Proteolytic Activation of Monocyte Chemoattractant Protein-1 by Plasmin Underlies Excitotoxic Neurodegeneration in Mice (original) (raw)
Abstract
Exposure of neurons to high concentrations of excitatory neurotransmitters causes them to undergo excitotoxic death via multiple synergistic injury mechanisms. One of these mechanisms involves actions undertaken locally by microglia, the CNS-resident macrophages. Mice deficient in the serine protease plasmin exhibit decreased microglial migration to the site of excitatory neurotransmitter release and are resistant to excitotoxic neurodegeneration. Microglial chemotaxis can be signaled by the chemokine monocyte chemoattractant protein-1 (MCP-1)/CCL2 (CC chemokine ligand 2). We show here that mice genetically deficient for MCP-1 phenocopy plasminogen deficiency by displaying decreased microglial recruitment and resisting excitotoxic neurodegeneration. Connecting these pathways, we demonstrate that MCP-1 undergoes a proteolytic processing step mediated by plasmin. The processing, which consists of removal of the C terminus of MCP-1, enhances the potency of MCP-1 in in vitro migration assays. Finally, we show that infusion of the cleaved form of MCP-1 into the CNS restores microglial recruitment and excitotoxicity in plasminogen-deficient mice. These findings identify MCP-1 as a key downstream effector in the excitotoxic pathway triggered by plasmin and identify plasmin as an extracellular chemokine activator. Finally, our results provide a mechanism that explains the resistance of plasminogen-deficient mice to excitotoxicity.
Keywords: MCP-1, plasmin, excitotoxicity, microglia, mice, neurodegeneration
Introduction
Excitotoxicity results from glutamate-triggered elevations in neuronal sodium or calcium (Rothman, 1985; Olney, 1986; Siman and Noszek, 1988; Lazarewicz et al., 1990; Koh and Choi, 1991; Lee et al., 1991; Strijbos et al., 1996), followed by intercellular signaling involving macroglia and microglia, ultimately resulting in neurodegeneration (Kreutzberg, 1996). Excitotoxicity is a component of several CNS pathologies (Dirnagl et al., 1999; Ferris, 2003; Srinivasan et al., 2005).
Mice deficient in the protease tissue plasminogen activator (tPA), or its substrate plasminogen (plg), are resistant to excitotoxicity (Tsirka et al., 1995, 1997). The plg-to-plasmin conversion by tPA results in laminin degradation (Chen and Strickland, 1997) and anoikis (Werb, 1997). Furthermore, the robust microglial activation that accompanies excitotoxicity and contributes to cell death is attenuated in tPA−/− and plg−/− mice.
Microglia normally perform surveillance functions, but when CNS homeostasis is disturbed, microglia activate by changes in gene expression, morphology, and proliferation, and migrate to injury sites (Andersson et al., 1991; Davalos et al., 2005; Nimmerjahn et al., 2005). Activated microglia are at lesion sites in neuropathological conditions, and mediate excitotoxicity; delay of microglial activation during excitotoxic injury reduces neurodegeneration (Rogove and Tsirka, 1998; Eljaschewitsch et al., 2006). Microglial increase at injury sites results from recruitment from other CNS regions or from the bloodborne monocyte population by chemotaxis. Chemokines, which are small, basic proteins produced during injury and infection, orchestrate chemotaxis as a protective response to injury (Hanisch, 2002). However, the recruited cells can also have harmful consequences by promoting toxicity (Nau, 2003). Monocyte chemoattractant protein-1 (MCP-1) is a potent chemoattractant for monocytes/microglia and is purported to promote monocyte infiltration into damaged CNS tissue (Gerard and Rollins, 2001). Studies using neonatal rats suggest that MCP-1 inhibition protects against NMDA-induced toxicity (Galasso et al., 2000a). Different forms of CNS injury (trauma, infection, hypoxia, peripheral nerve axotomy) elicit rapid upregulation of MCP-1 expression; neurons and astrocytes increase MCP-1 mRNA within hours after injury, before the accumulation of inflammatory mononuclear cells and with an intact blood–brain barrier (Mahad and Ransohoff, 2003). Full-length, pre-MCP-1 becomes secreted after proteolytic removal of its signal peptide to generate “mature” MCP-1 (Yoshimura et al., 1989). Like most chemokines, MCP-1 functions as a homodimer (Zhang et al., 1996). Dimeric MCP-1 activates the receptor CC chemokine receptor 2 (CCR2), which is expressed by microglia and astrocytes (Banisadr et al., 2002). Additionally, monomeric MCP-1 can bind to CCR2 (Paavola et al., 1998).
The chemoattractant activity of the mature form of several chemokines is increased after a second extracellular cleavage (van den Steen et al., 2000). Plasmin activates the chemokines cCAF (chicken chemotactic and angiogenic factor) and hemofiltrate CC chemokine (Martins-Green, 2001; Vakili et al., 2001). Lack of such activation could explain the decreased immune cell recruitment in plg−/− mice (Ploplis et al., 1998).
These observations suggested that a requisite chemoattractant is absent or dysfunctional in plg−/− mice. Herein, we investigated whether defects in microglia/monocyte recruitment during excitotoxicity in plg−/− mice could stem from decreased chemoattractant activation. We found that plasmin can potentiate MCP-1 attraction of microglia to the site of injury.
Materials and Methods
MCP-1 ELISA.
Brain homogenates at the level of the hippocampus were prepared from injected animals in 0.25% Triton X-100/PBS. Protein concentration was calculated using the Bio-Rad (Hercules, CA) DC Protein Assay. Samples were diluted to 10 mg/ml, of which 100 μl was assayed by mouse MCP-1 solid-phase sandwich ELISA with sensitivity <9 pg/ml, according to the manufacturers' instructions (Biosource, Camarillo, CA). Hippocampal extracts isolated from MCP-1-deficient mice were used as negative controls (MCP-1 at the injected side, −0.42 pg/mg of total protein; MCP-1 at the uninjected side, −1 pg/mg of total protein).
Cell culture.
N9 microglia were generously provided by Drs. Steve Barger (University of Arkansas, Fayetteville, AR) and Paola Ricciardi-Castagnoli (University of Milano-Bicocca, Milan, Italy). The cells were maintained in modified Eagle's medium supplemented with 10% fetal calf serum and 20 μm β-mercaptoethanol in a humidified 37°C/5% CO2 incubator. Primary microglia and hippocampal neurons were prepared from mixed cortical cultures (Siao and Tsirka, 2002).
The drop migration assays were performed as published with minor modifications (Milner et al., 1996). Briefly, microglia (primary or N9) were resuspended in the appropriate medium and mixed with cooled low melting point agarose. The cell/agarose mix was allowed to solidify and 25 μl of medium containing 2 ng of MCP-1, plasmin-processed MCP-1, and/or 6 ng of neutralizing anti-MCP-1 antibody was placed by the drop and allowed to dry. Then, the appropriate medium was placed over the cells and migration of microglia out of the drop was followed over time.
The chemotactic chamber (Boyden; NeuroProbe, Cabin John, MD) assays was performed with equal molar concentrations of full-length (FL), K104A, and K104Stop MCP-1. Microglia were separated by a 5.0 μm filter. Migration proceeded for 2 h at 37°C. Cells that did not migrate into the membrane were wiped off and cells that migrated into or through the membrane were stained with hematoxylin. The membrane was photographed at 100× magnification. Total migration was quantified by measuring stain intensity using Scion Image (Scion, Frederick, MD). The background for random cell movement (cells responding to buffer only) was subtracted; N = 3 per condition. Data are mean ± SD.
The agarose bead assay allows for a qualitative evaluation of migration also revealing microglial morphology along the way. The Boyden chemotactic chamber assay provides a more sensitive, unbiased, and accurate quantification of the cell migration. In combination, the two assays complement each other and further confirm the results observed.
In vivo studies (kainate).
