Th17 plasticity and its changes associated with inflammatory bowel disease (original) (raw)
Abstract
CD4 T helper (Th) cell differentiation into distinct T cell subsets is critical to the normal function of the immune system. Until recently, the paradigm held that naïve T cells differentiated into distinct subsets under the guidance of environmental cues (e.g., cytokines) and that once polarized, these cells were committed to a particular functional state. However, the existence of transdifferentiated T cell populations, which express signature transcription factors and cytokines associated with more than one Th subset, challenges the immutability of T helper subsets and suggests that plasticity is a feature of multifaceted immune responses. How this process impacts immune dysregulation in diseases such as inflammatory bowel diseases (IBD) and the machinery that underlies this process is far from fully understood. Interleukin (IL)-17 secreting helper T (Th17) cells have been heavily implicated in tissue-specific immune pathology including murine models of IBD, human Crohn’s disease and ulcerative colitis. Plasticity within this subset is suggested by the existence of IL-17 secreting cells, which, can also secrete interferon-γ, the signature cytokine for Th1 cells or, can co-express the anti-inflammatory transcription factor forkhead box p3, a signature transcription factor of regulatory T cells. In this review we mainly discuss evidence for Th17 plasticity, mechanisms, which govern it, and highlight the potential to therapeutically target this process in human IBD.
Keywords: Th7, Regulatory T cells, T cell plasticity, Inflammatory bowel disease
Core tip: Recently, two innovative clinical failures in inflammatory bowel disease which sought to manipulate T helper (Th) subsets via either transplantation of regulatory T cells or interleukin-17 blockade using secukinumab, suggest that altering the balance between inflammatory and regulatory subsets in inflammatory bowel diseases (IBD) may be more complex than previously thought. One reason may be the flexible nature of T helper subset commitment, otherwise referred to as plasticity. Here we discuss plasticity between regulatory and inflammatory subsets in T helper CD4+ cells, especially Th17 cell subset, and the potential to therapeutically target this process in human IBD.
INTRODUCTION
The spectrum of immune-mediated disease encompasses a wide variety of chronic inflammatory conditions such as inflammatory bowel disease (IBD) comprised of Crohn’s disease (CD), and ulcerative colitis (UC) as well as rheumatoid arthritis (RA), multiple sclerosis, and psoriasis. Although the underlying triggers remain poorly understood, it is now clear that T cells are essential to the development and perpetuation of these diseases[1-3]. Since the landmark description of functionally distinct T helper 1 (Th1) and Th2 CD4**+** effector subsets, each with unique cytokine profiles[4], much work has focussed on dissecting their roles in both health and disease. The spotlight has recently been drawn to a novel subset, the Th17 cell, so named due to its ability to secrete interleukin (IL)-17A, which has emerged as a major player in tissue-specific immune pathology. The initial emphasis on the detrimental effects of Th17 is reflected by the plethora of early literature supporting such a role in both human and murine studies[5-10]. However, a growing body of evidence now suggests essential protective roles, particularly in the context of mucosal integrity and defence against extracellular pathogens[5,11-16]. Th17 cells and their associated cytokines have been found to interact more closely with other adaptive immune cells than previously thought, raising interesting questions about how to select and design therapeutic strategies targeting this cell population[2,17]. New technologies such as transcriptome profiling, global epigenetic mapping and computerized simulation analysis[18,19] have captured a more accurate picture of this T cell subset revealing it to be more transient, complex and perhaps more reversible than previously imagined. In addition to a well-established role in extracellular pathogen clearance, Th17 cells can also participate in intracellular pathogen clearance via unconventional interferon (IFN)-γ secretion[20,21]. Human forkhead box p3 (Foxp3) + regulatory T cells (Treg) can differentiate into IL-17 promoting cells _in vitro_[22]. Flexibility within this subset may allow Th17 cells to embrace pro-inflammatory and protective roles in mucosal immunity by secreting a spectrum of cytokines without requiring de novo differentiation of naïve T cells. Likewise, Th17 cells may be generated quickly from Treg in order to defend from acute invasion of pathogens. This ability to transition between functional states is defined as T cell plasticity.
This review mainly focuses on human studies and outlines the major features of Th17 plasticity including the Treg/Th17 paradigm shift in the context of IBD and in the maintenance of intestinal homeostasis.
CHARACTERISATION OF TH17 AND TREG SUBSETS IN IBD
In the last 15 years or so, the focus of attention regarding T cell subsets has shifted from the classical Th1/Th2 paradigm to that of Th17/Treg. This has indeed been the case in IBD.
The discovery of extrathymic Treg development in the intestine has attracted enormous attention and highlights the importance of Treg cells in intestinal homeostasis. Hori et al[23], demonstrated that co-transfer of peripherally generated Foxp3 positive Treg cells could attenuate disease in the adoptive transfer model of mouse colitis. Shortly after this study, Makita et al[24], showed the intestinal prevalence of Treg in patients with IBD. Mucida et al[25], have since identified retinoic acid, derived from vitamin A and metabolized by dendritic cells, as a key signal regulating Foxp3 expression by naïve T cells in response to TGF-β. Overall, the digestive tract requires high levels of inducible Treg cells in order to preserve tolerance to the enormous antigenic burden comprised by commensal flora and dietary antigens[26].
At around the same time, Fujino et al[27], first reported on the prevalence of Th17 cells in patients with IBD. Patients with UC and CD show increased IL-17A levels in serum and mucosa[17] and an IL-17A gene polymorphism has been linked to UC susceptibility[28]. This cytokine, in addition to promoting barrier function, is a potent promoter of granulopoiesis and neutrophil chemotaxis and plays an important role in the clearance of extracellular bacterial and fungal infections[29]. Recently, Ciofani et al[30] have described an intracellular network regulating Th17 specification. Interestingly, genome-wide association studies linked at least 24 loci within this network to single nucleotide polymorphisms (SNPs) associated with ulcerative colitis and Crohn’s disease, highlighting the importance of this T cell subset towards the pathogenesis of IBD[30,31].
IL-17 secreting Th17 subset
Intestinal effector T cells arise from naïve lymphocytes derived from the thymus, which then undergo functional differentiation in the intestinal mucosa upon encountering their cognate antigen displayed by activated antigen-presenting cells (APCs). APCs and potentially other cells release cytokines, which act in combination with environmental cues, including bacterial and dietary products as well as salt concentration[32,33], thereby activating the Jak-STAT and other signalling pathways to exert their biological functions.
Differentiation of Th17 cells is exclusively dependent on signal transducer and activator of transcription 3 (STAT3)[34] and crucially requires the expression of the transcription factor retinoic acid receptor-related orphan receptor γ thymus in mouse (RORγt)[35]. Studies in the mouse and in vitro human cell cultures have revealed the critical roles of transforming growth factor β1 (TGF-β1) alongside IL-6[36-38], IL-1β[39] and IL-21[40] in Th17 polarization. IL-23, secreted mainly by innate myeloid cells including activated dendritic cells (DC), monocytes and macrophages, is critical for Th17 proliferation and maintenance, though dispensable for the initiation of Th17 development. Importantly, mutations within the il23r locus, which encodes a receptor subunit unique to IL-23, are associated with psoriasis[41], ankylosing spondylitis[42] and IBD[43,44].
Whilst Th17 cells are distinguished by IL-17A production, they are also capable of producing other cytokines, including but not limited to IL-17F, IL-21 and IL-22. IL-17F, a member of the IL-17 family, may also have dual roles in the context of mucosal disease. In the DSS model of IBD, IL-17F-/- mice show less severe disease than wild-types[28]. IL-17F evidently appears to have a distinct set of roles, despite structural similarity to IL-17A, and further research will be necessary to establish its specific importance. Substantial evidence exists for pathogenic roles of IL-22, particularly in the context of psoriasis[45]. However, IL-22 may play a critical role in the maintenance of the intestinal epithelial barrier and in mucosal healing[45]. It is thus important to emphasize that although Th17 cells are defined by their expression of IL-17 and RORc, human counterpart of RORγt, these cells likely express heterogeneous cytokine profiles, at times simultaneously expressing both protective (IL-22) and deleterious (IL-17A, IFN-γ) cytokines in a tissue-temporal specific manner[5]. In addition, the unexpected failure of secukinumab[46], a human anti-IL-17 monoclonal antibody, in the treatment of Crohn’s disease reflects the complex role of IL-17 and the heterogeneous causes of the disease[47].
