MeCP2 driven transcriptional repression in vitro: selectivity for methylated DNA, action at a distance and contacts with the basal transcription machinery (original) (raw)
Abstract
The pathways for selective transcriptional repression of methylated DNA templates by the methyl-CpG-binding protein MeCP2 have been investigated using a purified in vitro transcription system that does not assemble chromatin. MeCP2 selectively inhibits transcription complex assembly on methylated DNA but does not destabilize a pre-assembled transcription complex. MeCP2 functions to repress transcription at a distance of >500 bp from the transcription start site. The transcription repression domain (TRD) of MeCP2 will repress transcription in vitro when fused to a heterologous Gal4 DNA-binding domain. The TRD associates with TFIIB. Exogenous TFIIB does not relieve transcriptional repression established by either intact MeCP2 or a Gal4–TRD fusion protein under these in vitro conditions, nor does the addition of histone deacetylase inhibitors. We find that the transcriptional repression established by both MeCP2 and the Gal4–TRD fusion protein in vitro also correlates with selective assembly of large nucleoprotein complexes. The formation of such complexes reflects a local concentration of DNA-bound transcriptional repressor that may stabilize a state of repression even in the presence of exogenous transcriptional machinery.
INTRODUCTION
Mutations in the gene encoding the methyl-CpG-binding transcriptional repressor MeCP2 have been associated with Rett syndrome, a childhood neurodevelopmental disorder (1,2). MeCP2 is also essential for embryonic development in mice (3). These studies emphasize the potential importance of DNA methylation and methyl-CpG-binding proteins in gene control (see also 4–6).
MeCP2 is the archetypical methyl-CpG-binding protein defined in vertebrates (7) and is representative of a family of proteins containing similar methyl-CpG-binding domains (MBDs) (8–10). Within MeCP2, the N-terminal 85 amino acids comprise the MBD. This domain consists of a wedge-shaped structure with four antiparallel β-strands constituting one face of the wedge, of which the two longer β-strands are proposed to interact with the major groove of DNA (10). The MBD of MeCP2 can recognize a single symmetrically methylated CpG either as naked DNA (11) or when exposed in the major groove of nucleosomal DNA (12). MeCP2 can bind stably to methylated DNA in nucleosomes (12) and chromatin (13). In vivo MeCP2 is an abundant chromosomal protein that is concentrated in the centromeric heterochromatin of mouse cells (7,14). This localization requires both DNA methylation and the methyl-CpG-binding domain of MeCP2 (11,14). The available evidence indicates that MeCP2 has a high selectivity for association with methylated DNA within chromatin and chromosomes.
DNA methylation can repress transcription through multiple mechanisms (15–19). Pathways of repression include direct inhibition of transcription through the failure of transcription factors to associate with methylated recognition elements (20) and indirect pathways involving either occlusion of methylated sequences by transcriptional repressors that recognize methylated DNA (21) or the modification of chromatin structure targeted by methyl-CpG-specific transcriptional repressors (22,23). All of these mechanisms may operate in vivo, however, attention has recently focused on chromatin modification because the C-terminal transcription repression domain (TRD) of MeCP2 (13) can interact with Sin3A and recruit histone deacetylase to repress transcription (24,25). The inhibition of histone deacetylase activity using drugs such as Trichostatin A only partially relieves transcriptional repression established in vivo by the TRD (24,25). This partial relief raises questions concerning the existence of alternative pathways of transcriptional repression aside from the recruitment of histone deacetylase. The existence of alternative pathways for repression is further substantiated by in vivo observations in which inhibitors of DNA methyltransferase relieve transcriptional silencing at the Fragile X mental retardation gene 1 promoter, but not inhibitors of histone deacetylase (26).
Chromatin remodeling and histone modification undoubtedly have important roles in regulating transcription of methylated DNA (5,23–25); however, it is also important to identify other potential targets for transcriptional repression by MeCP2 that do not involve histone deacetylation. This is especially relevant because Rett syndrome mutations in MeCP2 occur in both the MBD and C-terminal sequences (1,2). Some of the C-terminal mutations lie inside the TRD while others are outside this domain, raising the possibility that these alterations influence functions of MeCP2 aside from the TRD-mediated contact with Sin3A. To explore this issue biochemically it is essential to use an experimental system that is devoid of chromatin components. Here we make use of highly purified transcription factors and recombinant MeCP2 to investigate interactions of this transcriptional repressor with the basal transcriptional machinery (27). We find that MeCP2 and the TRD will interact with TFIIB and that the local concentration of DNA-bound MeCP2 on methylated DNA may serve to stabilize the repressed state even in the presence of an excess of basal transcriptional machinery.
MATERIALS AND METHODS
Cloning of MeCP2 and construction of deletion mutants
A glutathione _S_-transferase (GST)–MeCP2 fusion was cloned in-frame using a _Sma_I–_Aat_II cDNA fragment from Xenopus MeCP2 (24) into the corresponding sites in pGex 4T-I (Pharmacia Biotech). The MBD (amino acids 79–163 relative to the first methionine) and the TRD (amino acids 204–310) were cloned into the same vector using PCR with primers containing engineered restriction sites (_Eco_RI and _Sal_I) and ligated in-frame into the corresponding sites of the vector. pGal4-TRD, encoding the yeast Gal4 DNA-binding domain (amino acids 1–147) fused to the MeCP2 TRD, was obtained by PCR amplification of the TRD and cloning of the fragment into the _Xba_I–_Bam_HI sites of pMSIIGal4. The Gal4–TRD fragment was subcloned into pGex 4T-I using PCR with primers containing _Eco_RI and _Sal_I engineered sites. All correct clones were expressed in BL21 DE3 pLysS cells. Cultures were grown to mid log phase and induced with 1 mM IPTG for 5 h at 30°C.
Protein purification
Recombinant transcription factors IIB, IIE (α and β), IIB, TBP, IIF (Rap30 and Rap74) and native IIH and RNA polymerase II were purified as previously described (27). MeCP2, Gal4–TRD and MBD were purified as described (12). Fractions were assayed for protein content by SDS–PAGE and then dialyzed against BC100 (20 mM HEPES, pH 7.9, 100 mM KCl, 10% glycerol, 2 mM DTT, 0.5 mM PMSF). For MeCP2, the purification was carried further over a 1 ml Heparin HiTrap (Pharmacia Biotech) column in a linear gradient from BC100 to BC1000. All fractions were analyzed by SDS–PAGE and the fractions containing the peak of MeCP2 were dialyzed against BC100.
GST pull-down assay and immunoblotting
GST–MeCP2, GST–TRD and GST–MBD (100–200 ng) together with 100–200 ng of the basal transcription factor of interest were incubated in a transcription reaction buffer (25 mM HEPES, pH 8.2, 0.5 mg/nl BSA, 100 mM KCl, 4 mM MgCl2, 5 mM DTT) for 30 min at room temperature. GST–agarose (Pharmacia Biotech) was preincubated in BC100 containing 0.2 mg/ml BSA, washed three times with BC100, added to the reactions and incubation was continued under constant agitation for an additional 2–4 h at 4°C. The beads were collected by centrifugation, washed three times with BC500 (500 mM KCl), 0.1% NP-40. The retained proteins were separated by SDS–PAGE, transferred to a nitrocellulose membrane, probed with an antibody against the protein of interest and visualized using an ECL protocol (Amersham).
