Amyloid-beta Alzheimer targets — protein processing, lipid rafts, and amyloid-beta pores (original) (raw)
. 2016 Mar 24;89(1):5–21.
Abstract
Amyloid beta (Aβ), the hallmark of Alzheimer’s Disease (AD), now appears to be deleterious in its low number aggregate form as opposed to the macroscopic Aβ fibers historically seen postmortem. While Alzheimer targets, such as the tau protein, amyloid precursor protein (APP) processing, and immune system activation continue to be investigated, the recent discovery that amyloid beta aggregates at lipid rafts and likely forms neurotoxic pores has led to a new paradigm regarding why past therapeutics may have failed and how to design the next round of compounds for clinical trials. An atomic resolution understanding of Aβ aggregates, which appear to exist in multiple conformations, is most desirable for future therapeutic development. The investigative difficulties, structures of these small Aβ aggregates, and current therapeutics are summarized in this review.
Keywords: Amyloid, amyloid-beta, Aß, Alzheimer, amyloid precursor protein, presenilin, secretase, plaque, dementia, pore, perforin, ß-barrel, ß-strand, dementia, lipid raft, cholesterol, ApoE, APBA, Mint, ganglioside, ß-secretase, γ-secretase
Introduction
For decades, the amyloid-beta (Aβ) and tau proteins have been the two leading targets thought to be the causative agents leading to Alzheimer’s disease (AD) [1-5]. These two proteins were intensively studied because of genetic and largely phenotypic observations, namely extracellular amyloid plaques and intracellular tau tangles. The fact that these two macroscopic aggregations of protein were observable in individuals suffering from AD now appears to have led the research field slightly astray.
Just over 100 years ago, Alois Alzheimer first described the pathology and symptoms of dementia that would come to be called AD. As life expectancy continues to increase, a growing percentage of the world’s population will get AD in their lifetime, with 7 percent of those ages 65 and older, and 40 percent of people 80 and older in industrialized countries being affected [6]. Despite decades of AD research, neither an effective therapy nor a vaccine has been developed. However, the wealth of molecular information now known about AD progression raises hope for therapeutic development in the next decade.
Multiple root causes of AD have been proposed that have varying levels of supporting data behind them. The amyloid theory — for decades the most widely accepted paradigm — initially posited that the extracellular amyloid plaques kill brain cells and are the causative agent leading to Alzheimer dementia. In 1995, genetic links leading to Early Onset Familial Alzheimer’s Disease (EOFAD) were discovered [7,8].
These mutations are in the presenilin gene, a component of γ-secretase that cleaves the amyloid precursor protein (APP), releasing the amyloid peptide to the cytosol, leading to the formation of amyloid plaques (Figure 1). In mouse models, mutation of the presenilin gene causes AD-like symptoms that could be alleviated through inactivation of presenilin [9,10]. Multiple methods have proven efficacious in reducing amyloid plaques, and some have been correlated with better cognitive outcomes [11-13], whereas others have not [14-16].
Figure 1.

**Classic Amyloid Precursor Protein modification and cleavage pathway:**The Amyloid Precursor Protein (APP) is cleaved by α, β, and γ-secretases. β-secretase cleavage of APP leads to amyloid plaques, whereas α-secretase results in a truncated amyloid protein, which does not aggregate
All of the available treatments for AD do not augment the molecular players that lead to AD, but instead target the downstream players associated with cognitive deficits. Three of the four drugs approved by the Federal Drug Administration are cholinesterase inhibitors intended to counter the loss of cholinergic neurons observed in AD patients. The fourth drug, memantine, is an NMDA receptor antagonist that is thought to work by blocking the cytotoxic effects of excess extracellular glutamate on neurons.
Although symptomatic control by these agents has been shown to be statistically significant, their therapeutic efficacy is far from robust, and the duration of their effects is limited [17]. The low efficacy of the existing treatments necessitates the development of agents with the ability to alter or halt the progression of the disease. Ongoing drug development has focused on several aspects of AD, and aims to modify the disease.
Current drug development falls into four major categories that target:
1. The metabolic changes and insulin resistance in neurons observed with AD.
2. The inflammation associated with certain regions of the brain that are affected in AD.
3. The production of pathological forms of Tau protein.
4. The production, plaque formation, and clearance of Aβ.
The best characterized of these approaches, and the agents that have shown the greatest promise, have been those that modify Aβ levels.
While both amyloid plaques and tau tangles are macroscopically visible phenomena that correlate with AD progression, recent research suggests they may not be the causative agents of dementia [18,19]. A new model to fit available data has been put forward in which it is Aβ pre-plaque monomers or small oligomers that cause neuronal death, not the aggregated plaque [20,21]. In this model, large amyloid plaques could even inhibit neuronal death by sequestering the smaller deleterious amyloid monomers and oligomers. In this light, previous conclusions about amyloid plaque may be reinterpreted as correlative instead of causative. Promising drugs have been shown to decrease amyloid plaque formation and delay cognitive decline; however, this could be due to decreased soluble amyloid, which in turn would decrease the formation of plaques. While there is not yet consensus on the causative agent leading to AD neuronal death and dementia, inhibiting Aβ aggregation has been attempted in numerous ways as a therapeutic target, with mixed results (Table 1). The structure of Aβ aggregates, their ability to form neurotoxic pores, and the mechanism and efficacy of how current compounds target Aβ will be addressed in this review.
Table 1. Table: Different approaches to lowering Aß levels.
| Approach | Therapeutic | How it lowers Aß levels | Clinical Trial Phase completed | Clinical Efficacy |
|---|---|---|---|---|
| Vaccine | CAD-106 | An antigen that mimics the amino acids 1-6 of Aß; produces anti-Aß antibodies | Phase II | Undetermined |
| Bapineuzumab | Binds to soluble and fibrillar forms of Aß | Phase III | Limited efficacy; discontinued because of side-effects | |
| Solanezumab | Binds to soluble Aß only | Phase III | Shows some efficacy in mild AD | |
| Antibodies | Crenezumab | Binds to soluble and fibrillar forms of Aß | Phase III | Shows some efficacy in mild AD |
| Aducanumab | Binds to fibrillar and plaque, but not soluble forms of Aß | Phase III | No efficacy in mild to moderate AD | |
| Gantenerumab | Binds to fibrillar and plaque, but not soluble forms of Aß | Phase II | Undetermined, phase III trial is ongoing | |
| MK-8931 | Blocks production of Aß by inhibiting ß-secretase enzyme | Phase II/III | Undetermined, phase III trial is ongoing | |
| ß-secretase Inhibitors | AZD3293 | Blocks production of Aß by inhibiting ß-secretase enzyme | Phase I | Undetermined, phase II/III trial is ongoing |
| E2609 | Blocks production of Aß by inhibiting ß-secretase enzyme | Phase I | Undetermined, phase II trial is ongoing |
Tau
Neurofibrillary tangles (NFTs) comprised of hyperphosphorylated tau protein (τ), in addition to Aß plaques, are the other hallmark associated with AD. Tau interactions have been reviewed well [22-24]. Although not the subject of this review, there are interactions between τ-tangles [25] and Aβ plaques and therefore these interactions will be cursorily covered. Tau -/- neurons were protected in vitro from Aβ-induced cell death with antibodies to tau delaying motor function impairment and weight loss in multiple transgenic mouse models [23]. The link between APP metabolism and tau proteostasis is further implicated with the evidence that familial forms of AD, which exhibit mutated APP or increased levels of APP, have marked increases in intracellular tau. Recently, there has been evidence suggesting Aβ and tau interact synergistically to result in the AD phenotype of neuronal death.
Jin et al. recently isolated Aβ dimers from Alzheimer cortex and showed at sub-nanomolar concentration, without the presence of Aβ fibrils, the Aβ dimers increased hyperphosphorylated tau, followed by microtubule cytoskeleton disruption and neuritic degeneration [26]. As the amyloid theory would predict, all of these effects were modified by: knocking down endogenous tau, which rescued neuron degeneration; overexpression of human tau, which intensified the neuronal degeneration; and administration of Aβ N-terminal antibodies, which prevented cytoskeletal disruption. Furthermore, synthetic dimers had a similar, albeit reduced, effect as the natural AD dimers [27,28].
The hyperphosphorylated tau forms the hallmark aggregates termed tau tangles, but has also been shown to have other effects that are deleterious such as binding to c-Jun N-terminal kinase-interacting protein 1 (JIP1), causing it to aggregate in the cell body and impair axonal transport in AD. JiP1 normally binds the kinesin light chain (KLC) and is involved in the linkage of cargos to the kinesin-i motor complex [1,29,30].
Amyloid Precursor Protein (APP
APP can be post-translationally processed to eventually create the deleterious Aβ peptides; therefore, therapeutic targeting of this protein processing is an attractive target (Figure 1). Full-length APP also has three isoforms — APP695, APP751, and APP770 — derived from alternative splicing, which has been reviewed well by Zhang et al. [31]. In summary, the latter two, APP751 and APP770, have a Kunitz Protease Inhibitor (KPI) domain on their intracellular side [32,33] and have been found at elevated levels in the brains of those with AD [34], along with increased Aß [35].
Interestingly, APP is part of a protein family in mammals with two homologous proteins, APP like protein 1 (APLP1) and 2 (APLP2), which appear to have overlapping function as the double deletion of APLP2 and either APP or APLP1 is lethal in mice [36-38]. APP itself is a transmembrane protein that has both cytoplasmic and extracellular cleavage products that are relevant in AD. APP is initially cleaved via α-secretase or β-secretase, which leads to normal or pathological products, respectively (Figure 1).
Therefore, both α-secretase agonists and β-secretase antagonists could act as Alzheimer therapeutics. Whether α-secretase or β-secretase cleaves APP, there is intracellular release of amyloid precursor protein intracellular domain (AICD). Following α or β cleavage, γ-secretase cleavage yields extracellular p3 or Aβ, respectively. The in vivo role of p3 has yet to be determined. While p3 can aggregate into fibers, it is not stable in smaller oligomers, which may explain why it does not have the neurotoxic effects of Aβ [39]. Two major isoforms of Aβ exist, namely Aβ40 and Aβ42. Aβ42 is the primary amyloidogenic form in AD and by far the most studied. Therefore, shifting processing toward Aβ40 is considered protective for AD [40-42]. Also, there is a rarer Aβ43 isoform that seems to be even more detrimental than Aβ42 [43].
The non-disease function of APP and its cleavage products remains a debated topic with evidence pointing to a variety of functions including neuron growth and synaptogenesis, protein trafficking in neurons, signal transduction across the membrane [44-46], cell adhesion [47-49], and calcium metabolism [31]. Despite the current debate of what the “good” function of non-amyloidogenic APP is, there has been more clear evidence of a benefit of shifting from amyloidogenic to non-amyloidogenic pathways. The understanding of molecular players involved in shifting toward non-amyloidogenic processing of APP has resulted in actionable behavioral modifications.
For example, green tea extract contains epigallocatechin gallate (EGCG), which has been shown to increase release of non-amyloidogenic APP six-fold [50]. One way the increased EGCG does this is by binding the estrogen receptor, which increases activation of an α-secretase, namely ADAM metallopeptidase domain 10 (ADAM10) [51]. ADAM10 increases the non-amyloidogenic pathway by cleaving APP via an ERα/PI3K/Akt dependent mechanism [50]. Numerous endogenous interactions also affect APP enzymatic cleavage. APP localizes to lipid rafts and its interactions with cholesterol, Apolipoprotein E (ApoE), and other proteins in the rafts such as the Amyloid beta (A4) Precusor protein-bvinding family A (APBA) proteins, affect APP processing. All of these interactions have been investigated as therapeutics targets and are discussed below.
β-secretase and γ-secretase
APP is processed into Aβ peptides by the actions of two proteolytic proteins, β-secreatse and γ-secretase. β-secretase, also known as β-site APP cleaving enzyme (BACE), is an aspartyl protease that initiates Aβ peptide production [52]. While β-secretase1 is the predominant form in neural tissue, β-secretase2 isoforms are present in lower levels and therefore inhibitors of both forms could be investigated as therapeutic targets [53]. β-secretase1 in particular has been found to have increased activity in Alzheimer’s patients, as well as those with mild cognitive impairment that went on to develop AD [54]. These enzymes are up-regulated in response to cellular stress such as oxidative stress, ischemia, and energy depletion [29,55]. β-secretase1 is in lipid raft domains of the cell membrane and requires glycosaminoglycans for effective cleavage [56,57].
γ-secretase is a protein complex comprised of at least four subunits: the enzymatic portion of the complex, presenilin (PSEN) 1 or 2; presenilin enhancer 2 (Pen2); nicastin (Nct); and anterior pharynx defective-1 (Aph-1) [58]. These subunits combine in a unique order to form functional γ-secretase: Nct and Aph-1 first form a dimer, followed by PSEN binding, and finally Pen2 binding, which may help in PSEN auto-cleavage [59,60]. More than 50 substrates (such as APP, Notch, ErbB4, and N-cadherin) can be cleaved due to recognition endowed via Nct and Aph-1 [60-62]. The fact that γ-secretase activates Notch is problematic in that nonselective inhibitors of γ-secretase would have detrimental side effects. Notch is involved in embryogenesis, so potentially women with EOFAD could be pregnant and unable to use nonspecific γ-secretase inhibitors. Notch is also involved in neurogenesis, so even older patients could have deleterious effects due to off-target inhibition [63].