Foxp3+ Treg subset
In contrast, anti-inflammatory Foxp3-expressing Treg cells play an important role in tissue homeostasis via controlling pro-inflammatory effector T cells. Treg cells were first described as “self”-recognising T cells which develop in the thymus. Nowadays this population of Treg cells has come to be referred to as naturally occurring Treg[48]. Initially, Treg cells were believed to differentiate along a distinct pathway to that of conventional T cells which arise from naïve T cells in the periphery (extra-thymically). It is now apparent however that naïve progenitor T cells can give rise to a population of anti-inflammatory Foxp3 expressing T cells extra-thymically. This population is referred to as inducible Treg (iTreg)[48]. The major residential and developmental organs of iTreg cells are in fact the gut mucosa and mesenteric lymph nodes. Differentiation of iTreg cells is dependent on STAT5 and requires activation of the lineage-specifying transcription factor Foxp3. In vitro studies using human cells have revealed the critical roles for TGF-β1, together with the cofactor retinoic acid in iTreg development from naïve Th cells[49].
Similarities and differences between Th17 and Treg development
Interestingly, both Th17 and iTreg cells share essential developmental cues, and thus similar developmental pathways, as both subsets can be generated under the influence of TGF-β1. However additional signals specify development of each subset. Similar to Th1 and Th2 subsets, Th17 and Treg can negatively influence each other. In mice, increasing concentrations of TGF-β1 are associated with increased Foxp3 levels and decreased IL-23R expression, leading to decreased IL-23-dependent maintenance of Th17 cells and resulting in impaired Th17 development[50]. In addition, Foxp3 was found to directly inhibit IL-17 expression. As a result, Foxp3 and RORγt double expressing T cells in the lamina propria produced lower levels of IL-17 compared to T cells expressing RORγt alone[50]. Smad 2, 3, and 4, which transduce the extracellular TGF-β1 signal to the nucleus, are pivotal to iTreg generation whilst dispensable for Th17 development[51,52].
Gut resident and pathogenic microbes contribute to gut homeostasis and disease in part by shaping Th subset polarization. Interestingly, Th17 cells are induced by components of the intestinal flora, as has been shown for segmented filamentous bacteria (SFB)[53]. This requires MHC class II-dependent presentation of SFB antigens by DCs. On the other hand, commensal bacteria are known to influence gut tolerance by generating Treg cells specific to themselves. Transplantation of specific Clostridia clusters into germ free mice induces increased colonic Treg cells[54,55]. Lactobacillus reuteri colonization is also associated with an increase in gut residential Treg cells[56]. Interestingly, bacterial fermentation products may also play an important role in Treg generation. The short-chain fatty acids can promote DC tolerance along with generation of colonic Treg cells[57,58]. Another bacterial product, polysaccharide A from Bacteroides fragilis can induce anti-inflammatory Foxp3+, IL-10 secreting Treg cells via TLR2 signalling[59]. Thus, intestinal dietary and commensal products can influence Treg and Th17 development.
MECHANISMS OF PLASTICITY AND RECIPROCAL REGULATION BETWEEN TREG AND TH17 SUBSET
There are at least four major pathways and/or factors which contribute to Th17 plasticity. The cytokine milieu directs T cell subset development and also induces plasticity via the activation of different and specific STAT molecules and multiple transcription factors. Furthermore, it has emerged that immune-regulatory microRNA (miR) plays a fundamental role in controlling gene expression, thus influencing T cell fate and plasticity. There is also the unique role of aryl hydrocarbon receptor (AhR) and its environmental and physiological ligands alongside histone methylation and epigenetic modifications which may fundamentally influence T cell plasticity. Figure 1 summarizes the concept of Th17 plasticity.
Figure 1.
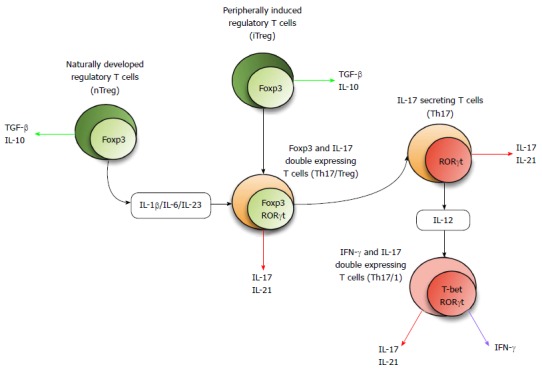
Concept of 17 secreting helper T plasticity. The Foxp3 expressing Treg subset either develops naturally (nTreg) or is peripherally induced from naïve T cells (iTreg). Th17 generating factors including cytokines, transcription factors, and other molecular cues can induce these cells to differentiate into transitional cells with co-expression of Foxp3 and RORc. Th17 cells may be converted into Th17/1-transitional cells with co-expression of RORc and TBX21. Th17: 17 secreting helper T; Foxp3: Forkhead box p3; RORγt: Related orphan receptor γ thymus in mouse; IL: Interleukin; TGF: Transforming growth factor; IFN: Interferon.
Cytokine pathways (main contributor to plasticity)
As described above, TGF-β signalling with the additional influence of IL-1β, IL-6 and IL-21 are critical to Th17 lineage development as well as the role of IL-23 signalling for the maintenance and function of this subset. High concentrations of exogenous Th17-generating cytokines are able to convert Foxp3+ Treg cells into IL-17 secreting Th cells in vitro. Since cytokine secretion is a final step in lineage differentiation, this population is considered to possess two signature features for two different subsets; Foxp3 for Treg, and IL-17 for Th17. We found increased levels of circulating IL-17 and Foxp3 double expressing T cells in IBD patients compared to healthy controls. Furthermore, we demonstrated the conversion of CD25**+** CD45RO- Treg cells from peripheral blood of IBD patients into IL-17 secreting Foxp3 expressing cells as well as RORc and Foxp3 expressing cells in the presence of combinations of the above mentioned cytokines[60]. Comparing the in vitro generation of double expressing cells in two types of IBD patients, CD patients have a higher prevalence of double expressing cells generated in the presence of IL-1β/TGF-β/IL-6 than UC while UC patients have an increased frequency of this population in response to IL-21/IL-23 than CD samples, suggesting disease-associated plasticity of Th cells in IBD[60]. A recent elegant study by Basu et al[61], revealed that IL-1 signalling represses SOCS3, an inhibitory molecule of STAT3 altering the STAT3/STAT5 balance resulting in Th17 generation. This model offers an explanation for why exogenous IL-1β converts Foxp3+ Treg cells into IL-17**+** Foxp3**+** double expressing Th cells[61].
Cytokines which induce Th17 development and plasticity are summarised in Table 1[62-69].
Table 1.
Cytokines related to 17 secreting helper T development and plasticity in human
| Cytokine | Effect | Ref. |
|---|---|---|
| IL-1β | Accelerate IL-17A secretion from IL-17 secreting Innate | [69] |
| Lymphoid cells and Th17 | ||
| IL-6 | Induce autocrine IL-21 and generation of Th17 | [20] |
| IL-10 | Suppress IL-17A and Th17 generation | [62] |
| IL-12 | Induce IFN-γ secretion from Th17 cells (Th1/Th17 plasticity) | [20] |
| IL-21 | Potentiates pathogenic effects of Th17 cells in the gut | [64,65] |
| IL-22 | Attenuate the development of intestinal pathology via Stat3-mediated effects on epithelial cells | [66,67] |
| IL-23 | Induce IL-22 and IL-17A secretions and maintain Th17 expansion | [68] |
| TGF-β1 | Initiate generation of Treg and Th17 populations dependent on the concentration | [63] |
Transcription factors
There is now vast literature detailing multiple transcription factors and their relationship to T cell plasticity. These transcription factors directly regulate and/or promote gene expression by binding to their promoter regions in order to contribute to the multifaceted nature of T cell subsets.
Ciofani et al[30] recently identified FOS-like antigen 2 (Fosl2) as a key determinant of Th17 plasticity in their remarkable study of Th17 cell specification[30]. in vitro differentiation of mouse CD4 T cells deficient in Fosl2 under Th17 polarizing conditions yielded IL-17 producing cells which co-expressed Foxp3. Interestingly, Fosl2 deficiency also enabled IFN-γ production in Th17 and Th2 cultures, particularly when Th17 cells were subsequently exposed to Th1-skewing conditions.