In vitro transcription reaction
In vitro transcription was carried out in a 25 µl reaction mixture containing 100 ng pG5ML (27), 100 ng methylated pMLΔ53 (27), 100 ng pGEM, 20 ng recombinant TFIIB, 10 ng recombinant TFIIE, 20 ng recombinant TBP, 10 ng recombinant TFIIF, 50 ng purified HeLa TFIIH and 50 ng purified HeLa RNA polymerase II in the presence or absence of other tested components specified in the figure legends using conditions previously described (27). Transcription was assayed using the adenovirus major late promoter fused to a G-free cassette. Reactions were incubated at 30°C for 60 min and then terninated by adding 200 µl of stop solution containing 1% SDS, 100 mM sodium acetate (pH 5.2) and 1 mg/ml tRNA. RNA was extracted with phenol/chloroform, precipitated and washed with ethanol and dissolved in 10 µl formamide containing 0.1% xylene cyanol, bromophenol blue. Samples were analyzed on a 6% polyacrylamide–7 M urea gel and visualized by autoradiography. Template pMLΔ63 was cytosine methylated in the CpG sequences with the CpG methylase _Sss_I (New England Biolabs). Methylation reactions were carried out until complete resistance to cleavage by _Hha_I was achieved (28), then the DNA was phenol/chloroform extracted and ethanol precipitated.
Electrophoretic mobility shift assay
Double-stranded templates were methylated as above and used as probes after radiolabeling with a random priming labeling kit (Amersham). Binding reactions were identical to transcription conditions, comprising 300 ng total DNA, including 10 ng radiolabeled probe, in 25 mM KCl, 10 mM MgCl2, 12.5 mM DTT mixed with protein in 10 µl before addition to 15 µl BC100 containing 0.5 mg/ml BSA. The DNA–protein complexes were resolved on a 0.7% agarose gel. The amount of DNA-binding protein (MeCP2, Gal4–TRD or histone H1) used is specified in the figure legends.
Microinjection of Xenopus oocytes
The preparation of Xenopus stage VI oocytes and the microinjection procedure were as previously described (29). pG5ML was injected in double-stranded form (1 ng/oocyte) into the nuclei (GV) of the oocytes. The injected oocytes were then incubated at 18°C for the time indicated in MBSH buffer supplemented with antibiotics (5 U/ml ampicillin and streptomycin) and then collected for DNA analysis as described below.
Supercoiling assay for chromatin assembly
The supercoiling assay was performed as previously described (30). To analyze DNA topology, 10 µl of each DNA sample was loaded onto a 1.2% agarose gel in 1× TPE (40 mM Tris, 30 mM NaH2PO4, 10 mM EDTA) containing 60 µg/ml freshly prepared chloroquine in both the gel and running buffer. The gels were run in the dark overnight at –3.5 V/cm. The gels were then washed with distilled water and the DNA was transferred to a Nytran Plus membrane. The DNA was probed with single-stranded DNA of plasmid pG5ML labeled with a random priming labeling kit (Amersham) as described by the manufacturer.
Micrococcal nuclease digestion assay for chromatin assembly
The micrococcal nuclease cleavage assay of chromatin structure was performed as previously described (31). Briefly, groups of 20–25 injected oocytes were collected after the indicated incubation times and homogenized in 300 µl of micrococcal nuclease buffer (10 mM HEPES, pH 8.0, 50 mM KCl, 5 mM MgCl2, 3 mM CaCl2, 1 mM DTT, 0.1% NP-40, 5% glycerol). The extract was divided into four fractions (60 µl each) and digested with 10, 5, 2.5 and 1.25 U/ml MNase (Worthington), respectively, at room temperature for 5 min. Digestion was stopped by addition of 200 µl of 20 mM EGTA, 1% SDS. The reactions were treated with RNase A (100 µg/ml) for 2–3 h at 55°C and then extracted twice with phenol/chloroform and precipitated with 0.7 vol of isopropanol. The DNA samples were resolved on a 1.5% agarose gel, blotted to a nylon membrane and probed as described above.
RESULTS
MeCP2 selectively represses transcription of methylated templates and can act at a distance of 500 bp
In an earlier work in vitro transcription reactions using crude nuclear extracts and individual templates in each reaction were used to demonstrate that MeCP2 could repress transcription for methylated and unmethylated templates in a process independent of DNA methylation status (21). Subsequent work established the capacity of recombinant MeCP2 to selectively repress transcription of methylated templates dependent on the presence of the MBD (13). We wished to confirm the selectivity of MeCP2 for repression of methylated templates using internally controlled reactions in which an unmethylated template was also present. We also wished to make use of a purified in vitro transcription system in which the roles of individual transcription factors in the repression process might be determined and from which contributions of nucleosomes could be excluded (27).
Mixtures of methylated pMLΔ53 and unmethylated pG5ML (100 ng of each template methylated using _Sss_I methyltransferase) were incubated with increasing amounts of MeCP2 before addition of the transcription factors and RNA polymerase. Transcription of the methylated pMLΔ53 template is selectively repressed as MeCP2 concentration is increased (Fig. 1A, lanes 2–6). We examined the selectivity of MeCP2 for association with methylated DNA using agarose gel shift assays. Supercoiled radiolabeled methylated pMLΔ53 was mixed with unlabeled supercoiled pG5ML under the exact transcription assay conditions (Materials and Methods). MeCP2 was titrated into the mixture and the nucleoprotein complexes resolved on 0.7% agarose gels (Fig. 1B, lanes 2–6). In a parallel set of binding reactions, pG5ML was radiolabeled and pMLΔ53 remained unlabeled (Fig. 1B, lanes 8–12). MeCP2 preferentially associates with the methylated pMLΔ53 template (Fig. 1B, lanes 2–6). Increasing amounts of MeCP2 led to the assembly of two complexes on methylated pMLΔ53: a nucleoprotein complex that migrates into the gel, albeit progressively more slowly as more MeCP2 is bound, and insoluble aggregates that do not enter the gel matrix. Under conditions of optimal selectivity for methylation-specific repression (Fig. 1A and B, lanes 5 and 6) densitometry (not shown) indicates that >80% of the methylated template does not enter the gel matrix. In contrast to the assembly of nucleoprotein complexes on methylated pMLΔ53, the vast majority of the unmethylated pG5ML template continues to migrate like free DNA at all excesses of MeCP2 (Fig. 1B, lanes 8–12). We conclude that MeCP2 selectively associates with methylated DNA templates and represses their transcription.
Figure 1.