However, because increased levels of γ-secretase have been shown to lead to events associated with the AD pathogenesis [64-67], it continues to be one of the main targets for the development of therapeutics. PSENs contain six to nine transmembrane regions with both ends in the cytoplasm, and the functional γ-secretase actually cuts APP in a transmembrane region. The combined actions of β-secretase1 and γ-secretase on APP lead to the release of Aβ peptides. β-secretase1 cleaves APP to release a soluble extracellular fragment and a membrane-bound fragment, C99 [68]. C99 is then cleaved by γ-secretase at several potential transmembrane locations [69], yielding a variety of Aβ peptides ranging from 39 to 43 amino acids in length.
Familial Alzheimer Disease (FAD)
Familial Alzheimer’s Disease (FAD) is an autosomal dominant condition that represents 5 percent of AD patients [70], resulting in the development of symptoms before age 65 [71]. FAD can be caused by mutations affecting three different genes APP, PSEN1, or PSEN2 on chromosomes 21, 14, and 1, respectively [72,73]. The mutations on APP all cause up-regulation of Aβ (Aβ40, Aβ42, or both) through one of three mechanisms: increased APP expression, α-secretase inhibition, or increased activity of β-secretase [71,74-76].
Aβ
It is important to note that both the amyloidogenic and non-amyloidogenic pathways are present in healthy individuals, with AD presumably being caused through increased amyloidogenic cleavage or decreased Aβ turnover [77]. In fact, healthy individuals have Aβ levels in their plasma and cerebrospinal fluid of 500pM and 3-8nM, respectively [78,79]. Clots formed in the presence of Aβ have an abnormal structure and are resistant to clearance. Aβ has been found to bind directly to fibrinogen with a dissociation constant (kd) of < 7nM, possibly modifying its tertiary structure and leading to clots [80]. The longer Aβ digestion fragments are more hydrophobic and associated with increased AD risk. Aβ 1-42 has been most extensively studied due to its abundance in patients predisposed to AD; however, Aβ 1-43 has been shown to be even more toxic in both cells and experimental animals [81]. Mutations in both APP and the PSENs can influence which Aβ peptides are formed, and missense mutations in APP and the presenilins are associated with increased release of Aβ42 and FAD [82].
Aβ Structure: When, where, and what
When does Aß matter — early
The hallmark of Alzheimer’s historically has been the large extracellular Aß plaques that are seen postmortem. If we rewind time from the point of an AD patient’s death and think about when the Aβ proteins are doing their damage, it now appears it is many years, even decades, earlier [83-85]. There is no therapeutic on the horizon that can reverse the neuronal damage associated with AD, and because it is such a protracted disease, drug development is now focused on early stages in the disease progression. For example, Eli Lilly is testing solanezumab in a Dominantly Inherited Alzheimer Network Trial on people ages 65 to 85 who are predisposed or have been diagnosed with AD, but who have yet to show clinical symptoms [86]. The recent Aβ structural insights detailed below suggest that late-stage large Aβ plaques are not the correct pharmacological target.
Where does Aß matter — lipid rafts
Historically, Aβ was measured as a single entity, not specifically by the conformation it was adopting. When measuring Aβ in aggregate, the spatial location of amyloid plaques has been found to correlate more strongly with cognitive impairment [87] than the total volume of plaque formation [88]. In particular, Aβ levels in the frontal cortex preceded cognitive decline, while Aβ levels in the entorhinal cortex correlated with measureable cognitive impairment. While levels of insoluble amyloid deposition can differentiate between disease state and control, only soluble amyloid correlated with measures of dementia [89].
Protein quantification by western blotting shows a three-fold increase in soluble Aβ of confirmed AD patients compared to controls [90]. Again, this fits with the model where amyloid fibrils are late-stage phenotypes of AD, but not necessarily causative of dementia, whereas soluble Aβ is directly deleterious and potentially the starting point of neuronal death observed in AD. PET/CT imaging has rapidly risen as a useful tool to measure Aβ in vivo with compounds such as monoclonal antibodies (mABs). However, PET/CT imagine can be inconsistent in certain organs, such as the kidney and the heart, and therefore origin of measurement is an important consideration when comparing to normal levels [91-94].
Currently, PET imaging modalities measure total amyloid. It remains to be seen how finely tuned compounds can detect and differentiate between different amyloid multimers. A growing area of research has shifted away from looking at Aβ in extracellular plaques [42] and instead turned toward the interaction of Aβ in the plasma membrane, namely direct interactions with lipid rafts and in β-sheet barrel pore formation [95-103]. Recent evidence has shown Aβ interactions with the plasma membrane are localized to high cholesterol lipid rafts domains where Aβ forms Ca+2 channels, thereby disrupting membrane function. The localization to lipid rafts has led to developing therapeutics that target other proteins localized to lipid rafts, such as those involved in processing of APP.
Cholesterol
The brain contains 20 percent of the body’s cholesterol, of which 70 percent is found in the myelin sheaths. Unlike the rest of the body, essentially all cholesterol in the brain is unesterified [104,105]. This is important because almost all of the cholesterol in the brain is synthesized in situ and not transferred from the rest of the body [106]; therefore, therapeutics targeting cholesterol for AD patients will have to cross the blood brain barrier to have their desired effect. The highly polarized nature of neurons causes cholesterol trafficking to be of particular importance as has been well-reviewed elsewhere [107].
Earlier studies suggested statin use, which inhibits cholesterol synthesis, was associated with decreased AD prevalence, but later studies did not support that finding [108-110]. It has recently been shown by Liu et al. that γ-secretase cleavage of APP, forming Aβ and AICD, downregulated low-density lipoprotein receptor-related protein 1 (LRP1) by AICD binding directly to the LRP1 promoter. LRP1, also known as apolipoprotein E receptor (APOER), regulates ApoE and cholesterol levels in the brain, and therefore could also be a target for AD treatment [111]. ApoE imports cholesterol into neurons via LRP1 and low activity of LRP1 damages neurons due to low levels of cholesterol [111]. APP knockouts increased ApoE activity and increased cholesterol levels.
The work by Liu et al. connects the two most significant genetic determinants of AD predisposition, namely APP and ApoE [112]. Cholesterol has been found to bind both Aβ and its precursor C99 via a cholesterol-binding domain (Aβ22-35) [113], with greater affinity for Aβ [113-115], suggesting another rationale for high cholesterol levels being deleterious. New therapeutics that mimic cholesterol, such as bexarotene, have been developed that can compete with cholesterol to bind Aβ and increase Aβ clearance, but do not initiate pore formation [115].
ApoE
ApoE is one of the main apolipoproteins in the brain and transports both lipids and Aβ. There are three ApoE variants (ApoE-ε2, ApoE-ε3, ApoE-ε4) with ApoE-ε2 being protective against AD while ApoE-ε4 allele is the greatest risk factor. Caucasians that are homozygous for the ApoE-ε4 allele are 15 times more likely to develop AD [116]. The mechanism for ApoE’s effect in AD is complex since ApoE is associated with numerous characteristics of AD, including Aβ plaque formation, τ-tangle formation, oxidative stress, inflammation, lipid homeostasis deregulation, synaptic plasticity loss and cholinergic dysfunction [117,118]. ApoE can remove Aβ plaques, with the ApoE-ε4, ApoE-ε3, and ApoE-ε2 alleles having a low, normal, and high ability to do so, respectively.
In patients with AD, ApoE has been found to localize to senile plaques (polymorphous beta-amyloid protein deposits), vascular amyloid, neurofibrillary tangles, and bind to APP [116,119-121]. Diets high in carbohydrates and low in essential fatty acids (EFA), especially ω-3 polyunsaturated fatty acids (PUFAs), increase the risk of developing AD [122]. One mechanistic hypothesis for the beneficial effects of PUFAs is that they bind ApoE, which is the primary ligand for the LDL-receptor and mediates delivery of lipids across the blood brain barrier (BBB). The relative efficacies of the ApoE-ε2 and ε4 alleles can therefore effect cholesterol homeostatis in the CNS [118]. While the BBB protects cholesterol in the brain from being sequestered by plasma ApoE; 24S-hydroxycholesterol can leave the brain crossing the BBB [123]. In fact, serum and cerebrospinal fluid (CSF) levels of 24S-hydroxycholesterol are increased in early AD, which could be due to increased cerebral cholesterol turnover during neurodegeneration [124]. While showing mixed results, there is some evidence for reducing 24S-hydroxycholesterol with statin treatment [123].
Most recently, both ε3 and ε4 isoforms of ApoE have been shown to leave the secretory pathway, enter the nucleus, and directly bind promoters and repress anti-inflammatory genes such as Sirt1 [125]. Chromatin immunoprecipitation sequencing (ChIP-seq) results suggest ApoE could interact with as many as ~1,700 gene promoter regions [125].
APBAs
The APBAs are a family of adapter proteins (APBA1, APBA2, and APBA3), previously known as MINT 1/2/3 or X11α/β/γ, which are in lipid rafts and involved in protein transport and synaptic function. APBAs have been found to bind to APP and, although mixed effects on Aβ production have been reported [126,127], are generally viewed as lowering deleterious Aβ production [128-131], which makes them possible AD therapeutic targets.
For example, APBA2 phosphorylation by SRC increases APP endocytosis leading to increased nondeleterious intracellular Aβ production, which was sequestered in vacuoles [132]. Conversely, APBA2 mutants resistant to phosphorylation increase APP entering the recycling pathway and were shuttled back to the cell surface, leading to increases in deleterious Aβ secretion [128,132]. APBAs consist of unique N-terminal regions specific for each isoform, a central phosphotyrosine binding (PTB) domain and two conserved, C-terminal postsynaptic density-95/discs large/zona occludens-1 (PDZ) domains [133]. The PTB domain interacts with a conserved NPTY (Asn-ProThr-Tyr) cytoplasmic motif of the APP that participates in APP trafficking and processing [134]. APBA1 and APBA2 interact with other proteins as well, including Pen1presenilin-1, which is required for γ-secretase activity, through the PDZ domain [135]. Surprisingly, transgenic mice that overproduce Aβ peptides were found to decrease amyloid plaque production when crossed with APBA knock-out mice [126].
Additional studies have shown that the N-terminus of APBA contains a Munc18s-interacting (MID) and Munc18a domain, that bind and retain APP, and can suppress Aβ secretion [136]. The role of APBAs with APP remains complex with APBAs being reported to increase β-secretase activity [126] and decrease γ-secretase activity [137], which are the first and second cleavage steps, respectively, in the amyloidogenic processing of APP to Aβ (Figure 1). Taken together, these studies suggest that regulation of APP processing by APBAs is achieved by interactions with both Pen1presenilin-1 and APP.
Gangliosides
Another enriched component of lipid rafts is gangliosides, a form of glycosphingolipid containing sialic acids on the sugar chain. Exogenous gangliosides have been found to elicit multiple neurotrophic effects [138] and not only help lipid rafts form, but stimulate Aβ production, directly bind Aβ, and also increase amyloid plaques [139-142]. GD3-synthase (GD3S) converts a-series ganglioside (GM3) to b-series ganglioside (GD3) via addition of a sialic acid and is thereby the branch point for a- and b-series gangliosides. This controls the level of a major b-series ganglioside in the brain, GM1, which greatly increases Aβ production [143].
A negative regulatory feedback loop has recently been elucidated by Grimm et al. in which GD3 increases APP cleavage forming Aβ and AICD, which work via two mechanisms to synergistically block production of GD3 [139]. Aβ directly binds GM3, reducing the GD3S reactant pool while increasing the ratio of GM3:GD3, which is an increase in the overall cell ratio of a-series:b-series gangliosides. AICD lowers GD3S at a transcriptional level (via binding the adaptor protein F365) [139]. It is worth noting that since Aβ is secreted, taken up by cells [144,145], and present in large concentrations in the cytosol of neurons [40,146,147], Aβ-GM3 binding could take place along the secretory pathway, in endosomes, by the cell lipid bilayer, as well as extracellularly [31]. In addition, while GM3 overall lowers Aβ production, one of its downstream a-series gangliosides, GM1, has been shown to have mixed effects by increasing Aβ generation and aggregation in vitro [148], while potentially being neuroprotective in vivo [149-151].
What conformation of Aβ matters
The investigation of Aβ aggregates has been greatly stymied by their lack of rigidity. The Aβ fibrils do not take on a single conformation, so there is not a single X-ray crystal structure that represents the only targetable conformation. For example, the Asp23-to-Asn mutant of Aβ (D23N-Aβ1–40), which is associated with EOFAD, can contain either parallel or antiparallel β-sheets [152,153]. Each Aβ peptide can form a β-sheet and two Aβ peptides can interact forming a β-sheet dimer, which has been termed a steric zipper. This dimer interaction can be classified structurally based on three variables: arrangement of the monomer β-strand internally (parallel or antiparallel), orientation of β-strands (same edge up for both sheets, or one up and one down), and orientation of the faces of the β-sheets (face-to-face [most common], or face-to-back). All eight of these combined possibilities have been observed experimentally with short peptides [154,155]. Initial investigations suggested the antiparallel fibrils are less stable and propagate less efficiently, but may nucleate more efficiently than parallel fibrils. Both antiparallel and parallel fibrils were found to be cytotoxic [156]. It should therefore not be surprising that several models for amyloid structure have been put forward [157].
For decades, the field had followed large Aβ and tau aggregates, which it continues to do. Indeed the aggregates are correlated with disease progress, with tau aggregates being especially positively correlated with worsening dementia. It has recently been shown that Aβ aggregates from two to 12 subunits may be the deleterious form [114]. In this paradigm, the large amyloids plaques seen postmortem historically could actually act as “sinks” to sequester the deleterious Aβ monomers. If the Aβ sequestering benefit is validated, it could lead to changes in therapeutic avenues. For example, breaking up amyloid plaques in an Alzheimer’s patient could be beneficial. This should only be the case if broken up into 13mers or larger since 3-12mers seem to form toxic pores. A single amyloid plaque 10,000 Aβ units long has at most two sequestering ends; if broken up into 100 Aβ units, it would have 100 times the sequestering power. In line with this concept, compounds have recently been discovered that increase Aβ aggregation, yet save cells, suggesting the Aβ plaques sequestered the more deleterious soluble Aβ multimer [18,19].
