Pathobiology of Helicobacter pylori-induced Gastric Cancer (original) (raw)
. Author manuscript; available in PMC: 2017 Jan 1.
Abstract
Colonization of the human stomach by Helicobacter pylori and its role in causing gastric cancer is one of the richest examples of complex relationship among human cells, microbes, and their environment. It is also a puzzle of enormous medical importance given the incidence and lethality of gastric cancer worldwide. We review recent findings that have changed how we view these relationships and affected the direction of gastric cancer research. For example, recent data indicate that subtle mismatches between host and microbe genetic traits greatly affect risk of gastric cancer. The ability of H pylori and its oncoprotein CagA to reprogram epithelial cells and activate properties of stemness demonstrates the sophisticated relationship among H pylori and progenitor cells in the gastric mucosa. The observation that cell-associated H pylori can colonize the gastric glands and directly affect precursor and stem cells supports these observations. The ability to mimic these interactions in human gastric organoid cultures as well as animal models will allow investigators to more fully unravel the extent of H pylori control on the renewing gastric epithelium. Finally, our realization that external environmental factors, such as dietary components and essential micronutrients, as well as the gastrointestinal microbiota, can change the balance between _H pylori_’s activity as a commensal or a pathogen has provided direction to studies aimed at defining the full carcinogenic potential of this organism.
Infection with Helicobacter pylori, a bacterial carcinogen, is the greatest risk factor for gastric cancer—a disease that claims hundreds of thousands of lives per year1. Approximately 75% of the global gastric cancer burden and 5.5% of malignancies worldwide are attributable to _H pylori_-induced inflammation and injury2, yet the precise mechanisms that regulate cancer development in response to this organism are less well defined. Investigations focused on understanding gastric carcinogenesis have compartmentalized risk factors into categories such as H pylori strain variation, host responses and genotypes, and environmental influences. However, recently defined interactions among these categories have increased our understanding of disease risk and progression in individuals with persistent colonization3. We review exciting new data from discovery-based approaches and innovative model systems that recapitulate the gastric niche; these have increased our understanding of mechanisms that promote gastric carcinogenesis, within the context of host–microbe interactions.
Interactions Between Microbial and Human Genetic Ancestries
H pylori strains are highly genetically diverse and thrive as freely recombinogenic populations within their cognate human hosts. One technique that has been used to broadly assess and compare the genetic composition of H pylori strains is multi-locus sequence typing. Using this technology, Linz et al. found that H pylori strains segregated into several major clades that reflected the phylogeographic origins of their corresponding human hosts4. These findings, in conjunction with previous data establishing a >100,000 year association between H pylori and humans, invoke a model of prolonged adaptation in which H pylori should become less virulent over time5–7. However, this organism remains the strongest known risk factor for gastric cancer, raising the possibility that disrupted co-evolution between H pylori and humans may affect pathogenesis.
In many regions of the world, rates of H pylori infection and gastric cancer are concordant; in Asia, high prevalence rates of H pylori mirror the high prevalence of gastric cancer. However, this association is not universal. For example, the prevalence of H pylori infection in Africa is high, but the frequency of gastric cancer is extremely low8. In Colombia, the prevalence of H pylori is also very high throughout the country (>90% of individuals are infected), but individuals residing in the mountains have high rates of gastric cancer (150 cases/100,000), whereas those on the coast have very low rates (6 cases/100,000)9. This disparity in the prevalence of gastric cancer, but not H pylori, has provided a unique opportunity to assess the effects of interactions between H pylori and human ancestry on gastric carcinogenesis.
Kodaman et al. recently used multi-locus sequence type and single nucleotide polymorphism analyses to assess genetic variations in H pylori and humans, respectively, in the Colombian population. Their goal was to determine how their co-evolutionary relationships affect development of intestinal-type gastric cancer6. Coastal Colombians comprised an admixture of African, European, and Amerindian genetic variation, whereas the mountain populations were of predominantly Amerindian ancestry, with only a minority of European genetic content. H pylori isolates collected from the same subjects contained genetic signatures of multiple ancestries; an ancestral African cluster predominated on the coast whereas a European cluster predominated in the mountains6, affirming previous results from studies of these populations10. Importantly, specific interactions between microbial and human genetic ancestries clearly predicted risk for intestinal-type gastric cancer.
All persons who fell within the lowest decile of African host ancestry content but who were infected with an H pylori strain containing >19.8% African ancestry (a genetic mismatch), had an intestinal-type gastric premalignancy 6. In terms of quantifiable risk, persons with high (95th percentile) Amerindian ancestry who are infected with H pylori strains containing high (95th percentile) African genetic content are predicted to have severe and extensive intestinal metaplasia (Figure 1)6. If the same individuals are instead infected with H pylori strains of the lowest levels (5th percentile) of African ancestry (genetic match), they are predicted to have only mild gastric atrophy6.
Figure 1.

Interactions between host and H pylori genetic ancestries and risk for developing intestinal-type gastric adenocarcinoma in Colombia. Persons with high (95th percentile) Amerindian ancestry who are infected with H pylori strains containing high (95th percentile) African genetic content, are predicted to have severe and extensive intestinal metaplasia6. If the same individuals are instead infected with H pylori strains harboring the lowest levels (5th percentile) of African ancestry (genetic match), the histologic prediction is mild gastric atrophy6, a lesion with much less risk of progression to cancer11. Similarly, persons harboring the lowest levels (5th percentile) of Amerindian ancestry but high African ancestry who are infected with H pylori strains of high African genetic content are predicted to have gastritis only6.
Gastric atrophy has lower risk of progression to cancer than intestinal metaplasia or dysplasia11. A recent study from Sweden predicted that 1/256 persons with normal mucosa, 1/85 persons with non-atrophic gastritis, 1/50 persons with atrophic gastritis, 1/39 persons with intestinal metaplasia, and 1/19 persons with dysplasia will develop gastric cancer within 20 years12. Similarly, persons in the lowest levels (5th percentile) of Amerindian ancestry but high African ancestry who are infected with H pylori strains of high African genetic content are predicted to have only gastritis (Figure 1)6. Interactions between host and pathogen ancestries therefore completely accounted for differences in the severity of gastric injury in these populations; human or H pylori genetic variation alone are not sufficient to determine disease susceptibility—a genetic mismatch is also required7. This finding implies that the constant genetic adaptation of individual H pylori strains to their particular hosts achieved a balance that was disrupted by the acquisition of mismatched strains as diverse human populations began to intermix. An important next step for co-evolutionary studies is to identify differential patterns of concerted selection in paired human and pathogenic loci obtained from other global populations, such as Asia, that differ in terms of cancer risk, human ancestry, and H pylori genetic variation.
There is also evidence that more granular interactions between host and pathogen genotypes can alter gastric cancer risk. From the bacterial side, the cag pathogenicity island (PAI) is a well-characterized and intensively studied H pylori virulence determinant; strains that contain the cag PAI increase the risk for distal gastric cancer compared to strains that lack this locus3. From the side of human genetics, specific polymorphisms in genes encoding inflammatory cytokines can greatly increase the risk of gastric cancer among _H pylori_-infected persons13. El-Omar et al. reported that the odds ratio estimates for distal gastric cancer conferred by polymorphisms in IL1, IL10, or TNF were increased in persons infected with cag+ strains compared to the total _H pylori_-infected population14. Persons infected with H pylori isolates that possess another strain-specific genetic locus, type s1/m1 vacA alleles, are more likely to develop hypochlorhydria—a phenotype linked to high-expression alleles of IL1β and gastric cancer. The combination of high-risk host genotypes and cancer-associated vacA alleles or cag genotype similarly markedly increases the risk for gastric cancer, up to 87-fold over baseline15. Evaluation of human genetic variation in conjunction with genetic analyses of infecting H pylori strains can therefore identify colonized persons at the highest risk for gastric cancer and who may be optimal candidates for antimicrobial intervention.
Virulence Factors
H pylori has evolved the capacity to colonize and persist in one of the harshest environments of the human body, the stomach, which is microbicidal to a large number of species. H pylori use their motility, chemotaxis, urease production, and other mechanisms to adapt to the acidic conditions of the lumen and colonize a narrow protected niche near the surface of epithelial cells. In gastric biopsy specimens and in animal models, most of the bacteria are observed to be free-swimming in the mucus gel, and some adhere directly to the surface of the epithelium, but none are found more than 25μm away from the surface of the stomach16, 17. The necessity to colonize this narrow space brings the bacteria in close proximity to the epithelium, where they are subject to recognition and regulation by the mucosal immune system. It also allows H pylori to deliver bacterial products to the epithelium that modulate its activity and inflammatory responses for its own benefit. The 2 best-studied bacterial factors associated with increased risk of cancer and peptic ulcer are CagA, with its associated T4SS, and particular alleles of the oligomeric toxin VacA. Both of these factors have a number of important effects on the epithelium and the mucosal immune system and can increase the risk of gastric pathology.
CagA is a large protein that varies in size from 120 to 140 kD; it is produced in the cytosol of the bacteria and then threaded through a molecular micro-syinge, the cag type IV secretion system (T4SS), across the 2 bacterial membranes and the host cell membrane into the host cell cytosol18–21. The cag TFSS also translocates peptidoglycan into host cells22, 23. The genes that encode CagA and the T4SS are located in a mobile region of the chromosome, the cag pathogenicity island (cag PAI), that is present in more virulent strains of H pylori24, 25. Once CagA reaches the host cell cytosol, tyrosines in EPIYA motifs are phosphorylated by host c-SRC and c-ABL kinases;26 CagA then functions as a eukaryotic signaling hub, creating a scaffold with multiple partners near the host cell membrane (reviewed in 27).