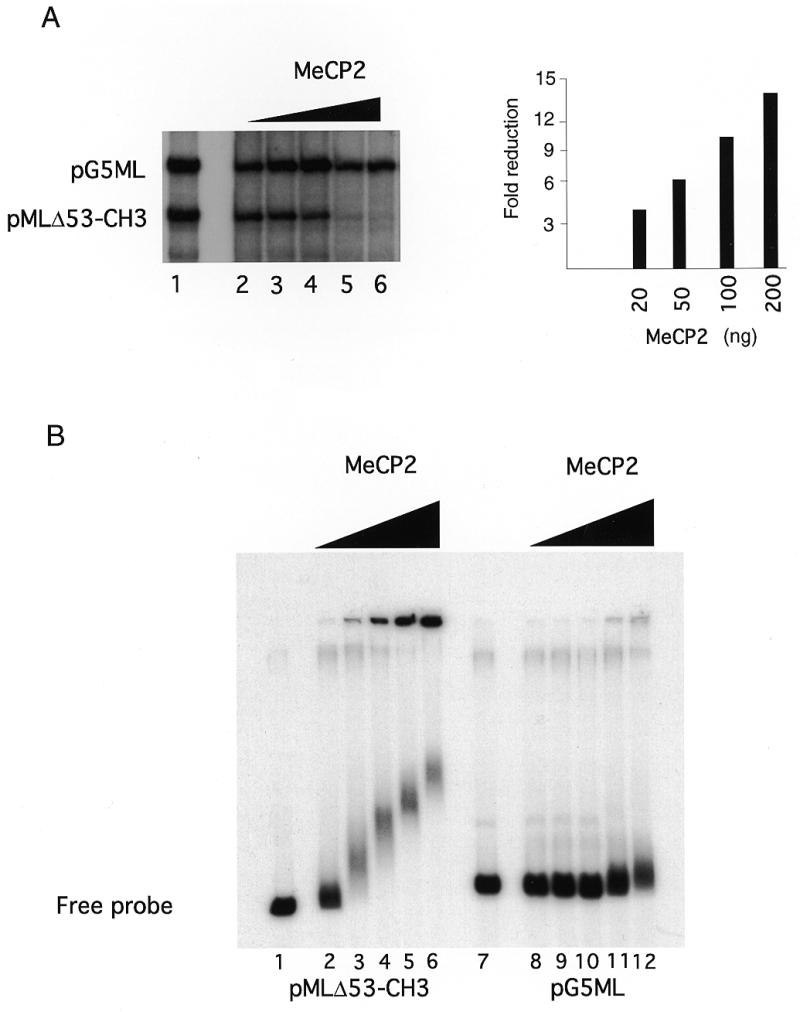
MeCP2-responsive in vitro transcription assay. (A) Transcription reactions were as described (28) except that one of the G-less cassette templates pMLΔ53-CH3 was _Sss_I methylated, whereas pG5ML remained unmethylated. Recombinant MeCP2 was preincubated with the templates for 20 min at room temperature prior to addition of the basal transcription factors (IIB, IIE, IIF, TBP, IIH and polymerase II). The reaction was allowed to continue for 60 min at 30°C. The RNAs were extracted and run on a denaturing gel. (Left) Lane 1, standard transcription reaction, no MeCP2; lanes 2–6, standard transcription reaction preincubated with 10, 20, 50, 100 and 200 ng of MeCP2, respectively. (Right) Quantitation of transcription from the pMLΔ53-CH3 template relative to pG5ML expressed as fold reduction relative to MeCP2 mass per reaction. (B) MeCP2 was preincubated with the DNA templates as in the transcription assay. The complexes were resolved on a 0.7% agarose gel. Lanes 1–6, methylated and labeled pMLΔ53 template; lanes 7–12, labeled pG5ML template; lanes 1 and 7, no MeCP2; lanes 2 and 8, 10 ng; lanes 3 and 9, 20 ng; lanes 4 and 10, 50 ng; lanes 5 and 11, 100 ng; lanes 6 and 12, 200 ng.
An issue when using in vitro transcription reactions is the context under which activation or repression occurs (32). The effect of DNA-binding proteins on the transcription process is often more pronounced if they are positioned such that they make direct contact with the basal transcriptional machinery bound near the transcription start site. In vitro Gal4 fusions with the TRD of MeCP2 repress transcription in nuclear extracts when positioned over 400 bp from the transcription start site (13). In order to examine the influence of the context of DNA methylation and MeCP2 on transcription from the adenovirus major late promoter under our conditions in the absence of chromatin, we cloned a methylatable DNA segment containing six sites for _Hha_I methyltransferase at 250 and 500 bp away from the promoter. We then methylated these constructs with _Hha_I methyltransferase. This strategy leaves the adenovirus major late promoter substantially unmethylated but places blocks of methylated DNA 250 and 500 bp away from the promoter (Fig. 2A). Addition of MeCP2 to mixtures of these templates with unmethylated pG5ML again led to the selective silencing of transcription from the methylated template (Fig. 2B and C). Analysis of MeCP2 binding to these radiolabeled templates again reveals the formation of two forms of nucleoprotein complex: a minor component that migrates into the gel and a major form that remains in the gel well (Fig. 2D). Under conditions of optimal selectivity for transcriptional silencing >90% of the methylated template remains in the gel well (Fig. 2D, lanes 3, 4, 7, 8, 11, 12, 15 and 16).
Figure 2.