Aβ aggregation
Amyloid plaques created in the lab form multiple structures dependent on the conditions set. An interesting parallel can be made to the field of crystallography, where scientists search for just the right buffers to force normally soluble proteins to form crystal lattices. In contrast, the amyloid protein is so prone to oligomerization that discerning the meaningful in vivo structure from the multitude of in vitro structures has been difficult. A plethora of structural methods have been used to investigate the dynamic Aβ aggregation including solution [158] and solid state nuclear magnetic resonance (NMR) [159-161], electron paramagnetic resonance (EPR) [162], cryo-electron microscopy (cryo-EM) [163,164], atomic force microscopy (AFM) [165-167], X-ray [155,168,169] and computational molecular dynamics [170-174] with implicit and explicit water. Investigating these in vitro amyloid aggregates is helpful in creating therapeutics.
For example, Agopian et al. [175] formed two significant amyloid formations of Aβ40, creating an untwisted conformation if agitated or twisted if not agitated. Both NMR [176] and EPR [177] work have shown that Aβ fibrils form a parallel in-register network in which the same residues from each monomer pack against each other. It is worth noting that only a couple of years ago, Liu et al. described, for the first time, an out-of-register amyloid fibril in which the full complement of hydrogen bonds between monomers was not formed [178]. Atomic resolution of the deleterious Aβ oligomer hydrogen bonding network is vital for rational development of compounds, which will disrupt this interaction. Since backbone hydrogen bonding of β-sheets always favors planar structure [179], understanding packing forces that may cause twists in fibrils is important. Using EPR spectroscopy and 14 spin-labeled positions, it was found that agitated (untwisted) fibrils had stronger inter-strand interactions. The two hydrophobic regions stretching from residues 17-20 and 31-36 had the strongest interactions with the inter-strand distance estimated to be 0.2Å closer in the untwisted compared to the twisted fibril [175].
Aβ pore formation causing membrane Ca+2 conductance
One of the most exciting amyloid discoveries, now two decades old, may be Aβ’s ability to form pores in neurons. Using electrophysiological techniques, Arispe et al. showed amyloid aggregates form channels in planar lipid bilayers [180,181]. While it has clearly been shown that Aβ at millimolar concentrations can cause ion conduction across the cell membrane [182-186] with a conductivity of 100pS/pore [187], the exact structural mechanism is debated. Multiple studies have shown Aβ allows Ca+2 influx, which can be blocked by Zn+2 ions [188-190]. Disruption of calcium channels can cause multisystem disorders collectively, termed “calciumopathies.”
In the brain, Ca+2 acts as a second messenger to regulate membrane potential, excitability [191-193], synaptic plasticity, and memory encoding [194-197]. Calcium release from the ER, which has Ca+2 levels multiple orders of magnitude greater than the cytosol, is triggered by both ryanodine and inositol triphosphate (IP3) receptors, which are activated by cytosolic calcium and IP3, respectively [198-200]. Increased Ca+2 influx into the cytosol via an Aβ pore leads to increased ER calcium release, activating ER and mitochondrial stress responses, which leads to apoptosis [201-205].
Interestingly, Aβ oligomers administered intracellularly have also been shown to activate ER Ca+2 release by increasing IP3 even when in a Ca+2 free solution, hence not related to pore formation [198]. The mechanism of IP3 stimulation is still being investigated. Several mechanisms have been presented for this membrane Ca+2 permeability [100], including tension induced poration [206,207] and disassembly of the lipid bilayer by both oligomers [96,100], as well as the interaction of the larger fibrils with the membrane [208,209]. Aβ pores have been measured with various numbers of subunits (3-6 Aβ monomers) by AFM. Most of the pores (two-thirds) are tetramers or pentamers, with one-third being trimers or hexamers [210]. Prangkio et al. developed controlled aggregation conditions and used partial least squares (PLS) to show that oligomers in the 4-13 range promoted both pore formation and cytotoxicity, while monomers, dimers, and trimers were negatively correlated with both of these deleterious outcomes.
Models of six-stranded β-barrels formed by the last third of Aβ42 show hydrophobic residues and a glycine in the core, which is shielded from bulk water by the central and N-terminus regions, which can adopt both sheet and helix conformations (Figure 2A-Figure 2B) [211]. Shafrir et al. predicted how these antiparallel six-stranded β-barrels interact to form larger fibrils as well without seeing any large energetic penalties for the transition (animations of this transition were made available as well) [211]. While the Aβ β-barrel structure is most easily investigated in silico, due to its dynamic and membrane bound structure, the Aβ β-barrel has been investigated at the atomic level by X-ray [157] and NMR [212].
Figure 2A: Amyloid ß-barrel .

2A is a cross-section view of the membrane. A six-strand ß-barrel crystal structure (pdb entry=3SGR), shown as a membrane cross-section and rotated 90 degrees looking though the pore, made of three ß-sheets. Colored by strand, each strand has a tighter ß-sheet partner on one side (teal with cyan, yellow with light orange, red with dark orange) and a weaker ß-sheet partner on the other side (teal with cyan, yellow with light orange, red with dark orange).
Figure 2B: Amyloid ß-barrel.
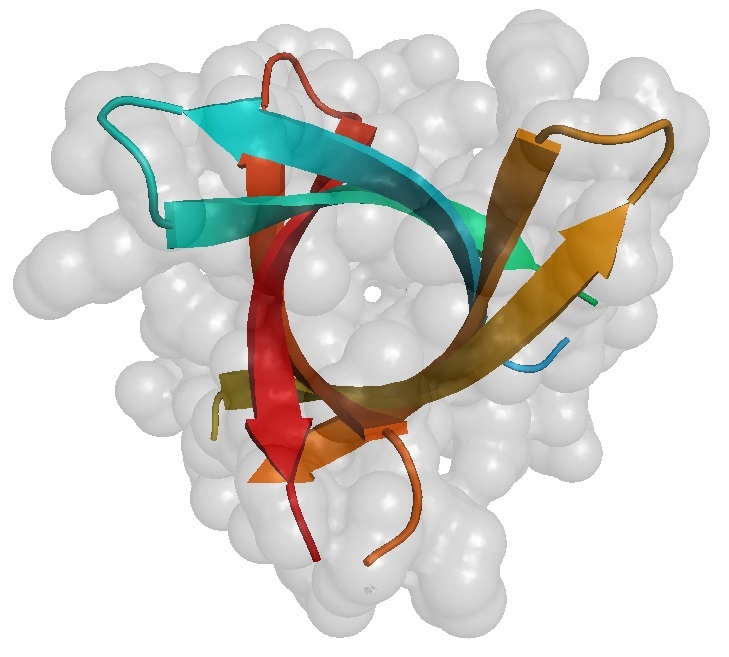
2B looks down the barrel of protein pore and through the membrane. A six-strand ß-barrel crystal structure (pdb entry=3SGR), shown as a membrane cross-section and rotated 90 degrees looking though the pore, made of three ß-sheets. Colored by strand, each strand has a tighter ß-sheet partner on one side (teal with cyan, yellow with light orange, red with dark orange) and a weaker ß-sheet partner on the other side (teal with cyan, yellow with light orange, red with dark orange).
In 2012, a β-barrel compatible with a sequence segment from Aβ protein was crystallized by Eisenberg et al. at 1.4Å resolution [157]. The structure could not be solved by molecular replacement with fiber-like structures and after bromine substitutions, the novel X-ray structure proved to be a six-stranded antiparallel β-barrel termed cylindrin. Each strand in cylidrin has a strong interface on one side made from 12 hydrogen bonds and a weak interface on the other side made by eight hydrogen bonds (Figure 2A-Figure 2B).
Computational simulations have been used to suggest how Aβ pore formation originates. Jang et al. used explicit solvent molecular dynamics simulations to model 12-, 16-, and 20-mer Aβ barrels and had results that were consistent with solids state NMR (ssNMR). For example, the 16mer barrel modeled had an outer diameter and pore diameter of ~7.2nm and ~1.6nm, respectively (Figure 2C-Figure 2D) [212]. It should be noted these measurements are similar to the 1-2nm pore size measured by ssNMR, which is physically wide enough for water to flow through. Unlike the cylindrin crystal structure, all β-sheet structures were parallel, meaning the pore had all β-loops on one side and all N- and C-terminal ends on the other side.
Figure 2C.

A 16 ß-strand ß-barrel, shown as a membrane cross-section and rotated 90 degrees looking though the pore, composed of eight Aß subunits, computational modeled by dynamics to match ssNMR data. The molecular surface is color coded by hydrophobicity (red = nonpolar, white = polar, green = cysteine). The images were made using pymol [231].
Figure 2B: Amyloid ß-barrel.
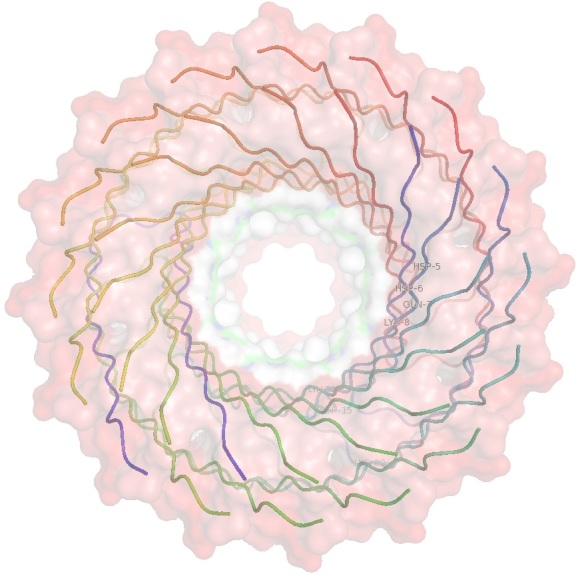
A 16 ß-strand ß-barrel, shown as a membrane cross-section and rotated 90 degrees looking though the pore, composed of eight Aß subunits, computational modeled by dynamics to match ssNMR data. The molecular surface is color coded by hydrophobicity (red = nonpolar, white = polar, green = cysteine). The images were made using pymol [231].
Two aspects to note are the pore size opening on each side and the channels hydrophobicity. The side of the pore with all β-turns has a smaller opening and a patch of residues (Glu14, Asp15, and Lys20) from each Aβ peptide, which forms a hydrophilic ring. On the opposite side of the pore, made of the N- and C-terminal ends, the residues Glu7 and Lys8 form a hydrophilic ring (shown as white in (Figure 2D). The peptide chain in these structures would be termed “coil” in a normal protein, but are in a pre-β-sheet layout and represent a possible early stage of pore formation (Figure 2C-Figure 2D) [212].
Regular antiparallel β-barrels come in two flavors as defined by their shear number (S) with a higher number indicating greater displacement between the edges of β-sheets, slope of strands in degrees (D) relative to the central axis of the barrel, and the number of hydrogen bonds (H) on each side of the β-strands, which may alternate. Normal β-barrels have either [S=12,D=56°,H=10,8] or [S=6,D=37°,H=10,10]. Cylindrin shares aspects from both of these groups with a topology [S=6,D=35°,H=12,8] [157]. While Aβ pores have been modeled with multiple sheets, and even with α-helical structures [113], the six-strand model X-ray model is useful for binding, folding, and modeling purposes.
Current therapeutic attempts, successes, and failures
Attempts to decrease Aβ levels have been attempted for over two decades, however even when effective, the side effects have proven too deleterious compared to little or no cognitive benefit. The first attempt at modifying Aβ levels was performed using immunization with a synthetic full-length peptide. Although this approach had shown robust effects in animal studies, and lowered total Aβ levels in the brains of human subjects, an accompanying T-lymphocyte mediated meningoencephalitis halted further development [213,214]. Current vaccination approaches have reduced T-lymphocyte activation by using an attenuated form of Aβ (Aβ-1-6) [215]. The development of many Aβ vaccines were terminated after phase II clinical trials because they showed low clinical efficacy. CAD106, which consists of multiple copies of Aβ1-6, has been shown to consistently increase Aβ-antibody titer, without inducing T-lymphocyte activation [216]. The safety profile of CAD106 suggests that it could be a potential therapeutic option for AD, but its clinical efficacy has yet to be determined.
The leading approach in developing agents that lower Aβ levels is passive immunity. Animal studies, in which systemic administration of anti-Aβ antibody was shown to cross the blood brain barrier and induce an immune response to Aβ were the first studies to show the effectiveness of passive immunity [217]. These initial studies lead to the development of an humanized anti-Aβ antibody, bapineuzumab [218]. Bapineuzumab binds to the soluble and fibrillar forms of Aβ and is well-tolerated at doses less than 2mg/Kg [219]. In phase III studies of mild to moderate dementia associated with AD, bapineuzumab was shown to decrease total brain Aβ load, compared with placebo control, using positron-emission tomography, but did not significantly improve cognitive or functional outcomes as measured by the Alzheimer’s Disease Assessment Scale and Disability Assessment for Dementia [220].
