogeneity of splenic natural suppressor cells induced in mice by treatment with cyclophosphamide. Immunopharm. 1993;25:117–129. doi: 10.1016/0162-3109(93)90015-i. [DOI] [PubMed] [Google Scholar]
- 7.Fidler IJ. Tumor heterogeneity and the biology of cancer invasion and metastasis. Cancer Res. 1978;38:2651–2660. [PubMed] [Google Scholar]
- 8.Porembka MR, et al. Pancreatic adenocarcinoma induces bone marrow mobilization of myeloid-derived suppressor cells which promote primary tumor growth. Cancer Immunol Immunother. 2012 doi: 10.1007/s00262-011-1178-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang L, et al. Increased myeloid-derived suppressor cells in gastric cancer correlate with cancer stage and plasma S100A8/A9 proinflammatory proteins. J Immunol. 2013;190:794–804. doi: 10.4049/jimmunol.1202088. [DOI] [PubMed] [Google Scholar]
- 10.Diaz-Montero CM, et al. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol Immunother. 2009;58:49–59. doi: 10.1007/s00262-008-0523-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Younos IH, Dafferner AJ, Gulen D, Britton HC, Talmadge JE. Tumor regulation of myeloid-derived suppressor cell proliferation and trafficking. Int Immunopharmacol. 2012;13:245–256. doi: 10.1016/j.intimp.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 12.Donkor MK, et al. Mammary tumor heterogeneity in the expansion of myeloid-derived suppressor cells. Int Immunopharmacol. 2009;9:937–948. doi: 10.1016/j.intimp.2009.03.021. [DOI] [PubMed] [Google Scholar]
- 13.Rashid OM, et al. Resection of the primary tumor improves survival in metastatic breast cancer by reducing overall tumor burden. Surgery. 2013 doi: 10.1016/j.surg.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Salvadori S, Martinelli G, Zier K. Resection of solid tumors reverses T cell defects and restores protective immunity. J Immunol. 2000;164:2214–2220. doi: 10.4049/jimmunol.164.4.2214. [DOI] [PubMed] [Google Scholar]
- 15.Bayne LJ, et al. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell. 2012;21:822–835. doi: 10.1016/j.ccr.2012.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Younos I, et al. Tumor- and organ-dependent infiltration by myeloid-derived suppressor cells. Int Immunopharmacol. 2011;11:814–826. doi: 10.1016/j.intimp.2011.02.021. [DOI] [PubMed] [Google Scholar]
- 17.Dolcetti L, et al. Hierarchy of immunosuppressive strength among myeloid-derived suppressor cell subsets is determined by GM-CSF. Eur J Immunol. 2010;40:22–35. doi: 10.1002/eji.200939903. [DOI] [PubMed] [Google Scholar]
- 18.Waight JD, Hu Q, Miller A, Liu S, Abrams SI. Tumor-Derived G-CSF Facilitates Neoplastic Growth through a Granulocytic Myeloid-Derived Suppressor Cell-Dependent Mechanism. PLoS One. 2011;6:e27690. doi: 10.1371/journal.pone.0027690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kowanetz M, et al. Granulocyte-colony stimulating factor promotes lung metastasis through mobilization of Ly6G+Ly6C+ granulocytes. Proc Natl Acad Sci U S A. 2010;107:21248–21255. doi: 10.1073/pnas.1015855107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pekarek LA, Starr BA, Toledano AY, Schreiber H. Inhibition of tumor growth by elimination of granulocytes. J Exp Med. 1995;181:435–440. doi: 10.1084/jem.181.1.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li H, Han Y, Guo Q, Zhang M, Cao X. Cancer-Expanded Myeloid-Derived Suppressor Cells Induce Anergy of NK Cells through Membrane-Bound TGF-{beta}1. J Immunol. 2009;182:240–249. doi: 10.4049/jimmunol.182.1.240. [DOI] [PubMed] [Google Scholar]
- 22.Abe F, et al. Myeloid-derived suppressor cells in mammary tumor progression in FVB Neu transgenic mice. Cancer Immunol Immunother. 2010;59:47–62. doi: 10.1007/s00262-009-0719-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lappat EJ, Cawein M. A Study of the Leukemoid Response to Transplantable a-280 Tumor in Mice. Cancer Res. 1964;24:302–311. [PubMed] [Google Scholar]
- 24.Lee MY, Rosse C. Depletion of lymphocyte subpopulations in primary and secondary lymphoid organs of mice by a transplanted granulocytosis-inducing mammary carcinoma. Cancer Res. 1982;42:1255–1260. [PubMed] [Google Scholar]
- 25.Tsuchiya Y, Igarashi M, Suzuki R, Kumagai K. Production of colony-stimulating factor by tumor cells and the factor-mediated induction of suppressor cells. J Immunol. 1988;141:699–708. [PubMed] [Google Scholar]
- 26.Bennett JA, Mitchell MS. Induction of suppressor cells by intravenous administration of Bacillus Calmette-Guerin and its modulation by cyclophosphamide. Biochem Pharmacol. 1979;28:1947–1952. doi: 10.1016/0006-2952(79)90649-x. [DOI] [PubMed] [Google Scholar]
- 27.Wren SM, Wepsic HT, Larson CH, De Silva MA, Mizushima Y. Inhibition of the graft-versus-host response by BCGcw-induced suppressor cells or prostaglandin E1. Cell Immunol. 1983;76:361–371. doi: 10.1016/0008-8749(83)90379-9. [DOI] [PubMed] [Google Scholar]
- 28.Predina JD, et al. Cytoreduction surgery reduces systemic myeloid suppressor cell populations and restores intratumoral immunotherapy effectiveness. J Hematol Oncol. 2012;5:34. doi: 10.1186/1756-8722-5-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Delano MJ, et al. MyD88-dependent expansion of an immature GR-1(+)CD11b(+) population induces T cell suppression and Th2 polarization in sepsis. J Exp Med. 2007;204:1463–1474. doi: 10.1084/jem.20062602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oseroff A, Okada S, Strober S. Natural suppressor (NS) cells found in the spleen of neonatal mice and adult mice given total lymphoid irradiation (TLI) express the null surface phenotype. J Immunol. 1984;132:101–110. [PubMed] [Google Scholar]
