Lost in Translation: Neuropsychiatric drug developments (original) (raw)
. Author manuscript; available in PMC: 2016 Dec 22.
Abstract
Recent studies have identified troubling method and practice lapses in neuropsychiatric drug developments. These problems have resulted in errors that are of sufficient magnitude to invalidate clinical trial data and interpretations. We identify two potential sources for these difficulties: investigators selectively choosing scientific practices for demonstrations of efficacy in human-testing phases of drug development and investigators failing to anticipate the needs of practitioners who must optimize treatment for the individual patient. When clinical investigators neglect to use clinical trials as opportunities to test hypotheses of disease mechanisms in humans, the neuropsychiatric knowledge base loses both credibility and scope. When clinical investigators do not anticipate the need to translate discoveries into applications, the practitioner cannot provide optimal care for the patient. We conclude from this evidence that clinical trials, and other aspects of neuropsychiatric drug development, must adopt more practices from basic science and show greater responsiveness to conditions of clinical practice. We feel that these changes are necessary to overcome current threats to the validity and utility of studies of neurological and psychiatric drugs.
Introduction
Clinical trials (CTs) are the cornerstone of clinical medicine. Besides providing definitive tests for drug efficacy and authoritative guidance for treatment, they reveal disease and drug mechanisms in humans and confirm (or refute) ideas derived from preclinical research (1, 2). Unfortunately, in neurology and psychiatry, CTs may not perform these tasks as well as they should. Their usefulness and validity are often compromised by methodological inadequacies: measurement errors (3–5), analysis and interpretation biases (6, 7), selective reporting and publication (8), inadequate preclinical and early clinical preparations (9, 10), and late-stage protocol modifications (6). These problems result in drug and CT failures, underestimations of effect sizes, and—probably the most unfortunate outcome—information of limited value to clinicians.
Drugs may inappropriately fail or succeed during development as a result of type I and II chance errors or from flawed methods. Unearned successes can result when designs, practices, methods, or analyses are mistaken, biased, or fraudulent. Needless failures will be almost inevitable if a study tests drug doses that are not adequate to affect the biochemical target. Methodological errors can result in unneeded studies during follow-up of falsely positive CT outcomes or flawed tests of truly effective drugs. Unearned successes and failures will compromise the clinical evidence base, undermine investigators’ confidence in preclinical models, and stifle innovation. The community’s attention can be misdirected and innovation stifled as academia and industry turns toward more promising lines of investigation. Left with a compromised evidence base, medical practitioners and patients seek guidance from next-best sources (1)—published CT reports of variable quality, expert opinions, commercial promotions, and personal experiences.
We are concerned that by relaxing the stricter scientific methodological standards applied during preclinical chemical and animal studies, CTs and neuropsychiatric drug development have become vulnerable to errors and biases. These problems do not self-correct because these errors and biases are not detected in time to alter the courses of studies and interpretations of outcomes (4, 9, 11, 12). Here, we have explored these issues further and conclude that industry and investigators rely too heavily on technologies designed primarily to meet regulatory standards for drug approvals, without adequate consideration given to advancing scientific knowledge and clinical practices.
The Problems
In general, we find evidence that investigators comply with scientific practice standards in preclinical non-animal and animal pharmacological research (13) and that neuropsychiatrists thoughtfully critique theories of disease (14, 15). Nevertheless, in drug development, we find frequent inadequate preclinical and initial clinical preparations for CTs, measurement errors, biases in interpretation and reporting, and other mistakes that undermine confidence in research on human subjects. Below, we discuss five factors that contribute to this situation (Fig. 1).
Figure 1.

Problems in neuropsychiatric drug discovery.
Multiple disease determinants and inadequate animal models cause difficulties for psychiatry
Psychiatric diseases are complicated and variable. The uniqueness of each patient and the situation introduces uncertainties into drug research and into recommendations for patient care. A single biochemical or physiological mechanism will not usually predict the pathological changes or progression of illness for an individual. Both genetic and environmental factors contribute, requiring the various perspectives to explain illness (14, 15). In brain disorders, interacting physical and social systems, behaviors, and personal histories influence data in unpredictable ways (2, 11, 14). As a further complication, drug efficacy and safety data, derived from group responses in CTs, cannot predict drug effects for the individual in clinical care.
Animal models are often inadequate for psychiatric diseases. They seldom show disease mechanisms, symptoms, or behaviors equivalent to those in humans (2, 11). Although animal models can mimic anatomical, metabolic, and behavioral aspects of brain diseases, psychiatric diagnosis depends heavily on the patient’s verbal history of illness, reports of subjective feelings, and cognitive performance, features that cannot be replicated in animals (15–20). In the absence of biomarkers that reflect disease presence, severity, and mechanisms, inferences about psychiatric disease from animal research remain limited in scope and often speculative (2, 11, 21, 22). Hence, too frequently, leaps of faith hide within the translation of preclinical work on psychiatric disease into bedside patient care (2, 11, 18, 19).
The community has insufficient access to research results
Full assessment of psychiatric drug development methods and practices is prevented by selective publication of data and the failure to publish negative CTs (7, 8, 23, 24). When information from drug developments and CTs go unreported, the predictive ability of preclinical models cannot be judged, and investigators do not learn from earlier failures how to improve methods and practices (2). Progress in science and its methods follows from overcoming errors through trial-and-error learning. Lack of access to apparently failed efforts ensures that others will repeat these mistakes and, in turn, not publish the results. In addition, the causes of drug failures are usually not obvious and often remain uncertain even after careful review and further investigation (10, 25). Companies tend to abandon studies of drugs considered to be without commercial benefits, leaving academia without access to the data needed to examine whether trials showing that a drug is ineffective may have resulted from flaws in development and design, not from lack of efficacy. This analysis cannot occur in academia without full access to information. To the discredit of medicine, incomplete publication of human research persists (8) even though Chalmers (23) identified underreporting as scientific misconduct in 1990 (10).