A number of other anti-Aβ antibodies have been developed that target different regions and forms of Aβ (Table 1) [221]. Unlike bapineuzumab, solanezumab is a monoclonal antibody that recognizes the soluble monomeric, but not the fibrillar, form of Aβ. By interacting with the soluble form of Aβ, solanezumab is thought to block the more neurotoxic form of Aβ [222]. In a multicenter phase III trial with 2,052 patients with mild to moderate AD, no improvement in cognition or functional ability was observed in subjects receiving 400mg of solanezumab every four weeks for 18 months when compared with the placebo group [223]. However, a subgroup analysis showed a reduction in decline of cognition in the group with mild AD [221]. Similarly, crenezumab, an anti-Aβ antibody that binds to both the soluble and fibrillar forms of Aβ, also seems to be efficacious, although not robust, in patients with mild AD, but not in those with moderate AD [224]. The clinical effectiveness of these agents in the early stages of AD, despite the reduction in Aβ levels, suggests that the effectiveness of disease-modifying agents may depend on when they are employed. An effective AD treatment will likely need to use strategies to identify early stages of the disease, potentially before symptoms of the disease begin to manifest, in addition to the disease-modifying agent.
Blocking the production of Aβ, by modifying the processing of APP, is another strategy for lowering Aβ levels. The target of this approach has been the enzyme β-secretase. Initially, most of the inhibitors were peptide-based, but ongoing work has focused on small molecule inhibitors [225]. Development of β-secretase inhibitors is in its early stage, and to date, only two drugs (MK-8931 and AZD3293) that inhibit β-secretase activity, and thus reduce the production of Aβ, have reached phase III clinical trials. MK-8931 and AZD3293 have been shown to be effective in lowering Aβ levels and are well tolerated [225,226]. The clinical efficacy of β-secretase inhibitors is still being determined, but like antibodies that lower Aβ, β-secretase inhibitors are likely to be more effective in the early stages of AD.
Conclusions and Outlook
The dynamic and numerous binding interactions of the Aβ peptide has slowed Alzheimer’s research for decades. While there is still not a single accepted causative, structurally characterized, and targetable Aβ protein structure, there are a number of leading candidates being pursued. The Aβ pore formed by six (or more) β-strands has been computationally predicted by multiple groups and fits experimental evidence, such as AFM images. Compounds that prevent the pore formation, followed by Ca+2 influx into the cytosol, followed by Ca+2 efflux from the ER into the cytosol, could halt neuronal death and associated cognitive decline. Likewise, new targets that disrupt lipid rafts, where Aβ localizes, could delay onset of disease (as has already been shown with increased ω-3 fatty acids [227-230]). Following the effect of new therapeutics on lipid raft formation and calcium flux should provide finer resolution of cellular progression during Alzheimer’s at the molecular level.
Unfortunately, despite all of this recent Aβ structural elucidation suggesting new avenues for targets, it needs to be noted that many historically failed therapeutics should have proven effective if the Aβ pore were the causative agent. The monoclonal antibodies to Aβ (e.g. solanezumab, bapineuzumab, and crenezumab) could have been detecting the wrong conformation of Aβ, namely monomers or fibrils. However, both β–secretase and γ–secretase drugs designed to lower the total amount of Aβ still seem like they should have proven effective. Developing compounds that can differentiate and detect the difference between multimers of Aβ will likely prove very difficult. Such compounds could inherently require large surface contacts to be able to differentiate. For example, if tetramers and octomers had the same repeating structure and only differed in their size, a compound to recognize the octomer as different would have to be at least a little bit longer than the tetramer to detect the higher multimer of Aβ.
The Alzheimer’s research field is therefore again left in a situation it has become all too familiar with: learning more about the disease without an overwhelming obvious best path forward.
Abbreviations
Aβ
Amyloid beta
AD
Alzheimer disease
ADAM10
A Disintegrin And Metalloproteinase domain-containing protein 16
AFM
Atomic Force Microscopy
AICD
Amyloid precursor protein IntraCellular Domain
Aph-1
Anterior pharynx-defective 1
APLP1 and 2
APP like protein 1 and 2, respectively
ApoE
Apolipoprotein E
APBA
Amyloid beta (A4) Precursor protein-Binding family A (also called MINTs or X11family)
APOER
apolipoprotein E receptor
APP
Amyloid Precusor Protein
BACE1
β-site APP cleaving enzyme 1 (also known as ß-secretase)
BBB
blood brain barrier
ChIP-seq
Chromatin immunoprecipitation sequencing
CNS
Central Nervous System
Cryo-EM
Cryo-Electron Microscopy
CSF
cerebrospinal fluid
EFA
essential fatty acids
EGCG
epigallocatechingallate
EOFAD
Early Onset Familial Alzheimer’s Disease
EPR
Electron paramagnetic resonance
FAD
Familial Alzheimer’s Disease
GD3
Disialoganglioside
GD3S
GD3-synthase
GM3
Monosialodihexosylganglioside
IP3
inositol triphosphate
JIP1
JNK-interacting protein
KLC
Kinesin Light Chain
KP1
Kunitz Protease Inhibitor
LRP1
Low density lipoprotein receptor-related protein 1
mAB
monoclonal antibodies
MID
Munc 18s-interacting
MINT
Munc 18-1 Interacting proteins (also called APBs or X11 family)
Nct
Nicastin
NFT
Neurofibrillary tangles
NMDA
N-methyl-D-aspartate receptor
NMR
Nuclear Magnetic Resonance
PDZ
Postsynaptic density-95/discs large/zona occludens-1 domain
PLS
partial least squares
Pen 2
Presenilin Enhancer 2
PSEN1 and PSEN2
Presenilin1 and 2, respectively
pS
picoSiemens
PTB
Phosphotyrosine binding domain
PUFA
polyunsaturated fatty acids
SRC
Sarcoma proto-oncogene
ssNMR
Solid state NMR
X11
a multidomain family of proteins containing one PTB and two PDZ domains (also called MINTs or APBs)
References
- Ittner LM, Götz J. Amyloid-β and tau--a toxic pas de deux in Alzheimer’s disease. Nat Rev Neurosci. 2011;12(2):65–72. doi: 10.1038/nrn2967. [DOI] [PubMed] [Google Scholar]
- Götz J, Lim YA, Ke YD, Eckert A, Ittner LM. Dissecting toxicity of tau and beta-amyloid. Neurodegener Dis. 2010;7(1-3):10–12. doi: 10.1159/000283475. [DOI] [PubMed] [Google Scholar]
- Blennow K, de Leon MJ, Zetterberg H. Alzheimer’s disease. Lancet. 2006;368(9533):387–403. doi: 10.1016/S0140-6736(06)69113-7. [DOI] [PubMed] [Google Scholar]
- Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH. et al. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987;325(6106):733–736. doi: 10.1038/325733a0. [DOI] [PubMed] [Google Scholar]
- Motter R, Vigo-Pelfrey C, Kholodenko D , Barbour R, Johnson-Wood K, Galasko D. et al. Reduction of beta-amyloid peptide42 in the cerebrospinal fluid of patients with Alzheimer’s disease. Ann Neurol. 1995;38(4):643–648. doi: 10.1002/ana.410380413. [DOI] [PubMed] [Google Scholar]
- Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140(6):918–934. doi: 10.1016/j.cell.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark RF, Hutton M, Fuldner M, Froelich S, Karran E, Talbot C. et al. The structure of the presenilin 1 (S182) gene and identification of six novel mutations in early onset AD families. Nat Genet. 1995;11(2):219–222. doi: 10.1038/ng1095-219. [DOI] [PubMed] [Google Scholar]
- Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M. et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature. 1995;375(6534):754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- Saura CA, Chen G, Malkani S, Choi S-Y, Takahashi RH, Zhang D. et al. Conditional inactivation of presenilin 1 prevents amyloid accumulation and temporarily rescues contextual and spatial working memory impairments in amyloid precursor protein transgenic mice. J Neurosci. 2005;25(29):6755–6764. doi: 10.1523/JNEUROSCI.1247-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saura CA, Choi S-Y, Beglopoulos V, Malkani S, Zhang D, Shankaranarayana Rao BS. et al. Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age-dependent neurodegeneration. Neuron. 2004;42(1):23–36. doi: 10.1016/s0896-6273(04)00182-5. [DOI] [PubMed] [Google Scholar]
- Adessi C, Frossard MJ, Boissard C, Fraga S, Bieler S, Ruckle T. et al. Pharmacological Profiles of Peptide Drug Candidates for the Treatment of Alzheimer’s Disease. J Biol Chem. 2003;278(16):13905–13911. doi: 10.1074/jbc.M211976200. [DOI] [PubMed] [Google Scholar]
- Lee DC, Rizer J, Hunt JB, Selenica ML, Gordon MN, Morgan D. Review: Experimental manipulations of microglia in mouse models of Alzheimer’s pathology: Activation reduces amyloid but hastens tau pathology. Neuropathol Appl Neurobiol. 2013;39(1):69–85. doi: 10.1111/nan.12002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YJ, Savtchenko R, Ostapchenko VG, Makarava N, Baskakov IV. Molecular Structure of Amyloid Fibrils Controls the Relationship between Fibrillar Size and Toxicity. PLoS ONE. 2011;6(5):e20244. doi: 10.1371/journal.pone.0020244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imbimbo BP, Panza F, Frisardi V, Solfrizzi V, D’Onofrio G, Logroscino G. et al. Therapeutic intervention for Alzheimer’s disease with γ-secretase inhibitors: still a viable option? Expert Opin Investig Drugs. 2011;20(3):325–341. doi: 10.1517/13543784.2011.550572. [DOI] [PubMed] [Google Scholar]
- Chakroborty S, Stutzmann GE. Calcium channelopathies and Alzheimer’s disease: insight into therapeutic success and failures. Eur J Pharmacol. 2014;739:83–95. doi: 10.1016/j.ejphar.2013.11.012. [DOI] [PubMed] [Google Scholar]
- Broadstock M, Ballard C, Corbett A. Latest treatment options for Alzheimer’s disease, Parkinson’s disease dementia and dementia with Lewy bodies. Expert Opin Pharmacother. 2014;15(13):1797–1810. doi: 10.1517/14656566.2014.936848. [DOI] [PubMed] [Google Scholar]
- Lanctôt PV, Rajaram RD, Herrmann N. Therapy for Alzheimer’s Disease: How Effective are Current Treatments? Ther Adv Neurol Disord. 2009;2(3):163–180. doi: 10.1177/1756285609102724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Armstrong AH, Koehler AN, Hecht MH. Small molecule microarrays enable the discovery of compounds that bind the Alzheimer’s Aβ peptide and reduce its cytotoxicity. J Am Chem Soc. 2010;132(47):17015–17022. doi: 10.1021/ja107552s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Liu Y, Gilthorpe J, van der Maarel JRC. MRP14 (S100A9) protein interacts with Alzheimer beta-amyloid peptide and induces its fibrillization. PLoS ONE. 2012;7(3):e32953. doi: 10.1371/journal.pone.0032953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I. et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 2008;14(8):837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvine GB, El-Agnaf OM, Shankar GM, Walsh DM. Protein aggregation in the brain: the molecular basis for Alzheimer’s and Parkinson’s diseases. Mol Med. 2008;14(7-8):451–464. doi: 10.2119/2007-00100.Irvine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Perry G, Moreira PI, Garrett MR, Liu Q, Zhu X. et al. Tau phosphorylation in Alzheimer’s disease: pathogen or protector? Trends Mol Med. 2005;11(4):164–169. doi: 10.1016/j.molmed.2005.02.008. [DOI] [PubMed] [Google Scholar]
- Chai X, Wu S, Murray TK, Kinley R, Cella CV, Sims H. et al. Passive immunization with anti-Tau antibodies in two transgenic models: reduction of Tau pathology and delay of disease progression. J Biol Chem. 2011;286(39):34457–34467. doi: 10.1074/jbc.M111.229633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapoport M, Dawson HN, Binder LI, Vitek MP, Ferreira A. Tau is essential to beta-amyloid-induced neurotoxicity. Proc Natl Acad Sci USA. 2002;99(9):6364–6369. doi: 10.1073/pnas.092136199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegmann S, Jung YJ, Chinnathambi S, Mandelkow E-M, Mandelkow E, Muller DJ. Human Tau isoforms assemble into ribbon-like fibrils that display polymorphic structure and stability. J Biol Chem. 2010;285(35):27302–27313. doi: 10.1074/jbc.M110.145318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn HJ, Zamolodchikov D, Cortes-Canteli M, Norris EH, Glickman JF, Strickland S. Alzheimer’s disease peptide beta-amyloid interacts with fibrinogen and induces its oligomerization. Proc Natl Acad Sci USA. 2010;107(50):21812–21817. doi: 10.1073/pnas.1010373107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neurodegeneration: Exploring Commonalities Across Diseases: Workshop Summary. National Academies Press [Internet] Available from:http://www.nap.edu/catalog.php?record_id=18341 . [PubMed]
- Vossel KA, Zhang K, Brodbeck J, Daub AC, Sharma P, Finkbeiner S. et al. Tau Reduction Prevents Aβ-Induced Defects in Axonal Transport. Science. 2010;330(6001):198. doi: 10.1126/science.1194653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor T, Sadleir KR, Maus E, Velliquette RA, Zhao J, Cole SL. et al. Phosphorylation of the translation initiation factor eIF2alpha increases BACE1 levels and promotes amyloidogenesis. Neuron. 2008;60(6):988–1009. doi: 10.1016/j.neuron.2008.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J. et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell. 2010;142(3):387–397. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Thompson R, Zhang H, Xu H. APP processing in Alzheimer’s disease. Mol Brain. 2011;4(3) doi: 10.1186/1756-6606-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohan de Silva HA, Jen A, Wickenden C, Jen LS, Wilkinson SL, Patel AJ. Cell-specific expression of beta-amyloid precursor protein isoform mRNAs and proteins in neurons and astrocytes. Brain Res Mol Brain Res. 1997;47(1-2):147–156. doi: 10.1016/s0169-328x(97)00045-4. [DOI] [PubMed] [Google Scholar]