- 31.Young MR, Wright MA. Myelopoiesis-associated immune suppressor cells in mice bearing metastatic Lewis lung carcinoma tumors: Gamma-interferon plus tumor necrosis factor-alpha synergistically reduces immune suppressor and tumor growth-promoting activities of bone marrow cells and diminishes tumor recurrence and metastasis. Cancer Res. 1992;52:6335–6340. [PubMed] [Google Scholar]
- 32.Kusmartsev SA, Li Y, Chen SH. Gr-1+ myeloid cells derived from tumor-bearing mice inhibit primary T cell activation induced through CD3/CD28 costimulation. J Immunol. 2000;165:779–785. doi: 10.4049/jimmunol.165.2.779. [DOI] [PubMed] [Google Scholar]
- 33.Gabrilovich D, et al. Vascular endothelial growth factor inhibits the development of dendritic cells and dramatically affects the differentiation of multiple hematopoietic lineages in vivo. Blood. 1998;92:4150–4166. [PubMed] [Google Scholar]
- 34.Watson GA, Fu YX, Lopez DM. Splenic macrophages from tumor-bearing mice co-expressing MAC-1 and MAC-2 antigens exert immunoregulatory functions via two distinct mechanisms. J Leukoc Biol. 1991;49:126–138. doi: 10.1002/jlb.49.2.126. [DOI] [PubMed] [Google Scholar]
- 35.Craddock CF, et al. Circulating stem cells in mice treated with cyclophosphamide. Blood. 1992;80:264–269. [PubMed] [Google Scholar]
- 36.Sy MS, Miller SD, Claman HN. Immune suppression with supraoptimal doses of antigen in contact sensitivity. I. Demonstration of suppressor cells and their sensitivity to cyclophosphamide. J Immunol. 1977;119:240–244. [PubMed] [Google Scholar]
- 37.Hooper DC, Hoskin DW, Gronvik KO, Murgita RA. Murine neonatal spleen contains natural T and non-T suppressor cells capable of inhibiting adult alloreactive and newborn autoreactive T-cell proliferation. Cell Immunol. 1986;99:461–475. doi: 10.1016/0008-8749(86)90254-6. [DOI] [PubMed] [Google Scholar]
- 38.Pak AS, et al. Mechanisms of immune suppression in patients with head and neck cancer: presence of CD34(+) cells which suppress immune functions within cancers that secrete granulocyte-macrophage colony-stimulating factor. Clin Cancer Res. 1995;1:95–103. [PubMed] [Google Scholar]
- 39.Young MRI, Young ME, Wright MA. Stimulation of immune-suppressive bone marrow cells by colony-stimulating factors. Exp Hematol. 1990;18:806–811. [PubMed] [Google Scholar]
- 40.Bronte V, et al. Unopposed production of granulocyte-macrophage colony-stimulating factor by tumors inhibits CD8+ T cell responses by dysregulating antigen-presenting cell maturation. J Immunol. 1999;162:5728–5737. [PMC free article] [PubMed] [Google Scholar]
- 41.Serafini P, et al. High-dose granulocyte-macrophage colony-stimulating factor-producing vaccines impair the immune response through the recruitment of myeloid suppressor cells. Cancer Res. 2004;64:6337–6343. doi: 10.1158/0008-5472.CAN-04-0757. [DOI] [PubMed] [Google Scholar]
- 42.Shojaei F, et al. G-CSF-initiated myeloid cell mobilization and angiogenesis mediate tumor refractoriness to anti-VEGF therapy in mouse models. Proc Natl Acad Sci USA. 2009;106:6742–6747. doi: 10.1073/pnas.0902280106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Talmadge JE. Pathways Mediating the Expansion and Immunosuppressive Activity of Myeloid-Derived Suppressor Cells and Their Relevance to Cancer Therapy. Clin Cancer Res. 2007;13:5243–5248. doi: 10.1158/1078-0432.CCR-07-0182. [DOI] [PubMed] [Google Scholar]
- 44.Boutte AM, McDonald WH, Shyr Y, Yang L, Lin PC. Characterization of the MDSC proteome associated with metastatic murine mammary tumors using label-free mass spectrometry and shotgun proteomics. PLoS One. 2011;6:e22446. doi: 10.1371/journal.pone.0022446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chornoguz O, et al. Proteomic pathway analysis reveals inflammation increases myeloid-derived suppressor cell resistance to apoptosis. Mol Cell Proteomics. 2011;10:M110 002980. doi: 10.1074/mcp.M110.002980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bronte V, Serafini P, Apolloni E, Zanovello P. Tumor-induced immune dysfunctions caused by myeloid suppressor cells. J Immunother. 2001;24:431–446. doi: 10.1097/00002371-200111000-00001. [DOI] [PubMed] [Google Scholar]
- 47.Gabrilovich DI, Velders MP, Sotomayor EM, Kast WM. Mechanism of immune dysfunction in cancer mediated by immature Gr-1+ myeloid cells. J Immunol. 2001;166:5398–5406. doi: 10.4049/jimmunol.166.9.5398. [DOI] [PubMed] [Google Scholar]
- 48.Bronte V, et al. Apoptotic death of CD8+ T lymphocytes after immunization: induction of a suppressive population of Mac-1+/Gr-1+ cells. J Immunol. 1998;161:5313–5320. [PMC free article] [PubMed] [Google Scholar]
- 49.Young MR, Wright MA, Matthews JP, Malik I, Prechel M. Suppression of T cell proliferation by tumor-induced granulocyte-macrophage progenitor cells producing transforming growth factor-beta and nitric oxide. J Immunol. 1996;156:1916–1922. [PubMed] [Google Scholar]