Adherence to current CT norms does not prevent threats to validity
It is easy to become overconfident that errors, biases, and other threats to trial validity are adequately controlled by practices such as randomization, investigator training, close monitoring, uses of reliability statistics, and so forth. Unfortunately, studies reveal that these practices do not always meet expectations. Such methods do not always prevent problems with dosing (2, 9, 25), biases (6, 7), ineptitude and deception (7, 26), protocol violations (6), or measurement errors (3–5, 25). Unfortunately, investigators cannot detect these troubling errors as they occur; rather, they must take preemptive actions (12, 25). Randomization, an effective support for blinding, will not necessarily also control biases (27). Even with extensive training, some clinicians do not develop adequate skills to participate in CTs (28, 29). Because investigators may not regularly evaluate the effectiveness of training or performance of staff that run CTs, imprecision, inaccuracy, bias, protocol violations, and other error sources can interfere with a study’s validity (4, 30). In two studies of antidepressants, only about half of all trained raters were able to detect drug effects on patients’ clinical status (3, 4). Even with two highly reliable rating scales for Alzheimer’s disease (AD), random measurement errors had sufficient magnitude to seriously interfere with CT power and clinical management of patients (5). These problems arise because tests of statistical reliability primarily evaluate whether repeated ratings maintain the original order of the subjects’ responses and not the variations present among the repeated ratings of each individual subject. Despite the use of rating scales that are highly reliable by these tests, imprecision and inaccuracy have been impugned as sources for failures in AD, depression, stroke, and other diseases (3–5, 30–32).
Consolidated Standards of Reporting Trials (CONSORT) were adopted to ensure that CT reports meet major journal editors’ standards for completeness of reporting (33), and CT registrations were undertaken to provide a record of human drug research (34). Nevertheless, these measures do not control fraudulent advertising (35), inadequate descriptions of methods in trial registrations (34), deviations from protocols (6, 24), failures to publish negative CT data (35 “spin” or misrepresentations of CT outcome implications (7), selective publications of data from a CT (8), measurement errors that undermine estimates of the magnitude of drug effects (3–6), and effects from inadequate preclinical preparations (2, 9, 10).
In laboratory research, investigators are expected to provide evidence that the conditions of the experiment, assumptions necessary to interpret outcomes, and other potentially influential factors will not bias the anticipated research outcomes and the investigators’ interpretations of these outcomes. Scientifically sound experiments and interpretations test for effects from independent variables and from the conditions and assumptions required to design and perform the experiments (36–39). These same stringent criteria must be applied for clinical research to realize laboratory levels of error controls. Experimental assumptions and conditions needed for testing or interpreting must be either compared to controls within the CT or empirically validated independent of the CT to claim that outcome effects or interpretive conclusions do not arise from the methods or assumptions.
Increases in power may be accompanied by increases in errors
Investigators commonly include more subjects in a CT to reach the statistical power required to see the effect of their drug (37). Regulators and statisticians encourage these practices to expose research hypotheses to diverse real-world conditions able to support generalizations of conclusions. Unfortunately, larger numbers of subjects also require additional locations for patient recruitment and hence additional monitoring and supervision of often inexperienced staff (30, 31). Investigators knowingly or unknowingly employ raters not experienced with the disease under study, not trained and familiar with the instruments used as outcome measures, and who use biased ratings and relaxed protocol conditions to meet quotas (28, 29, 32, 36, 40). One indicator that is consistent with an increased vulnerability of large CTs to errors is that placebo responses increase with industrial sponsorships and with added research sites (20, 41, 42).
Apparently valid studies contain hidden errors
Investigators apply the term validity to multiple aspects of drug developments: animal models, interpretations of outcome data, statistical analyses, generalizations beyond the subject sample, and so forth. Validity implies that the study can be replicated and the conclusions applied successfully in different settings and over time (39). Given the wide range of often silent errors and bias threats to psychiatric drug development and CTs, claims of validity are best regarded with suspicion. A process or result can be labeled as valid without specific evidence that errors or biases do not account for outcomes (36). Undetected error or bias effects may silently nullify an apparently valid study. CTs currently regarded as valid may in fact be invalid as a result of hidden or subtle methodological shortcomings (25).
Two Examples
Case 1: Tarenflurbil for AD: Insufficient preparations for a CT
AD is greatly in need of disease-modifying treatments, and several pharmaceutical firms have been pursuing various agents. In 2008, Myriad Genetics tested tarenflurbil, the _R_-enantiomer of an anti-inflammatory agent (flurbiprofen) with expected amyloid β (Aβ) peptide–lowering properties, in a phase 3 CT for its therapeutic effect in patients with AD.
The rationale for this trial rested on unreplicated early data and suspect post hoc assumptions: An initial report from transgenic mice showing that tarenflurbil lowered Aβ42 in brain by targeting γ-secretase (43, 44) could not be confirmed by others (45). In the phase 2 trial that formed the basis of the phase 3 trial, the results were analyzed by using post hoc criteria after an initial analysis of data (46). Specifically, dose-related effects of the drug on activities of daily living and global function ratings were used to draw conclusions, even though the patients in the phase 2 trial did not exhibit the cognitive changes required by the statistical analysis plan (6). With these post facto choices, the investigators accepted the following risky (statistically speaking) scenario strategy for developing the phase 3 trial: Because patients treated under the protocol conditions for the phase 2 trial failed to benefit from the drug, the investigators selected alternative outcomes (activities of daily living and global function ratings) that favored further tarenflurbil development. Drawing in this way on multiple comparisons (as a result of the post hoc analysis) increases the risk of positive outcomes as a result of chance and undermines the soundness of predictions based on the conclusions. The investigators may not have served themselves and their drug well by going into phase 3 trials without confirmation of the animal findings, with post hoc changes in statistical analyses, without confirming the phase 2 study, and with designs and analyses vulnerable to multiple comparisons.