CagA has multiple effects on epithelial cells. These broadly include stimulating cell proliferation through mitotic signaling pathways such as the PI3 kinase–AKT28, 29, SHP2, GRB2 and MEK–ERK,30–32 and β-catenin–WNT pathways. 33–35 CagA also reduces epithelial cell apoptosis by interfering with tumor suppressors such as p5336, 37 and RUNX338. CagA alters epithelial cell polarity through direct interactions with the polarity protein MAP/microtubule affinity-regulating kinase 2 (MARK2 or PAR1b) 39, 40, and disrupts assembly and signaling through the cell junctions41, 42. These direct effects of CagA on epithelial cells could promote cancer development, because transgenic mice and zebrafish engineered to express CagA develop carcinomas even in the absence of inflammation35, 43. In addition to its direct effects on epithelial cells, CagA and the T4SS activate inflammatory, NF-κB-dependent signaling22, 44–46 that leads to recruitment of inflammatory cells, reactive oxygen species-induced damage,47–49 and wound healing responses, which are all oncogenic. These findings and the epidemiological data linking CagA to gastric cancer risk, have led to the definition of CagA as a bacterial oncoprotein43.
CagA’s effects on epithelial cells are reversible and do not become permanent unless the target cells acquire mutations. CagA therefore induces cellular transformation only in special circumstances, by inducing accumulation of multiple genetic variants over time. One particularly intriguing emerging concept is that a combination of CagA’s signaling functions promotes cell de-differentiation or reprogramming of epithelial cells into more immature, stem-like cells that could be more prone to transformation.
Over the years, our understanding of CagA’s function has evolved from its discovery as a bacterial antigenic protein epidemiologically associated with disease to a sophisticated signaling molecule that controls fundamental aspects of epithelial biology. In recent years, several groups have begun to investigate the potential pathogenicity of CagA, along with the cellular context in which CagA exerts its effects in vivo. Researchers are asking question such as what determines where and when CagA is delivered into the host cell? How much CagA is delivered and what cell types are targets? Are there environmental or host factors that affect this process? Importantly, how does CagA benefit the bacterium?
The answers to these questions are not completely understood, but several avenues of investigation are providing insight. For example, CagA delivery to the host cell requires intimate contact between the T4SS and the host-cell membrane. The mechanisms of this interaction are being elucidated, but findings have reinforced the concept that bacterial adhesion to epithelial cells is a multi-step and complex process.
Several bacterial adhesins have been epidemiologically implicated in disease (reviewed in 50) and affect CagA delivery51. The T4SS, per se, has biologic activities independent of CagA. Several component of the TFSS needle, such as CagL, CagY and CagI, for example, bind β1 integrins to facilitate CagA delivery to epithelial cells52, 53. Since integrins are baso-lateral protein complexes, inaccessible at the lumenal surface of the epithelium, it not well understood where and when CagA is delivered. The bacterial protease HtrA might be able to disrupt the epithelial junctions to allow the bacteria to reach integrins54.
Conversely, CagA delivery might not always benefit the microbe, because it also induces inflammatory signals such as production of IL8 and recruitment of neutrophils. To counteract these responses, however, the interaction between bacteria and the epithelium can be modified by genetic molecular switches. For example, expression of CagY, an essential component of the T4SS, can be genetically switched on and off through immune selection, which alters the inflammatory response to promote long-term persistence55. The expression of other adhesins by H pylori, such as BabA and SabA, which bind glycosylated receptors on the gastric mucosa, are also controlled by molecular switches that finely regulate expression levels in vivo56, 57. CagA polymorphisms vary among H pylori strains isolated from different human populations; these may influence outcomes of disease. For example, the C-terminal region of CagA contains repeated amino acid motifs (EPIYA) flanked by distinct conserved sequences that vary in their host molecular partners and activity31. These tyrosine phosphorylation domains are classified as EPIYA- A, B, C, or D, and differ between Western strains (containing A, B, and C domains) and East Asian strains (containing A, B, and D domains)31. The number and types of repeats have been associated with differing risks of carcinogenesis58. Recently, it was reported that the EPIYA-B motif binds and activates PI3-kinase and AKT, but many Western strains of H pylori carry a single nucleotide polymorphism in this domain that reduces its association with PI3 kinase and also reduces the risk of gastric cancer59. The amount of CagA and its cellular target are also likely to have important roles in pathogenesis, but little is known about the precise location and timing CagA delivery.
VacA, a vacuolating cytotoxin, is asecreted by H pylori and can assemble into flower-shaped oligomers when added to various types of host cell membranes60, 61. The oligomers function as selective anion channels, so VacA is defined as a pore-forming cytotoxin (reviewed in 62). VacA pores have a number of effects, including changing the permeability of the plasma membrane63, causing paracellular leakage in epithelial monolayers64, affecting endosomal maturation65, and injuring mitochondria66. All H pylori strains contain vacA genes, but there are multiple alleles that confer various degrees of activities in different assays (reviewed in 67). The more pathogenic alleles of the toxin cause formation of large, late-endosomal vacuoles inside cells in vitro68. VacA also induces apoptosis in many cells, by interfering with mitochondrial function69. In addition to its cytotoxic features, VacA acts as a powerful immunosuppressant. It inhibits T-cell development70 and promotes differentiation of dendritic cells into a tolerogenic phenotype that induces development of regulatory T cells71.
The simultaneous presence of CagA and VacA in a cell can have important consequences, depending on cellular context and interactions between CagA and VacA72, 73. One striking recent example is that VacA reduces the half-life of CagA by stimulating its degradation through the autophagy pathway74. Interestingly, in CD44+ cells that have some characteristics of cancer stem cells, the half-life of CagA was increased and not affected by the presence of VacA. So, the pathogenic effects of CagA can be exacerbated in the context of immature precursor cells, transformed cells, or stem cells74.
New Systems for Studying _H pylori_-induced Carcinogenesis
Most studies of carcinogenic mechanisms of H pylori have been performed in cancer cell lines, short-term ex vivo primary cell cultures, and infected rodents. However, cell lines that have undergone many passages often contain mutations, and are derived from cancer specimens. In vivo models are expensive and can be time consuming to generate. Isolated ex vivo gastric gland systems are limited by their relatively short life span (e.g., 1 week) and a propensity to become heavily contaminated by fibroblasts75.
Gastroids effectively bridge in vitro and in vivo models by providing a replentishable culture system that can be readily generated from non-transformed gastric epithelium. Gastroids are 3-dimensional structures with central lumens that contain the major cell types found within gastric glands, and epithelial cells within gastroids are polarized76. Further, gastroids can survive for up to 9 months76.
However, there are limitations with this system. Although gastroids can be used to study direct interactions between H pylori and gastric epithelium (reflecting early responses to infection), inflammatory signals from infiltrating immune cells and paracrine signals from stromal or mesenchymal cells also influence pathogenesis—these are not represented in current systems. However, the potential to include inflammatory and stromal cells, such as myofibroblasts, in conjunction with microfluidic technology, in more complex organoid systems, should permit more detailed investigations into the role of chronic inflammation and injury on _H pylori_-induced gastric carcinogenesis in the future. Several groups have now developed and used gastroid models of H pylori infection originating from mouse and human tissues (Figure 2).
Figure 2.

Translational applications for using mouse or human gastroids to study _H pylori_-induced gastric carcinogenesis.
Wroblewski et al. used mouse gastroids to investigate the effects of H pylori strain-specific virulence factors on aberrant epithelial responses with carcinogenic potential42. This group demonstrated that gastroids can develop into a self-organizing, differentiating structure via expansion into single-layered epithelial spheroid structures that consist of mucus cells, parietal cells, G-cells, mucus neck cells, D-cells, and enterochromaffin-like (ECL) cells. Gastroids could be successfully infected with H pylori via microinjection (Figure 2), which resulted in mislocalization of occludin at the tight junction, a response identical to what had previously been demonstrated in _H pylori_-infected gastric epithelial cells in vitro and in vivo77. Infection of gastroids with H pylori cagA+ wild-type or isogenic mutant strains revealed that increased epithelial cell proliferation and β-catenin nuclear translocation was dependent upon CagA. The use of gastroids also identified a previously unreported mechanism through which H pylori may heighten the risk of carcinogenesis: increased snail expression, which was confirmed in human gastric tissue specimens42. Overall, these findings indicated that H pylori is effectively recognized by gastroids and that this system may be used as both a model for discovery as well as validation. Additionally, this system provides an important opportunity to study gastroids harvested from genetically deficient or transgenic mice in the future.
Other groups have extended these findings by developing human gastroids as a model of pathogenic interactions between H pylori and epithelial cells. McCracken et al. reported de novo generation of human gastroids derived from antral tissue via directed differentiation of stem cells78. Human gastroids could be successfully infected with H pylori, which resulted in an increase in CagA-dependent epithelial cell proliferation78, confirming previous findings from mouse gastroids. Importantly, intracellular CagA rapidly associated with the c-Met receptor, leading to its phosphorylation78, 79. Another group has recently shown that 3-dimensional human gastroids can be converted into a 2-dimensional planar polarized model system80. This innovation has added considerable versatility to gastroids as a model for studying interactions between mutant strains of H pylori as well as genetically manipulated ex vivo primary cell systems (Figure 2). Bartfeld et al. have generated gastroids from human gastric corpus tissue and shown that gastroids can be directed towards progenitor-like gastric gland lineages or well-differentiated gastric pit lineages via manipulation of WNT and nicotinamide concentrations81. Importantly, interactions between H pylori and epithelial cells increased when undifferentiated gastric gland organoids were used instead of differentiated gastric pit organoids81. These findings indicate that the ability of H pylori to interact selectively with stem or progenitor cells may lower the threshold for gastric carcinogenesis.
Effects on Gastric Stem Cells
Several potential models have been developed to investigate how H pylori infection and its inflammatory response contribute to the dysregulated growth of long-lived cells and eventually cancer. These include infection-induced de-differentiation of terminally differentiated epithelial cells into long lived, replicating cells; recruitment of mesenchymal stem cells to gastric glands during tissue damage and repair and subsequent transformation of these exogenous stem cells; and/or direct bacterial effects or inflammatory changes in the resident gastric progenitor and stem cells in the stomach.