- 50.Gabrilovich DI, et al. The Terminology Issue for Myeloid-Derived Suppressor Cells. Cancer Res. 2007;67:425–425. doi: 10.1158/0008-5472.CAN-06-3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang R, Roden RBS. The Terminology Issue for Myeloid-Derived Suppressor Cells. Cancer Res. 2007;67:426–426. doi: 10.1158/0008-5472.CAN-06-3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ribechini E, Greifenberg V, Sandwick S, Lutz MB. Subsets, expansion and activation of myeloid-derived suppressor cells. Medical Microbiology and Immunology. 2010;199:273–281. doi: 10.1007/s00430-010-0151-4. [DOI] [PubMed] [Google Scholar]
- 53.Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J.Immunol. 2008;181:5791–5802. doi: 10.4049/jimmunol.181.8.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Karsunky H, Merad M, Cozzio A, Weissman IL, Manz MG. Flt3 ligand regulates dendritic cell development from Flt3+ lymphoid and myeloid-committed progenitors to Flt3+ dendritic cells in vivo. J Exp Med. 2003;198:305–313. doi: 10.1084/jem.20030323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Movahedi K, et al. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res. 2010;70:5728–5739. doi: 10.1158/0008-5472.CAN-09-4672. [DOI] [PubMed] [Google Scholar]
- 56.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sawanobori Y, et al. Chemokine-mediated rapid turnover of myeloid-derived suppressor cells in tumor-bearing mice. Blood. 2008;111:5457–5466. doi: 10.1182/blood-2008-01-136895. [DOI] [PubMed] [Google Scholar]
- 58.Virtuoso LP, et al. Characterization of iNOS+ Neutrophil-like ring cell in tumor-bearing mice. J Transl Med. 2012;10:152. doi: 10.1186/1479-5876-10-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sasmono RT, et al. Mouse neutrophilic granulocytes express mRNA encoding the macrophage colony-stimulating factor receptor (CSF-1R) as well as many other macrophage-specific transcripts and can transdifferentiate into macrophages in vitro in response to CSF-1. J Leukoc Biol. 2007;82:111–123. doi: 10.1189/jlb.1206713. [DOI] [PubMed] [Google Scholar]
- 60.Movahedi K, et al. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood. 2008;111:4233–4244. doi: 10.1182/blood-2007-07-099226. [DOI] [PubMed] [Google Scholar]
- 61.Haile LA, Gamrekelashvili J, Manns MP, Korangy F, Greten TF. CD49d is a new marker for distinct myeloid-derived suppressor cell subpopulations in mice. J Immunol. 2010;185:203–210. doi: 10.4049/jimmunol.0903573. [DOI] [PubMed] [Google Scholar]
- 62.Brandau S, Moses K, Lang S. The kinship of neutrophils and granulocytic myeloid-derived suppressor cells in cancer: Cousins, siblings or twins? Semin Cancer Biol. 2013;23:171–182. doi: 10.1016/j.semcancer.2013.02.007. [DOI] [PubMed] [Google Scholar]
- 63.Mielcarek M, Martin PJ, Torok-Storb B. Suppression of alloantigen-induced T-cell proliferation by CD14+ cells derived from granulocyte colony-stimulating factor-mobilized peripheral blood mononuclear cells. Blood. 1997;89:1629–1634. [PubMed] [Google Scholar]
- 64.Talmadge J, et al. Immunologic attributes of cytokine mobilized peripheral blood stem cells and recovery following transplantation. Bone Marrow Transplantation. 1996;17:101–109. [PubMed] [Google Scholar]
- 65.Singh RK, et al. Fas-FasL-mediated CD4+ T-cell apoptosis following stem cell transplantation. Cancer Res. 1999;59:3107–3111. [PubMed] [Google Scholar]
- 66.Wanebo HJ, et al. Indomethacin sensitive suppressor-cell activity in head and neck cancer patients. The role of the adherent mononuclear cell. Cancer. 1988;61:462–474. doi: 10.1002/1097-0142(19880201)61:3<462::aid-cncr2820610310>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 67.Ko JS, et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin Cancer Res. 2009;15:2148–2157. doi: 10.1158/1078-0432.CCR-08-1332. [DOI] [PubMed] [Google Scholar]
- 68.Laoui D, et al. Mononuclear phagocyte heterogeneity in cancer: different subsets and activation states reaching out at the tumor site. Immunobiology. 2011;216:1192–1202. doi: 10.1016/j.imbio.2011.06.007. [DOI] [PubMed] [Google Scholar]
- 69.Youn JI, Gabrilovich DI. The biology of myeloid-derived suppressor cells: the blessing and the curse of morphological and functional heterogeneity. Eur J Immunol. 2010;40:2969–2975. doi: 10.1002/eji.201040895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lathers DM, Clark JI, Achille NJ, Young MR. Phase IB study of 25-hydroxyvitamin D(3) treatment to diminish suppressor cells in head and neck cancer patients. Hum Immunol. 2001;62:1282–1293. doi: 10.1016/s0198-8859(01)00317-2. [DOI] [PubMed] [Google Scholar]
- 71.Almand B, et al. Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J Immunol. 2001;166:678–689. doi: 10.4049/jimmunol.166.1.678. [DOI] [PubMed] [Google Scholar]
- 72.Zea AH, et al. Arginase-producing myeloid suppressor cells in renal cell carcinoma patients: a mechanism of tumor evasion. Cancer Res. 2005;65:3044–3048. doi: 10.1158/0008-5472.CAN-04-4505. [DOI] [PubMed] [Google Scholar]
- 73.Poschke I, Mougiakakos D, Hansson J, Masucci GV, Kiessling R. Immature immunosuppressive CD14+HLA-DR-/low cells in melanoma patients are Stat3hi and overexpress CD80, CD83, and DC-sign. Cancer Res. 2010;70:4335–4345. doi: 10.1158/0008-5472.CAN-09-3767. [DOI] [PubMed] [Google Scholar]
- 74.Filipazzi P, et al. Identification of a New Subset of Myeloid Suppressor Cells in Peripheral Blood of Melanoma Patients With Modulation by a Granulocyte-Macrophage Colony-Stimulation Factor-Based Antitumor Vaccine. J Clin Oncol. 2007;25:2546–2553. doi: 10.1200/JCO.2006.08.5829. [DOI] [PubMed] [Google Scholar]