Interferon regulatory factor 4 (IRF4) is a transcription factor expressed in hematopoietic cells and plays pivotal roles in the immune response. IRF4 levels were augmented in patients with active inflammatory bowel disease and correlated with enhanced production of IL-17 and IL-22 mRNA[70]. On the other hand, lack of IRF4 seems to cause resistance to Th17-mediated autoimmune diseases[71]. Basic leucine zipper transcription factor, BATF, along with IRF4, was recently proposed as a “pioneer factor” in T cells[30]. After T cell receptor ligation, BATF expression is rapidly induced in naïve T cells. The BATF-JUN heterodimer and IRF4 bind to the same regulatory regions to mediate chromatin remodelling and facilitate accessibility to regulatory elements by other Th cell subset-specific transcription factors including STAT3, MAF and RORγt in Th17. Interestingly, BATF and IRF4 are also necessary for Treg differentiation in visceral adipose tissue through direct regulation of IL-33 receptor, ST2 and PPAR-γ expression[72]. BATF**+** RORc- Th17 cells are found in gut tissues from UC but not CD patients[73]. Taken together BATF/IRF4 axis may direct Treg/Th17 balance and plasticity in the gut.
A member of the Ikaros family, IKZF3 (or Aiolos) is known to promote Th17 differentiation by supressing IL-2 production[74]. Interestingly, IKZF3 is also expressed in iTreg lacking the expression IKZF2, (or Helios). These IKZF2- IKZF3**+** Foxp3**+** Th cells express IL-17 and exert reduced regulatory functions in healthy human blood samples[75]. Furthermore, polymorphism in Ikzf3 locus shows an association with CD and UC[31]. Thus, IKZF3 may turn out to be an important regulator of Th17-Treg plasticity in IBD.
An environmental sensor, Hypoxia-inducible factor 1 (HIF-1), which is induced by Th17 cells to promote signalling in a Stat3-dependent manner, cooperates with RORγt to control expression of Th17 genes, such as IL-17A, IL-17F, and IL-23R. Furthermore, HIF-1 negatively regulates Treg development by mediating Foxp3 protein degradation[33].
On the other hand, Liu et al[76] recently reported that TGF-β and IL-6 regulate Th1 cell conversion into the Th17 subset via expression of Runx1. This is supported by the finding that siRNA mediated silencing of Runx1 inhibits this conversion. Furthermore, TGF-β enhanced histone H3K9 acetylation but inhibited H3K9 trimethylation of Runx1- and ROR-γt-binding sites on the IL-17 or RORc gene in Th1 cells in this disease model[76].
Transcription factors which are critical for Th17 development and plasticity are summarised in Table 2.
Table 2.
Transcription factors related to 17 secreting helper T development and plasticity
| Transcription factor | Effect | Ref. |
|---|---|---|
| Fosl2 | A key determinant of cellular plasticity | [30] |
| IRF4 | Th17 differentiation via RORγt dependent and independent pathways | [115] |
| Augmented in patients with IBD and correlated with enhanced production of IL-17 and IL-22 mRNA | [70] | |
| BATF | Required for differentiation of Th17 via induction of RORγt, and bound to IL-17, IL-21 and IL-22 promoters | [116] |
| BATF+ RORc- Th17 cells are found in gut tissues from UC but not CD patients. | [73] | |
| HIF-1 | A key transregulator of Th17 polarization and suppressor of Foxp3 in Treg | [33,81] |
| Reciprocal regulation between HIF-1 and miR210 | [81] | |
| Jmjd3 | H3K27 demethylase, important for Th1/Th17 plasticity | [101] |
| RORc (human) | Essential for Th17 differentiation induced by TGF-β1 and IL-6 or IL-21 | [35] |
| RORγt (mouse) | ||
| IKZF3 (Aiolos) | Promotes Th17 differentiation via silencing of the IL-2 locus | [74] |
| Aiolos+ iTreg respond to IL-1β and downregulate their suppressor functions | [75] |
Micro-RNA (a cause of plasticity)
miR are small fragments of non-coding RNA (mostly 17-22 nucleotides) that act as regulators of RNA expression through binding to the 3’-UTR of complementary mRNA resulting in repression/silencing of target RNAs. Several miRs have been reported to influence the differentiation of Th cell subsets[77] as well as plasticity and reciprocal regulation among Th cell subsets[78].
Targeting STAT1, which is required for optimal Th1 development, miR146a plays an essential role in Treg function and development[78]. Targeting suppressor of cytokine signalling 1 (SOCS1), an inhibitor of STAT1, 4, and 5, miR155 plays an unique role in the development of Th1, Th2, and Treg cells and conversely in the suppression of Th17 differentiation. Targeting of PIAS3, an antagonist of STAT3, miR301 influences Th17 expansion[78]. Interestingly, the miR146a and miR155 are induced by toll-like receptors, well-known bacterial sensors, suggesting a critical involvement of micro-organisms[79]. Moreover, NOD2, a bacterial sensor closely linked to the pathogenesis of Crohn’s disease, induces miR29 resulting in downregulation of IL-23 secretion by dendritic cells through targeting of IL-12p40 and IL-23p19. This, in turn, was shown to suppress RORγt expression in the DSS mouse model of colitis[80].
In activated T cells, miR-210, which targets HIF-1, is upregulated 100 fold. This is especially notable in Th17 cells, resulting in decreased HIF-1α expression. Deletion of Mir210 promotes Th17 differentiation under hypoxic conditions. In experimental colitis, miR210 reduced the abundance of Hif1a transcripts and the proportion of cells that produced inflammatory cytokines resulting in decreased disease severity[81]. Induced by natural and environmental ligands of the aryl hydrocarbon receptor, the miR132/212 cluster promotes Th17 development[82]. The mechanisms of Th plasticity in aryl hydrocarbon receptor signalling will be described in the next section of this review.
Targeting EST1 which is a negative regulator of Th17 cells, miR326 is critical for Th17 differentiation. Furthermore, miR10, which is selectively expressed in Treg and induced by TGF-β signalling together with retinoic acid, can limit Th17 differentiation and furthermore can convert conventional Th cells into iTreg. The contribution of this miRNA to Treg stability is highly dependent on Foxp3 expression yet not responsible for Foxp3 induction[78]. A regulator of the suppressive activity of Treg cells, miR-126 leads to enhanced Foxp3 expression by targeting PIK3R2, a regulatory component of PI3K which downregulates Foxp3 induction[83]. The cluster of miR17-92, a complex of 6 miRNAs, influences Treg function in vitro resulting in the generation of IL-10 secreting Foxp3+ T cells. Although miR17 specifically targets TGF-β receptor II and Creb1, the targets of other parts of this cluster are still unknown[78].
Interestingly, deletion of critical compounds for miR signalling, such as DICER or DROSHA which are found in the micro RNA biogenesis pathways, showed that Treg-specific micro RNA expression is required to suppress T effector cells and maintain tolerance, suggesting that lack of Treg-specific miR results in immune-dysregulation[78].
Since miRs directly regulate the expression of many genes essential to Treg and Th17 subsets, it is highly likely they contribute to Th17-Treg plasticity. However, this exciting field is still in its infancy and further studies are undeniably required.
MicroRNA which influence Th17 development and plasticity are summarised in Table 3.
Table 3.
MicroRNAs influencing 17 secreting helper T development and plasticity
| MicroRNA | Inducer | Target | Effect | Ref. |
|---|---|---|---|---|
| miR10 | TGF-β | Bcl-6 | Limit Th17, convert Th cells into Treg | [78,83] |
| miR17-92 cluster | Creb1, TGF-βRII, KZF4 (miR17), PTEN (miR19) | Accumulation of antigen-specific iTreg development, IL-10 production, and possibly Treg cell migration | [78,83] | |
| miR29 | NOD2 | IL-12p40, IL-23p19 | Inhibit IL-23R signalling | [80] |
| miR126 | PIK3R2 | Treg-mediated Immunosuppression | [83] | |
| miR132/212 cluster | TCDD, FICZ | Bcl-6 | Enhance Th17 development via AhR pathway | [82,83] |
| miR146a | TLR2-5 | STAT1 | Block Th1 development | [78,79] |
| miR155 | TLR2-4, TLR9 | SOCS1 | Unleash STAT1, 4, and 5 signals, and promote Th1, Th2, and Treg | [78,79] |
| miR210 | HIF-1 (counter regulator) | Control Foxp3 expression | [81,83] | |
| miR301 | PIAS3 | Unleash STAT3 signal, and generate Th17 | [78] | |
| miR326 | EST-1 | Critical for Th17 development | [78] |
Aryl hydrocarbon receptor (unique mechanistic role of a receptor with dual functions)
The AhR first came to attention1970-80s[84,85] as a receptor for a recognized carcinogen, 2,3,7,8-tetrachlorodibenzo-_p_-dioxin (TCDD). This receptor is a ligand-activated transcription factor, which is responsible for the regulation of several xenobiotic response genes, such as the cytochrome P450 family[86]. Soon, attention shifted to the contribution of this receptor to oncogenesis via the suppression of immune-surveillance in response to TCDD, suggesting a TCDD-induced immune regulatory function of AhR pathways[87]. Later, 6-formylindolo [3,2-b] carbazole (FICZ) was recognised as an endogenous ligand of AhR; however the function of the receptor bound to FICZ was not the same as that bound to TCDD, suggesting that this receptor has ligand specific functions[88]. In regard to T cell polarization, Quintana et al[89], discovered that activation of the AhR with TCDD leads to the generation of Tregs, and in the same issue of Nature, Veldhoen et al[90], reported alternative activation with FICZ leading to Th17 cell differentiation. The effect on Th17 generation has been confirmed by subsequent studies in vitro and _in vivo_[91,92], while the influence on Treg generation has been more controversial[93]. Recently, Mezrich et al[94], showed evidence for a missing link in AhR-derived Treg generation in that kynurenine, a metabolite in the indoleamine 2,3-dioxygenase (IDO) dependent tryptophan degradation pathway, is a key ligand of AhR[94,95]. IDO is an enzyme known to suppress effector T cells and is expressed in regulatory plasmacytoid dendritic cells in response to IFN-γ. This kynurenine bound AhR generates Treg via influencing the TGF-β signalling pathway[94]. Furthermore, Moura-Alves et al[96], has reported that AhR binds pathogen-associated molecular patterns (PAMPs), regulating immunity in response to bacteria. Although this observation was limited to myeloid and epithelial cells from lung in a murine infectious model, there is a possibility that bacterial activation of AhR may modulate development of Th subsets in a complex bacterial environment, such as the digestive tract.