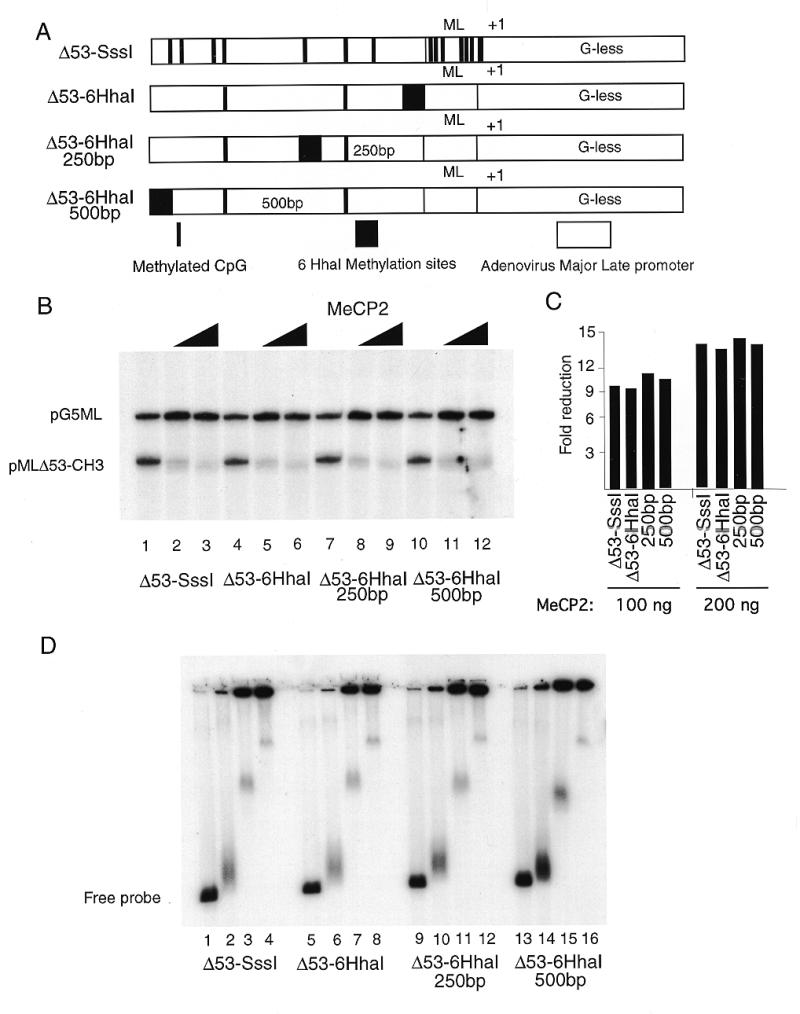
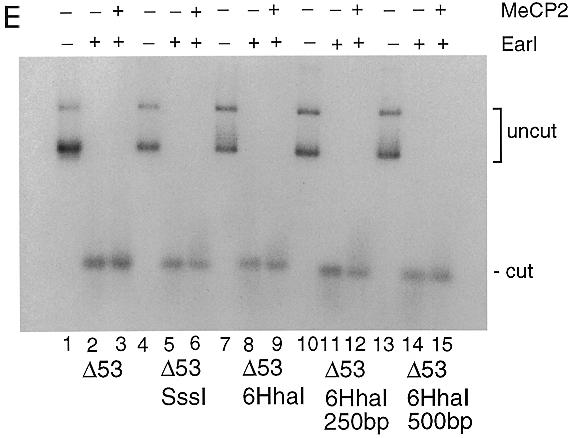
MeCP2 can repress transcription from a distance. (A) Six _Hha_I sites were cloned next to the adenovirus major late promoter, 250 and 500 bp away from the start site of transcription +1. The templates were methylated with _Hha_I which resulted in a methylation cluster (black box). Other methylated CpG islands are indicated by vertical bars. The templates were assayed in a standard transcription reaction. (B) Lanes 1–3, methylated _Sss_I template; lanes 4–6, methylated _Hha_I template with six _Hha_I sites next to the promoter; lanes 7–9, six _Hha_I sites 250 bp away; lanes 10–12, six _Hha_I sites 500 bp away; lanes 1, 4, 7 and 10, no MeCP2; lanes 2, 5, 8 and 11, 100 ng; lanes 3, 6, 9 and 12, 200 ng. (C) Quantitation of data shown in (A) as in Figure 1A. (D) Agarose gel shift with the same DNA templates as in (A). Lanes 1, 5, 9 and 13, no addition of MeCP2; lanes 2, 6, 10 and 14, 20 ng; lanes 3, 7, 11 and 15; 100 ng; lanes 4, 8, 12 and 16, 200 ng. (E) The methylated adenovirus major late promoter is accessible for restriction nuclease digestion in the presence of MeCP2. Methylated _Sss_I or _Hha_I template as indicated [labeled as in (A)] was incubated exactly as in the standard transcription reaction in the presence (+) or absence (–) of 200 ng MeCP2 for 15 min at room temperature as indicated. _Ear_I (2 U/reaction) was added and the incubation was continued for 45 min at 30°C. Reactions were deproteinated with phenol/chloroform, the DNA was ethanol precipitated and then the fragments were separated on a 1.2% agarose gel and the extent of digestion was analyzed by Southern blotting. The probe was ML64, a 64 bp oligonucleotide that spans the promoter region of the adenovirus major late promoter, end-labeled with T4 polynucleotide kinase. The positions of uncut and cut (restricted) DNA are indicated.
We considered the possibility that selective transcription inhibition under these reaction conditions might be due to the formation of these large nucleoprotein aggregates. To test this we made use of the restriction endonuclease _Ear_I, which has a recognition element 4 bp downstream from the start site of transcription in the adenovirus major late promoter. Under these conditions of aggregation in the presence of MeCP2 the promoter remains as accessible to restriction endonuclease _Ear_I cleavage as in the absence of the protein (Fig. 2E). This suggests that transcriptional repression under these conditions is not through occlusion of the promoter from access by DNA-binding proteins (see Discussion).
MeCP2 interferes with preinitiation complex assembly and interacts with TFIIB
Having established transcriptional repression dependent on the selective interaction of methylated DNA with MeCP2 (Figs 1 and 2), we next wished to examine the molecular mechanisms driving repression. We examined whether MeCP2 could repress in vitro transcription from a pre-assembled preinitiation complex. The pre-addition of MeCP2 to mixtures of methylated pMLΔ53 and unmethylated pG5ML will repress transcription of the methylated template (Fig. 3A, lanes 2 and 3, and B). In contrast, the pre-assembly of a preinitiation complex prevents transcriptional repression by MeCP2 (Fig. 3A, lanes 4–6, and B). Twenty minutes of preincubation of the transcriptional machinery with template DNA before addition of MeCP2 was sufficient to resist transcriptional silencing. We obtained similar results with the segmentally methylated templates used in Figure 2 (data not shown).
Figure 3.
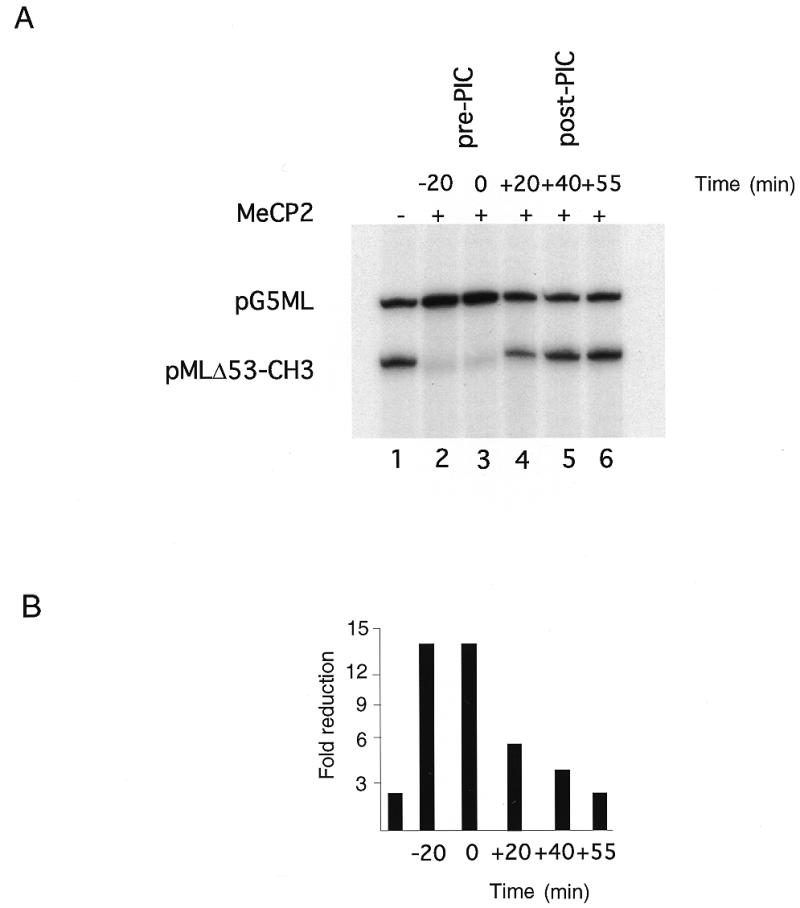
MeCP2 interferes with preinitiation complex (PIC) formation/function. (A) Lane 1, standard transcription reaction, no MeCP2; lane 2, 200 ng MeCP2 preincubated with the templates for 20 min prior to adding the transcription factors (–20 min); lane 3, 200 ng MeCP2 added at the same time as the transcription factors (0 min); lanes 4–6, 200 ng MeCP2 added 20, 40 and 55 min after addition of the transcription factors, respectively. (B) Quantitation of transcription from the pMLΔ53-CH3 template relative to pG5ML expressed as fold reduction relative to MeCP2 mass per reaction.