Adult wild-type (C57BL6; wt), MCP-1−/−, tPA−/−, or plg−/− mice were obtained from The Jackson Laboratory (Bar Harbor, ME) and bred in-house at Stony Brook. The MCP-1−/−, tPA−/−, or plg−/− animals have been backcrossed to the C57BL background for 10, 13, and 12 generations, respectively. The experiments were performed in accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals and the institutional guidelines established by the Division of Laboratory Animal Research at Stony Brook. All efforts were made to minimize the use and suffering of animals. Mice weighing 20–25 g were injected intraperitoneally with atropine (0.6 μg/g of body weight) and deeply anesthetized with 2.5% avertin (0.02 ml/g of body weight). At stereotaxic coordinates of bregma, −2.5 mm, and medial/lateral, 1.7 mm, a small burr hole was made. PBS or kainate (KA), purchased from either Sigma (St. Louis, MO) or Alexis (San Diego, CA) and tested for equivalence in potency, either at 1.0 or 0.75 nmol (Sigma) or 1.5 or 0.75 nmol (Alexis) was delivered in 300 nl of PBS at a depth of 1.6 mm over 60 s, and the needle remained in place for an extra 2 min to prevent reflux. Five days after injection, the mice were killed, and their brains were removed and fixed overnight in 4% paraformaldehyde. Coronal sections (25 μm) through the hippocampus were stained with cresyl violet to quantify neuronal survival (Tsirka et al., 1995). Five sections from the dorsal hippocampus of each genotype and treatment of mice were matched, and the linear distances of intact (completely spared), partially lost (few intact neurons present), and lost (completely eliminated) pyramidal cell layer were determined on each section by an evaluator blinded to the treatment/genotype. Distances were measured using the freeware NIH Image 1.62f/Scion Image. Briefly, the file was loaded into the software and the freehand tool used to outline the region(s) of neuronal loss, and measured as arbitrary length units. Then, the entire hippocampus is measured in the same manner, and the percentage loss or survival of neurons on the injected side is calculated, as described previously (Tsirka et al., 1995). Values are reported as the mean ± SD. Confirmation of cell death was additionally visualized using terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) assay (for example, see Fig. 6).
Figure 6.
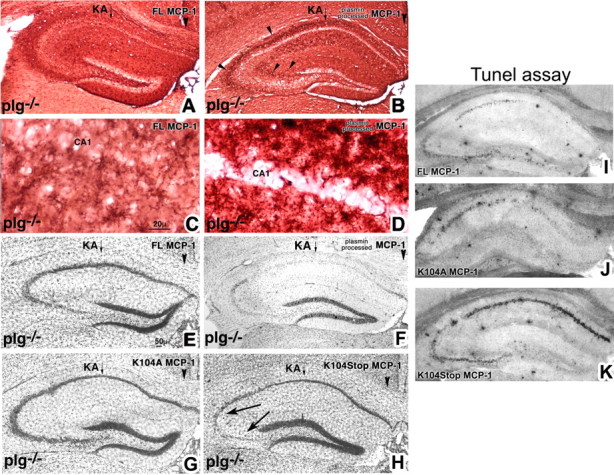
Plasmin-mediated cleavage of MCP-1 induces microglial migration and promotes excitotoxicity in plg−/− mice. A–H, FL, K104A, and K104Stop MCP-1 and plasmin-processed MCP-1 were delivered into plg−/− mice (n = 5) 2 d before intrahippocampal KA injection. Brains were processed 5 d later, using F4/80 to visualize microglia accumulating at the regions of cell death (A–D) or cresyl violet to determine the extent of cell death (E–H). There was no effect of the infusion or injection on the uninjected hippocampus. The panels display photomicrographs of coronal sections taken through the hippocampal injected side. The arrowheads in B point to the densely F4/80-immunopositive layers of microglia accumulated around the site of injury (CA1–CA3). Neuronal death was confirmed by TUNEL staining (I–K).
Neutralizing anti-MCP-1 antibody (nAb; 250 μg/ml; R&D Systems, Minneapolis, MN), normal goat serum (50% in PBS), mouse IgG1 (250 μg/ml), α2-antiplasmin (1 mg/ml), or recombinant MCP-1 [digested and full-length (2 μg/ml)] was delivered to the hippocampus via microosmotic pumps (Durect, Cupertino, CA) for 7 d delivering 0.5 μl/h to the following stereotactic coordinates: bregma, –2.5 mm; medial/lateral, 0.5 mm; dorsoventral, –1.6 mm. Two days later, the mice were injected with KA.
Generation of MCP-1.
For mass spectrometry and kinetic analysis, MCP-1 was subcloned without the signal peptide in a pET vector with an N-terminal His-tag. MCP-1, K104A MCP-1, and K104Stop MCP-1 were expressed in BL21 cells and purified using a cobalt affinity resin (Clontech, Cambridge, UK) according to the manufacturer's instructions. The fractions were analyzed on 16% Tris-Tricine SDS-PAGE. Fractions containing MCP-1 were pooled, loaded on MonoS cation exchanger, and eluted in an increasing gradient of KCl. Identity of the purified protein was confirmed by immunoblotting with 1:200 anti-MCP-1 (Santa Cruz Biotechnology, Santa Cruz, CA) or fluorescently labeled 1:10,000 anti-6His (Santa Cruz Biotechnology).
Recombinant MCP-1 (6His-MCP-1, with the signal peptide) was also generated by in vitro transcription (Ambion, Austin, TX)/translation (Promega, Madison, WI; wheat germ lysates). The [35S]methione-labeled MCP-1 was digested with 2.5 ng to 5 μg of tPA (Genentech, South San Francisco, CA) or 5 mU of plasmin (Sigma) for 30 min at 37°C. The digested products were separated on 12% SDS-PAGE and exposed to BioMax Film (Kodak, Rochester, NY).
In experiments involving functional studies of digested MCP-1, 400 ng of recombinant MCP-1 (made as above) was digested with 5 mU of plasmin for 30 min at 37°C. Plasmin activity was eliminated through reaction with a 10× excess of the irreversible inhibitor phenylmethylsulfonylfluoride (PMSF). Excess PMSF was removed from the mixture by centrifugation through a Microcon-3 filter (Millipore, Bedford, MA). The “undigested MCP-1” was subjected to the same procedure omitting plasmin. Absence of endotoxin was confirmed using the Pyrogent Plus kit (Siao and Tsirka, 2002). Delivery of PMSF-inactivated plasmin (at the concentration used to digest the MCP-1) had no effect on microglial migration or neurotoxicity (data not shown).
Mass spectometry.
Purified MCP-1 was mixed with 0.15 mU of plasmin (or buffer) and left at room temperature for 30 min. The digestion was terminated by addition of PMSF, and samples were prepared according to the Stony Brook Proteomics Facility website protocol. The samples were reduced, mixed with trifluoroacetic acid (Sigma), purified, concentrated, desalted using a C4 Zip Tip (Millipore), and eluted in 50% acetonitrile. They were mixed 1:5 with Sinapic Acid matrix and analyzed on a Voyager matrix-assisted laser desorption ionization (MALDI) machine.
Statistics.
Statistics were performed using one-way ANOVA followed by a Bonferroni–Dunn test for multiple comparisons within a group, or a two-tailed t test for comparisons between groups; p < 0.05 was considered significant. Error bars indicate the SEM.
Results
MCP-1 expression increases after excitotoxic challenge
MCP-1 expression levels increase rapidly and transiently in several paradigms of brain injury (Ghirnikar et al., 1996; Ivacko et al., 1997; Grzybicki et al., 1998; Szaflarski et al., 1998; Galasso et al., 2000a,b; Flugel et al., 2001; Little et al., 2002). We analyzed MCP-1 expression 1 d after excitotoxic KA injection and found similarly that it increased in this setting (Fig. 1A), returning to basal levels by day 5 (Fig. 1B). During the period of upregulated expression, MCP-1 was detected by immunofluorescence (Fig. 1C) in hippocampal astrocytes and neurons. The staining pattern was diffuse, possibly reflecting an extracellular rather than intracellular distribution. As determined using reverse transcription (RT)-PCR analysis on purified primary cells, no microglial expression was detected (Fig. 1D).