- 75.Rodriguez PC, et al. Arginase I-producing myeloid-derived suppressor cells in renal cell carcinoma are a subpopulation of activated granulocytes. Cancer Res. 2009;69:1553–1560. doi: 10.1158/0008-5472.CAN-08-1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dumitru CA, Moses K, Trellakis S, Lang S, Brandau S. Neutrophils and granulocytic myeloid-derived suppressor cells: immunophenotyping, cell biology and clinical relevance in human oncology. Cancer Immunol Immunother. 2012;61:1155–1167. doi: 10.1007/s00262-012-1294-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhao F, et al. S100A9 a new marker for monocytic human myeloid-derived suppressor cells. Immunology. 2012;136:176–183. doi: 10.1111/j.1365-2567.2012.03566.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mantovani A, Sica A. Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Curr Opin Immunol. 2010;22:231–237. doi: 10.1016/j.coi.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 79.Glasser L, Fiederlein RL. Functional differentiation of normal human neutrophils. Blood. 1987;69:937–944. [PubMed] [Google Scholar]
- 80.DuPre SA, Hunter KW., Jr Murine mammary carcinoma 4T1 induces a leukemoid reaction with splenomegaly: association with tumor-derived growth factors. Exp Mol Pathol. 2007;82:12–24. doi: 10.1016/j.yexmp.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 81.Sica A, Bronte V. Altered macrophage differentiation and immune dysfunction in tumor development. J Clin Invest. 2007;117:1155–1166. doi: 10.1172/JCI31422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sade-Feldman M, et al. Tumor Necrosis Factor-alpha Blocks Differentiation and Enhances Suppressive Activity of Immature Myeloid Cells during Chronic Inflammation. Immunity. 2013;38:541–554. doi: 10.1016/j.immuni.2013.02.007. [DOI] [PubMed] [Google Scholar]
- 83.Cheng P, et al. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J Exp Med. 2008;205:2235–2249. doi: 10.1084/jem.20080132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nagaraj S, Gabrilovich DI. Myeloid-derived suppressor cells in human cancer. Cancer J. 2010;16:348–353. doi: 10.1097/PPO.0b013e3181eb3358. [DOI] [PubMed] [Google Scholar]
- 85.Ostrand-Rosenberg S, Sinha P. Myeloid-Derived Suppressor Cells: Linking Inflammation and Cancer. J Immunol. 2009;182:4499–4506. doi: 10.4049/jimmunol.0802740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gibb DR, Saleem SJ, Kang DJ, Subler MA, Conrad DH. ADAM10 overexpression shifts lympho- and myelopoiesis by dysregulating site 2/site 3 cleavage products of Notch. J Immunol. 2011;186:4244–4252. doi: 10.4049/jimmunol.1003318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Song X, et al. CD11b+/Gr-1+ immature myeloid cells mediate suppression of T cells in mice bearing tumors of IL-1beta-secreting cells. J Immunol. 2005;175:8200–8208. doi: 10.4049/jimmunol.175.12.8200. [DOI] [PubMed] [Google Scholar]
- 88.Young MR, Wright MA, Young ME. Antibodies to colony-stimulating factors block Lewis lung carcinoma cell stimulation of immune-suppressive bone marrow cells. Cancer Immunol.Immunother. 1991;33:146–152. doi: 10.1007/BF01756134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Liu Y, et al. MicroRNA-494 Is Required for the Accumulation and Functions of Tumor-Expanded Myeloid-Derived Suppressor Cells via Targeting of PTEN. J Immunol. 2012;188:5500–5510. doi: 10.4049/jimmunol.1103505. [DOI] [PubMed] [Google Scholar]
- 90.Almand B, et al. Clinical significance of defective dendritic cell differentiation in cancer. Clin Cancer Res. 2000;6:1755–1766. [PubMed] [Google Scholar]
- 91.Melani C. Myeloid cell expansion elicited by the progression of spontaneous mammary carcinomas in c-erbB-2 transgenic BALB/c mice suppresses immune reactivity. Blood. 2003;102:2138–2145. doi: 10.1182/blood-2003-01-0190. [DOI] [PubMed] [Google Scholar]
- 92.Pan PY, et al. Reversion of immune tolerance in advanced malignancy: modulation of myeloid-derived suppressor cell development by blockade of stem-cell factor function. Blood. 2008;111:219–228. doi: 10.1182/blood-2007-04-086835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Solheim J, et al. Spleen but not tumor infiltration by dendritic and T cells is increased by intravenous adenovirus-Flt3 ligand injection. Cancer Gene Ther. 2007;14:364–371. doi: 10.1038/sj.cgt.7701018. [DOI] [PubMed] [Google Scholar]
- 94.Morales JK, Kmieciak M, Knutson KL, Bear HD, Manjili MH. GM-CSF is one of the main breast tumor-derived soluble factors involved in the differentiation of CD11b-Gr1- bone marrow progenitor cells into myeloid-derived suppressor cells. Breast Cancer Res Treat. 2010;123:39–49. doi: 10.1007/s10549-009-0622-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ko JS, et al. Direct and differential suppression of myeloid-derived suppressor cell subsets by sunitinib is compartmentally constrained. Cancer Res. 2010;70:3526–3536. doi: 10.1158/0008-5472.CAN-09-3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Priceman SJ, et al. Targeting distinct tumor-infiltrating myeloid cells by inhibiting CSF-1 receptor: combating tumor evasion of antiangiogenic therapy. Blood. 2010;115:1461–1471. doi: 10.1182/blood-2009-08-237412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.DeNardo DG, et al. Leukocyte Complexity Predicts Breast Cancer Survival and Functionally Regulates Response to Chemotherapy. Cancer Discov. 2011;1:54–67. doi: 10.1158/2159-8274.CD-10-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Fricke I, et al. Vascular endothelial growth factor-trap overcomes defects in dendritic cell differentiation but does not improve antigen-specific immune responses. Clin Cancer Res. 2007;13:4840–4848. doi: 10.1158/1078-0432.CCR-07-0409. [DOI] [PubMed] [Google Scholar]