This example also demonstrates consequences from failures to understand dosing in adequate detail (8, 36, 46). As we have discussed elsewhere, after CT successes with one dosing regimen, drugs can fail in subsequent CTs because investigators use inadequately tested alternative dosing (47). In the phase 2 study with tarenflurbil, the dose-response plots showed straight line slopes that continued to increase at the highest blood concentrations, an indication that the necessary or optimal dose may not have been reached. The dose used in the phase 3 study was drawn from within the dose range used in the phase 2 study, which was ineffective for the main outcome of cognitive changes. Therefore, a clinically effective tarenflurbil dose may not have been evaluated in phase 3, and so, this phase 3 trial does not provide a definitive test of the drug’s efficacy and postulated mechanism of therapeutic action (25, 47). Conflicting reports of tarenflurbil effects on Aβ42 in animal models (43–45) and failures to observe changes in Aβ42 in the cerebrospinal fluid (CSF) of normal elderly volunteers after 21 days of treatment (48) should have indicated to the investigators a need for special attention to ensure adequate concentrations of drug in the brain in the phase 3 trial. Because of these and other unresolved issues, this drug and its therapeutic mechanisms cannot be eliminated as potentially beneficial for patients with AD. Investigators may have lost a potential drug, a molecular backbone for further drug developments, and an opportunity to test human brain mechanisms possibly relevant to therapeutic response in AD.
Case 2: CATIE: Ad hoc assumptions
In a second example, the CATIE (Clinical Antipsychotic Trials of Intervention Effectiveness) CTs tested the effects of antipsychotic medications on schizophrenia but may have failed to yield clinically useful results because insufficient preparations necessitated that ad hoc assumptions be made to apply the trial outcomes in patient care (Fig. 2). The problems with this trial do not obviously conflict with good laboratory practices as described (13). Over the last half century, the results of commercially sponsored studies had shown that many different antipsychotic drugs were more effective than placebo treatments, but no direct drug-to-drug comparisons had been done. Exposed to heavy advertising by drug manufacturers, doctors developed preferences for a second generation of antipsychotics. In a much-needed test of these preferences, the U.S. National Institutes of Health sponsored the CATIE studies (49). Except for side-effect profiles that were useful to practitioners, these CATIE studies failed to confirm that the first- and second-generation antipsychotics differed in efficacy (50, 51).
Figure 2.

Inadequate options for neuropsychiatric patients.
In response to the CATIE studies, Lieberman (52) and others have argued that, given (i) the efficacy of individual drugs against placebo, (ii) the therapeutic equivalence among antipsychotics in CATIE studies, and (iii) clinician inclinations to match drugs with patients on the basis of their symptomatic responses, clinicians should be able to select among the widest array of treatment options. This argument combines untested assumptions (clinician inclinations) with CT-confirmed facts (equivalent efficacies) to construct expert clinical guidance (52). This guidance sets aside research-evidenced equivalent effectiveness of the antipsychotics to endorse the untested claim that the effectiveness of these drugs can be established in the clinic, as each clinician works with individual patients. If the interpretation of CATIE’s outcomes in fact depended on this assumption, then CATIE would more usefully have been designed to address the effects of antipsychotics in individual patients, how precisely and accurately these effects are quantified by clinicians, how practitioners use these estimates to optimize selections among antipsychotics for individual patients, and under what clinical care conditions antipsychotics are most effectively used with schizophrenic patients. CATIE investigated the symptomatic efficacies of antipsychotic drugs on the basis of group responses without consideration of how a practitioner would optimize drug management for an individual patient. As the resulting need for additional assumptions shows, group data and conditions divorced from the realities of patient care responsibilities do not predict outcome for a unique biological individual, the patient.
It is questionable whether clinicians can be successful arbiters of antipsychotic efficacy (53). Trained CT raters are prone to make precision and accuracy errors in measurement when assessing clinical severity in psychiatric patients (3–5), and the CATIE reports themselves undermine the assumption that practitioners can, with adequate precision and accuracy, assess and optimize patient benefits from antipsychotic drugs. Indeed, CATIE raters could find no evidence for an advantage in switching from a current to a new medication (54). Problems with measurement encountered by CT raters would be magnified for community practitioners, many of whom are not trained with and generally do not use rating scales. The difficulty of choosing the contexts in which antipsychotic treatments can be most effective with patients also complicates this CT’s utility and therefore its validity (39).
Another problem with the CATIE trial is that, consistent with current drug development practices, the CATIE investigators uncritically assumed that “psychosocial practices … augment pharmacotherapy” (52, 55). Rather, in treating schizophrenic patients, considerable research evidence shows that psychosocial factors, with necessary but adjunctive support from pharmacotherapies, play primary roles against disabilities from these psychiatric disorders. By testing for drug effects without regard to the psychosocial context, the CATIE results may have failed to inform clinicians how best to maximize efficacy, because both drug and environment influence the patient.
Anthony (56) argues from his research that neuropsychiatric drugs do not rehabilitate disabilities. Severely ill patients prescribed drugs and psychotherapies in hospitals and clinics show functional improvements only after moving to community-based psychosocial care (57–60). Indeed, CATIE investigators acknowledge that employment, not just antipsychotic drugs, can benefit symptoms (61). Studies may even underestimate the benefits of employment because socioeconomic factors undermine efforts by mentally ill persons to compete in work environments (55–60). As Carpenter and CATIE investigators concede, most people with schizophrenia are investigated and treated in settings that do not offer employment, a psychosocial intervention known to be effective (56–60). Thus, to understand the contributions that drugs can make to patients’ lives—overcoming disabilities, realizing improved quality of life, and reaching personal goals (62, 63)—CTs may need to provide their subjects with the psychosocial and vocational experiences needed for the patient to learn to live in these roles in society. Absent socially and vocationally enriched environments for drug testing, CATIE and similar neuropsychiatric trials may miss the characterizations of drug efficacy needed by clinicians: not just as effectiveness against symptoms but also as support for recovery from remediable disabilities.