Figure 1.

MCP-1 expression increases during excitotoxicity. A, Levels were determined by ELISA 24 h after PBS or KA injection unilaterally into the hippocampus of wt mice. Hippocampal homogenates were prepared from the injected and uninjected sides of the brains. Values of p are noted in the graph. B, Time course of MCP-1 increase on the injected side determined using ELISA. The day 1 (D1) levels of MCP-1 protein were set to 100. Data are the mean of results obtained from three mice ± SD. p < 0.0001 between uninjected and D1; p = 0.0001 between uninjected and D3; p = 0.28 between uninjected and D5. C, Colocalization of the astrocytic marker GFAP (green) with MCP-1 (red) in the CA3 hippocampal subfield of hippocampi from kainate-injected mice 3 d postinjection. D, RT-PCR amplification of MCP-1 from purified neurons and microglia (μglia). As controls, the purity of the cultures and quality of cDNA were confirmed using primers specific for neuronal [amyloid precursor protein (APP)] and microglial (F4/80) markers.
MCP-1 is critical for the microglial migration and subsequent neurodegeneration triggered by excitotoxic injury
Based on the literature and results above, we hypothesized that MCP-1 may function as a chemoattractant during excitotoxicity. We therefore investigated whether genetic ablation of MCP-1 altered microglial migration and excitotoxic injury progression. wt and MCP-1−/− mice were injected unilaterally in the hippocampus with KA, and injury was assessed 5 d later. Activated microglia were detected in wild-type mice as a dense band of cells surrounding the death-susceptible pyramidal neuron cell layer (Fig. 2A,C). In MCP-1−/− mice, activated microglial accumulation at these sites was diminished (Fig. 2B,D). This result is in agreement with reports indicating that the functional aspects of microglial activation in MCP-1−/− animals, measured by secretion of cytokines, are compromised after induction of activation (Rankine et al., 2006). In parallel, whereas CA1 and CA3 pyramidal neurons underwent neurodegeneration in wild-type mice (the extent of cell death reached 53 ± 9%) (Fig. 2E,G), these regions were spared in the MCP-1−/− mice (extent of cell death, 18 ± 6%) (Fig. 2F,H), consistent with our previous report that the presence of activated microglia is critical for the progression of neurodegeneration (Rogove and Tsirka, 1998). Similar results were obtained in wild-type mice after inhibiting endogenous MCP-1 by infusing neutralizing anti-MCP-1 antibody directly into the hippocampus (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), which further indicates that it is the acute lack of MCP-1 that underlies the recruitment defect in MCP-1−/− mice.
Figure 2.
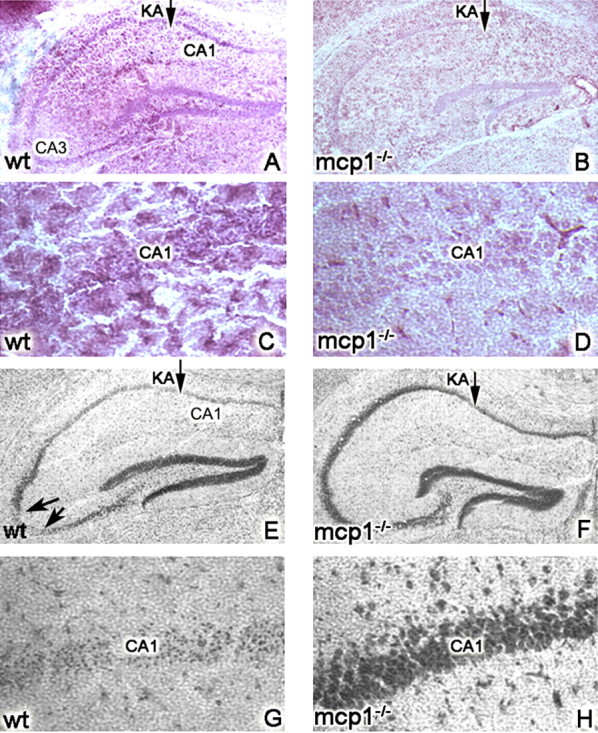
MCP-1 is required for excitotoxic death. wt (A, C, E, G) or MCP-1−/− (B, D, F, H) mice were injected unilaterally with 0.75 nmol of KA and killed at day 5. Panels display coronal sections of the injected hippocampus. A, B, E, and F are low-magnification images, and C, D, G, and H are high-magnification images. In A–D, microglia were visualized using F4/80 immunohistochemistry. In E–H, neuronal viability was visualized using cresyl violet and quantified (n = 8 mice per genotype; *p < 0.05). E, F, Representative images included in the quantification. The arrows in E point to the CA3 region, which undergoes neurodegeneration in wt animals but is spared in the MCP-1−/− mice. G, H, Extensive neurodegeneration is also observed in the CA1 region in wt mice but not in the MCP-1−/− mice.
MCP-1 is proteolytically processed by plasmin
Because MCP-1 is important for microglial recruitment during excitotoxicity, microglial migration is defective in plg−/− mice during excitotoxicity, and plasmin has been reported to activate several chemokines (Harter et al., 1994; Nufer et al., 1999; van den Steen et al., 2000; Martins-Green, 2001; Vakili et al., 2001), we explored the potential intersection between MCP-1 and plasmin. We first analyzed MCP-1 expression in mice deficient for tPA or plg. Similar to the wild-type mice, uninjected tPA−/− and plg−/− mice had very low levels of MCP-1 expression, and no MCP-1 expression was detected in MCP-1−/− mice. One day after KA injection, MCP-1 levels became elevated in tPA−/− and plg−/− mice and, in fact, were elevated higher than those observed for wild-type mice (Fig. 3A). The exaggerated upregulation of MCP-1 protein levels in tPA−/− and plg−/− mice appeared not to ensue from exaggerated increases in mRNA expression in the mutant animals (data not shown). We hypothesized, however, that the increase in MCP-1 protein might stem from a decrease in MCP-1 proteolytic processing, because it would not be an unexpected result that bioactive fragments produced from processing might have a shorter half-life than the full-length mature protein (i.e., in the absence of processing, full-length protein would accumulate). Accordingly, we tested whether MCP-1 is a direct target for tPA or plasmin processing. [35S]Met-labeled MCP-1 was generated by in vitro transcription/translation and subjected to proteolytic cleavage. The _in vitro_-translated MCP-1 migrated as a single band (Fig. 3B, lane 1) and remained intact in the presence of tPA (Fig. 3B, lane 2). However, incubation with plasmin resulted in the formation of two lower molecular weight bands accompanied by a decrease in full-length MCP-1 (arrowheads, lane 3), indicating that plasmin is capable of directly processing MCP-1. In hippocampal extracts prepared 3 d after the KA injection, MCP-1 appears degraded in wild-type KA-injected mice, but such processing is significantly decreased in injected plg−/− mice (Fig. 3C).
Figure 3.