Taken together one can speculate that a unique function of AhR may be to contribute to plasticity between the Treg and Th17 subsets via differential binding to environmental or physiological ligands including bacteria-derived metabolites and PAMPs.
Figure 2 illustrates the contributing factors to the plasticity between Treg and Th17.
Figure 2.
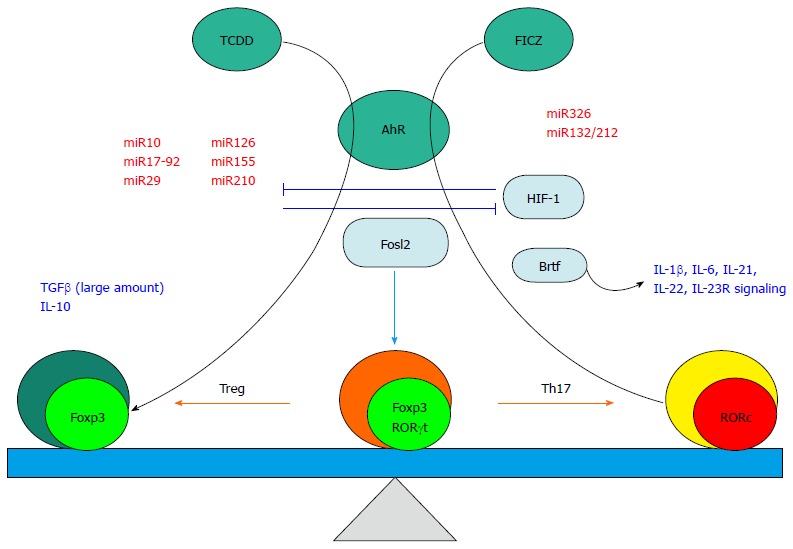
Factors influencing the plasticity between Treg and 17 secreting helper T subsets. Cytokines and growth factors (shown in blue letters) may trigger transdifferentiation of the pre-committed Foxp3 expressing Treg population to RORc expressing cells. Micro RNAs (miR, shown in red letters) play a pivotal role in differentially regulating Treg/Th17 plasticity. Transcription factors (aqua ovals) direct the plasticity by positively or negatively controlling Foxp3 and/or RORc expression. The aryl hydrocarbon receptor (AhR, green circle) can promote distinct differentiation pathways in response to two pathway-specific ligands (TCDD and FICZ, Green circles) resulting in either augmentation of Foxp3 or RORc, respectively. Th17: 17 secreting helper T; FICZ: 6-formylindolo [3,2-b] carbazole; TCDD: Tetrachlorodibenzo-_p_-dioxin; FICZ: Formylindolo [3,2-b] carbazole; AhR: Aryl hydrocarbon receptor; Treg: Regulatory T cells.
Histone methylation (monitoring the characteristics of plasticity)
Histone modifications including acetylation, methylation, and phosphorylation, are associated with gene expression or repression via alternations in chromatin structure[77]. In regard to Th1, Th2 and Th17 subset development, trimethylation of lysine 4(H3K4me3) and lysine 27(H3K27me3) play important roles in activation and repression, respectively, of gene expression downstream of transcription factors specific to Th subsets. Thus, monitoring H3K4me3 and H3K27me3 status can visualize the potential for plasticity in T cell subsets.
H3K4me3 marks the signature cytokine genes (Ifng for Th1, Il4 for Th2, Il17a) within the Th17 subset while repressive H3K27me3 is found on Th17 signature cytokine genes in Th1 (Il4, Il17a), Th2 (Ifng, Il17a) and Th17 (Il4, Ifng), suggesting lineage commitment to these particular subsets within these cells. Interestingly however, trimethylation on some transcription factor genes is bivalent (both H3K4 and H3K27) suggesting an element of reversibility in epigenetic status, particularly in Th17 cells. In these cells it is possible to find bivalent methylation in the Th1 transcription factor Tbx21 and Th2 transcription factor Gata3, in addition, Foxp3 may remain unmethylated suggesting a “neutral” state. Thus, histone methylation states may help explain why Th17 cells can swing to other subsets more easily than other T cell subsets[97-99]. Cytokines regulate the trimethylation status of Th17, resulting in subset conversion and plasticity. Also, the universal bivalent status of Tbx21 in all Th subsets except Th1, where you have only permissive H3K4me3, explains why plasticity towards the Th1 subset is dominant[99]. Thus, monitoring trimethylation status of H3K4 and H3K27 is useful in predicting the potential for, and direction of Th cell plasticity.
A histone modifying enzyme, JMJD3 is a histone H3K27 demethylase[100]. Li et al[101], reported that JMJD3 ablation promoted Th cell differentiation into Th2 and Th17 cells in the small and large intestines, and inhibited T-cell differentiation into Th1 cells in vitro and in a Th1-dependent colitis model. JMJD3 deficiency also restrains plasticity of Th2, Th17 and Treg cells towards Th1 cells. The skewing of T-cell differentiation is concomitant with changes in the expression of key transcription factors and cytokines via changes in H3K27me3 and H3K4me3 levels[101].
TH17 PLASTICITY IN CHRONIC INFLAMMATORY DISEASES INCLUDING IBD
IFN-γ+ IL-17+ double expressing cells are considered a cross-over transition of Th17 into Th1 lymphocytes[102]. This cell population represents an efficient local host defense system which shifts host defense from extracellular pathogens to intracellular microbial infections[103] and may contribute to autoimmune pathogeneses in both mouse models and in human diseases[104,105]. Globig et al[106], suggested that this population is indeed a subpopulation of Th17 cells and may be involved in IBD pathogenesis of both CD and UC. Interestingly, Weaver and colleagues recently have provided evidence that Th17 cells can act as precursors for IFN-γ secreting Th1 cells in a mouse of colitis, showing the indispensability of Th17/Th1 plasticity in the pathogenesis of colitis[21].
Likewise, IL-17+ Foxp3+ double expressing CD4+ T cells may differentiate into Th17 cells under pro-inflammatory conditions such as IL-1β, IL-6, IL-21, IL-23 and TGF-β**+**, having as yet largely unknown consequences for human disease initiation or progression[107]. On the other hand, plasticity from Th17 to Treg has not been frequently reported. However, a recent study from Yale University, suggests that this can in fact occur in the context of intestinal immune responses. According to their findings, Th17 cells generated during bacterial infection can be converted into IL-10high, Foxp3lo Tr1-like regulatory T cells. This was dependent on TGF-β1 and could be enhanced in vitro using an AhR ligand, FICZ[108]. This suggests that Th17 cells may alter their inflammatory status during infection, thus quenching inflammation and suggests possible avenues via which this mechanism could be harnessed therapeutically[108].