- 99.Panopoulos AD, Watowich SS. Granulocyte colony-stimulating factor: molecular mechanisms of action during steady state and 'emergency' hematopoiesis. Cytokine. 2008;42:277–288. doi: 10.1016/j.cyto.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rutella S, Zavala F, Danese S, Kared H, Leone G. Granulocyte colony-stimulating factor: a novel mediator of T cell tolerance. J Immunol. 2005;175:7085–7091. doi: 10.4049/jimmunol.175.11.7085. [DOI] [PubMed] [Google Scholar]
- 101.Kyo S, Kanaya T, Takakura M, Inoue M. A case of cervical cancer with aggressive tumor growth: possible autocrine growth stimulation by G-CSF and Il-6. Gynecol Oncol. 2000;78:383–387. doi: 10.1006/gyno.2000.5904. [DOI] [PubMed] [Google Scholar]
- 102.Okazaki T, et al. Granulocyte colony-stimulating factor promotes tumor angiogenesis via increasing circulating endothelial progenitor cells and Gr1+CD11b+ cells in cancer animal models. Int Immunol. 2006;18:1–9. doi: 10.1093/intimm/dxh334. [DOI] [PubMed] [Google Scholar]
- 103.Bronte V, Zanovello P. Regulation of immune responses by L-arginine metabolism. Nat Rev Immunol. 2005;5:641–654. doi: 10.1038/nri1668. [DOI] [PubMed] [Google Scholar]
- 104.Peranzoni E, et al. Myeloid-derived suppressor cell heterogeneity and subset definition. Curr Opin Immunol. 2010;22:238–244. doi: 10.1016/j.coi.2010.01.021. [DOI] [PubMed] [Google Scholar]
- 105.Young MR, Young ME, Wright MA. Myelopoiesis-associated suppressor-cell activity in mice with Lewis lung carcinoma tumors: interferon-gamma plus tumor necrosis factor-alpha synergistically reduce suppressor cell activity. Int.J.Cancer. 1990;46:245–250. doi: 10.1002/ijc.2910460217. [DOI] [PubMed] [Google Scholar]
- 106.Choi KL, Maier T, Holda JH, Claman HN. Suppression of cytotoxic T-cell generation by natural suppressor cells from mice with GVHD is partially reversed by indomethacin. Cell Immunol. 1988;112:271–278. doi: 10.1016/0008-8749(88)90297-3. [DOI] [PubMed] [Google Scholar]
- 107.Schlecker E, et al. Tumor-Infiltrating Monocytic Myeloid-Derived Suppressor Cells Mediate CCR5-Dependent Recruitment of Regulatory T Cells Favoring Tumor Growth. J Immunol. 2012;189:5602–5611. doi: 10.4049/jimmunol.1201018. [DOI] [PubMed] [Google Scholar]
- 108.Youn JI, Collazo M, Shalova IN, Biswas SK, Gabrilovich DI. Characterization of the nature of granulocytic myeloid-derived suppressor cells in tumor-bearing mice. J Leukoc Biol. 2012;91:167–181. doi: 10.1189/jlb.0311177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mirza N. All-trans-Retinoic Acid Improves Differentiation of Myeloid Cells and Immune Response in Cancer Patients. Cancer Res. 2006;66:9299–9307. doi: 10.1158/0008-5472.CAN-06-1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kusmartsev S, et al. All-trans-retinoic acid eliminates immature myeloid cells from tumor-bearing mice and improves the effect of vaccination. Cancer Res. 2003;63:4441–4449. [PubMed] [Google Scholar]
- 111.Young MRI, et al. 1-alpha,25-dihydroxyvitamin D3 plus gamma-interferon blocks lung tumor production of granulocyte-macrophage colony-stimulating factor and induction of immunosuppressor cells. Cancer Res. 1993;53:6006–6006. [PubMed] [Google Scholar]
- 112.Sinha P, et al. Proinflammatory s100 proteins regulate the accumulation of myeloid-derived suppressor cells. J Immunol. 2008;181:4666–4675. doi: 10.4049/jimmunol.181.7.4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yang L, et al. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell. 2004;6:409–421. doi: 10.1016/j.ccr.2004.08.031. [DOI] [PubMed] [Google Scholar]
- 114.Zon LI. Intrinsic and extrinsic control of haematopoietic stem-cell self-renewal. Nature. 2008;453:306–313. doi: 10.1038/nature07038. [DOI] [PubMed] [Google Scholar]
- 115.Si Y, Tsou CL, Croft K, Charo IF. CCR2 mediates hematopoietic stem and progenitor cell trafficking to sites of inflammation in mice. J Clin Invest. 2010;120:1192–1203. doi: 10.1172/JCI40310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Johns JL, Christopher MM. Extramedullary hematopoiesis: a new look at the underlying stem cell niche, theories of development, and occurrence in animals. Vet Pathol. 2012;49:508–523. doi: 10.1177/0300985811432344. [DOI] [PubMed] [Google Scholar]
- 117.Pruijt JF, Willemze R, Fibbe WE. Mechanisms underlying hematopoietic stem cell mobilization induced by the CXC chemokine interleukin-8. Curr Opin Hematol. 1999;6:152–158. doi: 10.1097/00062752-199905000-00005. [DOI] [PubMed] [Google Scholar]
- 118.Demetri GD, Griffin JD. Granulocyte colony-stimulating factor and its receptor. Blood. 1991;78:2791–2808. [PubMed] [Google Scholar]
- 119.Price TH, Chatta GS, Dale DC. Effect of recombinant granulocyte colony-stimulating factor on neutrophil kinetics in normal young and elderly humans. Blood. 1996;88:335–340. [PubMed] [Google Scholar]
