TGFβ3 inhibits E-cadherin gene expression in palate medial-edge epithelial cells through a Smad2-Smad4-LEF1 transcription complex (original) (raw)
. Author manuscript; available in PMC: 2009 Mar 23.
Published in final edited form as: J Cell Sci. 2007 May 1;120(Pt 9):1646–1653. doi: 10.1242/jcs.003129
Summary
Dissociation of medial-edge epithelium (MEE) during palate development is essential for mediating correct craniofacial morphogenesis. This phenomenon is initiated by TGFβ3 upon adherence of opposing palatal shelves, because loss of E-cadherin causes the MEE seam to break into small epithelial islands. To investigate the molecular mechanisms that cause this E-cadherin loss, we isolated and cultured murine embryonic primary MEE cells from adhered or non-adhered palates. Here, we provide the first evidence that lymphoid enhancer factor 1 (LEF1), when functionally activated by phosphorylated Smad2 (Smad2-P) and Smad4 (rather than β-catenin), binds with the promoter of the E-cadherin gene to repress its transcription in response to TGFβ3 signaling. Furthermore, we found that TGFβ3 signaling stimulates epithelial-mesenchymal transformation (EMT) and cell migration in these cells. LEF1 and Smad4 were found to be necessary for up-regulation of the mesenchymal markers vimentin and fibronectin, independently of β-catenin. We proved that TGFβ3 signaling induces EMT in MEE cells by forming activated transcription complexes of Smad2-P, Smad4 and LEF1 that directly inhibit E-cadherin gene expression.
Keywords: E-cadherin, LEF1, Smad, TGF-beta, Epithelial-m
Introduction
E-cadherin, the primary cell adhesion molecule within adherens junctions, is essential for maintaining apical-basal polarity in epithelial cells (Takeichi, 1988; Hay, 1995). During epithelial-mesenchymal transformation (EMT), loss of E-cadherin expression correlates with a transition to front-end to back-end polarity, leading to subsequent migration of the newly created mesenchymal cells (Thiery, 2003). This molecular mechanism is essential for correct development during embryogenesis and is a common initiator of tumor metastasis (Birchmeier et al., 1996; Thiery, 2002).
Many transcription factor proteins, such as Snail, Slug, E12/E47, SIP-1, ZEB-1 and Twist directly bind to the E-cadherin gene promoter to inhibit its transcription (Peinado et al., 2004). Lymphoid enhancer factor 1 (LEF1), a molecule typically associated with Wnt signaling (Behrens et al., 1996; Eastman and Grosschedl, 1999), has been proved do the same when activated by β-catenin (Jamora et al., 2003; Medici et al., 2006). A strong correlation has been made between β-catenin-LEF1 signaling and acquisition of the invasive morphology in colon carcinoma cells (Kim et al., 2002) and malignant melanomas (Murakami et al., 2001; Chen et al., 2003). LEF1 is also localized in most embryonic tissues that undergo EMT, including the neural crest, somites, primitive streak (Mohamed et al., 2004) and palate (Nawshad and Hay, 2003).
During embryogenesis, the formation of the palate occurs when two opposing palatal shelves grow beneath the nasal septum to the point of touching and adherence at their medial edges. When this occurs, the medial-edge epithelium (MEE) receives appropriate signals that cause its transformation to mesenchyme, thus forming one confluent palate tissue, rather than two palatal shelves. Failure of adherence (fusion) or EMT leads to cleft palate (Nawshad et al., 2004).
The transformation of palate medial-edge epithelium to mesenchyme has been well documented (Fitchett and Hay, 1989; Kaartinien et al., 1997; Nawshad et al., 2004). However, signaling mechanisms that promote EMT during palatogenesis have only recently been investigated in detail. Transforming growth factor-beta3 (TGFβ3) has been established to have an essential role in palate development, including the transformation of MEE cells to the mesenchymal morphology (Brunet et al., 1995; Kaartinen et al., 1997; Nawshad and Hay, 2003). Upon adherence of opposing palatal shelves and formation of the MEE seam, the basal MEE cells show increased expression of TGFβ3 (LaGamba et al., 2005), which remains until EMT is complete (Tudela et al., 2002). TGFβ3-knockout mice (Proetzel et al., 1995; Taya et al., 1999), as well as some naturally TGFβ3-null avian systems (Sun et al., 1998), always have cleft palate. Furthermore, treatment of palates from TGFβ3-knockout mice with exogenous TGFβ3 is sufficient to rescue palatal fusion (Taya et al., 1999).