- Kang J, Müller-Hill B. Differential splicing of Alzheimer’s disease amyloid A4 precursor RNA in rat tissues: PreA4(695) mRNA is predominantly produced in rat and human brain. Biochem Biophys Res Commun. 1990;166(3):1192–1200. doi: 10.1016/0006-291x(90)90992-v. [DOI] [PubMed] [Google Scholar]
- Menéndez-González M, Pérez-Pinera P, Martínez-Rivera M, Calatayud MT, Blázquez Menes B. APP processing and the APP-KPI domain involvement in the amyloid cascade. Neurodegener Dis. 2005;2(6):277–283. doi: 10.1159/000092315. [DOI] [PubMed] [Google Scholar]
- Bordji K, Becerril-Ortega J, Nicole O, Buisson A. Activation of extrasynaptic, but not synaptic, NMDA receptors modifies amyloid precursor protein expression pattern and increases amyloid-ß production. J Neurosci. 2010;30(47):15927–15942. doi: 10.1523/JNEUROSCI.3021-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Koch CS, Zheng H, Chen H, Trumbauer M, Thinakaran G, van der Ploeg LH. et al. Generation of APLP2 KO mice and early postnatal lethality in APLP2/APP double KO mice. Neurobiol Aging. 1997;18(6):661–669. doi: 10.1016/s0197-4580(97)00151-6. [DOI] [PubMed] [Google Scholar]
- Heber S, Herms J, Gajic V, Hainfellner J, Aguzzi A, Rülicke T. et al. Mice with combined gene knock-outs reveal essential and partially redundant functions of amyloid precursor protein family members. J Neurosci. 2000;20(21):7951–7963. doi: 10.1523/JNEUROSCI.20-21-07951.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herms J, Anliker J, Heber S, Ring S, Fuhrmann M, Kretzschmar H. et al. Cortical dysplasia resembling human type 2 lissencephaly in mice lacking all three APP family members. EMBO J. 2004;23(20):4106–4115. doi: 10.1038/sj.emboj.7600390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulin F, Léveillé F, Ortega JB, Mornon J-P, Buisson A, Callebaut I. et al. p3 peptide, a truncated form of Aβ devoid of synaptotoxic effect, does not assemble into soluble oligomers. FEBS Letters. 2008;582(13):1865–1870. doi: 10.1016/j.febslet.2008.05.002. [DOI] [PubMed] [Google Scholar]
- Cook DG, Forman MS, Sung JC, Leight S, Kolson DL, Iwatsubo T. et al. Alzheimer’s A beta(1-42) is generated in the endoplasmic reticulum/intermediate compartment of NT2N cells. Nat Med. 1997;3(9):1021–1023. doi: 10.1038/nm0997-1021. [DOI] [PubMed] [Google Scholar]
- Jankowsky JL, Fadale DJ, Anderson J, Xu GM, Gonzales V, Jenkins NA. et al. Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum Mol Genet. 2004;13(2):159–170. doi: 10.1093/hmg/ddh019. [DOI] [PubMed] [Google Scholar]
- Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A. et al. Long-term effects of Aβ42 immunisation in Alzheimer’s disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet. 2008;372(9634):216–223. doi: 10.1016/S0140-6736(08)61075-2. [DOI] [PubMed] [Google Scholar]
- Saito T, Suemoto T, Brouwers N, Sleegers K, Funamoto S, Mihira N. et al. Potent amyloidogenicity and pathogenicity of Aβ43. Nat Neurosci. 2011;14(8):1023–1032. doi: 10.1038/nn.2858. [DOI] [PubMed] [Google Scholar]
- Lorenzo A, Yuan M, Zhang Z, Paganetti PA, Sturchler-Pierrat C, Staufenbiel M. et al. Amyloid beta interacts with the amyloid precursor protein: a potential toxic mechanism in Alzheimer’s disease. Nat Neurosci. 2000;3(5):460–464. doi: 10.1038/74833. [DOI] [PubMed] [Google Scholar]
- Ho A, Südhof TC. Binding of F-spondin to amyloid-β precursor protein: A candidate amyloid-β precursor protein ligand that modulates amyloid-β precursor protein cleavage. Proc Natl Acad Sci U S A. 2004;101(8):2548–2553. doi: 10.1073/pnas.0308655100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lourenço F, Galvan V, Fombonne J, Corset V, Llambi F, Müller U. et al. Netrin-1 interacts with amyloid precursor protein and regulates amyloid-β production. Cell Death Differ. 2009;16(5):655–663. doi: 10.1038/cdd.2008.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki T, Koo EH, Selkoe DJ. Cell surface amyloid beta-protein precursor colocalizes with beta 1 integrins at substrate contact sites in neural cells. J Neurosci. 1997;17(3):1004–1010. doi: 10.1523/JNEUROSCI.17-03-01004.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Ha Y. The X-ray structure of an antiparallel dimer of the human amyloid precursor protein E2 domain. Mol Cell. 2004;15(3):343–353. doi: 10.1016/j.molcel.2004.06.037. [DOI] [PubMed] [Google Scholar]
- Soba P, Eggert S, Wagner K, Zentgraf H, Siehl K, Kreger S. et al. Homo- and heterodimerization of APP family members promotes intercellular adhesion. EMBO J. 2005;24(20):3624–3634. doi: 10.1038/sj.emboj.7600824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levites Y, Amit T, Mandel S, Youdim MBH. Neuroprotection and neurorescue against Aβtoxicity and PKC-dependent release of non-amyloidogenic soluble precursor protein by green tea polyphenol (-)- epigallocatechin-3-gallate. FASEB J [Internet] Available from:http://www.fasebj.org/content/early/2003/05/02/fj.02-0881fje . [DOI] [PubMed]
- Fernandez JW, Rezai-Zadeh K, Obregon D, Tan J. EGCG functions through estrogen receptor-mediated activation of ADAM10 in the promotion of non-amyloidogenic processing of APP. FEBS Lett. 2010;584(19):4259–4267. doi: 10.1016/j.febslet.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassar R. The beta-secretase, BACE: a prime drug target for Alzheimer’s disease. J Mol Neurosci. 2001;17(2):157–170. doi: 10.1385/JMN:17:2:157. [DOI] [PubMed] [Google Scholar]
- Laird FM, Cai H, Savonenko AV, Farah MH, He K, Melnikova T. et al. BACE1, a major determinant of selective vulnerability of the brain to amyloid-beta amyloidogenesis, is essential for cognitive, emotional, and synaptic functions. J Neurosci. 2005;25(50):11693–11709. doi: 10.1523/JNEUROSCI.2766-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetterberg H, Andreasson U, Hansson O. et al. Elevated cerebrospinal fluid bace1 activity in incipient alzheimer disease. Arch Neurol. 2008;65(8):1102–1107. doi: 10.1001/archneur.65.8.1102. [DOI] [PubMed] [Google Scholar]
- Guglielmotto M, Aragno M, Autelli R, Giliberto L, Novo E, Colombatto S. et al. The up-regulation of BACE1 mediated by hypoxia and ischemic injury: role of oxidative stress and HIF1alpha. J Neurochem. 2009;108(4):1045–1056. doi: 10.1111/j.1471-4159.2008.05858.x. [DOI] [PubMed] [Google Scholar]
- Omar A, Jovanovic K, Da Costa Dias B, Gonsalves D, Moodley K, Caveney R. et al. Patented biological approaches for the therapeutic modulation of the 37 kDa/67 kDa laminin receptor. Expert Opin Ther Pat. 2011;21(1):35–53. doi: 10.1517/13543776.2011.539203. [DOI] [PubMed] [Google Scholar]
- Mbazima V, Da Costa Dias B, Omar A, Jovanovic K, Weiss SFT. Interactions between PrP(c) and other ligands with the 37-kDa/67-kDa laminin receptor. Front Biosci. 2010;15:1150–1163. doi: 10.2741/3667. [DOI] [PubMed] [Google Scholar]
- Wolfe MS. Inhibition and modulation of gamma-secretase for Alzheimer’s disease. Neurotherapeutics. 2008;5(3):391–398. doi: 10.1016/j.nurt.2008.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Wolfe MS, Selkoe DJ. Toward structural elucidation of the gamma-secretase complex. Structure. 2009;17(3):326–334. doi: 10.1016/j.str.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow VW, Mattson MP, Wong PC, Gleichmann M. An overview of APP processing enzymes and products. Neuromolecular Med. 2010;12(1):1–12. doi: 10.1007/s12017-009-8104-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beel AJ, Sanders CR. Substrate specificity of gamma-secretase and other intramembrane proteases. Cell Mol Life Sci. 2008;65(9):1311–1334. doi: 10.1007/s00018-008-7462-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi T, De Strooper B. Presenilins: members of the gamma-secretase quartets, but part-time soloists too. Physiology (Bethesda) 2008;23:194–204. doi: 10.1152/physiol.00009.2008. [DOI] [PubMed] [Google Scholar]
- Zhong W, Jiang MM, Weinmaster G, Jan LY, Jan YN. Differential expression of mammalian Numb, Numblike and Notch1 suggests distinct roles during mouse cortical neurogenesis. Development. 1997;124(10):1887–1897. doi: 10.1242/dev.124.10.1887. [DOI] [PubMed] [Google Scholar]
- Marlow L, Canet RM, Haugabook SJ, Hardy JA, Lahiri DK, Sambamurti K. APH1, PEN2, and Nicastrin increase Abeta levels and gamma-secretase activity. Biochem Biophys Res Commun. 2003;305(3):502–509. doi: 10.1016/s0006-291x(03)00797-6. [DOI] [PubMed] [Google Scholar]
- Li R, Lindholm K, Yang L-B, Yue X, Citron M, Yan R. et al. Amyloid β peptide load is correlated with increased β-secretase activity in sporadic Alzheimer’s disease patients. Proc Natl Acad Sci U S A. 2004;101(10):3632–3637. doi: 10.1073/pnas.0205689101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holsinger RMD, McLean CA, Collins SJ, Masters CL, Evin G. Increased beta-Secretase activity in cerebrospinal fluid of Alzheimer’s disease subjects. Ann Neurol. 2004;55(6):898–899. doi: 10.1002/ana.20144. [DOI] [PubMed] [Google Scholar]
- Fukumoto H, Rosene DL, Moss MB, Raju S, Hyman BT, Irizarry MC. β-Secretase Activity Increases with Aging in Human, Monkey, and Mouse Brain. Am J Pathol. 2004;164(2):719–725. doi: 10.1016/s0002-9440(10)63159-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devi L, Ohno M. A combination Alzheimer’s therapy targeting BACE1 and neprilysin in 5XFAD transgenic mice. Molecular Brain. 2015;8(1):19. doi: 10.1186/s13041-015-0110-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley JC, Hudson M, Kanning KC, Schecterson LC, Bothwell M. Familial Alzheimer’s disease mutations inhibit gamma-secretase-mediated liberation of beta-amyloid precursor protein carboxy-terminal fragment. J Neurochem. 2005;94(5):1189–1201. doi: 10.1111/j.1471-4159.2005.03266.x. [DOI] [PubMed] [Google Scholar]
- Palmer AM. Neuroprotective therapeutics for Alzheimer’s disease: progress and prospects. Trends Pharmacol Sci. 2011;32(3):141–147. doi: 10.1016/j.tips.2010.12.007. [DOI] [PubMed] [Google Scholar]
- Bettens K, Sleegers K, Van Broeckhoven C. Current status on Alzheimer disease molecular genetics: from past, to present, to future. Hum Mol Genet. 2010;19(R1):R4–R11. doi: 10.1093/hmg/ddq142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy-Lahad E, Wijsman EM, Nemens E, Anderson L, Goddard KA, Weber JL. et al. A familial Alzheimer’s disease locus on chromosome 1. Science. 1995;269(5226):970–973. doi: 10.1126/science.7638621. [DOI] [PubMed] [Google Scholar]
- Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH. et al. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science. 1995;269(5226):973–977. doi: 10.1126/science.7638622. [DOI] [PubMed] [Google Scholar]
- Hardy J. The Alzheimer family of diseases: many etiologies, one pathogenesis? Proc Natl Acad Sci U S A. 1997;94(6):2095–2097. doi: 10.1073/pnas.94.6.2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J. Amyloid, the presenilins and Alzheimer’s disease. Trends Neurosci. 1997;20(4):154–159. doi: 10.1016/s0166-2236(96)01030-2. [DOI] [PubMed] [Google Scholar]
- Bettens K, Brouwers N, Van Miegroet H, Gil A, Engelborghs S, De Deyn PP. et al. Follow-up study of susceptibility loci for Alzheimer’s disease and onset age identified by genome-wide association. J Alzheimers Dis. 2010;19(4):1169–1175. doi: 10.3233/JAD-2010-1310. [DOI] [PubMed] [Google Scholar]
- Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC. et al. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science. 2010;330(6012):1774. doi: 10.1126/science.1197623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motter R, Vigo-Pelfrey C, Kholodenko D, Barbour R, Johnson-Wood K, Galasko D. et al. Reduction of beta-amyloid peptide42 in the cerebrospinal fluid of patients with Alzheimer’s disease. Ann Neurol. 1995;38(4):643–648. doi: 10.1002/ana.410380413. [DOI] [PubMed] [Google Scholar]
- Dias BDC, Jovanovic K, Gonsalves D, Weiss SFT. Structural and mechanistic commonalities of amyloid-β and the prion protein. Prion. 2011;5(3):126–137. doi: 10.4161/pri.5.3.17025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn HJ, Zamolodchikov D, Cortes-Canteli M, Norris EH, Glickman JF, Strickland S. Alzheimer’s disease peptide beta-amyloid interacts with fibrinogen and induces its oligomerization. Proc Natl Acad Sci U S A. 2010;107(50):21812–21817. doi: 10.1073/pnas.1010373107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svedružić ZM, Popović K, Smoljan I, Sendula-Jengić V. Modulation of γ-secretase activity by multiple enzyme-substrate interactions: implications in pathogenesis of Alzheimer’s disease. PLoS ONE. 2012;7(3):e32293. doi: 10.1371/journal.pone.0032293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001;81(2):741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- Rowe CC, Ellis KA, Rimajova M, Bourgeat P , Pike KE, Jones G. et al. Amyloid imaging results from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging. Neurobiol Aging. 2010;31(8):1275–1283. doi: 10.1016/j.neurobiolaging.2010.04.007. [DOI] [PubMed] [Google Scholar]
- Jack CR, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW. et al. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010;9(1):119. doi: 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Lowe VJ, Weigand SD, Wiste HJ, Senjem ML, Knopman DS. et al. Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer’s disease: implications for sequence of pathological events in Alzheimer’s disease. Brain. 2009;132(5):1355–1365. doi: 10.1093/brain/awp062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominantly Inherited Alzheimer Network Trial: An Opportunity to Prevent Dementia. A Study of Potential Disease Modifying Treatments in Individuals at Risk for or With a Type of Early Onset Alzheimer’s Disease Caused by a Genetic Mutation (Full Text View) ClinicalTrials.gov [Internet] Available from:https://clinicaltrials.gov/ct2/show/NCT01760005?term=solanezumab&&rank=4 .