Plasmin processes MCP-1 after excitotoxicity. A, ELISA showing the ratio of expression of MCP-1 in hippocampal lysates from tPA−/− and plg−/− mice in comparison with wild-type mice 1 d after KA injection. The experiment was performed in triplicate using three mice per genotype. The MCP-1 expression levels in wild-type mice at day 1 after KA injection were set to 100, as in Figure 1B, and the expression levels in tPA−/− and plg−/− are reported relative to those in wild-type hippocampi. Data are presented as mean ± SD. The p values between tPA−/− or plg−/− and wt samples are indicated on the graph. The p value between tPA−/− and plg−/− samples was 0.007. B, Incubation of [35S]methionine-labeled recombinant 6×-His-full-length-MCP-1 with tPA and plasmin. The reaction products were analyzed by 12% SDS-PAGE and subjected to autoradiography. 6×-His-FL-MCP-1 (−, first lane) is indicated by the uppermost arrowhead. The lower two arrowheads denote cleavage products observed after incubation with plasmin (pln). Numbers to the left indicate molecular weight markers. C, Western blot analysis of hippocampal extracts prepared 3 d after KA injection in wild-type (wt KA) and plg−/− (plg−/− KA) mice. Extracts from hippocampi from PBS-injected wt (wt PBS) and plg−/− (plg−/− PBS) mice were used to visualize the starting levels of MCP-1. Recombinant MCP-1-EGFP was used as a control for the MCP-1 antibody (Ctr) (R&D Systems). D, Time course of recombinant MCP-1 cleavage by plasmin. Reaction products were separated on a 16% Tris-Tricine gel and silver stained. Numbers on the left side denote the sizes of ladder bands. E, The bands corresponding to FL MCP-1 and the 13 kDa cleavage fragment shown in D were quantified using optical density, corrected for lane background, and normalized to time 0 value. N = 5.
To further analyze the processing, we generated recombinant MCP-1 (supplemental Fig. 2, available at www.jneurosci.org as supplemental material), which migrates on SDS-PAGE at a molecular weight of 17.5 kDa (Fig. 3D, left lane). MCP-1 incubated with plasmin was readily cleaved (Fig. 3D,E, lanes 2–5), generating fragments of ∼13 and 4 kDa, which appeared resistant to additional plasmin processing, although it should be noted that this does not address what the half-life of the fragments might be in vivo.
Plasmin processing enhances MCP-1 activity
Although plg−/− mice express more full-length MCP-1, they display defective microglial migration during excitotoxicity and defective leukocyte recruitment in general (Ploplis et al., 1998). To assess whether the diminished processing of MCP-1 by plasmin could explain this migration defect, plg−/− microglial cells were plated onto tissue culture dishes in an agarose drop adjacent to buffer solution, plasmin, MCP-1, or plasmin-processed MCP-1, overlaid with media, and cultured. Cells spotted adjacent to buffer (Fig. 4A) or plasmin (Fig. 4B) did not exhibit microglial migration (35 ± 10 and 38 ± 16 cells, respectively), nor did cells exposed to full-length MCP-1 (46 ± 13 cells) (Fig. 4C). However, when plasmin-processed MCP-1 was used, migration was readily observed (260 ± 16 cells) (Fig. 4D). These experiments demonstrate that plg−/− microglia do not have an inherent migration defect, and suggest that the decreased migration of plg−/− microglia during excitotoxicity is attributable to the absence of a chemotactic stimulus provided by plasmin-mediated cleavage of MCP-1 that leads to the activation or mobilization of MCP-1.
Figure 4.
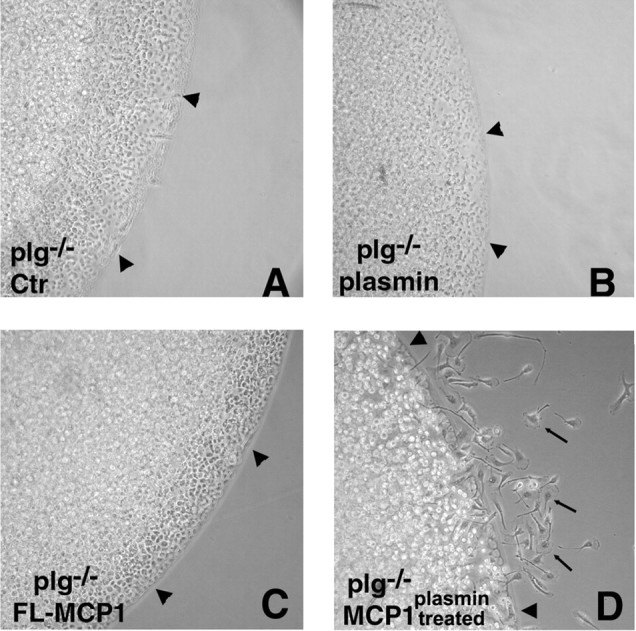
Plasmin-mediated cleavage of MCP-1 is necessary for microglial migration. Migration of plg−/− primary microglia was assessed using an in culture migration assay. The cultures were spotted adjacent to a point source of HBSS containing no addition, Ctr (A), plasmin (B), FL MCP-1 (C), or plasmin-processed MCP-1 (D), and then overlaid with medium. The arrowheads indicate the border of the agarose drop; the arrows point to migrating microglia. The number of migrating cells is reported as mean ± SD.
Plasmin processing of MCP-1 occurs at lysine 104
We set out to identify the precise cleavage site of MCP-1 by plasmin using MALDI mass spectrometry, which returned fragment sizes of 13,024 and 4405 Da (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). This finding indicated that cleavage occurs after lysine 104. To confirm that this specific site and the cleavage were important for MCP-1 chemotactic function, we generated two recombinant mutant MCP-1 proteins: a 13 kDa C-terminal deletion mutant lacking amino acids 105–137 (K104Stop MCP-1) and a 17 kDa K104A mutant allele (K104A MCP-1), in which the lysine at the cleavage site was mutated to alanine, thus eliminating cleavage by plasmin (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). If removal of the C terminus of MCP-1 via plasmin processing increases MCP-1 chemotactic potency, then the K104Stop MCP-1 mutant should be more potent than the full-length form. Conversely, the cleavage-resistant K104A MCP-1 mutant should fail to promote microglial migration. We compared the migration induced by equal molar concentrations of the FL and mutant proteins in a quantitative assay using a chemotactic Boyden chamber. Given that microglia do not produce plasmin(ogen) (Tsirka et al., 1997), no processing of recombinant wild-type MCP-1 would occur in this experiment. No migration was observed in the absence of MCP-1 (Fig. 5, Ctr). Addition of FL MCP-1 and K104A MCP-1 elicited modest and similar levels of migration. However, K104Stop MCP-1 potently increased migration. Thus, plasmin-mediated removal of the MCP-1 C terminus increases its bioactivity.
Figure 5.
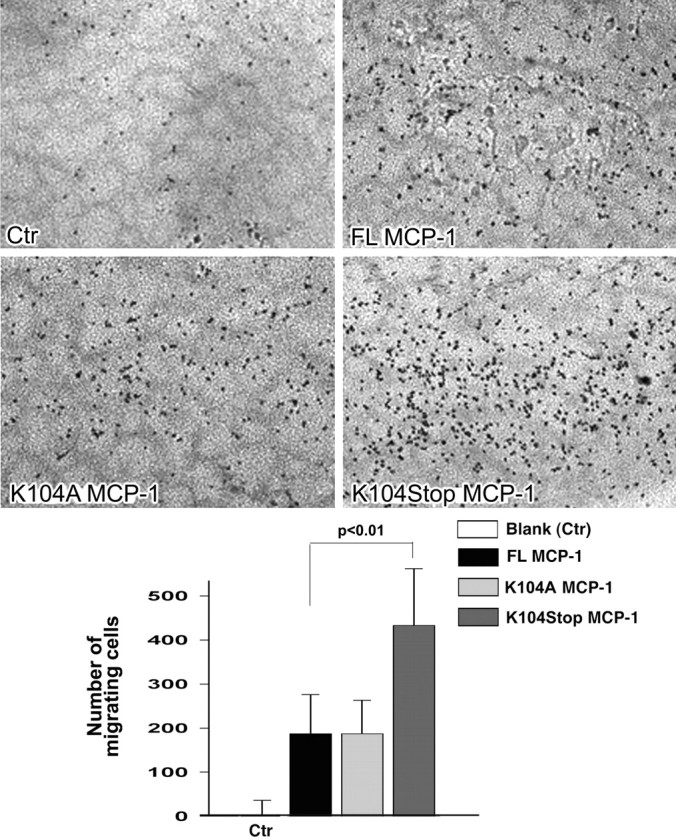
C-terminal truncation of MCP-1 increases its potency. Equal molar concentrations of FL, K104A, and K104Stop MCP-1 were tested for their ability to recruit microglia using a Boyden chamber. The cellular staining intensity of the migrating cells was quantified using Scion Image. Data are presented as the mean staining intensity ± SD (n = 3) after subtraction of background for random cell movement (cells responding to buffer only). p = 0.009 between K104Stp MCP-1 and FL MCP-1.