Unfused palatal seam cells lack the ability to undergo EMT because of insufficient levels of TGFβ signaling molecules. Upon adherence of palatal shelves (0-12 hours post initial contact) intracellular levels of TGFβ3, Smad anchor for receptor activation (SARA), Smad2 and Smad4 are all increased (LaGamba et al., 2005). TGFβ3 signaling has been shown to promote transcription of the LEF1 gene in these cells through a Smad-dependent mechanism. Surprisingly, it was found that β-catenin remained outside the nucleus during EMT of palate MEE in vivo. Furthermore, inhibition of β-catenin using antisense oligodeoxynucleotide (AS ODN) did not prevent EMT (Nawshad and Hay, 2003).
Based on prior findings of Smad proteins having the ability to bind and activate LEF1 - rather than the traditional activation by β-catenin (Labbe et al., 2000; Nishita et al., 2000), we previously suggested that TGFβ3 stimulates transformation of MEE cells by promoting both transcription and activation of LEF1 through a complex of dimerized phosphorylated Smad2 (Smad2-P) and Smad4. LEF1 then induces EMT, causing confluence of the palate (Nawshad and Hay, 2003). However, until now, this idea remained unproved. Here, we have provided evidence to support this hypothesis by demonstrating that this activated Smad2-_P_-Smad4-LEF1 transcription complex directly interacts with the promoter of the E-cadherin gene to repress its transcription, thus inducing EMT.
Results
TGFβ3 signaling forms Smad2-_P_-Smad4-LEF1 transcription complexes
MEE cells were isolated and cultured into primary cell lines from non-adhered (unfused, used as negative control) and adhered (fused, 12 hours post initial contact) palatal shelves. Whereas the term ‘fused’ is commonly used to describe confluent palate tissue, here we use it to describe pre-EMT MEE cells from adhered palatal shelves. We observed protein expression of TGFβ3 by immunoblotting in these cells, demonstrating no expression in unfused cells, but moderate expression in fused cells (Fig. 1A). Small traces of TGFβ3 have been described within in vivo MEE, however, upon culturing unfused cells, TGFβ3 expression was lost, allowing these cells to serve as an efficient negative control. Although some TGFβ3 signaling is seen in fused MEE cells, EMT does not occur at this stage in vivo. Increases in TGFβ3 expression are seen as palate development progresses (LaGamba et al., 2005). Also, the peak of LEF1 expression and EMT occurs 24 hours after adherence of palatal shelves is established (Nawshad and Hay, 2003). Since it was impossible to isolate MEE cells at this time point (owing to dissociation of the MEE seam) and because fused cells that were extracted for culture lose their continuous endogenous upregulation of TGFβ3, we added exogenous TGFβ3 for 24 hours to mimic the EMT observed in vivo.
Fig. 1.
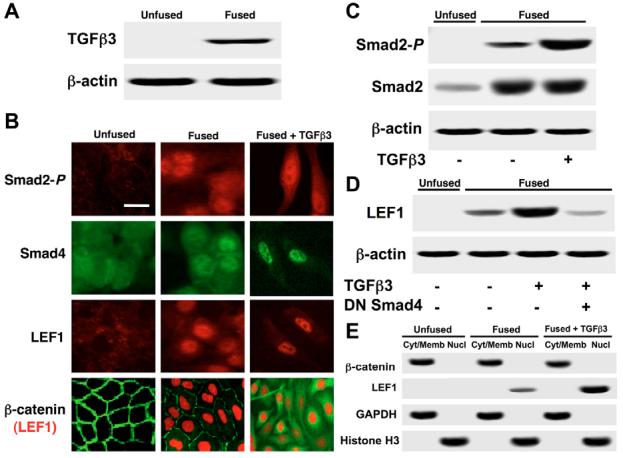
EMT is associated with nuclear localization of Smad2-P, Smad4 and LEF1, but not β-catenin. (A) Immunoblotting demonstrated that palatal adherence increases TGFβ3 expression. Unfused cells do not express TGFβ3, whereas fused cells show moderate protein expression. (B) Immunocytochemistry showed nuclear localization of Smad2-P, Smad4, and LEF1 in fused MEE cells and in fused cells treated with exogenous TGFβ3. Acquisition of the mesenchymal phenotype was observed when cells were treated with TGFβ3. β-catenin remained in the cytoplasm during EMT. Bar, 10 μm. (C) Smad2-P and total Smad2 expression was observed by immunoblotting. No expression was detected in unfused MEE cells for Smad2-P, whereas moderate levels were found in fused cells. Protein levels were heavily increased upon treatment with TGFβ3. Increased levels of total Smad2 were observed upon palatal adherence. (D) LEF1 protein expression was not detected in unfused cells, but showed steady increases in fused cells and in fused cells treated with TGFβ3. Addition of dominant-negative Smad4 prevented these increases. (E) Immunoblotting from cytoplasmic and/or membrane (Cyt/Memb) and nuclear (Nucl) protein fractions showed no evidence of nuclear β-catenin under any condition. LEF1 was observed in the nuclear fraction of fused cells, with increased levels in cells treated with TGFβ3. GAPDH and histone H3 were used as cytoplasmic and nuclear controls respectively.