- Cummings BJ, Pike CJ, Shankle R, Cotman CW. Beta-amyloid deposition and other measures of neuropathology predict cognitive status in Alzheimer’s disease. Neurobiol Aging. 1996;17(6):921–933. doi: 10.1016/s0197-4580(96)00170-4. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Cell biology of the amyloid beta-protein precursor and the mechanism of Alzheimer’s disease. Annu Rev Cell Biol. 1994;10:373–403. doi: 10.1146/annurev.cb.10.110194.002105. [DOI] [PubMed] [Google Scholar]
- Wang J, Dickson DW, Trojanowski JQ, Lee VM. The levels of soluble versus insoluble brain Abeta distinguish Alzheimer’s disease from normal and pathologic aging. Exp Neurol. 1999;158(2):328–337. doi: 10.1006/exnr.1999.7085. [DOI] [PubMed] [Google Scholar]
- McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K. et al. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann Neurol. 1999;46(6):860–866. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Wall JS, Kennel SJ, Williams A, Richey T, Stuckey A, Huang Y. et al. AL Amyloid Imaging and Therapy with a Monoclonal Antibody to a Cryptic Epitope on Amyloid Fibrils. PLoS ONE. 2012;7(12):e52686. doi: 10.1371/journal.pone.0052686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberghe R, Adamczuk K, Dupont P, Laere KV, Chételat G. Amyloid PET in clinical practice: Its place in the multidimensional space of Alzheimer’s disease. Neuroimage Clin. 2013;2:497–511. doi: 10.1016/j.nicl.2013.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberghe R, Adamczuk K, Van Laere K. The interest of amyloid PET imaging in the diagnosis of Alzheimer’s disease. [Miscellaneous Article] Curr Opin Neurol. 2013;26(6):646–655. doi: 10.1097/WCO.0000000000000036. [DOI] [PubMed] [Google Scholar]
- Leopoldo M, Contino M, Berardi F, Perrone R, Colabufo NA. PET Radiotracers for Imaging P-glycoprotein: The Challenge for Early Diagnosis in AD. ChemMedChem. 2014;9(1):38–42. doi: 10.1002/cmdc.201300362. [DOI] [PubMed] [Google Scholar]
- Sepúlveda FJ, Fierro H, Fernandez E, Castillo C, Peoples RW, Opazo C. et al. Nature of the neurotoxic membrane actions of amyloid-β on hippocampal neurons in Alzheimer’s disease. Neurobiol Aging. 2014;35(3):472–481. doi: 10.1016/j.neurobiolaging.2013.08.035. [DOI] [PubMed] [Google Scholar]
- Relini A, Marano N, Gliozzi A. Probing the interplay between amyloidogenic proteins and membranes using lipid monolayers and bilayers. Adv Colloid Interface Sci. 2014;207:81–92. doi: 10.1016/j.cis.2013.10.015. [DOI] [PubMed] [Google Scholar]
- Qiang W, Akinlolu RD, Nam M, Shu N. Structural Evolution and Membrane Interaction of the 40-Residue β Amyloid Peptides: Differences in the Initial Proximity between Peptides and the Membrane Bilayer Studied by Solid-State Nuclear Magnetic Resonance Spectroscopy. Biochem. 2014;53(48):7503–7514. doi: 10.1021/bi501003n. [DOI] [PubMed] [Google Scholar]
- Head BP, Patel HH, Insel PA. Interaction of membrane/lipid rafts with the cytoskeleton: impact on signaling and function. Biochim Biophys Acta. 2014;1838(2):532–545. doi: 10.1016/j.bbamem.2013.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dies H, Toppozini L, Rheinstädter MC. The Interaction between Amyloid-β Peptides and Anionic Lipid Membranes Containing Cholesterol and Melatonin. PLoS One. 2014;9(6):e99124. doi: 10.1371/journal.pone.0099124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Relini A, Marano N, Gliozzi A. Misfolding of Amyloidogenic Proteins and Their Interactions with Membranes. Biomolecules. 2013;4(1):20–55. doi: 10.3390/biom4010020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Ye S, Wei F, Ma S, Luo Y. In Situ Molecular-Level Insights into the Interfacial Structure Changes of Membrane-Associated Prion Protein Fragment [118-135] Investigated by Sum Frequency Generation Vibrational Spectroscopy. LANGMUIR. 2012;28(49):16979–16988. doi: 10.1021/la302655p. [DOI] [PubMed] [Google Scholar]
- Barrett PJ, Song Y, Van Horn WD, Hustedt EJ, Schafer JM, Hadziselimovic A. et al. The amyloid precursor protein has a flexible transmembrane domain and binds cholesterol. Science. 2012;336(6085):1168–1171. doi: 10.1126/science.1219988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa H, Peddi D, Osborne JM, Wilson BM, Pesaru RR, Kurva B. et al. Modeling the interface between islet amyloid polypeptide and insulin-based aggregation inhibitors: correlation to aggregation kinetics and membrane damage. J Chem Inf Model. 2012;52(5):1298–1307. doi: 10.1021/ci300119c. [DOI] [PubMed] [Google Scholar]
- Björkhem I, Meaney S. Brain Cholesterol: Long Secret Life Behind a Barrier. Arterioscler Thromb Vasc Biol. 2004;24(5):806–815. doi: 10.1161/01.ATV.0000120374.59826.1b. [DOI] [PubMed] [Google Scholar]
- Orth M, Bellosta S. Cholesterol: Its Regulation and Role in Central Nervous System Disorders. Cholesterol. 2012;2012:292598. doi: 10.1155/2012/292598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietschy JM, Turley SD. Cholesterol metabolism in the brain. Curr Opin Lipidol. 2001;12(2):105–112. doi: 10.1097/00041433-200104000-00003. [DOI] [PubMed] [Google Scholar]
- Chen X, Hui L, Soliman ML, Geiger JD. Altered Cholesterol Intracellular Trafficking and the Development of Pathological Hallmarks of Sporadic AD. J Parkinsons Dis Alzheimers Dis. 2014;1(1):8. doi: 10.13188/2376-922x.1000002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JK, Honea RA, Vidoni ED, Swerdlow RH, Burns JM. Is Alzheimer’s Disease a Systemic Disease? Biochim Biophys Acta. 2014;1842(9):1340–1349. doi: 10.1016/j.bbadis.2014.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daneschvar HL, Aronson MD, Smetana GW. Do statins prevent Alzheimer’s disease? A narrative review. Eur J Intern Med. 2015;26(9):666–669. doi: 10.1016/j.ejim.2015.08.012. [DOI] [PubMed] [Google Scholar]
- Shobab LA, Hsiung G-YR, Feldman HH. Cholesterol in Alzheimer’s disease. Lancet Neurol. 2005;4(12):841–852. doi: 10.1016/S1474-4422(05)70248-9. [DOI] [PubMed] [Google Scholar]
- Liu Q, Zerbinatti CV, Zhang J, Hoe H-S, Wang B, Cole SL. et al. Amyloid Precursor Protein Regulates Brain Apolipoprotein E and Cholesterol Metabolism through Lipoprotein Receptor LRP1. Neuron. 2007;56(1):66–78. doi: 10.1016/j.neuron.2007.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Liu Q. Cholesterol metabolism and homeostasis in the brain. Protein Cell. 2015;6(4):254–264. doi: 10.1007/s13238-014-0131-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Scala C, Troadec J-D, Lelièvre C, Garmy N, Fantini J, Chahinian H. Mechanism of cholesterol-assisted oligomeric channel formation by a short Alzheimer β-amyloid peptide. J Neurochem. 2014;128(1):186–195. doi: 10.1111/jnc.12390. [DOI] [PubMed] [Google Scholar]
- Di Scala C, Chahinian H, Yahi N, Garmy N, Fantini J. Interaction of Alzheimer’s β-Amyloid Peptides with Cholesterol: Mechanistic Insights into Amyloid Pore Formation. Biochem. 2014;53(28):4489–4502. doi: 10.1021/bi500373k. [DOI] [PubMed] [Google Scholar]
- Fantini J, Di Scala C, Yahi N, Troadec J-D, Sadelli K, Chahinian H. et al. Bexarotene Blocks Calcium-Permeable Ion Channels Formed by Neurotoxic Alzheimer’s β-Amyloid Peptides. ACS Chem Neurosci. 2014;5(3):216–224. doi: 10.1021/cn400183w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmechel DE, Saunders AM, Strittmatter WJ, Crain BJ, Hulette CM, Joo SH. et al. Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90(20):9649–9653. doi: 10.1073/pnas.90.20.9649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cedazo-Mínguez A. Apolipoprotein E and Alzheimer’s disease: molecular mechanisms and therapeutic opportunities. J Cell Mol Med. 2007;11(6):1227–1238. doi: 10.1111/j.1582-4934.2007.00130.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barberger-Gateau P, Samieri C, Féart C, Plourde M. Dietary omega 3 polyunsaturated fatty acids and Alzheimer’s disease: interaction with apolipoprotein E genotype. Curr Alzheimer Res. 2011;8(5):479–491. doi: 10.2174/156720511796391926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strittmatter WJ, Weisgraber KH, Huang DY, Dong LM, Salvesen GS, Pericak-Vance M. et al. Binding of human apolipoprotein E to synthetic amyloid beta peptide: isoform-specific effects and implications for late-onset Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90(17):8098–8102. doi: 10.1073/pnas.90.17.8098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS. et al. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90(5):1977–1981. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cras P, Kawai M, Lowery D, Gonzalez-DeWhitt P, Greenberg B, Perry G. Senile plaque neurites in Alzheimer disease accumulate amyloid precursor protein. Proc Natl Acad Sci U S A. 1991;88(17):7552–7556. doi: 10.1073/pnas.88.17.7552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane RM, Farlow MR. Lipid homeostasis and apolipoprotein E in the development and progression of Alzheimer’s disease. J Lipid Res. 2005;46(5):949–968. doi: 10.1194/jlr.M400486-JLR200. [DOI] [PubMed] [Google Scholar]
- Vega G, Weiner MF, Lipton AM. et al. Reduction in levels of 24s-hydroxycholesterol by statin treatment in patients with alzheimer disease. Arch Neurol. 2003;60(4):510–515. doi: 10.1001/archneur.60.4.510. [DOI] [PubMed] [Google Scholar]
- Lütjohann D, Papassotiropoulos A, Björkhem I, Locatelli S, Bagli M, Oehring RD. et al. Plasma 24S-hydroxycholesterol (cerebrosterol) is increased in Alzheimer and vascular demented patients. J Lipid Res. 2000;41(2):195–198. [PubMed] [Google Scholar]
- Theendakara V, Peters-Libeu CA, Spilman P, Poksay KS, Bredesen DE, Rao RV. Direct Transcriptional Effects of Apolipoprotein E. J Neurosci. 2016;36(3):685–700. doi: 10.1523/JNEUROSCI.3562-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho A, Liu X, Südhof TC. Deletion of Mint proteins decreases amyloid production in transgenic mouse models of Alzheimer’s disease. J Neurosci. 2008;28(53):14392–14400. doi: 10.1523/JNEUROSCI.2481-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan SE, Dillon GM, Sullivan JM, Ho A. Mint Proteins Are Required for Synaptic Activity-dependent Amyloid Precursor Protein (APP) Trafficking and Amyloid β Generation. J Biol Chem. 2014;289(22):15374–15383. doi: 10.1074/jbc.M113.541003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang S, Li Y, Zhang X, Bu G, Xu H, Zhang Y. Trafficking regulation of proteins in Alzheimer’s disease. Mol Neurodegener. 2014;9(1):6. doi: 10.1186/1750-1326-9-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saluja I, Paulson H, Gupta A, Turner RS. X11α haploinsufficiency enhances Aβ amyloid deposition in Alzheimer’s disease transgenic mice. Neurobiol Dis. 2009;36(1):162–168. doi: 10.1016/j.nbd.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borg J-P, Yang Y, Taddéo-Borg MD, Margolis B, Turner RS. The X11α Protein Slows Cellular Amyloid Precursor Protein Processing and Reduces Aβ40 and Aβ42 Secretion. J Biol Chem. 1998;273(24):14761–14766. doi: 10.1074/jbc.273.24.14761. [DOI] [PubMed] [Google Scholar]
- Sastre M, Turner RS, Levy E. X11 Interaction with β-Amyloid Precursor Protein Modulates Its Cellular Stabilization and Reduces Amyloid β-Protein Secretion. J Biol Chem. 1998;273(35):22351–22357. doi: 10.1074/jbc.273.35.22351. [DOI] [PubMed] [Google Scholar]