Since H pylori inhabit the superficial mucus layer overlying the stomach lumen, and adhere to mucus pit cells, it is possible that these terminally differentiated cells are the targets of oncogenic transformation. This would involve de-differentiation into replicating cells and acquisition of oncogenic mutations and cancer stem cell traits. Many studies have provided evidence that CagA has reprogramming potential that could convert somatic epithelial cells into a pluripotent, stem cell-like state, and facilitate the acquisition of mutations. For example, cells expressing CagA or infected with CagA-positive bacteria lose key features of epithelial differentiation and undergo phenotypic and molecular changes associated with stemness and epithelial–mesenchymal transition42, 82–86. Also, CagA can aberrantly activate WNT signaling to β-catenin and induce WNT target genes such as the transcription factor CDX133–35, 87. CDX1 can, in turn, induce the expression of several stemness-associated factors, such as SALL4 and KLF5, potentially making cells more pluripotent88. Consistent with this observation, H pylori infection has been shown to induce ectopic expression of KLF5 in mouse gastric glands89.
However, surface mucus cells are short lived, with a typical life span of only 1–2 days, so their interactions with H pylori or inflammatory factors are limited. H pylori causes significant inflammatory responses throughout the depths of the glands as well as hyperplasia during periods of chronic active gastritis, indicated by increased cell division and apoptosis of normal glands90. This expands the range of proliferating cells into the upper regions of the glands90, potentially bringing immature cells into contact with the bacteria.
It is well established that premalignant metaplastic lesions are preceded by multifocal-atrophic gastritis with loss of parietal cells and other differentiated cells91. Chronic atrophic gastritis is also accompanied by expansion of immature proliferating cells,92 which has been reproduced in a mouse model of atrophic gastritis in which parietal cells were genetically deleted93. When H pylori was introduced into this mouse model of atrophic gastritis, a direct interaction between the bacteria and gastric progenitor cells was observed, and some of the bacteria were internalized by progenitor cells94, 95. These studies were among the first to show that adult gastric stem cells could serve as a protective niche where a subpopulation of H pylori could reside to avoid clearance96.
More recently, direct interactions between H pylori and gastric precursor cells have been observed, in stomachs mice devoid of atrophy but infected with H pylori, and in samples from asymptomatic, infected patients with superficial gastritis. By reconstructing the gastric glands in 3-dimensions using confocal microscopy, these reports describe a subpopulation of H pylori that resides deep in the gastric glands. The gland-associated bacteria are distinct from the free-swimming bacteria in the surface mucus in that they grow as microcolonies that adhere directly to epithelial junctions of gastric precursor cells in the isthmus and in the base of the glands (Figure 3)97, 98.
Figure 3.

H pylori colonizes the gland base and interacts with gastric progenitor cells. H pylori are labeled green, actin red, and nuclei blue. Cells undergoing mitosis (labeled with ph-Histone) are marked by arrows.
In addition, localized inflammatory responses in susceptible niches might also be associated with disease progression. For example, neutrophil infiltration specifically localized to the pit proliferative zone has been described as a characteristic pathologic finding of _H pylori_-induced gastritis99. It is therefore plausible that direct interactions between H pylori and proliferative progenitor cells or longer-lived stem cells occurs throughout the life of the individual; these interactions may result in direct as well as indirect insults that lead to malignant transformation.
The precise markers that identify progenitor cells and stem cells in the stomach are under intense investigation and not fully understood. The gastric antrum shows similarities with the intestine in that the base of the glands contain stem cells marked by expression of the Lgr5 gene and controlled through WNT signaling76. LGR5 is a G-protein coupled receptor that regulates WNT signaling and marks stem cells in several tissues, including the small intestine, colon, hair follicles, and mammary glands (reviewed in 100). LGR5+ cells in the base of the antral glands give rise to rapidly dividing progenitor cells in the isthmus of the glands, which, in turn, proliferate and differentiate into all other cell types. Other potential stem cell markers in the antrum include rare cells marked by activity of the villin promoter101 and cells that express the transcription factor SOX2102. The identity of the stem cells in the corpus is less well understood. Similar to the antrum, the proliferative zone of the isthmus region below the gastric pits contains most of the mitotically active progenitor cells, and potential stem cells have been described in this region103–105.
Loss of parietal cells, which occurs in patients with atrophic gastritis, leads to expansion of a progenitor population in the isthmus marked by CD44 106. A subpopulation of mature chief cells can act as progenitor cells and give rise to metaplastic lesions in the corpus107. A recent study identified a subset of fully differentiated chief cells present at the base of the corpus glands that express the marker tumor necrosis factor receptor superfamily, member 19 (TNFRSF19 or TROY) and function as more slowly growing stem cells in the corpus108. Finally, there is evidence that LRIG1 identifies a distinct population of quiescent stem cells in the stomach, similar to findings in the colon; H pylori infection can lead to expansion of this subpopulation in vivo89, 109.
Stem cells and progenitor cells are important candidate tumor-initiating cells in gastric cancer, and are the source of gastric cancer stem cells. Mouse experiments in which the tumor suppressor gene APC was deleted from LGR5+ stem cells revealed rapidly developing adenomas76. Likewise, inactivation of the tumor suppressor gene, Klf4, in villin-positive gastric progenitor cells increased gastric tumor initiation and progression in mice110.
Several studies have correlated stem cell numbers or damage and progression of gastric carcinoma. For instance, an immunohistopathology study examined the numbers of LGR5+ stem cells in human gastric mucosa as well as associated DNA damage. It found that patients with gastric cancer and H pylori infection had an expanded pool of LGR5+ stem cells in the antrum, and higher amounts of DNA oxidative damage in these stem cells111. Several other studies have confirmed that levels of LGR5 mRNA and protein are increased in stomach tissues from patients with gastric cancer112, 113. Higher levels of LGR5 may be a marker of poor prognosis for patients with gastric cancer114.
In addition to transforming potential observed in normal gastric stem cells, in animal models (chimeras with inflammatory conditions), chronic damage can lead to recruitment of bone marrow-derived mesenchymal stem cells that incorporate and transdifferentiate into epithelial cells of the gastric mucosa. In mice with Helicobacter felis infection, chronic inflammation, gastric metaplasia, dysplasia, and high-grade intraepithelial neoplasia, inflamed glands were found to contain bone marrow-derived cells. Subsequent neoplastic lesions were derived from bone marrow precursors rather than resident gastric stem cells115. Similar findings were recently obtained in a mouse model of spasmolytic polypeptide-expressing metaplasia caused by H pylori, in which approximately 25% of metaplastic glands contained cells derived from bone marrow precursors116.
Regardless of whether or not H pylori infection progresses to neoplasia in humans, the bacteria appear to have evolved specialized mechanisms to interact and affect the stem and progenitor cells in the gastric glands. To avoid the gastric lumen, H pylori are able to reach the surface of the stomach, adhere to the epithelial cells, and even grow as attached micro-colonies directly on the epithelial junctions deep in the gastric glands97, 117. This gland-associated H pylori population is more prominent in the isthmus in areas rich in mitotic progenitor cells (Figure 3), and occurs early during colonization of mice and in asymptomatic individuals prior to the development of atrophic gastritis97, 98. In mice, LGR5+ stem cells can be labeled by expression of green fluorescent protein,76 and the effects of H pylori infection on these cells can be monitored (Figure 4). It was recently reported that, within 2 weeks of infection, and before the onset of chronic gastritis, H pylori infection of the gastric glands activated the antral LGR5+ stem cells, leading to a doubling of the number of stem cells per gland by 2 months of infection98. The activation and expansion of stem cells spatially correlates with glands occupied by gland-associated H pylori (Figure 4), and mutant H pylori unable to colonize the glands do not activate the stem cells, suggesting that direct interaction between the bacteria and these cells promotes this hyper-proliferation.
Figure 4.
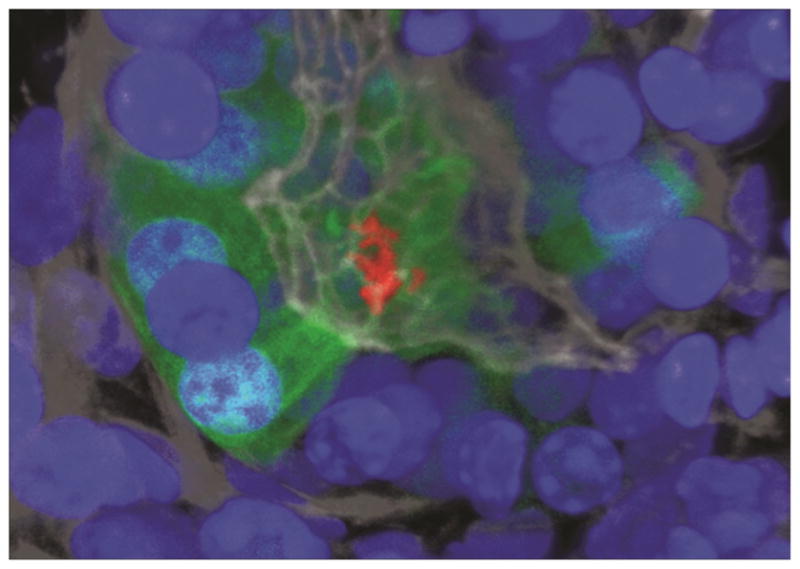
H pylori micro-colony interacting with LGR5 stem cells at the base of an antral gland. H pylori are labeled red, Lgr5+ cells green, and nuclei blue.
Overall, it appears that H pylori has evolved the capacity to colonize a specialized niche near precursor and stem cells, and that it manipulates these cells for its benefit94, 98. Microscopic localization and interactions between the microbe and the progenitor epithelium could therefore be an important variable in the pathogenesis of gastric cancer. A micro-niche could exist within gastric glandular units that is particularly vulnerable to the oncogenic effects of H pylori.
H pylori and Iron
Two important clinical observations highlight the relationship between iron and H pylori pathogenesis. One is increasing evidence that H pylori infection is associated with iron deficiency anemia (IDA), both in sporadic cases of individuals that present with iron-deficiency anemia refractory to iron supplementation118 as well as at a population level related to iron deficiency anemia of childhood119. The contribution of H pylori to IDA in childhood likely has a significant health impact in resource-poor settings, since IDA affects 30% of the human population and is associated with cognitive and developmental impairment120. A second observation is that markers of iron-deficiency are associated with increased risk of gastric cancer121. H pylori could therefore contribute to low iron states, which can worsen outcomes of H pylori infection. Research into the molecular mechanisms of these processes has made some connections between H pylori virulence factors and environmental risk factors for cancer.