We next examined whether MeCP2 was making protein–protein contacts with the basal transcriptional machinery and interfering with preinitiation complex assembly (for example 33–35). We performed GST pull-down assays with purified transcription factors. We find that MeCP2 interacts selectively with TFIIB (Fig. 4A), but not with TFIIA, TFIID, TFIIH, TFIIE or TFIIF. We next examined whether the interaction of TFIIB was with the TRD or MBD of MeCP2. Both the TRD and MBD have been shown to independently confer transcriptional silencing (13). We find that TFIIB interacts with the TRD of MeCP2, but not with the MBD (Fig. 4B). This result indicates that MeCP2 interaction with TFIIB does not require binding to DNA. The association of TFIIB with the TRD of MeCP2 led us to examine whether Gal4–TRD fusions would repress transcription in our in vitro system. We made use of the same mixture of the templates pG5ML and pMLΔ53, however, in these in vitro experiments both templates were unmethylated. In the pG5ML construct five Gal4-binding sites are fused immediately upstream of the adenovirus major late promoter (Fig. 5A). pMLΔ53 lacks Gal4-binding sites. We find that preincubation of recombinant Gal4–TRD (24) with the mixture of templates selectively represses transcription from the pG5ML promoter (Fig. 5B and C).
Figure 4.
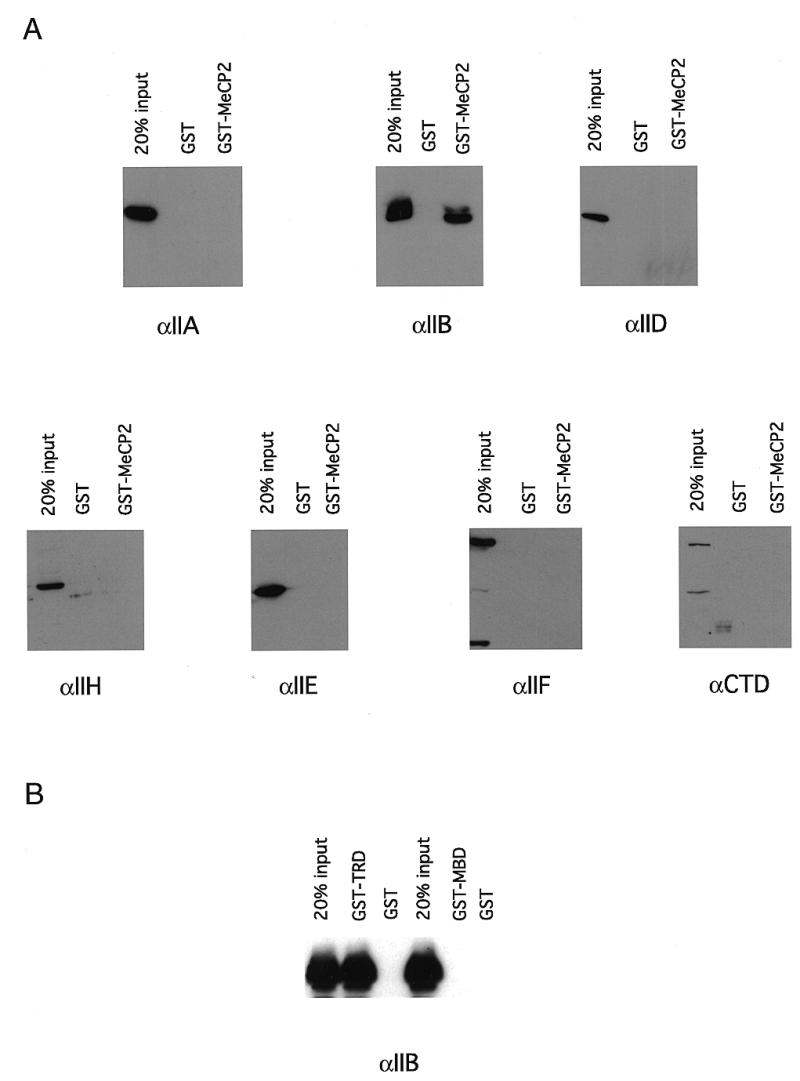
Contacts of MeCP2 with the basal transcription machinery. (A) MeCP2 interacts with TFIIB in a GST pull-down assay. GST–MeCP2 was incubated with recombinant IIA, IIB, IIEα, IIF and purified HeLa IID, IIH and RNA polymerase II in the same buffer conditions as in the transcription reaction (Materials and Methods). GST–agarose was added to the reactions and incubation was continued for another 2–4 h at 4°C. The beads were washed three times with a buffer containing 500 mM KCl, 0.1% NP-40 and the proteins retained by the agarose were separated by SDS–PAGE, transferred to nitrocellulose and probed with antibodies against the proteins of interest. The three lanes show 20% (one fifth) of input, the eluate from GST alone and the eluate from GST–MeCP2. (B) The TRD of MeCP2 but not the MBD interacts with TFIIB in a GST pull-down assay. As in (A) except that GST–TRD and GST–MBD (Materials and Methods) were used.
Figure 5.