The Th17-promoting cytokine, IL-23 is known to play an essential role in driving intestinal inflammation. This cytokine also plays an inhibitor role in augmentation of intestinal iTreg generation[109]. Furthermore, IL-23 together with IL-12 signalling promotes IFN-γ secretion from Th17 in intestine, leading Th17/Th1 plasticity[110]. IL-23 signalling pathway is considered as a plasticity initiator in regard of Th17 subset via decreasing Foxp3 expression and assisting increased IFN-γ secretion.
In 2011, a hallmark study of Hovhannisyan et al[111] showed evidence for Treg/Th17 plasticity in IBD by showing the presence of IL-17 producing, Foxp3 expressing Th cells in inflamed intestinal mucosa from Crohn’s disease patients. Importantly, this population showed suppressor activity in vitro. We have gone on to demonstrate the presence of this unique T cell subset in peripheral blood from IBD patients[60] and have found evidence for several types of plasticity including Treg/Th17, Th1/Th17 and Th22/Th17 within the lamina propria of lesions from IBD patients[112].
CONCLUSION
Plasticity between Treg and Th17 likely occurs in the context of dynamic changes in the inflammatory milieu. Thus, pro-inflammatory stimuli may promote conversion of immune-suppressive regulatory T cells into pro-inflammatory Th17 cells, while resolution of inflammation may trigger or even require the alternate shift from Th17 to Treg. This concept is just becoming appreciated and requires further study to correlate both causes and outcomes. Targeting plasticity may offer avenues to pharmacologically restore the Th17/Treg balance in the intestine for therapeutic benefit[113]. In addition, targeting plasticity may help improve upon future therapies. Clinical trials of Treg therapy of Crohn’s disease failed[114] perhaps in part due to Treg instability. Further understanding of Treg plasticity may help to augment Treg therapy in the future.
Many studies have implicitly suggested the possibility of plasticity during their experiments and observation of inflammatory processes. It is clear however that several pathways give rise to the heterogeneous populations referred to as Th17 and Treg in the intestine. The field is in part limited due to constraints inherent to the techniques used to analyse these cells. Most investigators rely upon multicolor flow cytometry, requiring the precise selection of surface and intracellular markers and setting of conditions and controls in order to provide limited functional and expression data. New technologies look to overcome these technical issues as well as to enhance the depth of analysis. Examples include deep immune-phenotyping using CYTOF 2 mass spectrometry which allows the analysis of over 100 parameters at the single cell level without dealing with spectral overlap, alone or in combination with single cell transcriptomics.
To conclude, unravelling the complexity that underlies plasticity between Th17 and Treg cells may be key to understanding the intricate pathogenesis of T cell-mediated immune disorders, such as IBD. However, novel approaches and their application to human IBD will be required to reach this objective.
Footnotes
Conflict-of-interest statement: The authors have no conflict of interest to report in this work.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: June 26, 2015
First decision: July 20, 2015
Article in press: October 26, 2015
P- Reviewer: Gardlik R, Kuo SM S- Editor: Yu J L- Editor: A E- Editor: Ma S
References
- 1.Feldmann M, Maini SR. Role of cytokines in rheumatoid arthritis: an education in pathophysiology and therapeutics. Immunol Rev. 2008;223:7–19. doi: 10.1111/j.1600-065X.2008.00626.x. [DOI] [PubMed] [Google Scholar]
- 2.Damsker JM, Hansen AM, Caspi RR. Th1 and Th17 cells: adversaries and collaborators. Ann N Y Acad Sci. 2010;1183:211–221. [Google Scholar]
- 3.Kunz M, Ibrahim SM. Cytokines and cytokine profiles in human autoimmune diseases and animal models of autoimmunity. Mediators Inflamm. 2009;2009:979258. doi: 10.1155/2009/979258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. 1986. J Immunol. 2005;175:5–14. [PubMed] [Google Scholar]
- 5.Ahern PP, Izcue A, Maloy KJ, Powrie F. The interleukin-23 axis in intestinal inflammation. Immunol Rev. 2008;226:147–159. doi: 10.1111/j.1600-065X.2008.00705.x. [DOI] [PubMed] [Google Scholar]
- 6.Annunziato F, Romagnani S. Do studies in humans better depict Th17 cells? Blood. 2009;114:2213–2219. doi: 10.1182/blood-2009-03-209189. [DOI] [PubMed] [Google Scholar]
- 7.Chen Z, O’Shea JJ. Th17 cells: a new fate for differentiating helper T cells. Immunol Res. 2008;41:87–102. doi: 10.1007/s12026-007-8014-9. [DOI] [PubMed] [Google Scholar]
- 8.Fouser LA, Wright JF, Dunussi-Joannopoulos K, Collins M. Th17 cytokines and their emerging roles in inflammation and autoimmunity. Immunol Rev. 2008;226:87–102. doi: 10.1111/j.1600-065X.2008.00712.x. [DOI] [PubMed] [Google Scholar]
- 9.Hue S, Ahern P, Buonocore S, Kullberg MC, Cua DJ, McKenzie BS, Powrie F, Maloy KJ. Interleukin-23 drives innate and T cell-mediated intestinal inflammation. J Exp Med. 2006;203:2473–2483. doi: 10.1084/jem.20061099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kullberg MC, Jankovic D, Feng CG, Hue S, Gorelick PL, McKenzie BS, Cua DJ, Powrie F, Cheever AW, Maloy KJ, et al. IL-23 plays a key role in Helicobacter hepaticus-induced T cell-dependent colitis. J Exp Med. 2006;203:2485–2494. doi: 10.1084/jem.20061082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Conti HR, Shen F, Nayyar N, Stocum E, Sun JN, Lindemann MJ, Ho AW, Hai JH, Yu JJ, Jung JW, et al. Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J Exp Med. 2009;206:299–311. doi: 10.1084/jem.20081463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Langrish CL, McKenzie BS, Wilson NJ, de Waal Malefyt R, Kastelein RA, Cua DJ. IL-12 and IL-23: master regulators of innate and adaptive immunity. Immunol Rev. 2004;202:96–105. doi: 10.1111/j.0105-2896.2004.00214.x. [DOI] [PubMed] [Google Scholar]
- 13.Lin Y, Ritchea S, Logar A, Slight S, Messmer M, Rangel-Moreno J, Guglani L, Alcorn JF, Strawbridge H, Park SM, et al. Interleukin-17 is required for T helper 1 cell immunity and host resistance to the intracellular pathogen Francisella tularensis. Immunity. 2009;31:799–810. doi: 10.1016/j.immuni.2009.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maloy KJ, Kullberg MC. IL-23 and Th17 cytokines in intestinal homeostasis. Mucosal Immunol. 2008;1:339–349. doi: 10.1038/mi.2008.28. [DOI] [PubMed] [Google Scholar]
- 15.O'Connor W, Kamanaka M, Booth CJ, Town T, Nakae S, Iwakura Y, Kolls JK, Flavell RA. A protective function for interleukin 17A in T cell-mediated intestinal inflammation. Nat Immunol. 2009;10:603–609. doi: 10.1038/ni.1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ogawa A, Andoh A, Araki Y, Bamba T, Fujiyama Y. Neutralization of interleukin-17 aggravates dextran sulfate sodium-induced colitis in mice. Clin Immunol. 2004;110:55–62. doi: 10.1016/j.clim.2003.09.013. [DOI] [PubMed] [Google Scholar]
- 17.Izcue A, Hue S, Buonocore S, Arancibia-Cárcamo CV, Ahern PP, Iwakura Y, Maloy KJ, Powrie F. Interleukin-23 restrains regulatory T cell activity to drive T cell-dependent colitis. Immunity. 2008;28:559–570. doi: 10.1016/j.immuni.2008.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee YK, Turner H, Maynard CL, Oliver JR, Chen D, Elson CO, Weaver CT. Late developmental plasticity in the T helper 17 lineage. Immunity. 2009;30:92–107. doi: 10.1016/j.immuni.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rebhahn JA, Deng N, Sharma G, Livingstone AM, Huang S, Mosmann TR. An animated landscape representation of CD4+ T-cell differentiation, variability, and plasticity: insights into the behavior of populations versus cells. Eur J Immunol. 2014;44:2216–2229. doi: 10.1002/eji.201444645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khader SA, Bell GK, Pearl JE, Fountain JJ, Rangel-Moreno J, Cilley GE, Shen F, Eaton SM, Gaffen SL, Swain SL, et al. IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nat Immunol. 2007;8:369–377. doi: 10.1038/ni1449. [DOI] [PubMed] [Google Scholar]