Discussion
We find that investigators in neuropsychiatric drug development too often use approaches that are designed to minimize the time to regulatory approval and focus too exclusively on acquiring evidence of drug efficacy. These methods, although necessary steps to develop drugs, are inadequate within themselves to support the scientifically rigorous translation of discoveries into practice benefits and expansions of scientific knowledge and methods. We have concluded from our accumulated evidence of the errors and limitations experienced by a focus on efficacy outcomes in current neuropsychiatric drug developments that incorporation of the methodological discipline exercised in fields of science outside neuropsychiatric drug development is needed to supplement current regulatory-oriented research. Below we outline recommendations toward this goal (Fig. 3).
Figure 3.

The way forward.
Develop research-based guidance to support personalized treatments
The CATIE investigations illustrate two perplexing problems faced by drug developers and clinical researchers in neuropsychiatry. Current statistical analyses cannot predict the success or failure of a treatment for an individual, which is after all the primary concern of the clinician caring for patients one at a time. Analyses of group or individual data can predict only the likely response, not the actual future response, of any individual in or outside the group (64). Comparative effectiveness trials, not regulatory-focused placebo superiority trials, will best inform the clinician about selection of one therapy from among a range of competing options. Statistical descriptions of individual CT subjects, for generalization to other individuals, depend on probabilities developed by using similarly behaved cases. Innovative individualized analyses that take into account the genetic and environmental uniqueness of each person are required to provide more useful applications of data from CTs. Until these analyses are available, the burden of disease monitoring inappropriately rests too exclusively with clinicians. We endorse the goal of drug development and CTs to move toward more extensive individual evaluation with the goal of determining how to effectively treat each patient.
Develop biomarkers
Rating scales for disease in CTs are inherently limited by imprecision, inaccuracy, biases, and uneven training. They require more extensive training, evaluation, monitoring, and protocol guidance than can be provided for many CTs and for clinicians in practice. In important ways, progress in research and patient care in neuropsychiatry seems dependent on the development of biomarkers and surrogate endpoints similar to those that have reduced the physician’s dependence on subjective forms of judgment in other areas of patient care (65).
Publish negative data
Translational medicine cannot approach its full potential if negative drug developments are unpublished or distorted in interpretation when published. As Reason’s (12) reviews of human reasoning and everyday experiences witness, human skills develop not de novo but by elimination of errors or mistakes through trial and error learning. If progress in psychiatric drug development depends on learning from errors, then lack of transparency into failed drug development efforts, a result of underreporting and distorted interpretation, will remain major impediments to progress (7, 23). To remove these impediments, as we and others have advocated, the standard in science and medicine should be that all human research and research with potential for human health benefits must be shared openly, publicly, completely, and in a timely manner (10).
Avoid errors
Preemptive interventions against errors need to become a priority for neuropsychiatric translational medicine. Our review of error sources provides little evidence that investigators can detect, manage, or correct effects from error intrusions once embarked on drug developments or data analyses (25). Early lapses in concentration during planning leave designs and methods vulnerable to inevitable human mistakes and the errors that result from these mistakes. Successes against undetected errors in CTs will depend on the introduction of preventive methods found effective in other areas of scientific practice (12, 25). Drug development researchers should work to develop community-wide standards for prevention of errors in CTs and avoidance of undetected CT invalidation.
Design CTs for optimal information, not just regulatory approval
Translational neurosciences will not optimally benefit human beings if drug developments, to meet commercial priorities, demonstrate efficacy but neglect the unique opportunities human clinical studies provide to test drug and disease mechanisms and to explore the conditions required for drug benefits in patient care. Corporate support for error-tainted or exclusively efficacy-focused drug development drains financial and human resources that could otherwise address important scientific and patient-care issues, such as dosing, drug-target interactions, drug-cellular and drug-metabolic dynamics, and so forth. These factors are not only important for investigators’ ultimate successes in demonstrating drug efficacy but also needed to realize the promise of neuroscience as a source of new knowledge and of motivations for investigators to address diseases of the brain and improve benefits to patients. Translational medicine as a whole must encourage neuropsychiatric clinical pharmacology that is scientifically sound and discourage drug development efforts not adequate to ensure certainties about outcomes.
Conclusions
This review points out how, by more effectively operationalizing the aim that scientific discoveries provide human benefits, translational medicine will strengthen current standards for validity of neuropsychiatric drug developments, their scientific adequacy, and their relevance to patient care. Braithwaite (39) states that, in science “effectiveness” is sufficient for “the adjective ‘valid’ to be applied to an inductive inference from known evidence.” We have discussed two CTs: one accepted as a valid test of its drug’s lack of efficacy and the second as valid to ground best-practice guidance for clinicians. We find that upon implementation, each fails to be sound, implying that the trials underpinning these inferences were not in fact valid.
We find these invalidities arising not from failures of investigators to comply with standards found in contemporary drug developments but from specific weaknesses in these contemporary methodologies (36) that result in lack of definitive tests for efficacy (25). Currently, neuropsychiatric drug development and its CTs are too single-mindedly directed toward technical demonstrations of efficacy, with insufficient concerns for the mechanisms through which the drug acts, the conditions required to extract from drugs optimal benefits for humans, and the resources a practitioner will need to realize these benefits in patients. We are concerned that the present selective uses of scientific methods do not provide the necessary discipline and objectivity required for a truly translational neuropsychiatric drug development science (Fig. 4).