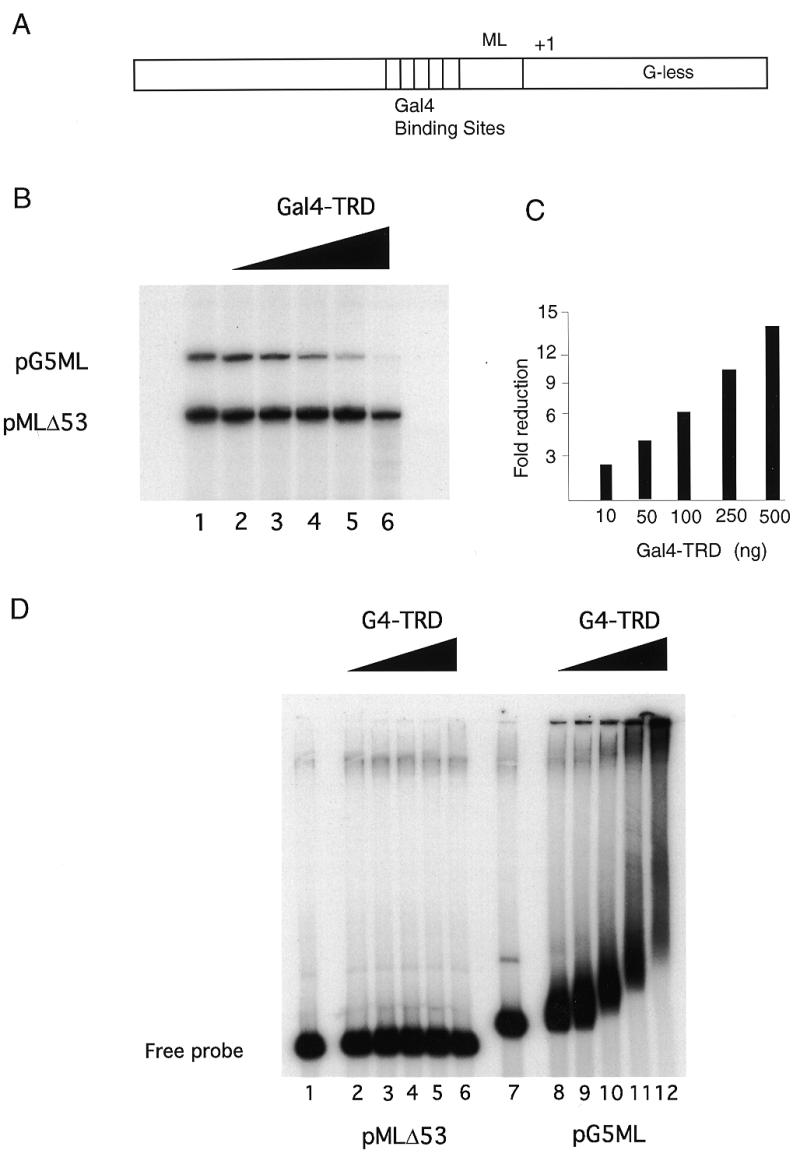
Gal4–TRD will repress transcription in cis. (A) The template used in these experiments was pG5ML in which five GAL4-binding sites are positioned 68 bp upstream from the start site of transcription (+1) of the adenovirus major late promoter (ML). (B) The TRD of MeCP2 when thethered to a Gal4-binding domain can repress transcription from a template containing Gal4-binding sites. Reaction conditions were as in Figure 1. Lane 1, standard transcription reaction; lanes 2–6, 50, 100, 200, 500 and 1000 ng of Gal4–TRD was preincubated with the templates for 30 min at room temperature, prior to adding the transcription factors. (C) Quantitation of transcription from the pG5ML template relative to pMLΔ53 template expressed as fold reduction relative to Gal4–TRD mass per reaction. (D) Gal4–TRD protein aggregates its target template. Gal4–TRD was preincubated with the DNA templates as in the transcription assay. The complexes were resolved in a 0.7% agarose gel. Lanes 1–6, labeled pMLΔ53 template; lanes 7–12, labeled pG5ML template; lanes 1 and 7, no addition of Gal4–TRD; lanes 2 and 8; 50 ng; lanes 3 and 9, 100 ng; lanes 4 and 10, 200 ng; lanes 5 and 11, 500 ng; lanes 6 and 12, 1000 ng.
We made use of selectively radiolabeled mixtures of template DNA to test by agarose gel shift for selective association of Gal4–TRD with the pG5ML template. We find that the pMLΔ53 template remains substantially unbound in the presence of Gal4–TRD (Fig. 5D, lanes 2–6), whereas the pG5ML template is assembled into large nucleoprotein complexes (Fig. 5D, lanes 8–12). Under conditions of optimal selectivity for transcriptional silencing (Fig. 5D, lanes 11 and 12) we find that the distribution of these complexes between insoluble aggregates and material that migrates into the gel maxtrix is very similar to that observed on methylated templates in the presence of MeCP2 (Fig. 1B, lanes 5 and 6).
In earlier work the TRD of MeCP2 has been shown to interact with Sin3A and recruit histone deacetylase (24,25). In this purified transcription system chromatin assembly, as assayed by DNA supercoiling and micrococcal nuclease digestion, does not occur (data not shown). In addition, in contrast to in vivo results (24,25), the addition of the histone deacetylase inhibitor Trichostatin A has no influence on transcriptional silencing by Gal4–TRD (data not shown). We conclude that Gal4–TRD is sufficient to confer transcriptional repression in the absence of chromatin assembly.
We next asked whether this repression might be relieved by the addition of additional TFIIB to the reaction. Under our reaction conditions TFIIB is not normally a limiting component (27,36). We found that neither MeCP2-mediated repression of methylated pMLΔ53 templates nor Gal4–TRD-mediated repression of pG5ML templates could be relieved by addition of excess TFIIB (data not shown). We next attempted to relieve repression using TFIIB in combination with other purified transcription factors and failed to recover transcription (data not shown). This failure could be due to the effects of Gal4–TRD or MeCP2, either bound to DNA or free in solution, that preclude the action of TFIIB at the promoter (see Discussion). Complementation of the purified system with crude HeLa cell nuclear extract relieves silencing mediated either by MeCP2 on methylated pMLΔ53 templates (Fig. 6A, lanes 3 and 4) or by Gal4–TRD on the pG5ML template (Fig. 6B, lanes 3 and 4). Note that addition of MeCP2 to the mixture of unmethylated pG5ML and methylated pMLΔ53 templates leads to the activation of unmethylated pG5ML transcription (Fig. 6A, compare lanes 1 and 2). This indicates that some component of the basal transcriptional machinery is limiting and that the two templates are competing for this component. Association of MeCP2 with the methylated template eliminates this competition. This result also suggests that MeCP2 is not titrating limiting transcription factors from solution, but is functioning to repress transcription only on the methylated DNA template to which it is bound. The relief of trancriptional repression by the HeLa nuclear extract occurs with the templates remaining in large nucleoprotein complexes (data not shown). We have not yet been able to identify the activity in the nuclear extract that alleviates repression in vitro. We conclude that the TRD of MeCP2 can repress transcription at least in part through interactions with a key component of the basal transcriptional machinery, TFIIB.
Figure 6.