Immunocytochemistry showed no expression of Smad2-P or LEF1 in unfused MEE cells. Upon fusion, these cells began to express TGFβ signaling molecules, which showed nuclear localization of Smad2-P, Smad4 and LEF1 in both fused cells, and fused cells treated with TGFβ3 for 24 hours (to stimulate EMT). A clear change in cellular morphology was observed upon exposure to TGFβ3 anatomically demonstrating EMT. Co-immunostaining for β-catenin and LEF1 showed that these two proteins appeared to remain in separate compartments [with β-catenin in the cytoplasm (green) and LEF1 in the nuclei (red)] during EMT (Fig. 1B). Expression levels of Smad2-P, total Smad2 (Fig. 1C) and LEF1 (Fig. 1D) were observed by immunoblotting. Levels of total Smad2 increased upon palatal adherence, consistent with previous findings (LaGamba et al., 2005). We found no expression of Smad2-P and LEF1 in unfused cells, moderate expression in fused cells, and high expression in fused cells treated with exogenous TGFβ3. LEF1 upregulation was prevented by the presence of a dominant-negative Smad4, which lacks a DNA binding domain, further supporting evidence that LEF1 expression is Smad-dependent in this system (Nawshad and Hay, 2003). To confirm compartmental localization of β-catenin, cytoplasmic and/or membrane and nuclear protein fractions were isolated under all three conditions. Immunoblotting was performed demonstrating that high levels of β-catenin were present in the cytoplasmic and/or membrane fractions, but no traces were found in the nuclear fractions. LEF1 was found in the nuclear fraction of fused cells, showing increased expression in cells exposed to TGFβ3. GAPDH was used as a cytoplasmic marker and histone H3 was used as a nuclear marker (Fig. 1E).
To demonstrate transcriptional activity Smad, a p3TP-Lux reporter gene construct was transfected into unfused (negative control), untreated fused and TGFβ3-treated fused MEE cells. Since unfused cells do not possess TGFβ3 signaling potential - as previously published (LaGamba et al., 2005), significant levels of Smad transcriptional activity were not detected. By contrast, fused cells demonstrated moderate levels of transcription, which increased upon stimulation with exogenous TGFβ3. The addition of a dominant negative Smad4 adenoviral construct significantly inhibited p3TP-Lux activity (Fig. 2A).
Fig. 2.
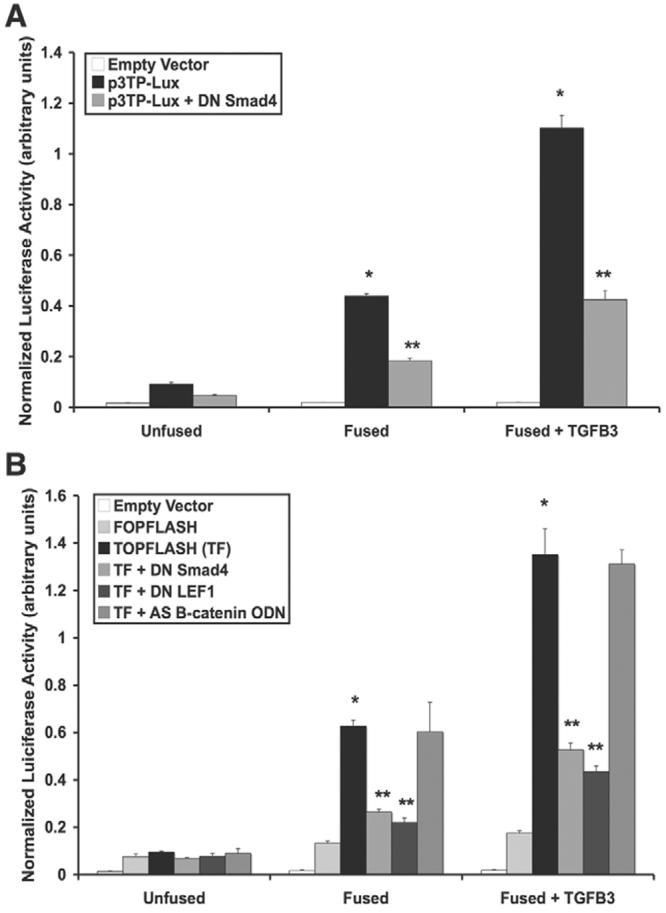
Confirmation of Smad and LEF1 transcriptional activities. (A) p3TP-Lux reporter gene assay demonstrated that Smad transcriptional activity increased from unfused to fused MEE cells, with a sharp increase in activity upon stimulation with TGFβ3. A dominant-negative Smad4 (DN Smad4) construct inhibited luciferase activity (mean ± s.d.; _n_=3; *P<0.05 compared with Unfused; **P<0.05 compared with p3TP-Lux). (B) pTOPFLASH-Lux (TF) reporter gene assay showed that LEF1 transcriptional activity continually increases as TGFβ3 signaling progresses. Mutated LEF1 binding sites of the pFOPFLASH-Lux reporter, as well as treatment with DN Smad4 (Smad4 being necessary for LEF1 gene expression) or dominant-negative LEF1 (DN LEF1) inhibited transcriptional activity whereas antisense β-catenin/γ-catenin oligodeoxynucleotide (AS B-catenin ODN) did not (mean ± s.d.; _n_=3; *P<0.001 compared with Unfused; **P<0.01 compared with TF).