Figure 4.
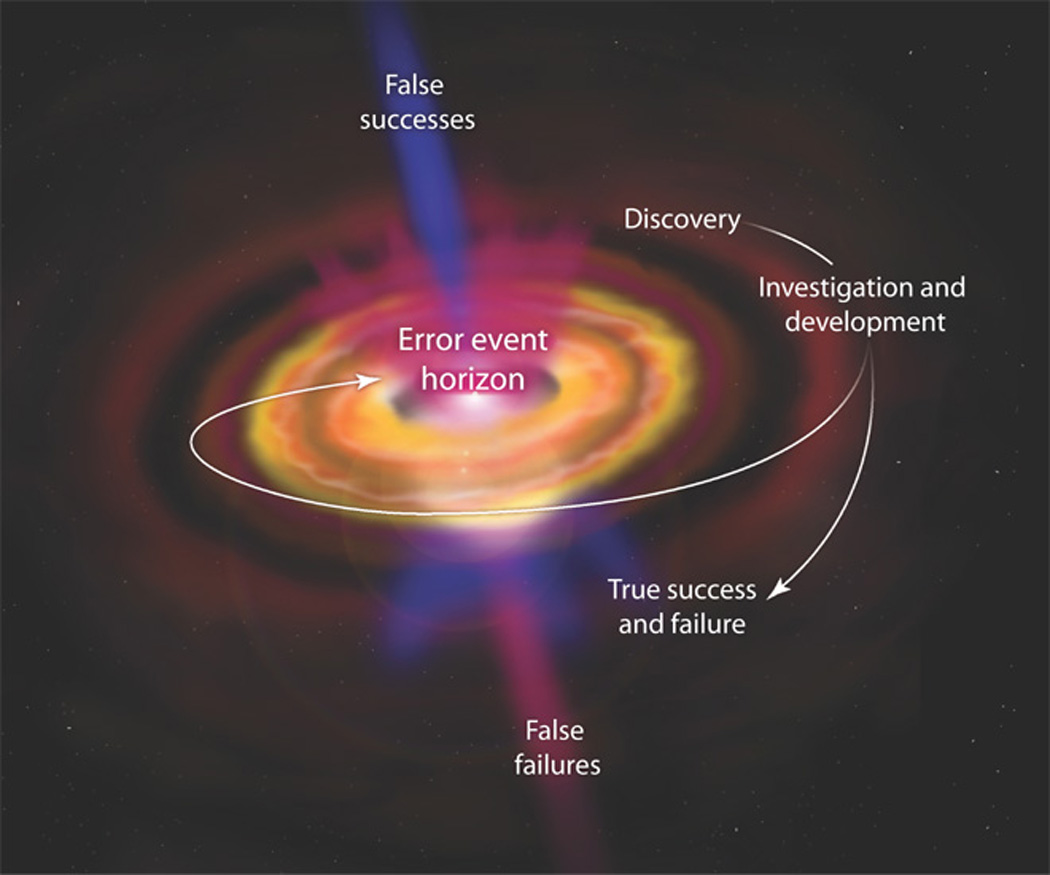
The black hole of neuropsychiatry drug development.
CTs that neglect to test mechanisms of drug actions in humans put only the drugs at risk, not scientific knowledge. Translationally adequate drug tests a hypothesis of how the drug interacts with the brain target, then tests how the brain changes as a result of those interactions, then demonstrates in humans the optimal drug-target conditions to ensure efficacy, and only then if, how, and under what conditions patient symptoms and functional impairments change. With these steps, translational medicine can best enhance scientific knowledge of brain functions, as well as provide clinically important information. The mechanistic hypothesis testing associated with a drug-target focus in drug development should have other benefits. Knowledge of the brain and its targets for intervention will be advanced. A CT outcome that does not confirm a brain mechanism threatens a sector of scientific theory and practice. This threat motivates investigators to assess with care whether the failure of prediction from current knowledge when tested in humans was due to methods, practices, intervening factors such as drug metabolism, or limits in what we know about the brain and its functions (38). With scientific knowledge at risk, investigators are energized to review how errors, biases, or other methodological conditions may mislead a CT’s interpretations.
Here, we describe how scientific practices may be needed to increase the certainty of conclusions drawn from neuropsychiatric drug developments and CTs. We find a critical role for science as medicine’s procedural barrier against the accrual of errors.
Acknowledgments
We thank Reviewers and Editors for critiques and Science Editors for editing that contributes greatly to the final manuscript.
Funding: This work was supported in part by the Intramural Research Program of the National Institute on Aging, NIH.
Footnotes
Author contributions: Both authors contributed equally to the conception and writing of the paper.
Competing interests: R.E.B. has an equity interest in Aristea Translational Medicine Corporation, which was formed as a platform for investigating and developing drugs.