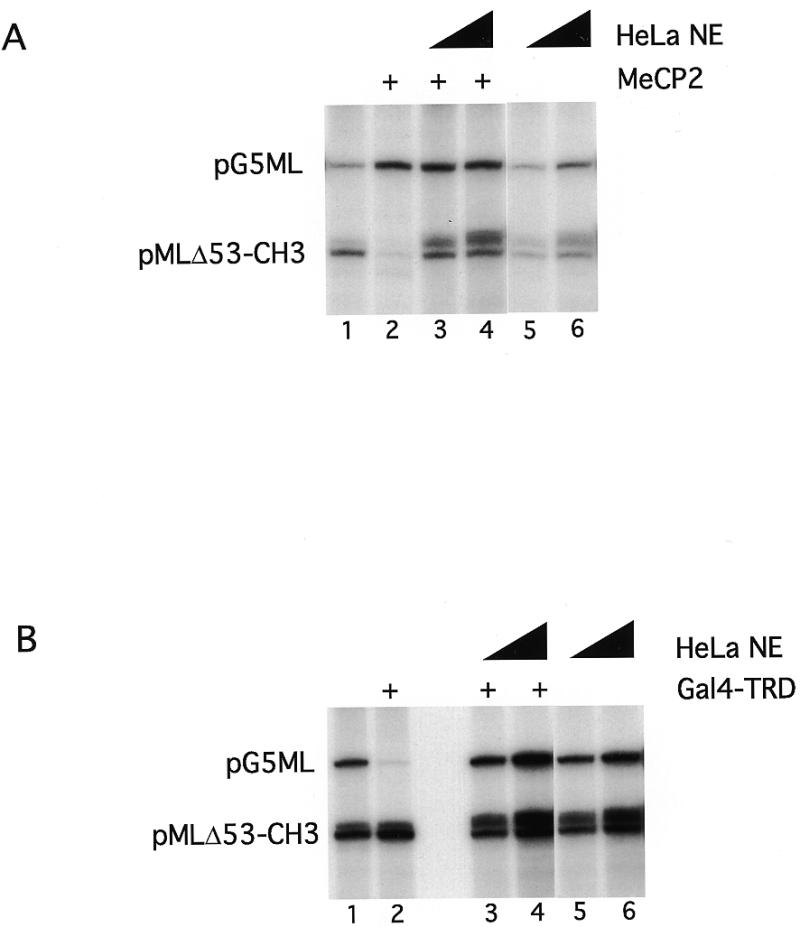
HeLa nuclear extract can rescue MeCP2- and Gal4–TRD-induced repression. (A) Increasing amounts of HeLa nuclear extract (1 and 5 µg, respectively) were added to the standard transcription reaction in the presence (lanes 3 and 4) or absence (lanes 5 and 6) of 200 ng MeCP2. As controls lane 1 contains no MeCP2 and no nuclear extract, lane 2 contains 200 ng MeCP2 and no nuclear extract. (B) As (A) except that Gal4–TRD (1 µg) was used as repressor.
DISCUSSION
Recent work strongly indicates that MeCP2 can function to repress transcription in vivo at least in part through the recruitment of histone deacetylase (24,25). However deacetylase inhibitors do not completely relieve transcriptional silencing of methylated DNA (24,25). In addition, several in vivo examples exist where inhibition of methyltransferase activity relieves transcriptional silencing of particular genes while inhibition of histone deacetylase in isolation is without effect (26,37). These observations clearly indicate that MeCP2, or indeed other MBD proteins such as MBD2 and MBD3 that also associate with histone deacetylase (38,39), may function to repress transcription through additional pathways (5,18).
In our experiments we have made use of a simplified system in which purified transcription factors, naked DNA and MeCP2 are used to examine the basis of the transcriptional repression of methylated DNA templates. The first significant observation is that MeCP2 will selectively repress transcription from methylated DNA in such a system (Figs 1–3, 5 and 6) and that this repression occurs during preinitiation complex assembly (Fig. 3). This indicates that MeCP2 directly prevents a component of the basal transcriptional machinery from functioning. In addition, this result suggests that neither chromatin assembly nor abundant quantities of complex co-repressors are necessarily required for MeCP2 to function. Previous experiments have made use of nuclear extracts in which both chromatin components and co-repressors could potentially contribute to the observed repression mediated by MeCP2 (see discussions in 18,19). The second significant observation is that the transcriptional repression domains of MeCP2 when tethered to a Gal4 DNA-binding domain can confer transcriptional repression in this in vitro system and that the TRD interacts with TFIIB, but not with other components of the basal machinery (Figs 4 and 5). This result indicates that MeCP2 may interfere with transcription through direct contact with the basal transcriptional machinery.
We have been unable to rescue transcriptional repression through the addition of additional TFIIB. However, this failure may be due to the large concentrations of either MeCP2 or Gal4–TRD that accumulate on the template DNA and that are necessary to achieve selective transcriptional repression (Figs 1, 2 and 5). Typically 500 ng of MeCP2 or Gal4–TRD need to be incubated with 200 ng of the template mixture to achieve selective transcriptional repression. We typically add 20 ng of recombinant TFIIB per reaction, however, even 200 ng of TFIIB does not rescue repression (data not shown). Our experience is that larger excesses of TFIIB have a non-specific inhibitory effect on the transcription process, potentially through the squelching of activities of other components of the basal machinery. For both MeCP2- and Gal4–TRD-mediated repression, transcription is inhibited under conditions where the proteins are interacting with many sites on a template DNA molecule in large nucleoprotein complexes. Thus it may be difficult to achieve TFIIB excess on the relevant promoter if many DNA-bound TRDs are competing for association with this key transcription factor. It is important to note that the transcriptional repression is specific for the template to which the repressor is bound and is not a general non-specific inhibitor of transcription factor function. This state of repression is reversible because the addition of HeLa nuclear extract can reactivate transcription from the MeCP2- or Gal4–TRD-bound template (Fig. 6). This result occurs with the template remaining in this large nucleoprotein complex, indicating that repression under these conditions is unlikely to occur solely through precipitation or aggregation of the template DNA. We have established through immunoblotting that the HeLa nuclear extract does not contain more TFIIB than is added to the reaction in recombinant form (data not shown). It seems probable that some other abundant component of the nuclear extract might compete with TFIIB for association with the TRD, thereby releasing the transcription factor and relieving repression. One candidate would be Sin3A itself, which is abundant and binds to the TRD (24,25). However Sin3A also interacts with TFIIB to inhibit transcription (34). Future experiments will examine other regulatory factors aside from TFIIB and Sin3A that interact with the TRD of MeCP2. It will also be important to investigate the consequence of the Rett syndrome mutations of MeCP2 for interaction with TFIIB and Sin3A and for the formation of these large nucleoprotein complexes. The formation of extended MeCP2–DNA complexes may reflect some measure of cooperativity in DNA binding, as for histone H1 (40,41). These interactions might also be influenced by Rett syndrome mutations.
The association of MeCP2-driven transcriptional repression with the selective enrichment of MeCP2 on methylated templates in the form of large nucleoprotein complexes may have biological relevance in vivo. It has been proposed that local concentrations of transcriptional repressors might reinforce a state of epigenetic silencing (42,43). MeCP2 accumulates in foci within the mammalian cell nucleus, reflecting such a local concentration (14). Since MeCP2 binds to nucleosomal DNA with comparable affinity as to naked DNA (12), such a local concentration of MeCP2 in the nucleus might lead to similar functional consequences as seen for the large MeCP2–DNA complexes that assemble in our experiments. Many MeCP2 molecules bound to methylated chromatin will serve to concentrate many transcriptional repression domains, thereby potentially presenting an impenetrable barrier to the assembly of a functional preinitiation complex.
Our results indicate that purified transcription systems will provide a useful tool in determining the functional properties of MeCP2 and for the future study of how these functions might be altered in Rett syndrome.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Drs Esteban Ballestar, Simon Chandler and, especially, Hui Ge for advice. We are grateful to Ms Kieu Pham for manuscript preparation.