Iron is an essential molecule for most living organisms, but it is a scarce resource for microbes colonizing the human body, because iron restriction is an ancient innate immune defense against infection. Interstitial and intracellular sources of iron are separated from the bacteria that colonize mucosal surfaces by the epithelial barrier and, within the body, iron is sequestered by high-affinity chelators such as transferrin, hemoglobin, and ferritin122. In the inflamed gastric mucosa, neutrophils secrete lactoferrin, which tightly binds free iron at the mucosal surface to starve bacteria123. In addition, inflammation induces upregulation of hepcidin, a central regulator of iron metabolism, which activates an iron reduction response to pathogens, by blocking iron uptake in the small intestine122.
So how does H pylori acquire iron in the stomach in the presence of such obstacles, and how is this related to the association between iron deficiency and carcinogenesis? H pylori has evolved multiple mechanisms of iron acquisition and each has effects that could contribute to gastric pathology. For example, H pylori express several membrane transporters that take up soluble forms of iron such as ferrous ions and ferric citrate molecules124. These forms of iron are usually insoluble, but the low pH in the stomach lumen solubilizes dietary ferric iron, and maintains it in solution as a complex with ascorbic acid125. Thus, the acidic gastric juice contains a source of soluble iron that can be directly used by H pylori. However, H pylori may have ready access this iron source, since it cannot survive in the low pH of the stomach lumen, instead remaining close to the epithelial surface where it produces urease to buffer its immediate surroundings. Long-term infection with H pylori often leads to a reduction in gastric acid and ascorbic acid secretion which, in turn, reduce the availability of soluble iron for H pylori and contribute to IDA126. In addition, lactoferrin further reduces the availability of free iron near the mucosa and may also contribute to IDA123.
The upregulation of hepicidin in response to H pylori infection might also reduce iron availability in the host and lead to IDA. For example, _H pylori_-associated iron-deficiency does not respond to oral iron therapy without eradication of the bacteria with antibiotics. Treatment of _H pylori_-infected children who have IDA with oral iron therapy did not reduce serum levels of hepcidin, indicating that eradication of the infection is required to restore normal iron homeostasis127. Hepcidin is expressed locally in parietal cells of the gastric glands, and gastric hepcidin increases during H pylori infection but normalizes following eradication128.
Since H pylori infection has effects on acid, hepcidin, and lactoferrin that lead to reduction of free iron, it is not surprising that H pylori also evolved alternative mechanisms of iron acquisition from the host. Unlike many other commensal and pathogenic bacteria that live at mucosal surfaces, H pylori does not produce siderophores—small molecules that can usurp iron from the host and other microbes. However, in vitro studies with defined media have shown that H pylori can actually utilize lactoferrin as a source of iron, but only if it is fully saturated with iron. The same studies showed that H pylori can also extract iron from saturated transferrin and from hemoglobin129. H pylori therefore uses on only dietary iron, but also internal host iron reserves that are sequestered across the epithelial barrier.
How does H pylori reach iron sources that are present inside the host across from the epithelium? One possibility is that inflammation leads to epithelial erosions or disruption of barrier function exposing H pylori to hemoglobin in red blood cells or serum transferrin. A recent study used elegant in vivo microscopy to show that H pylori use chemotaxis to swim to and concentrate at mucosal sites of injury, such as ulcers130. Other studies have identified a novel mechanism of iron acquisition across the intact epithelium that links iron uptake to the virulence factors CagA, the cag T4SS, and VacA. As noted, a subpopulation of H pylori adhere to and grow directly on the epithelial surface forming cell-associated microcolonies deep in the gastric glands97. In an in vitro model of infection using polarized epithelia, cell-associated H pylori grew on cell surfaces even under conditions of low iron availability that do not support the growth of free-swimming bacteria. This indicates that cell-associated H pylori can extract nutrients from or across polarized epithelial cells117.
CagA and VacA were each shown to be important for the ability to colonize the epithelial surface in conditions of low iron availability131. CagA, injected into the epithelium, stimulated the uptake of iron-saturated transferrin into the infected epithelial cells. It also perturbed cell polarity to alter the recycling of transferrin inducing its transcytosis across the epithelium (Figure 5). VacA also contributed to mislocalization of transferrin and its receptor by altering endosomal trafficking. These cellular effects raised the hypothesis that H pylori virulence factors have a role for bacterial survival in vivo in conditions of low iron availability. This hypothesis was tested in iron-deficient Mongolian gerbils fed iron-restricted diets before infection with H pylori. In an iron-deficient host, wild-type CagA+ H pylori were able to thrive, whereas the CagA-isogenic mutants were defective in gastric colonization131.
Figure 5.

Gastric epithelial cellular responses with carcinogenic potential that are induced by iron deficiency within the context of H pylori infection. The H pylori cag T4SS injects CagA into epithelial cells and this results in signaling that induces loss of cell polarity and effects on the epithelial junctions, pro-inflammatory phenotypes, and growth factor-like signaling that results in cellular proliferation. This also induces increased iron uptake and transcytosis. In the right panel, low levels of iron in the host induce increased adhesion and colonization of the glandular epithelium, increased number of T4SS pili, and an augmentation in levels of CagA injection and pathogenic signaling.
These observations were quickly followed by the surprising finding that iron-deficient Mongolian gerbils infected with CagA+ H pylori develop more severe inflammation, and accelerated pre-malignant and malignant lesions compared to infected animals fed an iron-replete diet121. Furthermore, H pylori isolated from iron-deficient animals or maintained in vitro in iron-deficient conditions have an increased ability to inject CagA and synthesize higher numbers of T4SS needles upon contact with host cells121, 132. The localization of H pylori in the mucosa was also different, with greater concentrations of bacteria migrating into the glands in iron-deficient animals. In human populations at risk of gastric cancer, H pylori strains isolated from patients with the lowest ferritin levels elicited the highest inflammatory responses when co-cultured with gastric epithelial cells. In summary, recent insights into the role of virulence factors in iron acquisition have provided a conceptual link between H pylori virulence, risk of gastric cancer, and iron-deficiency (Figure 5), and suggest that environmental factors such as micronutrient availability can significantly alter the outcome of the infection.
Salt and H pylori Virulence
A link between high salt consumption and increased gastric cancer risk has been reported from numerous human studies133, 134. Gene expression in several bacterial pathogens, including H pylori, can be regulated by salt concentrations135. Of interest, transcriptional and proteomic studies have revealed increased expression of cagA in response to high-salt conditions136, 137, but only in certain H pylori strains. Based on these findings, Loh et al. sequenced the cagA promoter in a population of clinical H pylori isolates and identified a unique DNA motif (TAATGA) that was present in either 1 or 2 copies. Salt-induced up-regulation of CagA was detected more commonly in strains containing 2 copies of the TAATGA motif than in strains containing 1 copy,138 and mutagenesis experiments confirmed that 2 copies of the TAATGA motif were required for salt-induced CagA expression.
The effects of salt on H pylori infection and gastric cancer have also been extended in vivo using mouse and gerbil models. One study in mice demonstrated that a high-salt diet increased levels of H pylori colonization in the stomach and resulted in increased parietal cell loss139. A study using gerbils reported that H pylori infection and a high-salt diet could independently induce atrophic gastritis and intestinal metaplasia140, whereas another investigation revealed that H pylori infection and high-salt diets have a synergistic effect on gastric carcinogenesis—but only when gerbils also received a chemical carcinogen141.
Gaddy et al. recently investigated the effects of a high-salt diet on microbe-induced cancer in gerbils using a unique carcinogenic strain of H pylori, 7.13142. Gastric adenocarcinoma was detected in a significantly higher proportion of infected animals on a high-salt diet than infected animals on a regular diet142. Infected animals fed a high-salt diet also developed more severe gastric inflammation142; hypochlorhydria, parietal cell loss, and high levels of gastric mucosal IL1B were detected in the animals that developed cancer. Animals infected with a cagA negative isogenic mutant strain and fed a high-salt diet had low levels of gastric inflammation and did not develop hypochlorhydria or gastric cancer. Similarly, a high-salt diet did not cause the development of gastric cancer in uninfected animals142. These results confirm that a high-salt diet potentiates the carcinogenic effects of cagA+ H pylori strains.
Based on these data, there are several potential mechanisms by which dietary elements may augment the development of gastric cancer. The direct effects of dietary constituents on gastric epithelium may damage the mucosa, thereby allowing an increased entry of carcinogens into gastric tissue. Certain dietary components interact with intestinal immune receptors and thereby regulate intestinal immunity143, and similar responses might occur in the stomach. Diet might influence the composition of the gastric microbiota, or promote expansion of H pylori variants with pathogenic properties. Dietary factors could also influence epigenetic alterations—folic acid supplementation protected against the loss of global DNA methylation and reduced the development of gastric inflammation and dysplasia in _Helicobacter_-infected mice144. The findings summarized here, however, raise another possibility—namely that dietary constituents directly influence the pathogenic potential of H pylori by augmenting the expression and function of cancer-associated microbial virulence determinants.
The Gastrointestinal Microbiota
The role of H pylori in gastric carcinogenesis is undisputed. However, other microbes in the gastric or intestinal niche could also affect transformation of gastric epithelial cells (for review, see 145). Studies demonstrating the effectiveness of anti-H pylori regimens on gastric cancer incidence raised this possibility by showing that the cancer-lowering effects of antibiotics may be mediated their effects on non-H pylori residents of the gastrointestinal tract146. Germ-free hypergastrinemic INS-GAS mice that are mono-colonized with H pylori develop pre-malignant lesions at a slower pace than _H pylori_-infected specific pathogen-free INS-GAS mice147. This accelerated phenotype was associated with an increase in the proportion of Firmicutes and a decrease in the numbers of Bacteroidetes within the stomach147. Rapid progression to gastric neoplasia could be restored in germ-free INS-GAS mice that were pre-infected with a restricted Altered Schaedler’s Flora before challenge with H pylori148.
In addition to the role of the microbiota within the stomach, extra-gastric constituents of the microbiome have been shown to affect gastric carcinogenesis in mouse models. Infection of C57BL/6 mice with H bilis or H muridarum before challenge with H pylori significantly reduced the severity of _H pylori_-induced gastric inflammation149, 150. In contrast, pre-colonization with a different entero-hepatic Helicobacter species, H hepaticus, increased _H pylori_-induced gastric injury150. These exciting results have provided an important framework for studies that can focus on defining fundamental relationships between H pylori and residents of the gastrointestinal microbiome as a means to understand gastric carcinogenesis.