References and Notes
- 1.Sackett DL, Strauss SE, Richardson WS, Rosenberg W, Haynes RB. Evidence-Based Medicine: How to Practice and Teach EBM. New York: Churchill Livingstone; 2000. [Google Scholar]
- 2.Markou A, Chiamulera C, Geyer MA, Tricklebank M, Steckler T. Removing obstacles in neuroscience drug discovery: The future path for animal models. Neuropsychopharmacology. 2009;34:74–89. doi: 10.1038/npp.2008.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Engelhardt N, Feiger AD, Cogger KO, Sikich D, DeBrota DJ, Lipsitz JD, Kobak KA, Evans KR, Potter WZ. Rating the raters: Assessing the quality of Hamilton Rating Scale for Depression clinical interviews in two industry-sponsored clinical drug trials. J. Clin. Psychopharmacol. 2006;26:71–74. doi: 10.1097/01.jcp.0000194621.61868.7c. [DOI] [PubMed] [Google Scholar]
- 4.Cogger KO. Rating rater improvement: A method for estimating increased effect size and reduction of clinical trial costs. J. Clin. Psychopharmacol. 2007;27:418–420. doi: 10.1097/01.jcp.0000280315.81366.f8. [DOI] [PubMed] [Google Scholar]
- 5.Becker RE, Markwell S. Problems arising from generalizing of treatment efficacy from clinical trials in Alzheimer’s disease. Clin. Drug Invest. 2000;19:33–41. [Google Scholar]
- 6.Chan AW, Hróbjartsson A, Jørgensen KJ, Gøtzsche PC, Altman DG. Discrepancies in sample size calculations and data analyses reported in randomised trials: Comparison of publications with protocols. BMJ. 2008;337:a2299. doi: 10.1136/bmj.a2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boutron I, Dutton S, Ravaud P, Altman DG. Reporting and interpretation of randomized controlled trials with statistically nonsignificant results for primary outcomes. JAMA. 2010;303:2058–2064. doi: 10.1001/jama.2010.651. [DOI] [PubMed] [Google Scholar]
- 8.Turner EH, Matthews AM, Linardatos E, Tell RA, Rosenthal R. Selective publication of antidepressant trials and its influence on apparent efficacy. N. Engl. J. Med. 2008;358:252–260. doi: 10.1056/NEJMsa065779. [DOI] [PubMed] [Google Scholar]
- 9.Pangalos MN, Schechter LE, Hurko O. Drug development for CNS disorders: Strategies for balancing risk and reducing attrition. Nat. Rev. Drug Discov. 2007;6:521–532. doi: 10.1038/nrd2094. [DOI] [PubMed] [Google Scholar]
- 10.Becker RE, Greig NH. Alzheimer’s disease drug development in 2008 and beyond: Problems and opportunities. Curr. Alzheimer Res. 2008;5:346–357. doi: 10.2174/156720508785132299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kola I, Landis J. Can the pharmaceutical industry reduce attrition rates? Nat. Rev. Drug Discov. 2004;3:711–715. doi: 10.1038/nrd1470. [DOI] [PubMed] [Google Scholar]
- 12.Reason J. Human Error. Cambridge: Cambridge University Press; 2001. [Google Scholar]
- 13.Swanson BN. Delivery of high-quality biomarker assays. Dis. Markers. 2002;18:47–56. doi: 10.1155/2002/212987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kendler KS. Explanatory models for psychiatric illness. Am. J. Psychiatry. 2008;165:695–702. doi: 10.1176/appi.ajp.2008.07071061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kendler KS. Toward a philosophical structure for psychiatry. Am. J. Psychiatry. 2005;162:433–440. doi: 10.1176/appi.ajp.162.3.433. [DOI] [PubMed] [Google Scholar]
- 16.McDonald MP, Overmier JB. Present imperfect: A critical review of animal models of the mnemonic impairments in Alzheimer’s disease. Neurosci. Biobehav. Rev. 1998;22:99–120. doi: 10.1016/s0149-7634(97)00024-9. [DOI] [PubMed] [Google Scholar]
- 17.Yamada K, Nabeshima T. Animal models of Alzheimer’s disease and evaluation of anti-dementia drugs. Pharmacol. Ther. 2000;88:93–113. doi: 10.1016/s0163-7258(00)00081-4. [DOI] [PubMed] [Google Scholar]
- 18.Götz J, Streffer JR, David D, Schild A, Hoerndli F, Pennanen L, Kurosinski P, Chen FJ. Transgenic animal models of Alzheimer’s disease and related disorders: Histopathology, behavior and therapy. Mol. Psychiatry. 2004;9:664–683. doi: 10.1038/sj.mp.4001508. [DOI] [PubMed] [Google Scholar]
- 19.Fuchs E, Flïugge G. Experimental animal models for the simulation of depression and anxiety. Dialogues Clin. Neurosci. 2006;8:323–333. doi: 10.31887/DCNS.2006.8.3/efuchs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Becker RE, Greig NH. Alzheimer’s disease drug development: Old problems require new priorities. CNS Neurol. Disord. Drug Targets. 2008;7:499–511. doi: 10.2174/187152708787122950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barrett JE, Witkin JM. Application of behavioral techniques to central nervous system (CNS) drug discovery and development. In: Bespalov A, Zvartau E , Beardsley P, Katz J, editors. Behavioral Pharmacology. St. Petersburg, Russia: PITER Publishing; 2009. pp. 430–455. [Google Scholar]
- 22.Willner P, Mitchell PJ. The validity of animal models of predisposition to depression. Behav. Pharmacol. 2002;13:169–188. doi: 10.1097/00008877-200205000-00001. [DOI] [PubMed] [Google Scholar]
- 23.Chalmers I. Underreporting research is scientific misconduct. JAMA. 1990;263:1405–1408. [PubMed] [Google Scholar]