Plasmin processing of MCP-1 is necessary for microglia migration and excitotoxic injury progression
We and others have demonstrated that activated microglia have neurotoxic properties during excitotoxic injury and that hippocampal pyramidal neurons in plg−/− mice are resistant to excitotoxic death. Moreover, infusion of plasmin into plg−/− mice restores neuronal susceptibility to KA (Chen and Strickland 1997). If plasmin-processing of MCP-1 is required in vivo for microglial migration in response to excitotoxic injury, then accumulation of activated microglia at the site of injury should be observed in plg−/− mice infused with the K104Stop MCP-1 or plasmin-processed MCP-1. Restoration of microglial migration should then restore susceptibility of plg−/− neurons to excitotoxins if this processing is the critical function performed by plasmin. We infused either nonprocessed or plasmin-processed MCP-1 into plg−/− mice before unilateral KA injection. In mice infused with plasmin-processed MCP-1 (Fig. 6B,D), bands of microglia surrounded the injured pyramidal cell layer (Fig. 6B, arrowheads). The pattern of microglial accumulation was similar to that observed in wild-type mice (Fig. 2A). In contrast, when nonprocessed FL MCP-1 was infused, no appreciable migration of microglia into the hippocampus and pyramidal layer was observed (Fig. 6A,C). The absence of microglial migration in these mice was similar to that observed in MCP-1−/− mice (Fig. 2B). However, in agreement with our previous reports, the morphological changes indicative of microglial activation were still apparent in microglia already in close proximity to the KA injection site (Fig. 6C). Mice that received the plasmin-processed MCP-1 exhibited degeneration in the hippocampus (Fig. 6F), whereas the mice that received the nonprocessed FL MCP-1 (Fig. 6E) did not. The outcome promoted by plasmin-processed MCP-1 was dependent on the initiation of excitotoxic injury, because in the absence of KA injection, plasmin-processed MCP-1 did not elicit any demonstrable effect on migration or neuronal survival.
We also delivered the noncleavable K104A MCP-1 and the truncated K104Stop MCP-1 into plg−/− mice before intrahippocampal injection of KA. Mice that received K104Stop MCP-1 exhibited degeneration (Fig. 6H), whereas the mice that received K104A did not (Fig. 6G). TUNEL assays confirmed the presence of dying neurons in the mice that received the K104Stop MCP-1 mutant (Fig. 6K), whereas the delivery of FL MCP-1 or K104A MCP-1 resulted in very limited presence of TUNEL-positive neurons (Fig. 6I–J). In Fig. 6K, the extent of TUNEL-positive neurons is larger than what is revealed by cresyl violet, presumably reflecting cells in the process of dying. This combination of in culture and in vivo data provide evidence that processing of MCP-1 by plasmin plays a crucial role in microglial migration in the brain after injury and the subsequent neurodegeneration.
Previous studies have shown that blockade of plasmin activity (through genetic deletion or by α2-antiplasmin) in the mouse hippocampus has a neuroprotective effect (Tsirka et al., 1997). We examined the mobilization of microglia after α2-antiplasmin infusion. Although there were many microglia present in the hippocampus of the inhibitor α2-antiplasmin-infused/KA-injected mice, few if any had migrated into the pyramidal cell layer, the site of excitotoxic death (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). This finding suggests that plasmin contributes to neuronal cell death by processing the chemokine MCP-1 into a potent bioactive peptide that recruits microglia to sites of injury.
Discussion
We have shown previously that the plg activator system promotes excitotoxic death (Tsirka et al., 1995, 1997). This action is dependent on the proteolytic activity of plasmin, which can cleave laminin (Chen and Strickland, 1997), an early step in the neuronal death pathway triggered by KA. However, this finding does not explain the altered microglial response in plg−/− mice, which are known to have diminished leukocyte recruitment (Ploplis et al., 1998), nor the important role microglia play in excitotoxic injury progression (Rogove et al., 1999). The findings herein, connecting the proteolytic function of plasmin with the chemokine MCP-1, provide a novel function for plasmin during excitotoxicity and a mechanism to explain why microglial recruitment is decreased in tPA−/− and plg−/− mice. The salient novel findings of this study are as follows: (1) the proteolytic action of plasmin on MCP-1 results in the removal of the C terminus of MCP-1; (2) cleaved MCP-1 exhibits increased chemotactic potency; and (3) enhanced microglial recruitment and extended neurodegeneration is observed in the presence of plasmin-cleaved MCP-1.
So what role might the tPA/plasmin(ogen) system mediate in the neuronal death process? Our findings, as well as those of many other investigators, suggest multiple roles for this system in the excitotoxic process. As shown previously, tPA, via the generation of plasmin, might promote anoikis (Chen and Strickland, 1997), affect neuronal function by interfering with NMDA receptor function (Nicole et al., 2001; Pawlak et al., 2005), or lead to the activation of nitric oxide synthases (Parathath et al., 2006). Moreover, in a nonproteolytic manner, tPA might directly affect the function of microglia by promoting their activation (Rogove et al., 1999; Siao and Tsirka, 2002), resulting in attenuated TNF-α (tumor necrosis factor α) release and NO (nitric oxide) and reactive oxygen species production. Finally, based on the current findings, the tPA/plasmin(ogen) system may direct the migration of active microglia to the site of injury.
Because neurons cultured in isolation from microglia are sensitive to excitotoxins (Amin and Pearce, 1997), the initial excitotoxic injury may be microglial-independent necrosis that is not amenable to treatment by microglial-modulating agents. However, delay of microglial activation by the IgG-derived tripeptide MIF (monocyte inhibitory factor) (tuftsin fragment 1–3) can protect neurons (Thanos et al., 1993; Rogove and Tsirka, 1998; Wang et al., 2003; Wang and Tsirka, 2004), suggesting that not all of the neuronal death that takes place ensues from the microglial-independent necrosis pathway. Microglial and astroglial activation could potentiate the injury either by attacking otherwise salvageable neurons near the injury site, or by interacting with neurons to induce the release of soluble inflammatory modulators such as chemokines and cytokines (Glabinski and Ransohoff, 1999) that decrease neuronal excitotoxin tolerance. Our findings that migration of activated microglia exacerbates excitotoxic neuronal injury extends previous reports that microglial migration involves the poly(ADP-ribose) polymerase (PARP-1)-mediated activation of microglial CD11a. Inhibition of microglial PARP-1 blocks the microglial migration and results in protection from excitotoxicity in organotypic hippocampal slice cultures (Ullrich et al., 2001). Here, we provide an additional factor in this setting: a signaling event that originates from injured neurons and reactive astrocytes that triggers MCP-1 activation and microglial migration.