Conclusions
Globally, gastric cancer leads to a high number of cancer-related deaths; increasing our understanding the risk factors for this disease is of utmost importance in identifying the individuals at greatest risk for developing gastric cancer. Infection with H pylori is extremely common and in some areas of the world, prevalence rates approach 100%, however, 97%–99% of colonized persons will never develop gastric cancer. The risk of developing gastric cancer is dependent on an opus of interacting components including H pylori strain-specific virulence factors, the host genotype, environmental factors such as diet as well as alternations in stem cell populations and the microbiome. Molecular interactions among these factors affect the outcome of long-term colonization of H pylori. It is therefore critical that results from mechanistic studies be used to identify persons who are at high risk of developing gastric cancer.
Acknowledgments
Grant support: NIH CA-116087, DK-58404, DK-58587, and CA-77955
Footnotes
Author contributions:
Manuel Amieva: analysis and interpretation of previous data, drafting of the manuscript, critical revision of manuscript for important intellectual content
Richard Peek: analysis and interpretation of previous data, drafting of the manuscript, critical revision of manuscript for important intellectual content
Disclosures/Conflict of interest: The authors declare there are no conflicts of interest
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hardbower DM, Peek RM, Jr, Wilson KT. At the Bench: Helicobacter pylori, dysregulated host responses, DNA damage, and gastric cancer. J Leukoc Biol. 2014;96:201–12. doi: 10.1189/jlb.4BT0214-099R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Parkin DM, Bray F, Ferlay J, et al. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 3.Polk DB, Peek RM., Jr Helicobacter pylori: gastric cancer and beyond. Nat Rev Cancer. 10:403–14. doi: 10.1038/nrc2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Linz B, Balloux F, Moodley Y, et al. An African origin for the intimate association between humans and Helicobacter pylori. Nature. 2007;445:915–8. doi: 10.1038/nature05562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moodley Y, Linz B, Bond RP, et al. Age of the association between Helicobacter pylori and man. PLoS Pathog. 2012;8:e1002693. doi: 10.1371/journal.ppat.1002693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kodaman N, Pazos A, Schneider BG, et al. Human and Helicobacter pylori coevolution shapes the risk of gastric disease. Proc Natl Acad Sci U S A. 2014;111:1455–60. doi: 10.1073/pnas.1318093111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kodaman N, Sobota RS, Mera R, et al. Disrupted human-pathogen co-evolution: a model for disease. Front Genet. 2014;5:290. doi: 10.3389/fgene.2014.00290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Holcombe C. Helicobacter pylori: the African enigma. Gut. 1992;33:429–31. doi: 10.1136/gut.33.4.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Correa P, Cuello C, Duque E, et al. Gastric cancer in Colombia. III. Natural history of precursor lesions. J Natl Cancer Inst. 1976;57:1027–35. doi: 10.1093/jnci/57.5.1027. [DOI] [PubMed] [Google Scholar]
- 10.de Sablet T, Piazuelo MB, Shaffer CL, et al. Phylogeographic origin of Helicobacter pylori is a determinant of gastric cancer risk. Gut. 2011;60:1189–95. doi: 10.1136/gut.2010.234468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Vries AC, van Grieken NC, Looman CW, et al. Gastric cancer risk in patients with premalignant gastric lesions: a nationwide cohort study in the Netherlands. Gastroenterology. 2008;134:945–52. doi: 10.1053/j.gastro.2008.01.071. [DOI] [PubMed] [Google Scholar]
- 12.Song H, Ekheden IG, Zheng Z, et al. Incidence of gastric cancer among patients with gastric precancerous lesions: observational cohort study in a low risk Western population. BMJ. 2015;351:h3867. doi: 10.1136/bmj.h3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Amieva MR, El-Omar EM. Host-bacterial interactions in Helicobacter pylori infection. Gastroenterology. 2008;134:306–23. doi: 10.1053/j.gastro.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 14.El-Omar EM, Rabkin CS, Gammon MD, et al. Increased risk of noncardia gastric cancer associated with proinflammatory cytokine gene polymorphisms. Gastroenterology. 2003;124:1193–201. doi: 10.1016/s0016-5085(03)00157-4. [DOI] [PubMed] [Google Scholar]
- 15.Figueiredo C, Machado JC, Pharoah P, et al. Helicobacter pylori and interleukin 1 genotyping: an opportunity to identify high-risk individuals for gastric carcinoma. J Natl Cancer Inst. 2002;94:1680–7. doi: 10.1093/jnci/94.22.1680. [DOI] [PubMed] [Google Scholar]
- 16.Schreiber S, Konradt M, Groll C, et al. The spatial orientation of Helicobacter pylori in the gastric mucus. Proc Natl Acad Sci U S A. 2004;101:5024–9. doi: 10.1073/pnas.0308386101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schreiber S, Bucker R, Groll C, et al. Rapid loss of motility of Helicobacter pylori in the gastric lumen in vivo. Infect Immun. 2005;73:1584–9. doi: 10.1128/IAI.73.3.1584-1589.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Segal ED, Cha J, Lo J, et al. Altered states: involvement of phosphorylated CagA in the induction of host cellular growth changes by Helicobacter pylori. Proc Natl Acad Sci U S A. 1999;96:14559–64. doi: 10.1073/pnas.96.25.14559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Backert S, Ziska E, Brinkmann V, et al. Translocation of the Helicobacter pylori CagA protein in gastric epithelial cells by a type IV secretion apparatus. Cell Microbiol. 2000;2:155–64. doi: 10.1046/j.1462-5822.2000.00043.x. [DOI] [PubMed] [Google Scholar]
- 20.Odenbreit S, Puls J, Sedlmaier B, et al. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science. 2000;287:1497–500. doi: 10.1126/science.287.5457.1497. [DOI] [PubMed] [Google Scholar]
- 21.Stein M, Rappuoli R, Covacci A. Tyrosine phosphorylation of the Helicobacter pylori CagA antigen after cag-driven host cell translocation. Proc Natl Acad Sci U S A. 2000;97:1263–8. doi: 10.1073/pnas.97.3.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Viala J, Chaput C, Boneca IG, et al. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat Immunol. 2004;5:1166–74. doi: 10.1038/ni1131. [DOI] [PubMed] [Google Scholar]
- 23.Suarez G, Romero-Gallo J, Piazuelo MB, et al. Modification of Helicobacter pylori peptidoglycan enhances NOD1 activation and promotes cancer of the stomach. Cancer Res. 2015;75:1749–59. doi: 10.1158/0008-5472.CAN-14-2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blaser MJ, Perez-Perez GI, Kleanthous H, et al. Infection with Helicobacter pylori strains possessing cagA is associated with an increased risk of developing adenocarcinoma of the stomach. Cancer Res. 1995;55:2111–5. [PubMed] [Google Scholar]
- 25.Censini S, Lange C, Xiang Z, et al. cag, a pathogenicity island of Helicobacter pylori, encodes type I- specific and disease-associated virulence factors. Proc Natl Acad Sci U S A. 1996;93:14648–53. doi: 10.1073/pnas.93.25.14648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mueller D, Tegtmeyer N, Brandt S, et al. c-Src and c-Abl kinases control hierarchic phosphorylation and function of the CagA effector protein in Western and East Asian Helicobacter pylori strains. J Clin Invest. 2012;122:1553–66. doi: 10.1172/JCI61143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hatakeyama M. Helicobacter pylori CagA and gastric cancer: a paradigm for hit-and-run carcinogenesis. Cell Host Microbe. 2014;15:306–16. doi: 10.1016/j.chom.2014.02.008. [DOI] [PubMed] [Google Scholar]
- 28.Nagy TA, Frey MR, Yan F, et al. Helicobacter pylori regulates cellular migration and apoptosis by activation of phosphatidylinositol 3-kinase signaling. J Infect Dis. 2009;199:641–51. doi: 10.1086/596660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Suzuki M, Mimuro H, Kiga K, et al. Helicobacter pylori CagA phosphorylation-independent function in epithelial proliferation and inflammation. Cell Host Microbe. 2009;5:23–34. doi: 10.1016/j.chom.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 30.Keates S, Keates AC, Warny M, et al. Differential activation of mitogen-activated protein kinases in AGS gastric epithelial cells by cag+ and cag- Helicobacter pylori. J Immunol. 1999;163:5552–9. [PubMed] [Google Scholar]
- 31.Higashi H, Tsutsumi R, Fujita A, et al. Biological activity of the Helicobacter pylori virulence factor CagA is determined by variation in the tyrosine phosphorylation sites. Proc Natl Acad Sci U S A. 2002;99:14428–33. doi: 10.1073/pnas.222375399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mimuro H, Suzuki T, Tanaka J, et al. Grb2 is a key mediator of Helicobacter pylori CagA protein activities. Mol Cell. 2002;10:745–55. doi: 10.1016/s1097-2765(02)00681-0. [DOI] [PubMed] [Google Scholar]
- 33.Franco AT, Israel DA, Washington MK, et al. Activation of B-catenin by carcinogenic Helicobacter pylori. Proc Natl Acad Sci U S A. 2005;102:10646–51. doi: 10.1073/pnas.0504927102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murata-Kamiya N, Kurashima Y, Teishikata Y, et al. Helicobacter pylori CagA interacts with E-cadherin and deregulates the beta-catenin signal that promotes intestinal transdifferentiation in gastric epithelial cells. Oncogene. 2007;26:4617–26. doi: 10.1038/sj.onc.1210251. [DOI] [PubMed] [Google Scholar]