- 120.Nefedova Y. Regulation of Dendritic Cell Differentiation and Antitumor Immune Response in Cancer by Pharmacologic-Selective Inhibition of the Janus-Activated Kinase 2/Signal Transducers and Activators of Transcription 3 Pathway. Cancer Res. 2005;65:9525–9535. doi: 10.1158/0008-5472.CAN-05-0529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Levesque JP, Hendy J, Takamatsu Y, Simmons PJ, Bendall LJ. Disruption of the CXCR4/CXCL12 chemotactic interaction during hematopoietic stem cell mobilization induced by GCSF or cyclophosphamide. J Clin Invest. 2003;111:187–196. doi: 10.1172/JCI15994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lévesque J-P, et al. Mobilization by either cyclophosphamide or granulocyte colony-stimulating factor transforms the bone marrow into a highly proteolytic environment. Exp Hematol. 2002;30:440–449. doi: 10.1016/s0301-472x(02)00788-9. [DOI] [PubMed] [Google Scholar]
- 123.Link DC. Neutrophil homeostasis: a new role for stromal cell-derived factor-1. Immunol Res. 2005;32:169–178. doi: 10.1385/IR:32:1-3:169. [DOI] [PubMed] [Google Scholar]
- 124.Nagasawa T, et al. Defects of B-cell lymphopoiesis and bone-marrow myelopoiesis in mice lacking the CXC chemokine PBSF/SDF-1. Nature. 1996;382:635–638. doi: 10.1038/382635a0. [DOI] [PubMed] [Google Scholar]
- 125.McQuibban GA. Matrix Metalloproteinase Activity Inactivates the CXC Chemokine Stromal Cell-derived Factor-1. J Biol Chem. 2001;276:43503–43508. doi: 10.1074/jbc.M107736200. [DOI] [PubMed] [Google Scholar]
- 126.Luster AD, Alon R, von Andrian UH. Immune cell migration in inflammation: present and future therapeutic targets. Nat Immunol. 2005;6:1182–1190. doi: 10.1038/ni1275. [DOI] [PubMed] [Google Scholar]
- 127.Ueno T, et al. Significance of macrophage chemoattractant protein-1 in macrophage recruitment, angiogenesis, and survival in human breast cancer. Clin Cancer Res. 2000;6:3282–3289. [PubMed] [Google Scholar]
- 128.Watanabe T, et al. GM-CSF-mobilized peripheral blood CD34+ cells differ from steady- state bone marrow CD34+ cells in adhesion molecule expression. Bone Marrow Transplant. 1997;19:1175–1181. doi: 10.1038/sj.bmt.1700814. [DOI] [PubMed] [Google Scholar]
- 129.Pruijt JF, et al. Anti-LFA-1 blocking antibodies prevent mobilization of hematopoietic progenitor cells induced by interleukin-8. Blood. 1998;91:4099–4105. [PubMed] [Google Scholar]
- 130.Jin H, Su J, Garmy-Susini B, Kleeman J, Varner J. Integrin α4β1 Promotes Monocyte Trafficking and Angiogenesis in Tumors. Cancer Res. 2006;66:2146–2152. doi: 10.1158/0008-5472.CAN-05-2704. [DOI] [PubMed] [Google Scholar]
- 131.Jin H, et al. A homing mechanism for bone marrow-derived progenitor cell recruitment to the neovasculature. J Clin Invest. 2006;116:652–662. doi: 10.1172/JCI24751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Jin F, et al. Degradation of BM SDF-1 by MMP-9: the role in G-CSF-induced hematopoietic stem/progenitor cell mobilization. Bone Marrow Transplant. 2008;42:581–588. doi: 10.1038/bmt.2008.222. [DOI] [PubMed] [Google Scholar]
- 133.Chavakis E, et al. Role of beta2-integrins for homing and neovascularization capacity of endothelial progenitor cells. J Exp Med. 2005;201:63–72. doi: 10.1084/jem.20041402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Youn JI, et al. Epigenetic silencing of retinoblastoma gene regulates pathologic differentiation of myeloid cells in cancer. Nat Immunol. 2013 doi: 10.1038/ni.2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Connolly MK, et al. Distinct populations of metastases-enabling myeloid cells expand in the liver of mice harboring invasive and preinvasive intra-abdominal tumor. J Leukoc Biol. 2010;87:713–725. doi: 10.1189/jlb.0909607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.De Santo C. Nitroaspirin corrects immune dysfunction in tumor-bearing hosts and promotes tumor eradication by cancer vaccination. Proc Natl Acad Sci. 2005;102:4185–4190. doi: 10.1073/pnas.0409783102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ellies LG, et al. Mammary tumor latency is increased in mice lacking the inducible nitric oxide synthase. Int J Cancer. 2003;106:1–7. doi: 10.1002/ijc.11178. [DOI] [PubMed] [Google Scholar]
- 138.Sun HL, et al. Increased frequency and clinical significance of myeloid-derived suppressor cells in human colorectal carcinoma. World J Gastroenterol. 2012;18:3303–3309. doi: 10.3748/wjg.v18.i25.3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Paget S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev. 1989;8:98–101. [PubMed] [Google Scholar]
- 140.Balwit JM, Hwu P, Urba WJ, Marincola FM. The iSBTc/SITC primer on tumor immunology and biological therapy of cancer: a summary of the 2010 program. J Transl Med. 2011;9:18. doi: 10.1186/1479-5876-9-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sumida K, et al. Anti-IL-6 receptor mAb eliminates myeloid-derived suppressor cells and inhibits tumor growth by enhancing T-cell responses. Eur J Immunol. 2012;42:2060–2072. doi: 10.1002/eji.201142335. [DOI] [PubMed] [Google Scholar]
- 142.Vincent J, et al. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. 2010;70:3052–3061. doi: 10.1158/0008-5472.CAN-09-3690. [DOI] [PubMed] [Google Scholar]