- 24.Mathew SJ, Charney DS. Publication bias and the efficacy of antidepressants. Am. J. Psychiatry. 2009;166:140–145. doi: 10.1176/appi.ajp.2008.08071102. [DOI] [PubMed] [Google Scholar]
- 25.Becker RE, Greig NH. Why so few drugs for Alzheimer’s disease? Are methods failing drugs? Curr. Alzheimer Res. 2010;7:642–651. doi: 10.2174/156720510793499075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baerlocher MO, O’Brien J, Newton M, Gautam T, Noble J. Data integrity, reliability and fraud in medical research. Eur. J. Intern. Med. 2010;21:40–45. doi: 10.1016/j.ejim.2009.11.002. [DOI] [PubMed] [Google Scholar]
- 27.Worrall J. Why there’s no cause to randomize. Br. J. Philos. Sci. 2007;58:451–488. [Google Scholar]
- 28.Demitrack MA, Faries D, Herrera JM, DeBrota D, Potter WZ. The problem of measurement error in multisite clinical trials. Psychopharmacol. Bull. 1998;34:19–24. [PubMed] [Google Scholar]
- 29.Kobak KA, Lipsitz JD, Williams JB, Engelhardt N, Bellew KM. A new approach to rater training and certification in a multicenter clinical trial. J. Clin. Psychopharmacol. 2005;25:407–412. doi: 10.1097/01.jcp.0000177666.35016.a0. [DOI] [PubMed] [Google Scholar]
- 30.Butcher J. The hunt for drugs to modify Alzheimer’s disease. Lancet Neurol. 2007;6:1038–1039. doi: 10.1016/S1474-4422(07)70280-6. [DOI] [PubMed] [Google Scholar]
- 31.Becker RE. Lessons from Darwin: 21st century designs for clinical trials. Curr. Alzheimer Res. 2007;4:458–467. doi: 10.2174/156720507781788828. [DOI] [PubMed] [Google Scholar]
- 32.Samsa GP, Matchar DB. Have randomized controlled trials of neuroprotective drugs been underpowered? An illustration of three statistical principles. Stroke. 2001;32:669–674. doi: 10.1161/01.str.32.3.669. [DOI] [PubMed] [Google Scholar]
- 33.Moher D, Jones A, Lepage L. CONSORT Group (Consolidated Standards for Reporting of Trials), Use of the CONSORT statement and quality of reports of randomized trials: A comparative before-and-after evaluation. JAMA. 2001;285:1992–1995. doi: 10.1001/jama.285.15.1992. [DOI] [PubMed] [Google Scholar]
- 34.De Angelis C, Drazen JM, Frizelle FA, Haug C, Hoey J, Horton R, Kotzin S, Laine C, Marusic A, Overbeke AJ, Schroeder TV, Sox HC, Van Der Weyden MB. International Committee of Medical Journal Editors, Clinical trial registration: A statement from the International Committee of Medical Journal Editors. N. Engl. J. Med. 2004;351:1250–1251. doi: 10.1056/NEJMe048225. [DOI] [PubMed] [Google Scholar]
- 35.Moncrieff J. The antidepressant debate. Br. J. Psychiatry. 2002;180:193–194. doi: 10.1192/bjp.180.3.193. [DOI] [PubMed] [Google Scholar]
- 36.Becker RE, Greig NH, Giacobini E. Why do so many drugs for Alzheimer’s disease fail in development? Time for new methods and new practices? J. Alzheimers Dis. 2008;15:303–325. doi: 10.3233/jad-2008-15213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cohen J. Things I have learned (so far) Am. Psychol. 1990;45:1304–1312. [Google Scholar]
- 38.Becker RE, Greig NH. Neuropsychiatric clinical trials: Should they accommodate real-world practices or set standards for clinical practices? J. Clin. Psychopharmacol. 2009;29:56–64. doi: 10.1097/JCP.0b013e318192e2fa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Braithwaite RB. Scientific Explanation. Bristol, UK: Thoemmes Press; 1994. pp. 270–271. [Google Scholar]
- 40.Kobak KA, DeBrota DJ, Engelhardt N, Williams JBW. Site vs. centralized raters in a clinical depression trial. paper presented at the 46th Annual Meeting of the National Institute of Mental Health, New Clinical Drug Evaluation Unit; Boca Raton, FL. 2006. [Google Scholar]
- 41.Bridge JA, Birmaher B, Iyengar S, Barbe RP, Brent DA. Placebo response in randomized controlled trials of antidepressants for pediatric major depressive disorder. Am. J. Psychiatry. 2009;166:42–49. doi: 10.1176/appi.ajp.2008.08020247. [DOI] [PubMed] [Google Scholar]
- 42.Emslie GJ. Understanding placebo response in pediatric depression trials. Am. J. Psychiatry. 2009;166:1–3. doi: 10.1176/appi.ajp.2008.08101541. [DOI] [PubMed] [Google Scholar]
- 43.Eriksen JL, Sagi SA, Smith TE, Weggen S, Das P, McLendon DC, Ozols VV, Jessing KW, Zavitz KH, Koo EH, Golde TE. NSAIDs and enantiomers of flurbiprofen target γ-secretase and lower Aβ42 in vivo. J. Clin. Invest. 2003;112:440–449. doi: 10.1172/JCI18162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weggen S, Eriksen JL, Das P, Sagi SA, Wang R, Pietrzik CU, Findlay KA, Smith TE, Murphy MP, Bulter T, Kang DE, Marquez-Sterling N, Golde TE, Koo EH. A subset of NSAIDs lower amyloidogenic Aβ42 independently of cyclooxygenase activity. Nature. 2001;414:212–216. doi: 10.1038/35102591. [DOI] [PubMed] [Google Scholar]
- 45.Lan TA, Fici GJ, Merchant KM. Lack of specific amyloid-β(1–42) suppression by nonsteroidal anti-inflammatory drugs in young, plaque-free Tg2576 mice and in guinea pig neuronal cultures. J. Pharmacol. Exp. Ther. 2005;312:399–406. doi: 10.1124/jpet.104.073965. [DOI] [PubMed] [Google Scholar]