- 35.Neal JT, Peterson TS, Kent ML, et al. H pylori virulence factor CagA increases intestinal cell proliferation by Wnt pathway activation in a transgenic zebrafish model. Dis Models Mech. 2013;6:802–10. doi: 10.1242/dmm.011163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Buti L, Spooner E, Van der Veen AG, et al. Helicobacter pylori cytotoxin-associated gene A (CagA) subverts the apoptosis-stimulating protein of p53 (ASPP2) tumor suppressor pathway of the host. Proc Natl Acad Sci U S A. 2011;108:9238–43. doi: 10.1073/pnas.1106200108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wei J, Noto JM, Zaika E, et al. Bacterial CagA protein induces degradation of p53 protein in a p14ARF-dependent manner. Gut. 2015;64:1040–8. doi: 10.1136/gutjnl-2014-307295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsang YH, Lamb A, Romero-Gallo J, et al. Helicobacter pylori CagA targets gastric tumor suppressor RUNX3 for proteasome-mediated degradation. Oncogene. 2010;29:5643–50. doi: 10.1038/onc.2010.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saadat I, Higashi H, Obuse C, et al. Helicobacter pylori CagA targets PAR1/MARK kinase to disrupt epithelial cell polarity. Nature. 2007;447:330–3. doi: 10.1038/nature05765. [DOI] [PubMed] [Google Scholar]
- 40.Zeaiter Z, Cohen D, Musch A, et al. Analysis of detergent-resistant membranes of Helicobacter pylori infected gastric adenocarcinoma cells reveals a role for MARK2/Par1b in CagA-mediated disruption of cellular polarity. Cell Microbiol. 2008;10:781–94. doi: 10.1111/j.1462-5822.2007.01084.x. [DOI] [PubMed] [Google Scholar]
- 41.Amieva MR, Vogelmann R, Covacci A, et al. Disruption of the epithelial apical-junctional complex by Helicobacter pylori CagA. Science. 2003;300:1430–4. doi: 10.1126/science.1081919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wroblewski LE, Piazuelo MB, Chaturvedi R, et al. Helicobacter pylori targets cancer-associated apical-junctional constituents in gastroids and gastric epithelial cells. Gut. 2015;64:720–30. doi: 10.1136/gutjnl-2014-307650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ohnishi N, Yuasa H, Tanaka S, et al. Transgenic expression of Helicobacter pylori CagA induces gastrointestinal and hematopoietic neoplasms in mouse. Proc Natl Acad Sci U S A. 2008;105:1003–8. doi: 10.1073/pnas.0711183105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brandt S, Kwok T, Hartig R, et al. NF-kB activation and potentiation of proinflammatory responses by the Helicobacter pylori CagA protein. Proc Natl Acad Sci U S A. 2005;102:9300–5. doi: 10.1073/pnas.0409873102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lamb A, Yang XD, Tsang YH, et al. Helicobacter pylori CagA activates NF-kappaB by targeting TAK1 for TRAF6-mediated Lys 63 ubiquitination. EMBO Rep. 2009;10:1242–9. doi: 10.1038/embor.2009.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gorrell RJ, Guan J, Xin Y, et al. A novel NOD1- and CagA-independent pathway of interleukin-8 induction mediated by the Helicobacter pylori type IV secretion system. Cell Microbiol. 2013;15:554–70. doi: 10.1111/cmi.12055. [DOI] [PubMed] [Google Scholar]
- 47.Danese S, Cremonini F, Armuzzi A, et al. Helicobacter pylori CagA-positive strains affect oxygen free radicals generation by gastric mucosa. Scand J Gastroenterol. 2001;36:247–50. doi: 10.1080/003655201750074474. [DOI] [PubMed] [Google Scholar]
- 48.Ding SZ, O’Hara AM, Denning TL, et al. Helicobacter pylori and H2O2 increase AP endonuclease-1/redox factor-1 expression in human gastric epithelial cells. Gastroenterology. 2004;127:845–58. doi: 10.1053/j.gastro.2004.06.017. [DOI] [PubMed] [Google Scholar]
- 49.Chaturvedi R, Asim M, Romero-Gallo J, et al. Spermine oxidase mediates the gastric cancer risk associated with Helicobacter pylori CagA. Gastroenterology. 2011;141:1696–708. doi: 10.1053/j.gastro.2011.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Backert S, Clyne M, Tegtmeyer N. Molecular mechanisms of gastric epithelial cell adhesion and injection of CagA by Helicobacter pylori. Cell Commun Signal. 2011;9:28. doi: 10.1186/1478-811X-9-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Belogolova E, Bauer B, Pompaiah M, et al. Helicobacter pylori outer membrane protein HopQ identified as a novel T4SS-associated virulence factor. Cell Microbiol. 2013;15:1896–912. doi: 10.1111/cmi.12158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kwok T, Zabler D, Urman S, et al. Helicobacter exploits integrin for type IV secretion and kinase activation. Nature. 2007;449:862–6. doi: 10.1038/nature06187. [DOI] [PubMed] [Google Scholar]
- 53.Jimenez-Soto LF, Kutter S, Sewald X, et al. Helicobacter pylori type IV secretion apparatus exploits beta1 integrin in a novel RGD-independent manner. PLoS Pathog. 2009;5:e1000684. doi: 10.1371/journal.ppat.1000684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hoy B, Lower M, Weydig C, et al. Helicobacter pylori HtrA is a new secreted virulence factor that cleaves E-cadherin to disrupt intercellular adhesion. EMBO Rep. 2010;11:798–804. doi: 10.1038/embor.2010.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Barrozo RM, Cooke CL, Hansen LM, et al. Functional plasticity in the type IV secretion system of Helicobacter pylori. PLoS Pathogens. 2013;9:e1003189. doi: 10.1371/journal.ppat.1003189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Solnick JV, Hansen LM, Salama NR, et al. Modification of Helicobacter pylori outer membrane protein expression during experimental infection of rhesus macaques. Proc Natl Acad Sci U S A. 2004;101:2106–11. doi: 10.1073/pnas.0308573100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Aberg A, Gideonsson P, Vallstrom A, et al. A repetitive DNA element regulates expression of the Helicobacter pylori sialic acid binding adhesin by a rheostat-like mechanism. PLoS Pathog. 2014;10:e1004234. doi: 10.1371/journal.ppat.1004234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Beltran-Anaya FO, Poblete TM, Roman-Roman A, et al. The EPIYA-ABCC motif pattern in CagA of Helicobacter pylori is associated with peptic ulcer and gastric cancer in Mexican population. BMC Gastroenterol. 2014;14:223. doi: 10.1186/s12876-014-0223-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang XS, Tegtmeyer N, Traube L, et al. A specific A/T polymorphism in Western tyrosine phosphorylation B-motifs regulates Helicobacter pylori CagA epithelial cell interactions. PLoS Pathog. 2015;11:e1004621. doi: 10.1371/journal.ppat.1004621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pagliaccia C, Wang XM, Tardy F, et al. Structure and interaction of VacA of Helicobacter pylori with a lipid membrane. Eur J Biochem. 2000;267:104–9. doi: 10.1046/j.1432-1327.2000.00970.x. [DOI] [PubMed] [Google Scholar]
- 61.Chambers MG, Pyburn TM, Gonzalez-Rivera C, et al. Structural analysis of the oligomeric states of Helicobacter pylori VacA toxin. J Mol Biol. 2013;425:524–35. doi: 10.1016/j.jmb.2012.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cover TL, Blanke SR. Helicobacter pylori VacA, a paradigm for toxin multifunctionality. Nat Rev Microbiol. 2005;3:320–32. doi: 10.1038/nrmicro1095. [DOI] [PubMed] [Google Scholar]
- 63.Szabo I, Brutsche S, Tombola F, et al. Formation of anion-selective channels in the cell plasma membrane by the toxin VacA of Helicobacter pylori is required for its biological activity. EMBO J. 1999;18:5517–27. doi: 10.1093/emboj/18.20.5517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Papini E, Satin B, Norais N, et al. Selective increase of the permeability of polarized epithelial cell monolayers by Helicobacter pylori vacuolating toxin. J Clin Invest. 1998;102:813–20. doi: 10.1172/JCI2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Papini E, de Bernard M, Milia E, et al. Cellular vacuoles induced by Helicobacter pylori originate from late endosomal compartments. Proc Natl Acad Sci U S A. 1994;91:9720–4. doi: 10.1073/pnas.91.21.9720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Willhite DC, Cover TL, Blanke SR. Cellular vacuolation and mitochondrial cytochrome c release are independent outcomes of Helicobacter pylori vacuolating cytotoxin activity that are each dependent on membrane channel formation. J Biol Chem. 2003;278:48204–9. doi: 10.1074/jbc.M304131200. [DOI] [PubMed] [Google Scholar]
- 67.Kim IJ, Blanke SR. Remodeling the host environment: modulation of the gastric epithelium by the Helicobacter pylori vacuolating toxin (VacA) Front Cell Infect Microbiol. 2012;2:37. doi: 10.3389/fcimb.2012.00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pagliaccia C, de Bernard M, Lupetti P, et al. The m2 form of the Helicobacter pylori cytotoxin has cell type-specific vacuolating activity. Proc Natl Acad Sci U S A. 1998;95:10212–7. doi: 10.1073/pnas.95.17.10212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cover TL, Krishna US, Israel DA, et al. Induction of gastric epithelial cell apoptosis by Helicobacter pylori vacuolating cytotoxin. Cancer Res. 2003;63:951–7. [PubMed] [Google Scholar]
- 70.Gebert B, Fischer W, Weiss E, et al. Helicobacter pylori vacuolating cytotoxin inhibits T lymphocyte activation. Science. 2003;301:1099–102. doi: 10.1126/science.1086871. [DOI] [PubMed] [Google Scholar]
- 71.Oertli M, Noben M, Engler DB, et al. Helicobacter pylori gamma-glutamyl transpeptidase and vacuolating cytotoxin promote gastric persistence and immune tolerance. Proc Natl Acad Sci U S A. 2013;110:3047–52. doi: 10.1073/pnas.1211248110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Argent RH, Hale JL, El-Omar EM, et al. Differences in Helicobacter pylori CagA tyrosine phosphorylation motif patterns between Western and East Asian strains, and influences on interleukin-8 secretion. J Med Microbiol. 2008;57:1062–7. doi: 10.1099/jmm.0.2008/001818-0. [DOI] [PubMed] [Google Scholar]