To confirm LEF1 transcriptional activity, pTOPFLASH-Lux (containing LEF1-binding sites) and pFOPFLASH-Lux (containing mutated LEF1-binding sites) constructs were transfected into MEE cells. As expected, unfused MEE cells demonstrated no transcriptional activity, whereas fused cells showed that LEF1 is promoting transcription of the reporter gene. Addition of exogenous TGFβ3 significantly increased LEF1 transcriptional activity. Since LEF1 gene expression in MEE cells is dependent upon Smad signaling (Nawshad and Hay, 2003), addition of dominant-negative Smad4 greatly reduced luciferase activity. The addition of a dominant-negative LEF1 construct, producing a negative competitor protein that lacks DNA-binding potential, also inhibited TOPFLASH luciferase activity, whereas antisense oligodeoxynucleotide against β-catenin/γ-catenin (AS β-catenin ODN) did not (Fig. 2B). Control immunoblots were conducted to demonstrate loss of β-catenin and γ-catenin in the presence of antisense oligodeoxynucleotide (supplementary material Fig. S1).
Co-immunoprecipitation was performed to demonstrate the formation of a Smad2-_P_-Smad4-LEF1 protein complex. Protein extracts were immunoprecipitated from unfused, fused and fused MEE cells treated with TGFβ3 using antibodies against Smad2-P, Smad4 or LEF1. Each precipitate was then immunoblotted with antibodies against β-catenin, Smad4, Smad3-P, Smad2-P and LEF1 proteins. Unfused extracts showed no protein binding because LEF1 is not expressed in these cells. We found that complexes of Smad2-_P_-Smad4-LEF1 were present in fused cells, and were heavily increased upon treatment with TGFβ3. No β-catenin or Smad3-P was detected in the protein complex (Fig. 3A). Immunoprecipitation and immunoblotting for β-actin served as an internal control (Fig. 3B).
Fig. 3.
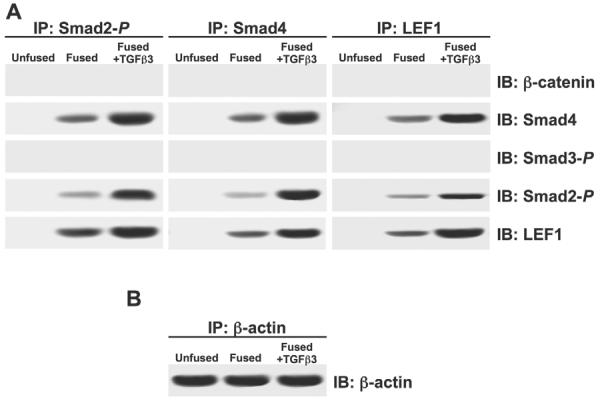
TGFβ3 signaling promotes formation of a Smad2-_P_-Smad4-LEF1 transcription complex. (A) Co-immunoprecipitation (IP) of cell extracts from MEE cells was performed using antibodies against Smad2-P, Smad4 or LEF1. Immunoblot (IB) analysis showed that Smad4, Smad2-P and LEF1 are bound together in the extracts of fused MEE cells; increased levels were observed when cells were exposed to TGFβ3. Smad3-P and β-catenin were not found in this complex. Unfused MEE cells showed no traces of interaction because LEF1 is not expressed in these cells. (B) Immunoprecipitation and immunoblotting for β-actin was used as an internal control.