- Chaufty J, Sullivan SE, Ho A. Intracellular APP Sorting and Aβ Secretion are Regulated by Src-mediated Phosphorylation of Mint2. J Neurosci. 2012;32(28):9613–9625. doi: 10.1523/JNEUROSCI.0602-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butz S, Okamoto M, Sädhof TC. A Tripartite Protein Complex with the Potential to Couple Synaptic Vesicle Exocytosis to Cell Adhesion in Brain. Cell. 1998;94(6):773–782. doi: 10.1016/s0092-8674(00)81736-5. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Lee C-H, Mandiyan V, Borg J-P, Margolis B, Schlessinger J. et al. Sequence-specific recognition of the internalization motif of the Alzheimer’s amyloid precursor protein by the X11 PTB domain. EMBO J. 1997;16(20):6141–6150. doi: 10.1093/emboj/16.20.6141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau KF, McLoughlin DM, Standen C, Miller CC. X11 alpha and x11 beta interact with presenilin-1 via their PDZ domains. Mol Cell Neurosci. 2000;16(5):557–565. doi: 10.1006/mcne.2000.0898. [DOI] [PubMed] [Google Scholar]
- Ho CS, Marinescu V, Steinhilb ML, Gaut JR, Turner RS, Stuenkel EL. Synergistic effects of Munc18a and X11 proteins on amyloid precursor protein metabolism. J Biol Chem. 2002;277(30):27021–27028. doi: 10.1074/jbc.M201823200. [DOI] [PubMed] [Google Scholar]
- Wang X, Huang T, Bu G, Xu H. Dysregulation of protein trafficking in neurodegeneration. Mol Neurodegener. 2014;9:31. doi: 10.1186/1750-1326-9-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mocchetti I. Exogenous gangliosides, neuronal plasticity and repair, and the neurotrophins. Cell Mol Life Sci. 2005;62(19-20):2283–2294. doi: 10.1007/s00018-005-5188-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm MOW, Zinser EG, Grösgen S, Hundsdörfer B, Rothhaar TL, Burg VA. et al. Amyloid precursor protein (APP) mediated regulation of ganglioside homeostasis linking Alzheimer’s disease pathology with ganglioside metabolism. PLoS One. 2012;7(3):e34095. doi: 10.1371/journal.pone.0034095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kracun I, Rosner H, Drnovsek V, Heffer-Lauc M, Cosović C, Lauc G. Human brain gangliosides in development, aging and disease. Int J Dev Biol. 1991;35(3):289–295. [PubMed] [Google Scholar]
- Molander-Melin M, Blennow K, Bogdanovic N, Dellheden B, Månsson J-E, Fredman P. Structural membrane alterations in Alzheimer brains found to be associated with regional disease development; increased density of gangliosides GM1 and GM2 and loss of cholesterol in detergent-resistant membrane domains. J Neurochem. 2005;92(1):171–182. doi: 10.1111/j.1471-4159.2004.02849.x. [DOI] [PubMed] [Google Scholar]
- Barrier L, Ingrand S, Damjanac M, Rioux Bilan A, Hugon J, Page G. Genotype-related changes of ganglioside composition in brain regions of transgenic mouse models of Alzheimer’s disease. Neurobiol Aging. 2007;28(12):1863–1872. doi: 10.1016/j.neurobiolaging.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Lahiri S, Futerman AH. The metabolism and function of sphingolipids and glycosphingolipids. Cell Mol Life Sci.: CMLS. 2007;64(17):2270–2284. doi: 10.1007/s00018-007-7076-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C, Koo EH, Teplow DB, Selkoe DJ. Polarized secretion of beta-amyloid precursor protein and amyloid beta-peptide in MDCK cells. Proc Natl Acad Sci U S A. 1994;91(4):1564–1568. doi: 10.1073/pnas.91.4.1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Crick SL, Bu G, Frieden C, Pappu RV, Lee J-M. Amyloid seeds formed by cellular uptake, concentration, and aggregation of the amyloid-beta peptide. Proc Natl Acad Sci U S A. 2009;106(48):20324–20329. doi: 10.1073/pnas.0911281106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann T, Bieger SC, Brühl B, Tienari PJ, Ida N, Allsop D. et al. Distinct sites of intracellular production for Alzheimer’s disease A beta40/42 amyloid peptides. Nat Med. 1997;3(9):1016–1020. doi: 10.1038/nm0997-1016. [DOI] [PubMed] [Google Scholar]
- Grimm HS, Beher D, Lichtenthaler SF, Shearman MS, Beyreuther K, Hartmann T. gamma-Secretase cleavage site specificity differs for intracellular and secretory amyloid beta. J Biol Chem. 2003;278(15):13077–13085. doi: 10.1074/jbc.M210380200. [DOI] [PubMed] [Google Scholar]
- Oikawa N, Yamaguchi H, Ogino K, Taki T, Yuyama K, Yamamoto N. et al. Gangliosides determine the amyloid pathology of Alzheimerʼs disease. Neuroreport. 2009;20(12):1043–1046. doi: 10.1097/WNR.0b013e32832e4b9d. [DOI] [PubMed] [Google Scholar]
- Bernardo A, Harrison FE, McCord M, Zhao J, Bruchey A, Davies SS. et al. Elimination of GD3 synthase improves memory and reduces amyloid-beta plaque load in transgenic mice. Neurobiol Aging. 2009;30(11):1777–1791. doi: 10.1016/j.neurobiolaging.2007.12.022. [DOI] [PubMed] [Google Scholar]
- Chatr-aryamontri A, Breitkreutz B-J, Heinicke S, Boucher L, Winter A, Stark C. et al. The BioGRID interaction database: 2013 update. Nucleic Acids Res. 2013;41(D1):D816–D823. doi: 10.1093/nar/gks1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Lu Z-H, Wang J, Wang Y, Xie X, Meyenhofer MF. et al. Enhanced Susceptibility to Kainate-Induced Seizures, Neuronal Apoptosis, and Death in Mice Lacking Gangliotetraose Gangliosides: Protection with LIGA 20, a Membrane-Permeant Analog of GM1. J Neurosci. 2005;25(47):11014–11022. doi: 10.1523/JNEUROSCI.3635-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignaud H, Bobo C, Lascu I, Sörgjerd KM, Zako T, Maeda M. et al. A Structure-Toxicity Study of Aß42 Reveals a New Anti-Parallel Aggregation Pathway. PLoS One. 2013;8(11):e80262. doi: 10.1371/journal.pone.0080262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiang W, Yau YM, Luo Y, Mattson MP, Tycko R. Antiparallel β-sheet architecture in Iowa-mutant β-amyloid fibrils. Proc Natl Acad Sci U S A. 2012;109(12):4443–4448. doi: 10.1073/pnas.1111305109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yau J, Sharpe S. Structures of amyloid fibrils formed by the prion protein derived peptides PrP(244-249) and PrP(245-250) J Struct Biol. 2012;180(2):290–302. doi: 10.1016/j.jsb.2012.08.002. [DOI] [PubMed] [Google Scholar]
- Eisenberg D, Jucker M. The amyloid state of proteins in human diseases. Cell. 2012;148(6):1188–1203. doi: 10.1016/j.cell.2012.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiang W, Yau W-M, Luo Y, Mattson MP, Tycko R. Antiparallel β-sheet architecture in Iowa-mutant β-amyloid fibrils. Proc Natl Acad Sci U S A. 2012;109(12):4443–4448. doi: 10.1073/pnas.1111305109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colletier J-P, Laganowsky A, Landau M, Zhao M, Soriaga AB, Goldschmidt L. et al. Molecular basis for amyloid-beta polymorphism. Proc Natl Acad Sci U S A. 2011;108(41):16938–16943. doi: 10.1073/pnas.1112600108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenman DJ, Connors C, Chen W, Wang C, García AE. Aβ Monomers Transiently Sample Oligomer and Fibril-like Configurations: Ensemble Characterization Using a Combined MD/NMR Approach. J Mol Biol. 2013;425(18):3338–3359. doi: 10.1016/j.jmb.2013.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheidt HA, Morgado I, Rothemund S, Huster D. Dynamics of Amyloid β Fibrils Revealed by Solid-state NMR. J Biol Chem. 2012;287(3):2017–2021. doi: 10.1074/jbc.M111.308619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheidt HA, Morgado I, Rothemund S, Huster D, Fändrich M. Solid-State NMR Spectroscopic Investigation of Aβ Protofibrils: Implication of a β-Sheet Remodeling upon Maturation into Terminal Amyloid Fibrils. Angew Chem Int Ed. 2011;50(12):2837–2840. doi: 10.1002/anie.201007265. [DOI] [PubMed] [Google Scholar]
- Ahmed M, Davis J, Aucoin D, Sato T, Ahuja S, Aimoto S. et al. Structural conversion of neurotoxic amyloid-β(1-42) oligomers to fibrils. Nat Struct Mol Biol. 2010;17(5):561–567. doi: 10.1038/nsmb.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu L, Liu C, Guo Z. Structural Insights into Aβ42 Oligomers Using Site-directed Spin Labeling. J Biol Chem. 2013;288(26):18673–18683. doi: 10.1074/jbc.M113.457739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debelouchina GT, Bayro MJ, Fitzpatrick AW, Ladizhansky V, Colvin MT, Caporini MA. et al. Higher Order Amyloid Fibril Structure by MAS NMR and DNP Spectroscopy. J Am Chem Soc. 2013;135(51):19237–19247. doi: 10.1021/ja409050a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick AWP, Debelouchina GT, Bayro MJ, Clare DK, Caporini MA, Bajaj VS. et al. Atomic structure and hierarchical assembly of a cross-β amyloid fibril. Proc Natl Acad Sci U S A. 2013;110(14):5468–5473. doi: 10.1073/pnas.1219476110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono K, Condron MM, Teplow DB. Structure-neurotoxicity relationships of amyloid beta-protein oligomers. Proc Natl Acad Sci U S A. 2009;106(35):14745–14750. doi: 10.1073/pnas.0905127106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glabe CG. Structural Classification of Toxic Amyloid Oligomers. J Biol Chem. 2008;283(44):29639–29643. doi: 10.1074/jbc.R800016200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campioni S, Mannini B, Zampagni M, Pensalfini A, Parrini C, Evangelisti E. et al. A causative link between the structure of aberrant protein oligomers and their toxicity. Nat Chem Biol. 2010;6(2):140–147. doi: 10.1038/nchembio.283. [DOI] [PubMed] [Google Scholar]
- Nelson R, Sawaya M, Balbirnie M, Madsen A, Riekel C, Grothe R. et al. Structure of the cross-beta spine of amyloid-like fibrils. Nature. 2005;435(7043):773–778. doi: 10.1038/nature03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawaya MR, Sambashivan S, Nelson R, Ivanova MI, Sievers SA, Apostol MI. et al. Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature. 2007;447(7143):453–457. doi: 10.1038/nature05695. [DOI] [PubMed] [Google Scholar]
- Zheng X, Wu C, Ponder JW, Marshall GR. Molecular Dynamics of β-Hairpin Models of Epigenetic Recognition Motifs. J Am Chem Soc. 2012;134(38):15970–15978. doi: 10.1021/ja306803v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Su H, Zhang JZH, Mu Y. Molecular Dynamics Simulation Study on the Molecular Structures of the Amylin Fibril Models. J Phys Chem B. 2012;116(48):13991–13999. doi: 10.1021/jp308708h. [DOI] [PubMed] [Google Scholar]
- Lapidus LJ. Understanding protein aggregation from the view of monomer dynamics. Mol Biosyst. 2013;9(1):29–35. doi: 10.1039/c2mb25334h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty S, Chatterjee B, Basu S. A mechanistic insight into the amyloidogenic structure of hIAPP peptide revealed from sequence analysis and molecular dynamics simulation. Biophys Chem. 2012;168-169:1–9. doi: 10.1016/j.bpc.2012.05.003. [DOI] [PubMed] [Google Scholar]
- Berhanu WM, Masunov AE. Alternative packing modes leading to amyloid polymorphism in five fragments studied with molecular dynamics. Biopolymers. 2012;98(2):131–144. doi: 10.1002/bip.21731. [DOI] [PubMed] [Google Scholar]
- Agopian A, Guo Z. Structural origin of polymorphism for Alzheimer’s amyloid-β fibrils. Biochem J. 2012;447(1):43–50. doi: 10.1042/BJ20120034. [DOI] [PubMed] [Google Scholar]
- Tycko R. Solid-state NMR studies of amyloid fibril structure. Annu Rev Phys Chem. 2011;62:279–299. doi: 10.1146/annurev-physchem-032210-103539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Török M, Milton S, Kayed R, Wu P, McIntire T, Glabe CG. et al. Structural and dynamic features of Alzheimer’s Abeta peptide in amyloid fibrils studied by site-directed spin labeling. J Biol Chem. 2002;277(43):40810–40815. doi: 10.1074/jbc.M205659200. [DOI] [PubMed] [Google Scholar]
- Liu C, Zhao M, Jiang L, Cheng P-N, Park J, Sawaya MR. et al. Out-of-register β-sheets suggest a pathway to toxic amyloid aggregates. Proc Natl Acad Sci U S A. 2012;109(51):20913–20918. doi: 10.1073/pnas.1218792109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salemme FR. Structural properties of protein beta-sheets. Prog Biophys Mol Biol. 1983;42(2-3):95–133. doi: 10.1016/0079-6107(83)90005-6. [DOI] [PubMed] [Google Scholar]