- 46.Wilcock GK, Black SE, Hendrix SB, Zavitz KH, Swabb EA, Laughlin MA. Tarenflurbil Phase II Study investigators, Efficacy and safety of tarenflurbil in mild to moderate Alzheimer’s disease: A randomised phase II trial. Lancet Neurol. 2008;7:483–493. doi: 10.1016/S1474-4422(08)70090-5. [DOI] [PubMed] [Google Scholar]
- 47.Becker RE, Unni LK, Greig NH. Resurrecting clinical pharmacology as a context for Alzheimer disease drug development. Curr. Alzheimer Res. 2009;6:79–81. doi: 10.2174/156720509787313916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Galasko DR, Graff-Radford N, May S, Hendrix S, Cottrell BA, Sagi SA, Mather G, Laughlin M, Zavitz KH, Swabb E, Golde TE, Murphy MP, Koo EH. Safety, tolerability, pharmacokinetics, and Aβ levels after short-term administration of R-flurbiprofen in healthy elderly individuals. Alzheimer Dis. Assoc. Disord. 2007;21:292–299. doi: 10.1097/WAD.0b013e31815d1048. [DOI] [PubMed] [Google Scholar]
- 49.Lieberman JA, Stroup TS, McEvoy JP, Swartz MS, Rosenheck RA, Perkins DO, Keefe RS, Davis SM, Davis CE, Lebowitz BD, Severe J, Hsiao JK. Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) Investigators, Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N. Engl. J. Med. 2005;353:1209–1223. doi: 10.1056/NEJMoa051688. [DOI] [PubMed] [Google Scholar]
- 50.Swartz MS, Stroup TS, McEvoy JP, Davis SM, Rosenheck RA, Keefe RS, Hsiao JK, Lieberman JA. What CATIE found: Results from the schizophrenia trial. Psychiatr. Serv. 2008;59:500–506. doi: 10.1176/ps.2008.59.5.500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stroup TS, Lieberman JA, McEvoy JP, Davis SM, Swartz MS, Keefe RS, Miller AL, Rosenheck RA, Hsiao JK. CATIE Investigators, Results of phase 3 of the CATIE schizophrenia trial. Schizophr. Res. 2009;107:1–12. doi: 10.1016/j.schres.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lieberman JA. What the CATIE study means for clinical practice. Psychiatr. Serv. 2006;57:1075. doi: 10.1176/ps.2006.57.8.1075. [DOI] [PubMed] [Google Scholar]
- 53.Heerdink ER, Stolker JJ, Meijer WE, Hugenholtz GW, Egberts AC. Need for medicine-based evidence in pharmacotherapy. Br. J. Psychiatry. 2004;184:452. doi: 10.1192/bjp.184.5.452. [DOI] [PubMed] [Google Scholar]
- 54.Rosenheck RA, Davis S, Covell N, Essock S, Swartz M, Stroup S, McEvoy J, Lieberman J. Does switching to a new antipsychotic improve outcomes? Data from the CATIE Trial. Schizophr. Res. 2009;107:22–29. doi: 10.1016/j.schres.2008.09.031. [DOI] [PubMed] [Google Scholar]
- 55.Rosenheck R, Leslie D, Keefe R, McEvoy J, Swartz M, Perkins D, Stroup S, Hsiao JK, Lieberman J. CATIE Study Investigators Group, Barriers to employment for people with schizophrenia. Am. J. Psychiatry. 2006;163:411–417. doi: 10.1176/appi.ajp.163.3.411. [DOI] [PubMed] [Google Scholar]
- 56.Anthony WA. Psychiatric rehabilitation: A key to prevention. Psychiatr. Serv. 2009;60:3. doi: 10.1176/ps.2009.60.1.3. [DOI] [PubMed] [Google Scholar]
- 57.Becker RE. The challenge of rehabilitation. In: Cancro R, Fox N, Shapiro LE, editors. Strategic Intervention in Schizophrenia: Current Development in Treatment. New York: Behavioral Publications; 1974. pp. 261–278. [Google Scholar]
- 58.Becker RE. An evaluation of a rehabilitation program for chronically hospitalized psychiatric patients. Soc. Psychiatry Psychiatr. Epidemiol. 1967;2:32–38. [Google Scholar]
- 59.Stein LI, Test MA, Marx AJ. Alternative to the hospital: A controlled study. Am. J. Psychiatry. 1975;132:517–522. doi: 10.1176/ajp.132.5.517. [DOI] [PubMed] [Google Scholar]
- 60.Becker RE, Meisler N, Stormer G, Brondino MJ. Employment outcomes for clients with severe mental illness in a PACT model replication. Program for Assertive Community Treatment. Psychiatr. Serv. 1999;50:104–106. doi: 10.1176/ps.50.1.104. [DOI] [PubMed] [Google Scholar]
- 61.Swartz MS, Perkins DO, Stroup TS, Davis SM, Capuano G, Rosenheck RA, Reimherr F, McGee MF, Keefe RS, McEvoy JP, Hsiao JK, Lieberman JA. CATIE Investigators, Effects of antipsychotic medications on psychosocial functioning in patients with chronic schizophrenia: Findings from the NIMH CATIE study. Am. J. Psychiatry. 2007;164:428–436. doi: 10.1176/ajp.2007.164.3.428. [DOI] [PubMed] [Google Scholar]
- 62.Carpenter WT., Jr Targeting schizophrenia research to patient outcomes. Am. J. Psychiatry. 2006;163:353–355. doi: 10.1176/appi.ajp.163.3.353. [DOI] [PubMed] [Google Scholar]
- 63.Becker RE. Psychiatry: The importance of understanding persons while treating diagnoses. Sci. Culture. 2008;17:317–334. [Google Scholar]
- 64.Becker RE. Modifying clinical trial designs to test treatments for clinical significance in individual patients. Clin. Drug Invest. 2001;21:727–733. [Google Scholar]
- 65.Thal LJ, Kantarci K, Reiman EM, Klunk WE, Weiner MW, Zetterberg H, Galasko D, Praticò D, Griffin S, Schenk D, Siemers E. The role of biomarkers in clinical trials for Alzheimer disease. Alzheimer Dis. Assoc. Disord. 2006;20:6–15. doi: 10.1097/01.wad.0000191420.61260.a8. [DOI] [PMC free article] [PubMed] [Google Scholar]