- 21.Harbour SN, Maynard CL, Zindl CL, Schoeb TR, Weaver CT. Th17 cells give rise to Th1 cells that are required for the pathogenesis of colitis. Proc Natl Acad Sci USA. 2015;112:7061–7066. doi: 10.1073/pnas.1415675112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koenen HJ, Smeets RL, Vink PM, van Rijssen E, Boots AM, Joosten I. Human CD25highFoxp3pos regulatory T cells differentiate into IL-17-producing cells. Blood. 2008;112:2340–2352. doi: 10.1182/blood-2008-01-133967. [DOI] [PubMed] [Google Scholar]
- 23.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–1061. [PubMed] [Google Scholar]
- 24.Makita S, Kanai T, Oshima S, Uraushihara K, Totsuka T, Sawada T, Nakamura T, Koganei K, Fukushima T, Watanabe M. CD4+CD25bright T cells in human intestinal lamina propria as regulatory cells. J Immunol. 2004;173:3119–3130. doi: 10.4049/jimmunol.173.5.3119. [DOI] [PubMed] [Google Scholar]
- 25.Mucida D, Park Y, Kim G, Turovskaya O, Scott I, Kronenberg M, Cheroutre H. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science. 2007;317:256–260. doi: 10.1126/science.1145697. [DOI] [PubMed] [Google Scholar]
- 26.Gratz IK, Campbell DJ. Organ-specific and memory treg cells: specificity, development, function, and maintenance. Front Immunol. 2014;5:333. doi: 10.3389/fimmu.2014.00333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fujino S, Andoh A, Bamba S, Ogawa A, Hata K, Araki Y, Bamba T, Fujiyama Y. Increased expression of interleukin 17 in inflammatory bowel disease. Gut. 2003;52:65–70. doi: 10.1136/gut.52.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang XO, Chang SH, Park H, Nurieva R, Shah B, Acero L, Wang YH, Schluns KS, Broaddus RR, Zhu Z, et al. Regulation of inflammatory responses by IL-17F. J Exp Med. 2008;205:1063–1075. doi: 10.1084/jem.20071978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blaschitz C, Raffatellu M. Th17 cytokines and the gut mucosal barrier. J Clin Immunol. 2010;30:196–203. doi: 10.1007/s10875-010-9368-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ciofani M, Madar A, Galan C, Sellars M, Mace K, Pauli F, Agarwal A, Huang W, Parkurst CN, Muratet M, et al. A validated regulatory network for Th17 cell specification. Cell. 2012;151:289–303. doi: 10.1016/j.cell.2012.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–124. doi: 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu C, Yosef N, Thalhamer T, Zhu C, Xiao S, Kishi Y, Regev A, Kuchroo VK. Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature. 2013;496:513–517. doi: 10.1038/nature11984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, Bordman Z, Fu J, Kim Y, Yen HR, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell. 2011;146:772–784. doi: 10.1016/j.cell.2011.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.de Beaucoudrey L, Puel A, Filipe-Santos O, Cobat A, Ghandil P, Chrabieh M, Feinberg J, von Bernuth H, Samarina A, Jannière L, et al. Mutations in STAT3 and IL12RB1 impair the development of human IL-17-producing T cells. J Exp Med. 2008;205:1543–1550. doi: 10.1084/jem.20080321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 36.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 37.Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 38.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 39.Sutton C, Brereton C, Keogh B, Mills KH, Lavelle EC. A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. J Exp Med. 2006;203:1685–1691. doi: 10.1084/jem.20060285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nurieva R, Yang XO, Martinez G, Zhang Y, Panopoulos AD, Ma L, Schluns K, Tian Q, Watowich SS, Jetten AM, et al. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature. 2007;448:480–483. doi: 10.1038/nature05969. [DOI] [PubMed] [Google Scholar]
- 41.Cargill M, Schrodi SJ, Chang M, Garcia VE, Brandon R, Callis KP, Matsunami N, Ardlie KG, Civello D, Catanese JJ, et al. A large-scale genetic association study confirms IL12B and leads to the identification of IL23R as psoriasis-risk genes. Am J Hum Genet. 2007;80:273–290. doi: 10.1086/511051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Burton PR, Clayton DG, Cardon LR, Craddock N, Deloukas P, Duncanson A, Kwiatkowski DP, McCarthy MI, Ouwehand WH, Samani NJ, et al. Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat Genet. 2007;39:1329–1337. doi: 10.1038/ng.2007.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, Steinhart AH, Abraham C, Regueiro M, Griffiths A, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461–1463. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li LJ, Gong C, Zhao MH, Feng BS. Role of interleukin-22 in inflammatory bowel disease. World J Gastroenterol. 2014;20:18177–18188. doi: 10.3748/wjg.v20.i48.18177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hueber W, Sands BE, Lewitzky S, Vandemeulebroecke M, Reinisch W, Higgins PD, Wehkamp J, Feagan BG, Yao MD, Karczewski M, et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut. 2012;61:1693–1700. doi: 10.1136/gutjnl-2011-301668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.O'Shea JJ, Kanno Y, Chan AC. In search of magic bullets: the golden age of immunotherapeutics. Cell. 2014;157:227–240. doi: 10.1016/j.cell.2014.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol. 2012;30:531–564. doi: 10.1146/annurev.immunol.25.022106.141623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kang SG, Lim HW, Andrisani OM, Broxmeyer HE, Kim CH. Vitamin A metabolites induce gut-homing FoxP3+ regulatory T cells. J Immunol. 2007;179:3724–3733. doi: 10.4049/jimmunol.179.6.3724. [DOI] [PubMed] [Google Scholar]
- 50.Zhou L, Lopes JE, Chong MM, Ivanov II, Min R, Victora GD, Shen Y, Du J, Rubtsov YP, Rudensky AY, et al. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature. 2008;453:236–240. doi: 10.1038/nature06878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang KL, Chang WT, Hung KC, Li EI, Chuang CC. Inhibition of transforming growth factor-beta-induced liver fibrosis by a retinoic acid derivative via the suppression of Col 1A2 promoter activity. Biochem Biophys Res Commun. 2008;373:219–223. doi: 10.1016/j.bbrc.2008.05.192. [DOI] [PubMed] [Google Scholar]
- 52.Martinez GJ, Zhang Z, Reynolds JM, Tanaka S, Chung Y, Liu T, Robertson E, Lin X, Feng XH, Dong C. Smad2 positively regulates the generation of Th17 cells. J Biol Chem. 2010;285:29039–29043. doi: 10.1074/jbc.C110.155820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Goto Y, Panea C, Nakato G, Cebula A, Lee C, Diez MG, Laufer TM, Ignatowicz L, Ivanov II. Segmented filamentous bacteria antigens presented by intestinal dendritic cells drive mucosal Th17 cell differentiation. Immunity. 2014;40:594–607. doi: 10.1016/j.immuni.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, Fukuda S, Saito T, Narushima S, Hase K, et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature. 2013;500:232–236. doi: 10.1038/nature12331. [DOI] [PubMed] [Google Scholar]
- 55.Geuking MB, Cahenzli J, Lawson MA, Ng DC, Slack E, Hapfelmeier S, McCoy KD, Macpherson AJ. Intestinal bacterial colonization induces mutualistic regulatory T cell responses. Immunity. 2011;34:794–806. doi: 10.1016/j.immuni.2011.03.021. [DOI] [PubMed] [Google Scholar]
- 56.Livingston M, Loach D, Wilson M, Tannock GW, Baird M. Gut commensal Lactobacillus reuteri 100-23 stimulates an immunoregulatory response. Immunol Cell Biol. 2010;88:99–102. doi: 10.1038/icb.2009.71. [DOI] [PubMed] [Google Scholar]