- Arispe N, Pollard HB, Rojas E. Giant multilevel cation channels formed by Alzheimer disease amyloid beta-protein [A beta P-(1-40)] in bilayer membranes. Proc Natl Acad Sci U S A. 1993;90(22):10573–10577. doi: 10.1073/pnas.90.22.10573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard HB, Rojas E, Arispe N. A new hypothesis for the mechanism of amyloid toxicity, based on the calcium channel activity of amyloid beta protein (A beta P) in phospholipid bilayer membranes. Ann N Y Acad Sci. 1993;695:165–168. doi: 10.1111/j.1749-6632.1993.tb23046.x. [DOI] [PubMed] [Google Scholar]
- Nelson O, Supnet C, Tolia A, Horré K, De Strooper B, Bezprozvanny I. Mutagenesis Mapping of the Presenilin 1 Calcium Leak Conductance Pore. J Biol Chem. 2011;286(25):22339–22347. doi: 10.1074/jbc.M111.243063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz JC, Linnehan J, Pollard H, Arispe N. Histidines 13 and 14 in the Abeta sequence are targets for inhibition of Alzheimer’s disease Abeta ion channel and cytotoxicity. Biol Res. 2006;39(3):447–460. doi: 10.4067/s0716-97602006000300007. [DOI] [PubMed] [Google Scholar]
- Hirakura Y, Lin M-C, Kagan BL. Alzheimer amyloid aβ1–42 channels: Effects of solvent, pH, and congo red. J Neurosci Res. 1999;57(4):458–466. [PubMed] [Google Scholar]
- Lin MA, Kagan BL. Electrophysiologic properties of channels induced by Abeta25-35 in planar lipid bilayers. Peptides. 2002;23(7):1215–1228. doi: 10.1016/s0196-9781(02)00057-8. [DOI] [PubMed] [Google Scholar]
- Sanderson KL, Butler L, Ingram VM. Aggregates of a beta-amyloid peptide are required to induce calcium currents in neuron-like human teratocarcinoma cells: relation to Alzheimer’s disease. Brain Res. 1997;744(1):7–14. doi: 10.1016/s0006-8993(96)01060-8. [DOI] [PubMed] [Google Scholar]
- Schauerte JA, Wong PT, Wisser KC, Ding H, Steel DG, Gafni A. Simultaneous Single Molecule Fluorescence and Conductivity Studies Reveal Distinct Classes of Aβ Species on Lipid Bilayers. Biochem. 2010;49(14):3031–3039. doi: 10.1021/bi901444w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayed R, Pensalfini A, Margol L, Sokolov Y, Sarsoza F, Head J. et al. Annular protofibrils are a structurally and functionally distinct type of amyloid oligomer. J Biol Chem. 2009;284(7):4230–4237. doi: 10.1074/jbc.M808591200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demuro A, Smith M, Parker I. Single-channel Ca2+ imaging implicates Aβ1-42 amyloid pores in Alzheimer’s disease pathology. J Cell Biol. 2011;195(3):515–524. doi: 10.1083/jcb.201104133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang H, Arce FT, Ramachandran S, Capone R, Azimova R, Kagan BL. et al. Truncated beta-amyloid peptide channels provide an alternative mechanism for Alzheimer’s Disease and Down syndrome. Proc Natl Acad Sci U S A. 2010;107(14):6538–6543. doi: 10.1073/pnas.0914251107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagenston AM, Fitzpatrick JS, Yeckel MF. MGluR-Mediated Calcium Waves that Invade the Soma Regulate Firing in Layer V Medial Prefrontal Cortical Pyramidal Neurons. Cereb Cortex. 2008;18(2):407–423. doi: 10.1093/cercor/bhm075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutzmann GE, Smith I, Caccamo A, Oddo S, LaFerla FM, Parker I. Enhanced Ryanodine Receptor Recruitment Contributes to Ca2+ Disruptions in Young, Adult, and Aged Alzheimer’s Disease Mice. J Neurosci. 2006;26(19):5180–5189. doi: 10.1523/JNEUROSCI.0739-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhratsky A. The endoplasmic reticulum and neuronal calcium signalling. Cell Calcium. 2002;32(5-6):393–404. doi: 10.1016/s0143416002001896. [DOI] [PubMed] [Google Scholar]
- Fitzjohn SM, Collingridge GL. Calcium stores and synaptic plasticity. Cell Calcium. 2002;32(5-6):405–411. doi: 10.1016/s0143416002001999. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44(1):5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA. Long-term potentiation—a decade of progress? Science. 1999;285(5435):1870–1874. doi: 10.1126/science.285.5435.1870. [DOI] [PubMed] [Google Scholar]
- Mulkey RM, Malenka RC. Mechanisms underlying induction of homosynaptic long-term depression in area CA1 of the hippocampus. Neuron. 1992;9(5):967–975. doi: 10.1016/0896-6273(92)90248-c. [DOI] [PubMed] [Google Scholar]
- Demuro A, Parker I. Cytotoxicity of intracellular Aβ42 amyloid oligomers involves Ca2+ release from the ER by stimulated production of inositol trisphosphate. J Neurosci. 2013;33(9):3824–3833. doi: 10.1523/JNEUROSCI.4367-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki-Mann M, Demuro A, Parker I. Cytosolic [Ca2+] regulation of InsP3-evoked puffs. Biochem J. 2013;449(1):167–173. doi: 10.1042/BJ20121271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen RA, Shtifman A, Allen PD, Lopez JR, Querfurth HW. Calcium Dyshomeostasis in β-Amyloid and Tau-bearing Skeletal Myotubes. J Biol Chem. 2004;279(51):53524–53532. doi: 10.1074/jbc.M408473200. [DOI] [PubMed] [Google Scholar]
- Alberdi E, Sánchez-Gómez MV, Cavaliere F, Pérez-Samartín A, Zugaza JL, Trullas R. et al. Amyloid beta oligomers induce Ca2+ dysregulation and neuronal death through activation of ionotropic glutamate receptors. Cell Calcium. 2010;47(3):264–272. doi: 10.1016/j.ceca.2009.12.010. [DOI] [PubMed] [Google Scholar]
- Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13(3):184–190. doi: 10.1038/ncb0311-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shore GC, Papa FR, Oakes SA. Signaling Cell Death from the Endoplasmic Reticulum Stress Response. Curr Opin Cell Biol. 2011;23(2):143–149. doi: 10.1016/j.ceb.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra JD, Kaufman RJ. ER Stress and Its Functional Link to Mitochondria: Role in Cell Survival and Death. Cold Spring Harb Perspect Biol. 2011;3(9):a004424. doi: 10.1101/cshperspect.a004424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo R, Vicencio JM, Parra V, Troncoso R, Munoz JP, Bui M. et al. Increased ER–mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J Cell Sci. 2011;124(13):2143–2152. doi: 10.1242/jcs.080762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M-T, Chen F-Y, Huang HW. Energetics of Pore Formation Induced by Membrane Active Peptides. Biochem. 2004;43(12):3590–3599. doi: 10.1021/bi036153r. [DOI] [PubMed] [Google Scholar]
- Huang HW, Chen F-Y, Lee MT. Molecular mechanism of Peptide-induced pores in membranes. Phys Rev Lett. 2004;92(19):198304. doi: 10.1103/PhysRevLett.92.198304. [DOI] [PubMed] [Google Scholar]
- Sparr E, Engel MFM, Sakharov DV, Sprong M, Jacobs J, de Kruijff B. et al. Islet amyloid polypeptide-induced membrane leakage involves uptake of lipids by forming amyloid fibers. FEBS Letters. 2004;577(1-2):117–120. doi: 10.1016/j.febslet.2004.09.075. [DOI] [PubMed] [Google Scholar]
- Pieri L, Madiona K, Bousset L, Melki R. Fibrillar α-Synuclein and Huntingtin Exon 1 Assemblies Are Toxic to the Cells. Biophys J. 2012;102(12):2894–2905. doi: 10.1016/j.bpj.2012.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connelly L, Jang H, Teran Arce F, Ramachandran S, Kagan BL, Nussinov R. et al. Effects of Point Substitutions on the Structure of Toxic Alzheimer’s β-Amyloid Channels: Atomic Force Microscopy and Molecular Dynamics Simulations. Biochem. 2012;51(14):3031–3038. doi: 10.1021/bi300257e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafrir Y, Durell SR, Anishkin A, Guy HR. Beta-barrel models of soluble amyloid beta oligomers and annular protofibrils. Proteins. 2010;78(16):3458–3472. doi: 10.1002/prot.22832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang H, Arce FT, Ramachandran S, Capone R, Lal R, Nussinov R. β-Barrel topology of Alzheimer’s β-amyloid ion channels. J Mol Biol. 2010;404(5):917–934. doi: 10.1016/j.jmb.2010.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenk D, Barbour R, Dunn W, Gordon G. et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400(6740):173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- Nicoll JAR, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO. Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: a case report. Nat Med. 2003;9(4):448–452. doi: 10.1038/nm840. [DOI] [PubMed] [Google Scholar]
- Lambracht-Washington D, Rosenberg RN. Advances in the Development of Vaccines for Alzheimer’s Disease. Discov Med. 2013;15(84):319–326. [PMC free article] [PubMed] [Google Scholar]
- Farlow MR, Andreasen N, Riviere M-E, Vostiar I, Vitaliti A, Sovago J. et al. Long-term treatment with active Aβ immunotherapy with CAD106 in mild Alzheimer’s disease. Alzheimers Res Ther. 2015;7(1):23. doi: 10.1186/s13195-015-0108-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H. et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6(8):916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- Lemere CA. Immunotherapy for Alzheimer’s disease: hoops and hurdles. Mol Neurodegener. 2013;8:36. doi: 10.1186/1750-1326-8-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black RS, Sperling RA, Safirstein B, Motter RN, Pallay A, Nichols A. et al. Single Ascending Dose Study of Bapineuzumab in Patients With Alzheimer Disease. Alzheimer Dis Assoc Disord. 2010;24(2):198–203. doi: 10.1097/WAD.0b013e3181c53b00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salloway S, Sperling R, Fox NC, Blennow K, Klunk W, Raskind M. et al. Two Phase 3 Trials of Bapineuzumab in Mild-to-Moderate Alzheimer’s Disease. N Engl J Med. 2014;370(4):322–333. doi: 10.1056/NEJMoa1304839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafii MS, Aisen PS. Advances in Alzheimer’s Disease Drug Development. BMC Med. 2015;13:62. doi: 10.1186/s12916-015-0297-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samadi H, Sultzer D. Solanezumab for Alzheimer’s disease. Expert Opin Biol Ther. 2011;11(6):787–798. doi: 10.1517/14712598.2011.578573. [DOI] [PubMed] [Google Scholar]
- Doody RS, Thomas RG, Farlow M, Iwatsubo T, Vellas J, Joffe S. et al. Phase 3 Trials of Solanezumab for Mild-to-Moderate Alzheimer’s Disease. N Engl J Med. 2014;370(4):311–321. doi: 10.1056/NEJMoa1312889. [DOI] [PubMed] [Google Scholar]
- Cummings J, Cho W, Ward M, Friesenhahn M, Brunstein F, Honigberg L. et al. A Randomized, Double-Blind, Placebo-Controlled Phase 2 Study To Evaluate The Efficacy And Safety Of Crenezumab In Patients With Mild To Moderate Alzheimer’s Disease. Alzheimers Dement: J Alzheimers Assoc. 10(4):P275. [Google Scholar]
- Vassar R. BACE1 inhibitor drugs in clinical trials for Alzheimer’s disease. Alzheimers Res Ther. 2014;6:89. doi: 10.1186/s13195-014-0089-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman M, Kleijn H-J, Dockendorf M, Palcza J, Tseng J, Canales C. et al. The novel BACE inhibitor MK-8931 dramatically lowers CSF beta-amyloid in patients with mild-to-moderate Alzheimer’s disease. Alzheimers Dement: J Alzheimers Assoc. 9(4):P139. [Google Scholar]
- Cooper DJL. Dietary Lipids in the Aetiology of Alzheimer’s Disease. Drugs Aging. 2012;20(6):399–418. doi: 10.2165/00002512-200320060-00001. [DOI] [PubMed] [Google Scholar]
- Dannenberger D, Nuernberg G, Renne U, Nuernberg K, Langhammer M, Huber K. et al. High-fat diets rich in ω-3 or ω-6 polyunsaturated fatty acids have distinct effects on lipid profiles and lipid peroxidation in mice selected for either high body weight or leanness. Nutrition. 2013;29(5):765–771. doi: 10.1016/j.nut.2012.10.010. [DOI] [PubMed] [Google Scholar]
- Wassall SR, Stillwell W. Polyunsaturated fatty acid-cholesterol interactions: domain formation in membranes. Biochim Biophys Acta. 2009;1788(1):24–32. doi: 10.1016/j.bbamem.2008.10.011. [DOI] [PubMed] [Google Scholar]
- Soni SP, LoCascio DS, Liu Y, Williams JA, Bittman R, Stillwell W. et al. Docosahexaenoic Acid Enhances Segregation of Lipids between Raft and Nonraft Domains: 2H-NMR Study. Biophys J. 2008;95(1):203–214. doi: 10.1529/biophysj.107.123612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PyMOL. pyMol.org [Internet] Available from:http://pymol.org/