The recruited cells could derive from three separate populations: division and activation of resident microglia at the site of injury, recruitment of resident microglia from other regions of the CNS, and the recruitment of bloodborne monocytes/macrophages. Our methods do not distinguish among these subpopulations. We know of no reports attributing direct microglial mitogenic properties to MCP-1, and therefore hypothesize that the function of MCP-1 is to recruit more distant resident microglia and bloodborne monocytes. Supporting this, both chemotactic migration of resident microglia (Davalos et al., 2005; Nimmerjahn et al., 2005) and integration of blood-derived immune cells into the CNS (Massengale et al., 2005; Newman et al., 2005) have been reported, including in response to MCP-1 (Fuentes et al., 1995). It is possible that these two populations of microglia, although morphologically indistinguishable, react to the injury very differently. For example, microglia have been shown to secrete neurotrophic factors (Batchelor et al., 1999); in Alzheimer's disease, microglial activation may reduce disease severity by reducing plaque burden (Streit, 2005; Simard et al., 2006); additionally, activation of microglia with zymosan resulted in axon growth of injured retina ganglion cells (Filbin, 2006; Yin et al., 2006); however, microglia have been demonstrated to be toxic in multiple disease models (Block et al., 2006; Cardona et al., 2006; Lai and Todd, 2006), including our own studies of excitotoxicity (Rogove and Tsirka, 1998). Nevertheless, in this study, we found that the population(s) of microglia recruited, although possibly of diverse origin and exerting opposite effects on neuronal survival, had a net negative contribution.
Additional studies are underway to understand how cleavage of the C terminus from MCP-1 generates a chemotactically more potent peptide. Possibilities include increased binding or activation of its receptor, CCR2, and altered downstream signaling; altered dimerization, because chemokines can exist as dimers in solution; and/or more potent gradient formation, possibly by altering the binding of MCP-1 to the extracellular matrix (ECM). The C terminus of mouse MCP-1, approximately corresponding to the 4 kDa region removed by plasmin, although not well characterized, is heavily _O_-glycosylated (Zhang et al., 1996). It has been speculated that the glycosylation may confer stability to MCP-1 (by protecting it from proteolysis) or tether it to an immobilized surface. It has also been suggested that chemotactic gradients could be formed when needed by detaching MCP-1 from the matrix by removing the GAG tethering. Our findings demonstrate that C-terminal truncated MCP-1 is more active in vitro and in vivo; yet altering the association of MCP-1 with the ECM does not affect its chemotactic ability in vitro (Proudfoot et al., 2003; Handel et al., 2005). Therefore, this cleavage likely alters more than just the affinity of MCP-1 for the ECM, suggesting the C terminus may have additional affects on mouse MCP-1 activity. An intriguing possibility is a potential function for the 4 kDa C-terminal fragment.
Coinjection of recombinant MCP-1 into postnatal day 7 rats with NMDA exacerbates excitotoxicity injury; conversely, inhibition of MCP-1 activity with a neutralizing antibody decreases the injury (Galasso et al., 2000a). We have extended these observations into adult mice using MCP-1-deficient mice, and find that they resist excitotoxicity, supporting a neurotoxic role for MCP-1 in this injury process. Although MCP-1 functions primarily to chemoattract monocytic cells, it has also been shown to promote apoptosis when applied at higher concentrations either directly to cultured rat neurons or in vivo in the absence of any excitotoxin (Kalehua et al., 2004). However, we did not detect neuronal toxicity when we infused MCP-1 in the absence of kainate; such discrepancy could stem from differences in the MCP-1 concentrations used. In other reports, cleavage of the chemokine SDF-1 (stromal cell-derived factor 1) by matrix metalloprotease resulted in a new peptide directly toxic to neurons (Zhang et al., 2003). This does not appear to be the case in our experiments, because the delivery of K104Stop MCP-1 in the absence of KA did not cause degeneration of hippocampal neurons (Fig. 6).
Footnotes
This work was supported by an Investigator Award established by the American Heart Association and National Institutes of Health Grants 5R01NS04216804 (S.E.T.) and 5R01ES01203904 (D.F.B.). We thank Drs. Michael Frohman, Lisa Sheehan, Howard Crawford, and members of the Tsirka Laboratory for critical discussions.
References
- Amin N, Pearce B. Glutamate toxicity in neuron-enriched and neuron-astrocyte co-cultures: effect of the glutamate uptake inhibitor l-trans-pyrrolidine-2,4-dicarboxylate. Neurochem Int. 1997;30:271–276. doi: 10.1016/s0197-0186(96)00092-7. [DOI] [PubMed] [Google Scholar]
- Andersson P, Perry V, Gordon S. The kinetics and morphological characteristics of the macrophage-microglial response to kainic acid-induced neuronal degeneration. Neuroscience. 1991;42:201–214. doi: 10.1016/0306-4522(91)90159-l. [DOI] [PubMed] [Google Scholar]
- Banisadr G, Queraud-Lesaux F, Boutterin M, Pelaprat D, Zalc B, Rostene W, Haour F, Parsadaniantz S. Distribution, cellular localization and functional role of CCR2 chemokine receptors in adult rat brain. J Neurochem. 2002;81:257–269. doi: 10.1046/j.1471-4159.2002.00809.x. [DOI] [PubMed] [Google Scholar]
- Batchelor P, Liberatore G, Wong J, Porritt M, Frerichs F, Donnan G, Howells D. Activated macrophages and microglia induce dopaminergic sprouting in the injured striatum and express brain-derived neurotrophic factor and glial cell line-derived neurotrophic factor. J Neurosci. 1999;19:1708–1716. doi: 10.1523/JNEUROSCI.19-05-01708.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block M, Li G, Qin L, Wu X, Pei Z, Wang T, Wilson B, Yang J, Hong J. Potent regulation of microglia-derived oxidative stress and dopaminergic neuron survival: substance P vs. dynorphin. FASEB J. 2006;20:251–258. doi: 10.1096/fj.05-4553com. [DOI] [PubMed] [Google Scholar]
- Cardona A, Pioro E, Sasse M, Kostenko V, Cardona S, Dijkstra I, Huang D, Kidd G, Dombrowski S, Dutta R, Lee J-C, Cook D, Jung S, Lira S, Littman D, Ransohoff R. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci. 2006;9:917–924. doi: 10.1038/nn1715. [DOI] [PubMed] [Google Scholar]
- Chen Z-L, Strickland S. Neuronal death in the hippocampus is promoted by plasmin-catalyzed degradation of laminin. Cell. 1997;91:917–925. doi: 10.1016/s0092-8674(00)80483-3. [DOI] [PubMed] [Google Scholar]
- Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan WB. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8:752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- Eljaschewitsch E, Witting A, Mawrin C, Lee T, Schmidt P, Wolf S, Hoertnagl H, Raine C, Schneider-Stock R, Nitsch R, Ullrich O. The endocannabinoid anandamide protects neurons during CNS inflammation by induction of MKP-1 in microglial cells. Neuron. 2006;49:67–79. doi: 10.1016/j.neuron.2005.11.027. [DOI] [PubMed] [Google Scholar]