- 143.Kodumudi KN, et al. A novel chemoimmunomodulating property of docetaxel: suppression of myeloid-derived suppressor cells in tumor bearers. Clin Cancer Res. 2010;16:4583–4594. doi: 10.1158/1078-0432.CCR-10-0733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Serafini P, et al. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J Exp Med. 2006;203:2691–2702. doi: 10.1084/jem.20061104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Suzuki E. Gemcitabine Selectively Eliminates Splenic Gr-1+/CD11b+ Myeloid Suppressor Cells in Tumor-Bearing Animals and Enhances Antitumor Immune Activity. Clin Cancer Res. 2005;11:6713–6721. doi: 10.1158/1078-0432.CCR-05-0883. [DOI] [PubMed] [Google Scholar]
- 146.Antonia SJ, et al. Combination of p53 Cancer Vaccine with Chemotherapy in Patients with Extensive Stage Small Cell Lung Cancer. Clin Cancer Res. 2006;12:878–887. doi: 10.1158/1078-0432.CCR-05-2013. [DOI] [PubMed] [Google Scholar]
- 147.Mukherjee P, et al. Progression of Pancreatic Adenocarcinoma Is Significantly Impeded with a Combination of Vaccine and COX-2 Inhibition. J Immunol. 2009;182:216–224. [PMC free article] [PubMed] [Google Scholar]
- 148.Stenvold H, et al. Overexpression of matrix metalloproteinase-7 and-9 in NSCLC tumor and stromal cells: correlation with a favorable clinical outcome. Lung Cancer. 2012;75:235–241. doi: 10.1016/j.lungcan.2011.06.010. [DOI] [PubMed] [Google Scholar]
- 149.Corzo CA, et al. HIF-1alpha regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med. 2010;207:2439–2453. doi: 10.1084/jem.20100587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Du R, et al. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell. 2008;13:206–220. doi: 10.1016/j.ccr.2008.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Yang L, et al. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell. 2008;13:23–35. doi: 10.1016/j.ccr.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Forrester E, et al. Effect of conditional knockout of the type II TGF-beta receptor gene in mammary epithelia on mammary gland development and polyomavirus middle T antigen induced tumor formation and metastasis. Cancer Res. 2005;65:2296–2302. doi: 10.1158/0008-5472.CAN-04-3272. [DOI] [PubMed] [Google Scholar]
- 153.Fidler IJ. Metastasis: Quantitative Analysis of Distribution and Fate of Tumor Emboli Labeled With 1251-5-lodo-2'-deoxyuridine1,2,3. J Natl Cancer Inst. 1970;45:773–782. [PubMed] [Google Scholar]
- 154.Fidler IJ, Gersten DM, Hart IR. The biology of cancer invasion and metastasis. Adv Cancer Res. 1978;28:149–250. doi: 10.1016/s0065-230x(08)60648-x. [DOI] [PubMed] [Google Scholar]
- 155.Hart IR, Fidler IJ. Role of organ selectivity in the determination of metastatic patterns of B16 melanoma. Cancer Res. 1980;40:2281–2287. [PubMed] [Google Scholar]
- 156.Psaila B, Lyden D. The metastatic niche: adapting the foreign soil. Nat Rev Cancer. 2009;9:285–293. doi: 10.1038/nrc2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kaplan RN, Rafii S, Lyden D. Preparing the"Soil": The Premetastatic Niche. Cancer Research. 2006;66:11089–11093. doi: 10.1158/0008-5472.CAN-06-2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Talmadge JE, Fidler IJ. AACR centennial series: the biology of cancer metastasis: historical perspective. Cancer Res. 2010;70:5649–5669. doi: 10.1158/0008-5472.CAN-10-1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Nishie A, et al. Macrophage infiltration and heme oxygenase-1 expression correlate with angiogenesis in human gliomas. Clinical Cancer Research. 1999;5:1107–1113. [PubMed] [Google Scholar]
- 160.Asahara T, et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275:964–967. doi: 10.1126/science.275.5302.964. [DOI] [PubMed] [Google Scholar]
- 161.Mauro E, et al. Mobilization of endothelial progenitor cells in patients with hematological malignancies after treatment with filgrastim and chemotherapy for autologous transplantation. Eur J Haematol. 2007;78:374–380. doi: 10.1111/j.1600-0609.2007.00831.x. [DOI] [PubMed] [Google Scholar]
- 162.Nolan DJ, et al. Bone marrow-derived endothelial progenitor cells are a major determinant of nascent tumor neovascularization. Genes Dev. 2007;21:1546–1558. doi: 10.1101/gad.436307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Davidoff AM, et al. Bone marrow-derived cells contribute to tumor neovasculature and, when modified to express an angiogenesis inhibitor, can restrict tumor growth in mice. Clin Cancer Res. 2001;7:2870–2879. [PubMed] [Google Scholar]
- 164.Dome B. Identification and Clinical Significance of Circulating Endothelial Progenitor Cells in Human Non-Small Cell Lung Cancer. Cancer Res. 2006;66:7341–7347. doi: 10.1158/0008-5472.CAN-05-4654. [DOI] [PubMed] [Google Scholar]
- 165.Young MR, Kolesiak K, Wright MA, Gabrilovich DI. Chemoattraction of femoral CD34+ progenitor cells by tumor-derived vascular endothelial cell growth factor. Clin Exp Metastasis. 1999;17:881–888. doi: 10.1023/a:1006708607666. [DOI] [PubMed] [Google Scholar]
- 166.Mulligan JK, Rosenzweig SA, Young MR. Tumor secretion of VEGF induces endothelial cells to suppress T cell functions through the production of PGE2. J Immunother. 2010;33:126–135. doi: 10.1097/CJI.0b013e3181b91c9c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Madlambayan GJ, et al. Bone marrow stem and progenitor cell contribution to neovasculogenesis is dependent on model system with SDF-1 as a permissive trigger. Blood. 2009;114:4310–4319. doi: 10.1182/blood-2009-03-211342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Li B, et al. Low levels of tumor necrosis factor alpha increase tumor growth by inducing an endothelial phenotype of monocytes recruited to the tumor site. Cancer Res. 2009;69:338–348. doi: 10.1158/0008-5472.CAN-08-1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Asahara T, et al. VEGF contributes to postnatal neovascularization by mobilizing bone marrow-derived endothelial progenitor cells. EMBO J. 1999;18:3964–3972. doi: 10.1093/emboj/18.14.3964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Lyden D, et al. Impaired recruitment of bone-marrow-derived endothelial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nat Med. 2001;7:1194–1201. doi: 10.1038/nm1101-1194. [DOI] [PubMed] [Google Scholar]