- 57.Singh N, Gurav A, Sivaprakasam S, Brady E, Padia R, Shi H, Thangaraju M, Prasad PD, Manicassamy S, Munn DH, et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity. 2014;40:128–139. doi: 10.1016/j.immuni.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, Liu H, Cross JR, Pfeffer K, Coffer PJ, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature. 2013;504:451–455. doi: 10.1038/nature12726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Round JL, Lee SM, Li J, Tran G, Jabri B, Chatila TA, Mazmanian SK. The Toll-like receptor 2 pathway establishes colonization by a commensal of the human microbiota. Science. 2011;332:974–977. doi: 10.1126/science.1206095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ueno A, Jijon H, Chan R, Ford K, Hirota C, Kaplan GG, Beck PL, Iacucci M, Fort Gasia M, Barkema HW, et al. Increased prevalence of circulating novel IL-17 secreting Foxp3 expressing CD4+ T cells and defective suppressive function of circulating Foxp3+ regulatory cells support plasticity between Th17 and regulatory T cells in inflammatory bowel disease patients. Inflamm Bowel Dis. 2013;19:2522–2534. doi: 10.1097/MIB.0b013e3182a85709. [DOI] [PubMed] [Google Scholar]
- 61.Basu R, Whitley SK, Bhaumik S, Zindl CL, Schoeb TR, Benveniste EN, Pear WS, Hatton RD, Weaver CT. IL-1 signaling modulates activation of STAT transcription factors to antagonize retinoic acid signaling and control the TH17 cell-iTreg cell balance. Nat Immunol. 2015;16:286–295. doi: 10.1038/ni.3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang L, Yuan S, Cheng G, Guo B. Type I IFN promotes IL-10 production from T cells to suppress Th17 cells and Th17-associated autoimmune inflammation. PLoS One. 2011;6:e28432. doi: 10.1371/journal.pone.0028432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang L, Anderson DE, Baecher-Allan C, Hastings WD, Bettelli E, Oukka M, Kuchroo VK, Hafler DA. IL-21 and TGF-beta are required for differentiation of human T(H)17 cells. Nature. 2008;454:350–352. doi: 10.1038/nature07021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fantini MC, Rizzo A, Fina D, Caruso R, Becker C, Neurath MF, Macdonald TT, Pallone F, Monteleone G. IL-21 regulates experimental colitis by modulating the balance between Treg and Th17 cells. Eur J Immunol. 2007;37:3155–3163. doi: 10.1002/eji.200737766. [DOI] [PubMed] [Google Scholar]
- 65.Peluso I, Fantini MC, Fina D, Caruso R, Boirivant M, MacDonald TT, Pallone F, Monteleone G. IL-21 counteracts the regulatory T cell-mediated suppression of human CD4+ T lymphocytes. J Immunol. 2007;178:732–739. doi: 10.4049/jimmunol.178.2.732. [DOI] [PubMed] [Google Scholar]
- 66.Sugimoto K, Ogawa A, Mizoguchi E, Shimomura Y, Andoh A, Bhan AK, Blumberg RS, Xavier RJ, Mizoguchi A. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J Clin Invest. 2008;118:534–544. doi: 10.1172/JCI33194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Stevens S, Flavell RA. Innate and adaptive interleukin-22 protects mice from inflammatory bowel disease. Immunity. 2008;29:947–957. doi: 10.1016/j.immuni.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Awasthi A, Riol-Blanco L, Jäger A, Korn T, Pot C, Galileos G, Bettelli E, Kuchroo VK, Oukka M. Cutting edge: IL-23 receptor gfp reporter mice reveal distinct populations of IL-17-producing cells. J Immunol. 2009;182:5904–5908. doi: 10.4049/jimmunol.0900732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Coccia M, Harrison OJ, Schiering C, Asquith MJ, Becher B, Powrie F, Maloy KJ. IL-1β mediates chronic intestinal inflammation by promoting the accumulation of IL-17A secreting innate lymphoid cells and CD4(+) Th17 cells. J Exp Med. 2012;209:1595–1609. doi: 10.1084/jem.20111453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mudter J, Yu J, Zufferey C, Brüstle A, Wirtz S, Weigmann B, Hoffman A, Schenk M, Galle PR, Lehr HA, et al. IRF4 regulates IL-17A promoter activity and controls RORγt-dependent Th17 colitis in vivo. Inflamm Bowel Dis. 2011;17:1343–1358. doi: 10.1002/ibd.21476. [DOI] [PubMed] [Google Scholar]
- 71.Huber M, Lohoff M. IRF4 at the crossroads of effector T-cell fate decision. Eur J Immunol. 2014;44:1886–1895. doi: 10.1002/eji.201344279. [DOI] [PubMed] [Google Scholar]
- 72.Vasanthakumar A, Moro K, Xin A, Liao Y, Gloury R, Kawamoto S, Fagarasan S, Mielke LA, Afshar-Sterle S, Masters SL, et al. The transcriptional regulators IRF4, BATF and IL-33 orchestrate development and maintenance of adipose tissue-resident regulatory T cells. Nat Immunol. 2015;16:276–285. doi: 10.1038/ni.3085. [DOI] [PubMed] [Google Scholar]
- 73.Punkenburg E, Vogler T, Büttner M, Amann K, Waldner M, Atreya R, Abendroth B, Mudter J, Merkel S, Gallmeier E, et al. Batf-dependent Th17 cells critically regulate IL-23 driven colitis-associated colon cancer. Gut. 2015:Epub ahead of print. doi: 10.1136/gutjnl-2014-308227. [DOI] [PubMed] [Google Scholar]
- 74.Quintana FJ, Jin H, Burns EJ, Nadeau M, Yeste A, Kumar D, Rangachari M, Zhu C, Xiao S, Seavitt J, et al. Aiolos promotes TH17 differentiation by directly silencing Il2 expression. Nat Immunol. 2012;13:770–777. doi: 10.1038/ni.2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Raffin C, Pignon P, Celse C, Debien E, Valmori D, Ayyoub M. Human memory Helios- FOXP3+ regulatory T cells (Tregs) encompass induced Tregs that express Aiolos and respond to IL-1β by downregulating their suppressor functions. J Immunol. 2013;191:4619–4627. doi: 10.4049/jimmunol.1301378. [DOI] [PubMed] [Google Scholar]
- 76.Liu HP, Cao AT, Feng T, Li Q, Zhang W, Yao S, Dann SM, Elson CO, Cong Y. TGF-β converts Th1 cells into Th17 cells through stimulation of Runx1 expression. Eur J Immunol. 2015;45:1010–1018. doi: 10.1002/eji.201444726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nakayamada S, Takahashi H, Kanno Y, O’Shea JJ. Helper T cell diversity and plasticity. Curr Opin Immunol. 2012;24:297–302. doi: 10.1016/j.coi.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Baumjohann D, Ansel KM. MicroRNA-mediated regulation of T helper cell differentiation and plasticity. Nat Rev Immunol. 2013;13:666–678. doi: 10.1038/nri3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.O'Neill LA, Sheedy FJ, McCoy CE. MicroRNAs: the fine-tuners of Toll-like receptor signalling. Nat Rev Immunol. 2011;11:163–175. doi: 10.1038/nri2957. [DOI] [PubMed] [Google Scholar]
- 80.Brain O, Owens BM, Pichulik T, Allan P, Khatamzas E, Leslie A, Steevels T, Sharma S, Mayer A, Catuneanu AM, et al. The intracellular sensor NOD2 induces microRNA-29 expression in human dendritic cells to limit IL-23 release. Immunity. 2013;39:521–536. doi: 10.1016/j.immuni.2013.08.035. [DOI] [PubMed] [Google Scholar]
- 81.Wang H, Flach H, Onizawa M, Wei L, McManus MT, Weiss A. Negative regulation of Hif1a expression and TH17 differentiation by the hypoxia-regulated microRNA miR-210. Nat Immunol. 2014;15:393–401. doi: 10.1038/ni.2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nakahama T, Hanieh H, Nguyen NT, Chinen I, Ripley B, Millrine D, Lee S, Nyati KK, Dubey PK, Chowdhury K, et al. Aryl hydrocarbon receptor-mediated induction of the microRNA-132/212 cluster promotes interleukin-17-producing T-helper cell differentiation. Proc Natl Acad Sci USA. 2013;110:11964–11969. doi: 10.1073/pnas.1311087110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kroesen BJ, Teteloshvili N, Smigielska-Czepiel K, Brouwer E, Boots AM, van den Berg A, Kluiver J. Immuno-miRs: critical regulators of T-cell development, function and ageing. Immunology. 2015;144:1–10. doi: 10.1111/imm.12367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kouri RR, Ratrie H, Atlas SA, Niwa A, Nebert DW. Aryl hydrocarbon hydroxylase induction in human lymphocyte cultures by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Life Sci. 1974;15:1585–1595. doi: 10.1016/0024-3205(74)90324-5. [DOI] [PubMed] [Google Scholar]