- 73.Oldani A, Cormont M, Hofman V, et al. Helicobacter pylori counteracts the apoptotic action of its VacA toxin by injecting the CagA protein into gastric epithelial cells. PLoS Pathog. 2009;5:e1000603. doi: 10.1371/journal.ppat.1000603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tsugawa H, Suzuki H, Saya H, et al. Reactive oxygen species-induced autophagic degradation of Helicobacter pylori CagA is specifically suppressed in cancer stem-like cells. Cell Host Microbe. 2012;12:764–77. doi: 10.1016/j.chom.2012.10.014. [DOI] [PubMed] [Google Scholar]
- 75.Wroblewski LE, Noble PJ, Pagliocca A, et al. Stimulation of MMP-7 (matrilysin) by Helicobacter pylori in human gastric epithelial cells: role in epithelial cell migration. J Cell Sci. 2003;116:3017–26. doi: 10.1242/jcs.00518. [DOI] [PubMed] [Google Scholar]
- 76.Barker N, Huch M, Kujala P, et al. Lgr5(+ve) stem cells drive self-renewal in the stomach and build long-lived gastric units in vitro. Cell Stem Cell. 2010;6:25–36. doi: 10.1016/j.stem.2009.11.013. [DOI] [PubMed] [Google Scholar]
- 77.Wroblewski LE, Shen L, Ogden S, et al. Helicobacter pylori dysregulation of gastric epithelial tight junctions by urease-mediated myosin II activation. Gastroenterology. 2009;136:236–46. doi: 10.1053/j.gastro.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.McCracken KW, Cata EM, Crawford CM, et al. Modelling human development and disease in pluripotent stem-cell-derived gastric organoids. Nature. 2014;516:400–4. doi: 10.1038/nature13863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bertaux-Skeirik N, Feng R, Schumacher MA, et al. CD44 plays a functional role in Helicobacter pylori-induced epithelial cell proliferation. PLoS Pathog. 2015;11:e1004663. doi: 10.1371/journal.ppat.1004663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schlaermann P, Toelle B, Berger H, et al. A novel human gastric primary cell culture system for modelling Helicobacter pylori infection in vitro. Gut. 2014 doi: 10.1136/gutjnl-2014-307949. Epub December 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bartfeld S, Bayram T, van de Wetering M, et al. In vitro expansion of human gastric epithelial stem cells and their responses to bacterial infection. Gastroenterology. 2015;148:126–136. doi: 10.1053/j.gastro.2014.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bagnoli F, Buti L, Tompkins L, et al. Helicobacter pylori CagA induces a transition from polarized to invasive phenotypes in MDCK cells. Proc Natl Acad Sci U S A. 2005;102:16339–44. doi: 10.1073/pnas.0502598102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Baud J, Varon C, Chabas S, et al. Helicobacter pylori initiates a mesenchymal transition through ZEB1 in gastric epithelial cells. PLoS One. 2013;8:e60315. doi: 10.1371/journal.pone.0060315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bessede E, Staedel C, Acuna Amador LA, et al. Helicobacter pylori generates cells with cancer stem cell properties via epithelial-mesenchymal transition-like changes. Oncogene. 2014;33:4123–31. doi: 10.1038/onc.2013.380. [DOI] [PubMed] [Google Scholar]
- 85.Saito Y, Murata-Kamiya N, Hirayama T, et al. Conversion of Helicobacter pylori CagA from senescence inducer to oncogenic driver through polarity-dependent regulation of p21. J Exp Med. 2010;207:2157–74. doi: 10.1084/jem.20100602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yin Y, Grabowska AM, Clarke PA, et al. Helicobacter pylori potentiates epithelial:mesenchymal transition in gastric cancer: links to soluble HB-EGF, gastrin and matrix metalloproteinase-7. Gut. 2010;59:1037–45. doi: 10.1136/gut.2009.199794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nagy TA, Wroblewski LE, Wang D, et al. beta-Catenin and p120 mediate PPARdelta-dependent proliferation induced by Helicobacter pylori in human and rodent epithelia. Gastroenterology. 2011;141:553–64. doi: 10.1053/j.gastro.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fujii Y, Yoshihashi K, Suzuki H, et al. CDX1 confers intestinal phenotype on gastric epithelial cells via induction of stemness-associated reprogramming factors SALL4 and KLF5. Proc Natl Acad Sci U S A. 2012;109:20584–9. doi: 10.1073/pnas.1208651109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Noto JM, Khizanishvili T, Chaturvedi R, et al. Helicobacter pylori promotes the expression of Kruppel-like factor 5, a mediator of carcinogenesis, in vitro and in vivo. PLoS One. 2013;8:e54344. doi: 10.1371/journal.pone.0054344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Panella C, Ierardi E, Polimeno L, et al. Proliferative activity of gastric epithelium in progressive stages of Helicobacter pylori infection. Dig Dis Sci. 1996;41:1132–8. doi: 10.1007/BF02088228. [DOI] [PubMed] [Google Scholar]
- 91.Correa P. Helicobacter pylori and gastric carcinogenesis. Am J Surg Pathol. 1995;19:S37–43. [PubMed] [Google Scholar]
- 92.Cahill RJ, Kilgallen C, Beattie S, et al. Gastric epithelial cell kinetics in the progression from normal mucosa to gastric carcinoma. Gut. 1996;38:177–81. doi: 10.1136/gut.38.2.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Li Q, Karam SM, Gordon JI. Diphtheria toxin-mediated ablation of parietal cells in the stomach of transgenic mice. J Biol Chem. 1996;271:3671–6. [PubMed] [Google Scholar]
- 94.Oh JD, Karam SM, Gordon JI. Intracellular Helicobacter pylori in gastric epithelial progenitors. Proc Natl Acad Sci U S A. 2005;102:5186–91. doi: 10.1073/pnas.0407657102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Giannakis M, Chen SL, Karam SM, et al. Helicobacter pylori evolution during progression from chronic atrophic gastritis to gastric cancer and its impact on gastric stem cells. Proc Natl Acad Sci U S A. 2008;105:4358–63. doi: 10.1073/pnas.0800668105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Oh JD, Kling-Backhed H, Giannakis M, et al. Interactions between gastric epithelial stem cells and Helicobacter pylori in the setting of chronic atrophic gastritis. Curr Opin Microbiol. 2006;9:21–7. doi: 10.1016/j.mib.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 97.Howitt MR, Lee JY, Lertsethtakarn P, et al. ChePep controls Helicobacter pylori Infection of the gastric glands and chemotaxis in the Epsilonproteobacteria. MBio. 2011:2. doi: 10.1128/mBio.00098-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sigal M, Rothenberg ME, Logan CY, et al. Helicobacter pylori activates and expands Lgr5(+) stem cells through direct colonization of the gastric glands. Gastroenterology. 2015;148:1392–1404. doi: 10.1053/j.gastro.2015.02.049. [DOI] [PubMed] [Google Scholar]
- 99.Lee I, Lee H, Kim M, et al. Ethnic difference of Helicobacter pylori gastritis: Korean and Japanese gastritis is characterized by male- and antrum-predominant acute foveolitis in comparison with American gastritis. World J Gastroenterol. 2005;11:94–8. doi: 10.3748/wjg.v11.i1.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Koo BK, Clevers H. Stem cells marked by the R-spondin receptor LGR5. Gastroenterology. 2014;147:289–302. doi: 10.1053/j.gastro.2014.05.007. [DOI] [PubMed] [Google Scholar]
- 101.Qiao XT, Ziel JW, McKimpson W, et al. Prospective identification of a multilineage progenitor in murine stomach epithelium. Gastroenterology. 2007;133:1989–98. doi: 10.1053/j.gastro.2007.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Arnold K, Sarkar A, Yram MA, et al. Sox2(+) adult stem and progenitor cells are important for tissue regeneration and survival of mice. Cell Stem Cell. 2011;9:317–29. doi: 10.1016/j.stem.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Karam SM, Leblond CP. Dynamics of epithelial cells in the corpus of the mouse stomach. I. Identification of proliferative cell types and pinpointing of the stem cell. Anat Rec. 1993;236:259–79. doi: 10.1002/ar.1092360202. [DOI] [PubMed] [Google Scholar]
- 104.Mills JC, Andersson N, Hong CV, et al. Molecular characterization of mouse gastric epithelial progenitor cells. Proc Natl Acad Sci U S A. 2002;99:14819–24. doi: 10.1073/pnas.192574799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Karam SM, Straiton T, Hassan WM, et al. Defining epithelial cell progenitors in the human oxyntic mucosa. Stem Cells. 2003;21:322–36. doi: 10.1634/stemcells.21-3-322. [DOI] [PubMed] [Google Scholar]
- 106.Khurana SS, Riehl TE, Moore BD, et al. The hyaluronic acid receptor CD44 coordinates normal and metaplastic gastric epithelial progenitor cell proliferation. J Biol Chem. 2013;288:16085–97. doi: 10.1074/jbc.M112.445551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Nam KT, Lee HJ, Sousa JF, et al. Mature chief cells are cryptic progenitors for metaplasia in the stomach. Gastroenterology. 2010;139:2028–2037. doi: 10.1053/j.gastro.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Stange DE, Koo BK, Huch M, et al. Differentiated Troy+ chief cells act as reserve stem cells to generate all lineages of the stomach epithelium. Cell. 2013;155:357–68. doi: 10.1016/j.cell.2013.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Powell AE, Wang Y, Li Y, et al. The pan-ErbB negative regulator Lrig1 is an intestinal stem cell marker that functions as a tumor suppressor. Cell. 2012;149:146–58. doi: 10.1016/j.cell.2012.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Li Q, Jia Z, Wang L, et al. Disruption of Klf4 in villin-positive gastric progenitor cells promotes formation and progression of tumors of the antrum in mice. Gastroenterology. 2012;142:531–42. doi: 10.1053/j.gastro.2011.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Uehara T, Ma D, Yao Y, et al. H pylori infection is associated with DNA damage of Lgr5-positive epithelial stem cells in the stomach of patients with gastric cancer. Dig Dis Sci. 2013;58:140–9. doi: 10.1007/s10620-012-2360-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yamanoi K, Fukuma M, Uchida H, et al. Overexpression of leucine-rich repeat-containing G protein-coupled receptor 5 in gastric cancer. Pathol Int. 2013;63:13–9. doi: 10.1111/pin.12013. [DOI] [PubMed] [Google Scholar]