- 171.Whiteside TL. The tumor microenvironment and its role in promoting tumor growth. Oncogene. 2008;27:5904–5912. doi: 10.1038/onc.2008.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Whiteside TL. Tricks tumors use to escape from immune control. Oral Oncol. 2009;45:e119–e123. doi: 10.1016/j.oraloncology.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 173.Montero AJ, Diaz-Montero CM, Kyriakopoulos CE, Bronte V, Mandruzzato S. Myeloid-derived suppressor cells in cancer patients: a clinical perspective. J Immunother. 2012;35:107–115. doi: 10.1097/CJI.0b013e318242169f. [DOI] [PubMed] [Google Scholar]
- 174.Poschke I, Kiessling R. On the armament and appearances of human myeloid-derived suppressor cells. Clin Immunol. 2012;144:250–268. doi: 10.1016/j.clim.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 175.Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer. 2009;9:239–252. doi: 10.1038/nrc2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Mazzoni A, et al. Myeloid suppressor lines inhibit T cell responses by an NO-dependent mechanism. J Immunol. 2002;168:689–695. doi: 10.4049/jimmunol.168.2.689. [DOI] [PubMed] [Google Scholar]
- 177.Rodriguez PC, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. 2004;64:5839–5849. doi: 10.1158/0008-5472.CAN-04-0465. [DOI] [PubMed] [Google Scholar]
- 178.Nagaraj S, Schrum AG, Cho HI, Celis E, Gabrilovich DI. Mechanism of T cell tolerance induced by myeloid-derived suppressor cells. J Immunol. 2010;184:3106–3116. doi: 10.4049/jimmunol.0902661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Wu L, et al. Inhibition of PPARgamma in myeloid-lineage cells induces systemic inflammation, immunosuppression, and tumorigenesis. Blood. 2012;119:115–126. doi: 10.1182/blood-2011-06-363093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Wolf AM, et al. The effect of zoledronic acid on the function and differentiation of myeloid cells. Haematologica. 2006;91:1165–1171. [PubMed] [Google Scholar]
- 181.Talmadge JE, et al. Chemoprevention by cyclooxygenase-2 inhibition reduces immature myeloid suppressor cell expansion. Int Immunopharmacol. 2007;7:140–151. doi: 10.1016/j.intimp.2006.09.021. [DOI] [PubMed] [Google Scholar]
- 182.van Cruijsen H, et al. Sunitinib-Induced Myeloid Lineage Redistribution in Renal Cell Cancer Patients: CD1c+ Dendritic Cell Frequency Predicts Progression-Free Survival. Clin Cancer Res. 2008;14:5884–5892. doi: 10.1158/1078-0432.CCR-08-0656. [DOI] [PubMed] [Google Scholar]
- 183.Shojaei F, et al. Bv8 regulates myeloid-cell-dependent tumour angiogenesis. Nature. 2007;450:825–831. doi: 10.1038/nature06348. [DOI] [PubMed] [Google Scholar]
- 184.Huang D, et al. Interleukin-8 mediates resistance to antiangiogenic agent sunitinib in renal cell carcinoma. Cancer Res. 2010;70:1063–1071. doi: 10.1158/0008-5472.CAN-09-3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Shojaei F, et al. Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells. Nature Biotechnol. 2007;25:911–920. doi: 10.1038/nbt1323. [DOI] [PubMed] [Google Scholar]
- 186.Finke J, et al. MDSC as a mechanism of tumor escape from sunitinib mediated anti-angiogenic therapy. Int Immunopharmacol. 2011 doi: 10.1016/j.intimp.2011.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Fischer C, Mazzone M, Jonckx B, Carmeliet P. FLT1 and its ligands VEGFB and PlGF: drug targets for anti-angiogenic therapy? Nat Rev Cancer. 2008;8:942–956. doi: 10.1038/nrc2524. [DOI] [PubMed] [Google Scholar]
- 189.Xu L, et al. Direct Evidence that Bevacizumab, an Anti-VEGF Antibody, Up-regulates SDF1α, CXCR4, CXCL6, and Neuropilin 1 in Tumors from Patients with Rectal Cancer. Cancer Res. 2009;69:7905–7910. doi: 10.1158/0008-5472.CAN-09-2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Filipazzi P, Huber V, Rivoltini L. Phenotype, function and clinical implications of myeloid-derived suppressor cells in cancer patients. Cancer Immunol Immunother. 2012;61:255–263. doi: 10.1007/s00262-011-1161-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Greten TF, Manns MP, Korangy F. Myeloid derived suppressor cells in human diseases. Int Immunopharmacol. 2011 doi: 10.1016/j.intimp.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Zhang J, Patel L, Pienta KJ. CC chemokine ligand 2 (CCL2) promotes prostate cancer tumorigenesis and metastasis. Cytokine & Growth Factor Reviews. 2010;21:41–48. doi: 10.1016/j.cytogfr.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Melani C, Sangaletti S, Barazzetta FM, Werb Z, Colombo MP. Amino-Biphosphonate Mediated MMP-9 Inhibition Breaks the Tumor-Bone Marrow Axis Responsible for Myeloid-Derived Suppressor Cell Expansion and Macrophage Infiltration in Tumor Stroma. Cancer Res. 2007;67:11438–11446. doi: 10.1158/0008-5472.CAN-07-1882. [DOI] [PMC free article] [PubMed] [Google Scholar]