- 85.Mason ME, Okey AB. Aryl hydrocarbon hydroxylase activity in mouse, rat, and human mammary tumors. Cancer Res. 1981;41:2778–2782. [PubMed] [Google Scholar]
- 86.Okey AB, Riddick DS, Harper PA. The Ah receptor: mediator of the toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) and related compounds. Toxicol Lett. 1994;70:1–22. doi: 10.1016/0378-4274(94)90139-2. [DOI] [PubMed] [Google Scholar]
- 87.Marshall NB, Kerkvliet NI. Dioxin and immune regulation: emerging role of aryl hydrocarbon receptor in the generation of regulatory T cells. Ann N Y Acad Sci. 2010;1183:25–37. [Google Scholar]
- 88.Esser C, Rannug A, Stockinger B. The aryl hydrocarbon receptor in immunity. Trends Immunol. 2009;30:447–454. doi: 10.1016/j.it.2009.06.005. [DOI] [PubMed] [Google Scholar]
- 89.Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, Caccamo M, Oukka M, Weiner HL. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature. 2008;453:65–71. doi: 10.1038/nature06880. [DOI] [PubMed] [Google Scholar]
- 90.Veldhoen M, Hirota K, Westendorf AM, Buer J, Dumoutier L, Renauld JC, Stockinger B. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature. 2008;453:106–109. doi: 10.1038/nature06881. [DOI] [PubMed] [Google Scholar]
- 91.Kimura A, Naka T, Nohara K, Fujii-Kuriyama Y, Kishimoto T. Aryl hydrocarbon receptor regulates Stat1 activation and participates in the development of Th17 cells. Proc Natl Acad Sci USA. 2008;105:9721–9726. doi: 10.1073/pnas.0804231105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Veldhoen M, Hirota K, Christensen J, O’Garra A, Stockinger B. Natural agonists for aryl hydrocarbon receptor in culture medium are essential for optimal differentiation of Th17 T cells. J Exp Med. 2009;206:43–49. doi: 10.1084/jem.20081438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hauben E, Gregori S, Draghici E, Migliavacca B, Olivieri S, Woisetschläger M, Roncarolo MG. Activation of the aryl hydrocarbon receptor promotes allograft-specific tolerance through direct and dendritic cell-mediated effects on regulatory T cells. Blood. 2008;112:1214–1222. doi: 10.1182/blood-2007-08-109843. [DOI] [PubMed] [Google Scholar]
- 94.Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, Bradfield CA. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol. 2010;185:3190–3198. doi: 10.4049/jimmunol.0903670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nguyen NT, Nakahama T, Le DH, Van Son L, Chu HH, Kishimoto T. Aryl hydrocarbon receptor and kynurenine: recent advances in autoimmune disease research. Front Immunol. 2014;5:551. doi: 10.3389/fimmu.2014.00551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Moura-Alves P, Faé K, Houthuys E, Dorhoi A, Kreuchwig A, Furkert J, Barison N, Diehl A, Munder A, Constant P, et al. AhR sensing of bacterial pigments regulates antibacterial defence. Nature. 2014;512:387–392. doi: 10.1038/nature13684. [DOI] [PubMed] [Google Scholar]
- 97.Lee BM, Mahadevan LC. Stability of histone modifications across mammalian genomes: implications for ‘epigenetic’ marking. J Cell Biochem. 2009;108:22–34. doi: 10.1002/jcb.22250. [DOI] [PubMed] [Google Scholar]
- 98.Wei G, Wei L, Zhu J, Zang C, Hu-Li J, Yao Z, Cui K, Kanno Y, Roh TY, Watford WT, et al. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity. 2009;30:155–167. doi: 10.1016/j.immuni.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rowell E, Wilson CB. Programming perpetual T helper cell plasticity. Immunity. 2009;30:7–9. doi: 10.1016/j.immuni.2008.12.012. [DOI] [PubMed] [Google Scholar]
- 100.Xiang Y, Zhu Z, Han G, Lin H, Xu L, Chen CD. JMJD3 is a histone H3K27 demethylase. Cell Res. 2007;17:850–857. doi: 10.1038/cr.2007.83. [DOI] [PubMed] [Google Scholar]
- 101.Li Q, Zou J, Wang M, Ding X, Chepelev I, Zhou X, Zhao W, Wei G, Cui J, Zhao K, et al. Critical role of histone demethylase Jmjd3 in the regulation of CD4+ T-cell differentiation. Nat Commun. 2014;5:5780. doi: 10.1038/ncomms6780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Esposito M, Ruffini F, Bergami A, Garzetti L, Borsellino G, Battistini L, Martino G, Furlan R. IL-17- and IFN-γ-secreting Foxp3+ T cells infiltrate the target tissue in experimental autoimmunity. J Immunol. 2010;185:7467–7473. doi: 10.4049/jimmunol.1001519. [DOI] [PubMed] [Google Scholar]
- 103.Zhou L, Chong MM, Littman DR. Plasticity of CD4+ T cell lineage differentiation. Immunity. 2009;30:646–655. doi: 10.1016/j.immuni.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 104.Nistala K, Adams S, Cambrook H, Ursu S, Olivito B, de Jager W, Evans JG, Cimaz R, Bajaj-Elliott M, Wedderburn LR. Th17 plasticity in human autoimmune arthritis is driven by the inflammatory environment. Proc Natl Acad Sci USA. 2010;107:14751–14756. doi: 10.1073/pnas.1003852107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 106.Globig AM, Hennecke N, Martin B, Seidl M, Ruf G, Hasselblatt P, Thimme R, Bengsch B. Comprehensive intestinal T helper cell profiling reveals specific accumulation of IFN-γ+IL-17+coproducing CD4+ T cells in active inflammatory bowel disease. Inflamm Bowel Dis. 2014;20:2321–2329. doi: 10.1097/MIB.0000000000000210. [DOI] [PubMed] [Google Scholar]
- 107.Singh K, Gatzka M, Peters T, Borkner L, Hainzl A, Wang H, Sindrilaru A, Scharffetter-Kochanek K. Reduced CD18 levels drive regulatory T cell conversion into Th17 cells in the CD18hypo PL/J mouse model of psoriasis. J Immunol. 2013;190:2544–2553. doi: 10.4049/jimmunol.1202399. [DOI] [PubMed] [Google Scholar]
- 108.Gagliani N, Vesely MC, Iseppon A, Brockmann L, Xu H, Palm NW, de Zoete MR, Licona-Limón P, Paiva RS, Ching T, et al. Th17 cells transdifferentiate into regulatory T cells during resolution of inflammation. Nature. 2015;523:221–225. doi: 10.1038/nature14452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Barnes MJ, Powrie F. Regulatory T cells reinforce intestinal homeostasis. Immunity. 2009;31:401–411. doi: 10.1016/j.immuni.2009.08.011. [DOI] [PubMed] [Google Scholar]
- 110.Morrison PJ, Ballantyne SJ, Kullberg MC. Interleukin-23 and T helper 17-type responses in intestinal inflammation: from cytokines to T-cell plasticity. Immunology. 2011;133:397–408. doi: 10.1111/j.1365-2567.2011.03454.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hovhannisyan Z, Treatman J, Littman DR, Mayer L. Characterization of interleukin-17-producing regulatory T cells in inflamed intestinal mucosa from patients with inflammatory bowel diseases. Gastroenterology. 2011;140:957–965. doi: 10.1053/j.gastro.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Li J, Ueno A, Fort Gasia M, Hirota C, Deane M, Chan R, Iacucci M, Kaplan G, Panaccione R, Luider J, et al. Distinctive Th17 Lymphocyte Plasticity in Intestinal Lamina Propria of IBD Patients Compared With Healthy Controls. Gastoroenterology. 2015;148:S323. [Google Scholar]
- 113.Eisenstein EM, Williams CB. The T(reg)/Th17 cell balance: a new paradigm for autoimmunity. Pediatr Res. 2009;65:26R–31R. doi: 10.1203/PDR.0b013e31819e76c7. [DOI] [PubMed] [Google Scholar]
- 114.Desreumaux P, Foussat A, Allez M, Beaugerie L, Hébuterne X, Bouhnik Y, Nachury M, Brun V, Bastian H, Belmonte N, et al. Safety and efficacy of antigen-specific regulatory T-cell therapy for patients with refractory Crohn’s disease. Gastroenterology. 2012;143:1207–1217.e1-e2. doi: 10.1053/j.gastro.2012.07.116. [DOI] [PubMed] [Google Scholar]
- 115.Brüstle A, Heink S, Huber M, Rosenplänter C, Stadelmann C, Yu P, Arpaia E, Mak TW, Kamradt T, Lohoff M. The development of inflammatory T(H)-17 cells requires interferon-regulatory factor 4. Nat Immunol. 2007;8:958–966. doi: 10.1038/ni1500. [DOI] [PubMed] [Google Scholar]
- 116.Schraml BU, Hildner K, Ise W, Lee WL, Smith WA, Solomon B, Sahota G, Sim J, Mukasa R, Cemerski S, et al. The AP-1 transcription factor Batf controls T(H)17 differentiation. Nature. 2009;460:405–409. doi: 10.1038/nature08114. [DOI] [PMC free article] [PubMed] [Google Scholar]