- Ferris SH. Evaluation of memantine for the treatment of Alzheimer's disease. Expert Opin Pharmacother. 2003;4:2305–2313. doi: 10.1517/14656566.4.12.2305. [DOI] [PubMed] [Google Scholar]
- Filbin M. How inflammation promotes regeneration. Nat Neurosci. 2006;9:715–717. doi: 10.1038/nn0606-715. [DOI] [PubMed] [Google Scholar]
- Flugel A, Hager G, Horvat A, Spitzer C, Singer GM, Graeber MB, Kreutzberg GW, Schwaiger FW. Neuronal MCP-1 expression in response to remote nerve injury. J Cereb Blood Flow Metab. 2001;21:69–76. doi: 10.1097/00004647-200101000-00009. [DOI] [PubMed] [Google Scholar]
- Fuentes ME, Durham SK, Swerdel MR, Lewin AC, Barton DS, Megill JR, Bravo R, Lira SA. Controlled recruitment of monocytes and macrophages to specific organs through transgenic expression of monocyte chemoattractant protein-1. J Immunol. 1995;155:5769–5776. [PubMed] [Google Scholar]
- Galasso JM, Liu Y, Szaflarski J, Warren JS, Silverstein FS. Monocyte chemoattractant protein-1 is a mediator of acute excitotoxic injury in neonatal rat brain. Neuroscience. 2000a;101:737–744. doi: 10.1016/s0306-4522(00)00399-7. [DOI] [PubMed] [Google Scholar]
- Galasso JM, Miller MJ, Cowell RM, Harrison JK, Warren JS, Silverstein FS. Acute excitotoxic injury induces expression of monocyte chemoattractant protein-1 and its receptor, CCR2, in neonatal rat brain. Exp Neurol. 2000b;165:295–305. doi: 10.1006/exnr.2000.7466. [DOI] [PubMed] [Google Scholar]
- Gerard C, Rollins BJ. Chemokines and disease. Nat Immunol. 2001;2:108–115. doi: 10.1038/84209. [DOI] [PubMed] [Google Scholar]
- Ghirnikar RS, Lee YL, He TR, Eng LF. Chemokine expression in rat stab wound brain injury. J Neurosci Res. 1996;46:727–733. doi: 10.1002/(SICI)1097-4547(19961215)46:6<727::AID-JNR9>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Glabinski AR, Ransohoff RM. Sentries at the gate: chemokines and the blood-brain barrier. J Neurovirol. 1999;5:623–634. doi: 10.3109/13550289909021291. [DOI] [PubMed] [Google Scholar]
- Grzybicki D, Moore S, Schelper R, Glabinski A, Ransohoff R, Murphy S. Expression of monocyte chemoattractant protein (MCP-1) and nitric oxide synthase-2 following cerebral trauma. Acta Neuropathol. 1998;95:98–103. doi: 10.1007/s004010050770. [DOI] [PubMed] [Google Scholar]
- Handel T, Johnson Z, Crown S, Lau E, Sweeney M, Proudfoot A. Regulation of protein function by glycosaminoglycans as exemplified by chemokines. Annu Rev Biochem. 2005;74:385–410. doi: 10.1146/annurev.biochem.72.121801.161747. [DOI] [PubMed] [Google Scholar]
- Hanisch UK. Microglia as a source and target of cytokines. Glia. 2002;40:140–155. doi: 10.1002/glia.10161. [DOI] [PubMed] [Google Scholar]
- Harter L, Petersen F, Flad H, Brandt E. Connective tissue-activating peptide III desensitizes chemokine receptors on neutrophils—requirement for proteolytic formation of the neutrophil-activating peptide 2. J Immunol. 1994;153:5698–5708. [PubMed] [Google Scholar]
- Ivacko J, Szaflarski J, Malinak C, Flory C, Warren J, Silverstein F. Hypoxic-ischemic injury induces monocyte chemoattractant protein-1 expression in neonatal rat brain. J Cereb Blood Flow Metab. 1997;17:759–770. doi: 10.1097/00004647-199707000-00006. [DOI] [PubMed] [Google Scholar]
- Kalehua AN, Nagel JE, Whelchel LM, Gides JJ, Pyle RS, Smith RJ, Kusiak JW, Taub DD. Monocyte chemoattractant protein-1 and macrophage inflammatory protein-2 are involved in both excitotoxin-induced neurodegeneration and regeneration. Exp Cell Res. 2004;297:197–211. doi: 10.1016/j.yexcr.2004.02.031. [DOI] [PubMed] [Google Scholar]
- Koh JY, Choi DW. Selective blockade of non-NMDA receptors does not block rapidly triggered glutamate-induced neuronal death. Brain Res. 1991;548:318–321. doi: 10.1016/0006-8993(91)91140-v. [DOI] [PubMed] [Google Scholar]
- Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- Lai A, Todd K. Hypoxia-activated microglial mediators of neuronal survival are differentially regulated by tetracyclines. Glia. 2006;53:809–816. doi: 10.1002/glia.20335. [DOI] [PubMed] [Google Scholar]
- Lazarewicz JW, Wroblewski JT, Costa E. N-methyl-d-aspartate-sensitive glutamate receptors induce calcium-mediated arachidonic acid release in primary cultures of cerebellar granule cells. J Neurochem. 1990;55:1875–1881. doi: 10.1111/j.1471-4159.1990.tb05771.x. [DOI] [PubMed] [Google Scholar]
- Lee VM, Balin BJ, Otvos L, Jr, Trojanowski JQ. A68: a major subunit of paired helical filaments and derivatized forms of normal Tau. Science. 1991;251:675–678. doi: 10.1126/science.1899488. [DOI] [PubMed] [Google Scholar]
- Little A, Benkovic S, Miller D, O'Callaghan J. Chemically induced neuronal damage and gliosis: enhanced expression of the proinflammatory chemokine, monocyte chemoattractant protein (MCP)-1, without a corresponding increase in proinflammatory cytokines. Neuroscience. 2002;115:307–320. doi: 10.1016/s0306-4522(02)00359-7. [DOI] [PubMed] [Google Scholar]
- Mahad D, Ransohoff R. The role of MCP-1 (CCL2) and CCR2 in multiple sclerosis and experimental autoimmune encephalomyelitis (EAE) Semin Immunol. 2003;15:23–32. doi: 10.1016/s1044-5323(02)00125-2. [DOI] [PubMed] [Google Scholar]
- Martins-Green M. The chicken chemotactic and angiogenic factor (cCAF), a CXC chemokine. Int J Biochem Cell Biol. 2001;33:427–432. doi: 10.1016/s1357-2725(01)00029-2. [DOI] [PubMed] [Google Scholar]
- Massengale M, Wagers A, Vogel H, Weissman I. Hematopoietic cells maintain hematopoietic fates upon entering the brain. J Exp Med. 2005;201:1579–1589. doi: 10.1084/jem.20050030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner R, Edwards G, Streuli C, Ffrench-Constant C. A role in migration for the αVβ1 integrin expressed on oligodendrocyte precursors. J Neurosci. 1996;16:7240–7252. doi: 10.1523/JNEUROSCI.16-22-07240.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nau R. Pathophysiology of neuronal injury in bacterial meningitis: concepts and implications (in Spanish) Neurologia. 2003;18:47–53. [PubMed] [Google Scholar]
- Newman T, Galea I, van Rooijen N, Perry V. Blood-derived dendritic cells in an acute brain injury. J Neuroimmunol. 2005;166:167–172. doi: 10.1016/j.jneuroim.2005.04.026. [DOI] [PubMed] [Google Scholar]
- Nicole O, Docagne F, Ali C, Margaill I, Carmeliet P, MacKenzie E, Vivien D, Buisson A. The proteolytic activity of tissue-plasminogen activator enhances NMDA receptor-mediated signaling. Nat Med. 2001;7:59–64. doi: 10.1038/83358. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- Nufer O, Corbett M, Walz A. Amino-terminal processing of chemokine ENA-78 regulates biological activity. Biochemistry. 1999;38:636–642. doi: 10.1021/bi981294s. [DOI] [PubMed] [Google Scholar]
- Olney J. Inciting excitotoxic cytoside among central neurons. Adv Exp Med Biol. 1986;203:631–645. doi: 10.1007/978-1-4684-7971-3_48. [DOI] [PubMed] [Google Scholar]
- Paavola C, Hemmerich S, Grunberger D, Polsky I, Bloom A, Freedman R, Mulkins M, Bhakta S, McCarley D, Wiesent L, Wong B, Jarnagin K, Handel T. Monomeric monocyte chemoattractant protein-1 (MCP-1) binds and activates the MCP-1 receptor CCR2B. J Biol Chem. 1998;273:33157–33168. doi: 10.1074/jbc.273.50.33157. [DOI] [PubMed] [Google Scholar]