- 113.Zheng ZX, Sun Y, Bu ZD, et al. Intestinal stem cell marker LGR5 expression during gastric carcinogenesis. World J Gastroenterol. 2013;19:8714–21. doi: 10.3748/wjg.v19.i46.8714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Xi HQ, Cai AZ, Wu XS, et al. Leucine-rich repeat-containing G-protein-coupled receptor 5 is associated with invasion, metastasis, and could be a potential therapeutic target in human gastric cancer. Br J Cancer. 2014;110:2011–20. doi: 10.1038/bjc.2014.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Houghton J, Stoicov C, Nomura S, et al. Gastric cancer originating from bone marrow-derived cells. Science. 2004;306:1568–71. doi: 10.1126/science.1099513. [DOI] [PubMed] [Google Scholar]
- 116.Varon C, Dubus P, Mazurier F, et al. Helicobacter pylori infection recruits bone marrow-derived cells that participate in gastric preneoplasia in mice. Gastroenterology. 2012;142:281–91. doi: 10.1053/j.gastro.2011.10.036. [DOI] [PubMed] [Google Scholar]
- 117.Tan S, Tompkins LS, Amieva MR. Helicobacter pylori usurps cell polarity to turn the cell surface into a replicative niche. PLoS Pathog. 2009;5:e1000407. doi: 10.1371/journal.ppat.1000407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yamanouchi J, Azuma T, Yakushijin Y, et al. Dramatic and prompt efficacy of Helicobacter pylori eradication in the treatment of severe refractory iron deficiency anemia in adults. Ann Hematol. 2014;93:1779–80. doi: 10.1007/s00277-014-2052-x. [DOI] [PubMed] [Google Scholar]
- 119.Queiroz DM, Harris PR, Sanderson IR, et al. Iron status and Helicobacter pylori infection in symptomatic children: an international multi-centered study. PLoS One. 2013;8:e68833. doi: 10.1371/journal.pone.0068833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Queiroz DM, Rocha AM, Crabtree JE. Unintended consequences of Helicobacter pylori infection in children in developing countries: iron deficiency, diarrhea, and growth retardation. Gut Microbes. 2013;4:494–504. doi: 10.4161/gmic.26277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Noto JM, Gaddy JA, Lee JY, et al. Iron deficiency accelerates Helicobacter pylori-induced carcinogenesis in rodents and humans. J Clin Invest. 2013;123:479–92. doi: 10.1172/JCI64373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Cassat JE, Skaar EP. Iron in infection and immunity. Cell Host Microbe. 2013;13:509–19. doi: 10.1016/j.chom.2013.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Choe YH, Oh YJ, Lee NG, et al. Lactoferrin sequestration and its contribution to iron-deficiency anemia in Helicobacter pylori-infected gastric mucosa. J Gastroenterol Hepatol. 2003;18:980–5. doi: 10.1046/j.1440-1746.2003.03098.x. [DOI] [PubMed] [Google Scholar]
- 124.Velayudhan J, Hughes NJ, McColm AA, et al. Iron acquisition and virulence in Helicobacter pylori: a major role for FeoB, a high-affinity ferrous iron transporter. Mol Microbiol. 2000;37:274–86. doi: 10.1046/j.1365-2958.2000.01987.x. [DOI] [PubMed] [Google Scholar]
- 125.Conrad ME, Schade SG. Ascorbic acid chelates in iron absorption: a role for hydrochloric acid and bile. Gastroenterology. 1968;55:35–45. [PubMed] [Google Scholar]
- 126.Annibale B, Capurso G, Lahner E, et al. Concomitant alterations in intragastric pH and ascorbic acid concentration in patients with Helicobacter pylori gastritis and associated iron deficiency anaemia. Gut. 2003;52:496–501. doi: 10.1136/gut.52.4.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Azab SF, Esh AM. Serum hepcidin levels in Helicobacter pylori-infected children with iron-deficiency anemia: a case-control study. Ann Hematol. 2013;92:1477–83. doi: 10.1007/s00277-013-1813-2. [DOI] [PubMed] [Google Scholar]
- 128.Schwarz P, Kubler JA, Strnad P, et al. Hepcidin is localised in gastric parietal cells, regulates acid secretion and is induced by Helicobacter pylori infection. Gut. 2012;61:193–201. doi: 10.1136/gut.2011.241208. [DOI] [PubMed] [Google Scholar]
- 129.Senkovich O, Ceaser S, McGee DJ, et al. Unique host iron utilization mechanisms of Helicobacter pylori revealed with iron-deficient chemically defined media. Infect Immun. 2010;78:1841–9. doi: 10.1128/IAI.01258-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Aihara E, Closson C, Matthis AL, et al. Motility and chemotaxis mediate the preferential colonization of gastric injury sites by Helicobacter pylori. PLoS Pathog. 2014;10:e1004275. doi: 10.1371/journal.ppat.1004275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Tan S, Noto JM, Romero-Gallo J, et al. Helicobacter pylori perturbs iron trafficking in the epithelium to grow on the cell surface. PLoS Pathogens. 2011;7:e1002050. doi: 10.1371/journal.ppat.1002050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Noto JM, Lee JY, Gaddy JA, et al. Regulation of Helicobacter pylori virulence within the context of iron deficiency. J Infect Dis. 2015;211:1790–4. doi: 10.1093/infdis/jiu805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lee SA, Kang D, Shim KN, et al. Effect of diet and Helicobacter pylori infection to the risk of early gastric cancer. J Epidemiol. 2003;13:162–8. doi: 10.2188/jea.13.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Tsugane S. Salt, salted food intake, and risk of gastric cancer: epidemiologic evidence. Cancer Sci. 2005;96:1–6. doi: 10.1111/j.1349-7006.2005.00006.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Cameron A, Frirdich E, Huynh S, et al. Hyperosmotic stress response of Campylobacter jejuni. J Bacteriol. 2012;194:6116–30. doi: 10.1128/JB.01409-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Loh JT, Torres VJ, Cover TL. Regulation of Helicobacter pylori cagA expression in response to salt. Cancer Res. 2007;67:4709–15. doi: 10.1158/0008-5472.CAN-06-4746. [DOI] [PubMed] [Google Scholar]
- 137.Loh JT, Friedman DB, Piazuelo MB, et al. Analysis of Helicobacter pylori cagA promoter elements required for salt-induced upregulation of CagA expression. Infection and Immunity. 2012;80:3094–106. doi: 10.1128/IAI.00232-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Loh JT, Shaffer CL, Piazuelo MB, et al. Analysis of cagA in Helicobacter pylori strains from Colombian populations with contrasting gastric cancer risk reveals a biomarker for disease severity. Cancer Epid Biomark Prev. 2011;20:2237–49. doi: 10.1158/1055-9965.EPI-11-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Fox JG, Dangler CA, Taylor NS, et al. High-salt diet induces gastric epithelial hyperplasia and parietal cell loss, and enhances Helicobacter pylori colonization in C57BL/6 mice. Cancer Res. 1999;59:4823–8. [PubMed] [Google Scholar]
- 140.Bergin IL, Sheppard BJ, Fox JG. Helicobacter pylori infection and high dietary salt independently induce atrophic gastritis and intestinal metaplasia in commercially available outbred Mongolian gerbils. Dig Dis Sci. 2003;48:475–85. doi: 10.1023/a:1022524313355. [DOI] [PubMed] [Google Scholar]
- 141.Nozaki K, Shimizu N, Inada K, et al. Synergistic promoting effects of Helicobacter pylori infection and high-salt diet on gastric carcinogenesis in Mongolian gerbils. Jpn J Can Res. 2002;93:1083–9. doi: 10.1111/j.1349-7006.2002.tb01209.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Gaddy JA, Radin JN, Loh JT, et al. High dietary salt intake exacerbates Helicobacter pylori-induced gastric carcinogenesis. Infection and Immunity. 2013;81:2258–67. doi: 10.1128/IAI.01271-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Tilg H. Diet and intestinal immunity. N Engl J Med. 2012;366:181–3. doi: 10.1056/NEJMcibr1113158. [DOI] [PubMed] [Google Scholar]
- 144.Gonda TA, Kim YI, Salas MC, et al. Folic acid increases global DNA methylation and reduces inflammation to prevent Helicobacter-associated gastric cancer in mice. Gastroenterology. 2012;142:824–833. doi: 10.1053/j.gastro.2011.12.058. [DOI] [PubMed] [Google Scholar]
- 145.Abreu MT, Peek RM., Jr Gastrointestinal malignancy and the microbiome. Gastroenterology. 2014;146:1534–1546. e3. doi: 10.1053/j.gastro.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ma JL, Zhang L, Brown LM, et al. Fifteen-year effects of Helicobacter pylori, garlic, and vitamin treatments on gastric cancer incidence and mortality. J Natl Can Inst. 2012;104:488–92. doi: 10.1093/jnci/djs003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Lofgren JL, Whary MT, Ge Z, et al. Lack of commensal flora in Helicobacter pylori-infected INS-GAS mice reduces gastritis and delays intraepithelial neoplasia. Gastroenterology. 2011;140:210–20. doi: 10.1053/j.gastro.2010.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Lertpiriyapong K, Whary MT, Muthupalani S, et al. Gastric colonisation with a restricted commensal microbiota replicates the promotion of neoplastic lesions by diverse intestinal microbiota in the Helicobacter pylori INS-GAS mouse model of gastric carcinogenesis. Gut. 2013;63:54–63. doi: 10.1136/gutjnl-2013-305178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Lemke LB, Ge Z, Whary MT, et al. Concurrent Helicobacter bilis infection in C57BL/6 mice attenuates proinflammatory H pylori-induced gastric pathology. Infect Immun. 2009;77:2147–58. doi: 10.1128/IAI.01395-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ge Z, Feng Y, Muthupalani S, et al. Coinfection with Enterohepatic Helicobacter species can ameliorate or promote Helicobacter pylori-induced gastric pathology in C57BL/6 mice. Infection and Immunity. 2011;79:3861–71. doi: 10.1128/IAI.05357-11. [DOI] [PMC free article] [PubMed] [Google Scholar]