REFERENCES
- 1.Amir R.E., Van den Veyver,I.B., Wan,M., Tran,C.Q., Francke,U. and Zoghbi,H.Y. (1999) Nature Genet., 23, 185–188. [DOI] [PubMed] [Google Scholar]
- 2.Wan M., Lee,S.S., Zhang,X., Houwink-Manville,I., Song,H.R., Amir,R.E., Budden,S., Naidu,S., Pereira,J.L., Lo,I.F., Zoghbi,H.Y., Schanen,N.C. and Francke,U. (1999) Am. J. Hum. Genet., 65, 1520–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tate P., Skarnes,W. and Bird,A. (1996) Nature Genet., 12, 205–208. [DOI] [PubMed] [Google Scholar]
- 4.Baylin S.B. (1999) Semin. Cancer Biol., 9, 327–328. [DOI] [PubMed] [Google Scholar]
- 5.Bird A.P. and Wolffe,A.P. (1999) Cell, 99, 451–454. [DOI] [PubMed] [Google Scholar]
- 6.Jones P.A. and Laird,P.W. (1999) Nature Genet., 21, 163–167. [DOI] [PubMed] [Google Scholar]
- 7.Lewis J.D., Meehan,R.R., Henzel,W.J., Maurer-Fogy,I., Jeppesen,P., Klein,F. and Bird,A. (1992) Cell, 69, 904–914. [DOI] [PubMed] [Google Scholar]
- 8.Hendrich B. and Bird,A. (1998) Mol. Cell. Biol., 18, 6538–6547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ohki I., Shimotake,N., Fujita,N., Nakao,M. and Shirakawa,M. (1999) EMBO J., 18, 6653–6661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wakefield R.I., Smith,B.O., Nan,X., Free,A., Steriou,A., Uhrin,D., Bird,A.P. and Barlow,P.N. (1999) J. Mol. Biol., 291, 1055–1065. [DOI] [PubMed] [Google Scholar]
- 11.Nan X., Meehan,R.R. and Bird,A. (1993) Nucleic Acids Res., 21, 4886–4892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chandler S.P., Guschin,D., Landsberger,N. and Wolffe,A.P. (1999) Biochemistry, 38, 7008–7018. [DOI] [PubMed] [Google Scholar]
- 13.Nan X., Champoy,F.J. and Bird,A. (1997) Cell, 88, 471–481. [DOI] [PubMed] [Google Scholar]
- 14.Nan X., Tate,P., Li,E. and Bird,A.P. (1996) Mol. Cell. Biol., 16, 414–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bird A.P. (1995) Trends Genet., 11, 94–100. [DOI] [PubMed] [Google Scholar]
- 16.Kass S.U., Pruss,D. and Wolffe,A.P. (1997) Trends Genet., 13, 444–449. [DOI] [PubMed] [Google Scholar]
- 17.Razin Z. (1998) EMBO J., 17, 4905–4908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nan X., Cross,S. and Bird,A. (1998) Novartis Found. Symp., 214, 6–16. [DOI] [PubMed] [Google Scholar]
- 19.Kass S.U. and Wolffe,A.P. (1998) Novartis Found. Symp., 214, 22–35. [DOI] [PubMed] [Google Scholar]
- 20.Iguchi-Arigan S.M.M. and Schaffner,W. (1989) Genes Dev., 3, 612–619. [DOI] [PubMed] [Google Scholar]
- 21.Meehan R.R., Lewis,J.D. and Bird,A.P. (1992) Nucleic Acids Res., 20, 5085–5092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buschhausen G., Wittig,B., Graessmann,M. and Graessmann,A. (1987) Proc. Natl Acad. Sci. USA, 84, 1177–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kass S.U., Landsberger,N. and Wolffe,A.P. (1997) Curr. Biol., 7, 157–165. [DOI] [PubMed] [Google Scholar]
- 24.Jones P.L., Veenstra,G.J., Wade,P.A., Vermaak,D., Kass,S.U., Landsberger,N., Strouboulis,J. and Wolffe,A.P. (1998) Nature Genet., 19, 187–191. [DOI] [PubMed] [Google Scholar]
- 25.Nan X., Ng,N.H., Johnson,C.A., Laherty,C.D., Turner,B.M., Eisenman,R.N. and Bird,A. (1998) Nature, 393, 386–389. [DOI] [PubMed] [Google Scholar]
- 26.Coffee B., Zhang,F., Warren,S.T. and Reines,D. (1999) Nature Genet., 22, 98–101. [DOI] [PubMed] [Google Scholar]
- 27.Ge H., Martinez,E., Chiang,C.M. and Roeder,R.G. (1996) Methods Enzymol., 274, 41–57. [DOI] [PubMed] [Google Scholar]
- 28.Nightingale K. and Wolffe,A.P. (1995) J. Biol. Chem., 270, 4197–4200. [DOI] [PubMed] [Google Scholar]
- 29.Almouzni G. and Wolffe,A.P. (1993) Genes Dev., 7, 2033–2047. [DOI] [PubMed] [Google Scholar]
- 30.Clark D.J. and Wolffe,A.P. (1991) EMBO J., 10, 3419–3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lansberger N. and Wolffe,A.P. (1995) Mol. Cell. Biol., 15, 6013–6024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carey M., Leatherwood,J. and Ptashne,M. (1990) Science, 247, 710–712. [DOI] [PubMed] [Google Scholar]
- 33.Li C. and Manley,J.L. (1998) Mol. Cell. Biol., 18, 3771–3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wong C.W. and Manley,J.L. (1998) Mol. Cell. Biol., 18, 5500–5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Muscat G.E., Burke,L.J. and Downes,M. (1998) Nucleic Acids Res., 26, 2899–2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Imhof A., Yang,X.J., Ogryzko,V.V., Nakatani,Y., Wolffe,A.P. and Ge,H. (1997) Curr. Biol., 7, 689–692. [DOI] [PubMed] [Google Scholar]
- 37.Cameron E.E., Bachman,K.E., Myhanen,S., Herman,J.G. and Baylin,S.B. (1999) Nature Genet., 21, 103–107. [DOI] [PubMed] [Google Scholar]
- 38.Ng H.H., Zhang,Y., Hendrich,B., Johnson,C.A., Turner,B.M., Erdjument-Bromage,H., Tempst,P., Reinberg,D. and Bird,A. (1999) Nature Genet., 23, 58–61. [DOI] [PubMed] [Google Scholar]
- 39.Wade P.A., Gegonne,A., Jones,P.L., Ballestar,E., Aubry,F. and Wolffe,A.P. (1999) Nature Genet., 23, 62–66. [DOI] [PubMed] [Google Scholar]
- 40.Draves P.H., Lowary,P.T. and Widom,J. (1992) J. Mol. Biol., 225, 1105–1121. [DOI] [PubMed] [Google Scholar]
- 41.Thomas J.P., Rees,C. and Finch,J.T. (1992) Nucleic Acids Res., 20, 187–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wolffe A.P. and Matzke,M.-A. (1999) Science, 286, 481–486. [DOI] [PubMed] [Google Scholar]
- 43.Gasser S.M., Gotta,M., Renauld,H., Laroche,T. and Cockell,M. (1998) Novartis Symp., 214, 114–126. [DOI] [PubMed] [Google Scholar]