- Parathath S, Parathath S, Tsirka S. Nitric oxide mediates neurodegeneration and breakdown of the blood brain barrier in tPA-dependent excitotoxic injury in mice. J Cell Sci. 2006;119:339–349. doi: 10.1242/jcs.02734. [DOI] [PubMed] [Google Scholar]
- Pawlak R, Melchor J, Matys T, Skrzypiec A, Strickland S. Ethanol-withdrawal seizures are controlled by tissue plasminogen activator via modulation of NR2B-containing NMDA receptors. Proc Natl Acad Sci USA. 2005;102:443–448. doi: 10.1073/pnas.0406454102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ploplis VA, French EL, Carmeliet P, Collen D, Plow EF. Plasminogen deficiency differentially affects recruitment of inflammatory cell populations in mice. Blood. 1998;91:2005–2009. [PubMed] [Google Scholar]
- Proudfoot A, Handel T, Johnson Z, Lau E, LiWang P, Clark-Lewis I, Borlat F, Wells T, Kosco-Vilbois M. Glycosaminoglycan binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc Natl Acad Sci USA. 2003;100:1885–1890. doi: 10.1073/pnas.0334864100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rankine E, Hughes P, Botham M, Perry V, Felton L. Brain cytokine synthesis induced by an intraparenchymal injection of LPS is reduced in MCP-1-deficient mice prior to leucocyte recruitment. Eur J Neurosci. 2006;24:77–86. doi: 10.1111/j.1460-9568.2006.04891.x. [DOI] [PubMed] [Google Scholar]
- Rogove A, Siao C, Keyt B, Strickland S, Tsirka S. Activation of microglia reveals a non-proteolytic cytokine function for tissue plasminogen activator in the central nervous system. J Cell Sci. 1999;112:4007–4016. doi: 10.1242/jcs.112.22.4007. [DOI] [PubMed] [Google Scholar]
- Rogove AD, Tsirka SE. Neurotoxic responses by microglia elicited by excitotoxic injury in the mouse hippocampus. Curr Biol. 1998;8:19–25. doi: 10.1016/s0960-9822(98)70016-8. [DOI] [PubMed] [Google Scholar]
- Rothman SM. The neurotoxicity of excitatory amino acids is produced by passive chloride influx. J Neurosci. 1985;5:1483–1489. doi: 10.1523/JNEUROSCI.05-06-01483.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siao C-J, Tsirka S. Tissue plasminogen activator mediates microglial activation via its finger domain through annexin II. J Neurosci. 2002;22:3352–3358. doi: 10.1523/JNEUROSCI.22-09-03352.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siman R, Noszek JC. Excitatory amino acids activate calpain I and induce structural protein breakdown in vivo. Neuron. 1988;1:279–287. doi: 10.1016/0896-6273(88)90076-1. [DOI] [PubMed] [Google Scholar]
- Simard A, Soulet D, Gowing G, Julien J-P, Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer's disease. Neuron. 2006;49:489–502. doi: 10.1016/j.neuron.2006.01.022. [DOI] [PubMed] [Google Scholar]
- Srinivasan R, Sailasuta N, Hurd R, Nelson S, Pelletier D. Evidence of elevated glutamate in multiple sclerosis using magnetic resonance spectroscopy at 3 T. Brain. 2005;128:1016–1025. doi: 10.1093/brain/awh467. [DOI] [PubMed] [Google Scholar]
- Streit W. Microglia and neuroprotection: implications for Alzheimer's disease. Brain Res Rev. 2005;48:234–239. doi: 10.1016/j.brainresrev.2004.12.013. [DOI] [PubMed] [Google Scholar]
- Strijbos PJ, Leach MJ, Garthwaite J. Vicious cycle involving Na+ channels, glutamate release, and NMDA receptors mediates delayed neurodegeneration through nitric oxide formation. J Neurosci. 1996;16:5004–5013. doi: 10.1523/JNEUROSCI.16-16-05004.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szaflarski J, Ivacko J, Liu XH, Warren JS, Silverstein FS. Excitotoxic injury induces monocyte chemoattractant protein-1 expression in neonatal rat brain. Brain Res Mol Brain Res. 1998;55:306–314. doi: 10.1016/s0169-328x(98)00013-8. [DOI] [PubMed] [Google Scholar]
- Thanos S, Mey J, Wild M. Treatment of the adult retina with microglia-suppressing factors retards axotomy-induced neuronal degradation and enhances axonal regeneration in vivo and in vitro. J Neurosci. 1993;13:455–466. doi: 10.1523/JNEUROSCI.13-02-00455.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsirka S, Gualandris A, Amaral D, Strickland S. Excitotoxin induced neuronal degeneration and seizure are mediated by tissue-type plasminogen activator. Nature. 1995;377:340–344. doi: 10.1038/377340a0. [DOI] [PubMed] [Google Scholar]
- Tsirka S, Rogove A, Bugge T, Degen J, Strickland S. An extracellular proteolytic cascade promotes neuronal degeneration in the mouse hippocampus. J Neurosci. 1997;17:543–552. doi: 10.1523/JNEUROSCI.17-02-00543.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullrich O, Diestel A, Eyüpoglu I, Nitsch R. Regulation of microglial expression of integrins by poly(ADP-ribose) polymerase-1. Nat Cell Biol. 2001;3:1035–1042. doi: 10.1038/ncb1201-1035. [DOI] [PubMed] [Google Scholar]
- Vakili J, Standker L, Detheux M, Vassart G, Forssmann WG, Parmentier M. Urokinase plasminogen activator and plasmin efficiently convert hemofiltrate CC chemokine 1 into its active. J Immunol. 2001;167:3406–3413. doi: 10.4049/jimmunol.167.6.3406. [DOI] [PubMed] [Google Scholar]
- van den Steen PE, Proost P, Wuyts A, Van Damme J, Opdenakker G. Neutrophil gelatinase B potentiates interleukin-8 tenfold by aminoterminal processing, whereas it degrades CTAP-III, PF-4, and GRO-alpha and leaves RANTES and MCP-2 intact. Blood. 2000;96:2673–2681. [PubMed] [Google Scholar]
- Wang J, Tsirka S. Tuftsin fragment 1–3 (MIF) is beneficial when delivered after the induction of intracerebral hemorrhage. Stroke. 2004;36:613–618. doi: 10.1161/01.STR.0000155729.12931.8f. [DOI] [PubMed] [Google Scholar]
- Wang J, Rogove A, Tsirka A, Tsirka S. Protective role of MIF in an animal model of intracerebral hemorrhage. Ann Neurol. 2003;54:655–664. doi: 10.1002/ana.10750. [DOI] [PubMed] [Google Scholar]
- Werb Z. ECM and cell surface proteolysis: regulating cellular ecology. Cell. 1997;91:439–442. doi: 10.1016/s0092-8674(00)80429-8. [DOI] [PubMed] [Google Scholar]
- Yin Y, Henzl M, Lorber B, Nakazawa T, Thomas T, Jiang F, Langer R, Benowitz L. Oncomodulin is a macrophage-derived signal for axon regeneration in retinal ganglion cells. Nat Neurosci. 2006;9:843–852. doi: 10.1038/nn1701. [DOI] [PubMed] [Google Scholar]
- Yoshimura T, Yuhki N, Moore S, Appella E, Lerman M, Leonard E. Human monocyte chemoattractant protein-1 (MCP-1). Full-length cDNA cloning, expression in mitogen-stimulated blood mononuclear leukocytes, and sequence similarity to mouse competence gene JE. FEBS Lett. 1989;244:487–493. doi: 10.1016/0014-5793(89)80590-3. [DOI] [PubMed] [Google Scholar]
- Zhang K, McQuibban G, Silva C, Butler G, Johnston J, Holden J, Clark-Lewis I, Overall C, Power C. HIV-induced metalloproteinase processing of the chemokine stromal cell derived factor-1 causes neurodegeneration. Nat Neurosci. 2003;6:1064–1071. doi: 10.1038/nn1127. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Ernst CA, Rollins BJ. MCP-1: structure/activity analysis. Methods. 1996;10:93–103. doi: 10.1006/meth.1996.0083. [DOI] [PubMed] [Google Scholar]