Smad2-_P_-Smad4-LEF1 inhibits E-cadherin gene expression to promote EMT
Since the most common molecular change used to characterize and promote EMT is the loss of E-cadherin expression, we performed sequence analysis of the promoter region of the murine E-cadherin gene. We identified one distinct LEF1-binding region separated from the traditional E-pal (E-box 1/2) TCF and/or LEF1 binding site (Peinado et al., 2004). We also found that a Smad binding element (SBE; Smad4-binding site) flanks the E-pal LEF1 site (Fig. 4A). Binding of the Smad2-_P_-Smad4-LEF1 protein complex to the endogenous loci of these regions was confirmed by chromatin immunoprecipitation (ChIP). ChIP was performed using antibodies against IgG (negative control), LEF1, Smad4, Smad2-P and β-catenin. PCR was then performed with primers specific for the LEF1-binding (E-pal and non-E-pal) and SBE-binding sites. Unfused cells showed no signal with any antibodies, whereas fused cells showed moderate signal with antibodies against LEF1, Smad4 and Smad2-P. These signals were increased upon treatment of cells with exogenous TGFβ3. β-catenin was not found to be associated with these binding regions. LEF1 and Smad4 bind to their respective sites and, although Smad2 lacks a DNA-binding domain (Derynk et al., 1998; Xu et al., 2000; Ten Dijke and Hill, 2004), it still appears in the complex with respect to all three binding sites (Fig. 4B). To avoid the possibility of the observed PCR signals being the result of over-amplification, we conducted real-time PCR for a quantitative assessment of ChIP signals. Similar results to those found using standard gel-based PCR were observed (supplementary material Fig. S2).
Fig. 4.

Smad2-_P_-Smad4-LEF1 directly bind to the promoter of the E-cadherin gene. (A) Promoter analysis of the murine E-cadherin gene revealed one unique LEF1-binding site (independent of the standard TCF and/or LEF site located in the E-pal) and a Smad-binding element (SBE), both within close proximity to the E-pal LEF1-binding region. (B) Binding of the Smad2-_P_-Smad4-LEF1 protein complex to the endogenous loci was confirmed by chromatin immunoprecipitation. Chromatin was immunoprecipitated with antibodies against IgG (negative control), LEF1, Smad4, Smad2-P or β-catenin. PCR analysis using precipitated DNA showed binding of LEF1, Smad4 and Smad2-P to LEF1 (non-E-pal), LEF1 (E-pal) and SBE regions of the E-cadherin promoter. Whereas LEF1 and Smad4 interact with their respective binding regions, all three proteins are observed because they form a transcription complex. No signal was observed from unfused MEE cells whereas a moderate signal was detected from fused MEE cells that was heavily increased upon treatment with exogenous TGFβ3. β-catenin was not detected for interaction with these binding sites.
Reporter gene assays were conducted to determine whether this Smad2-_P_-Smad4-LEF1 transcription complex is responsible for repressing E-cadherin gene transcription during MEE cell EMT. E-cadherin gene promoter activity was assessed using a pGL3-E-cad-Lux vector. Reporter gene activity was detected at high levels in unfused MEE cells, moderate levels in fused MEE cells, and was almost fully inhibited upon treatment of fused MEE cells with TGFβ3. Presence of dominant-negative Smad4 or dominant-negative LEF1 constructs maintained promoter activity. Addition of AS β-catenin/γ-catenin ODN did not affect loss of promoter activity (Fig. 5A). E-cadherin promoter activity was also determined using another set of pGL3-E-cad-Lux reporter plasmids, with the relative LEF1 or Smad4 binding sites mutated. Interestingly, site-directed mutagenesis of the LEF1 (E-pal)-binding, LEF1 (non E-pal)-binding, or Smad4 (SBE)-binding regions prevented loss of E-cadherin gene promoter activity, suggesting that anchoring of the Smad2-_P_-Smad4-LEF1 complex at all three binding sites is essential for loss of E-cadherin (Fig. 5B). Real-time quantitative PCR was performed to assess E-cadherin gene expression relative to the unfused negative control. Fused cells showed moderately less mRNA, whereas fused cells treated with TGFβ3 showed a major reduction in gene expression. Addition of dominant-negative LEF1 or dominant-negative Smad4 prevented loss of E-cadherin gene expression, but AS β-catenin/γ-catenin ODN had no effect (Fig. 5C).
Fig. 5.
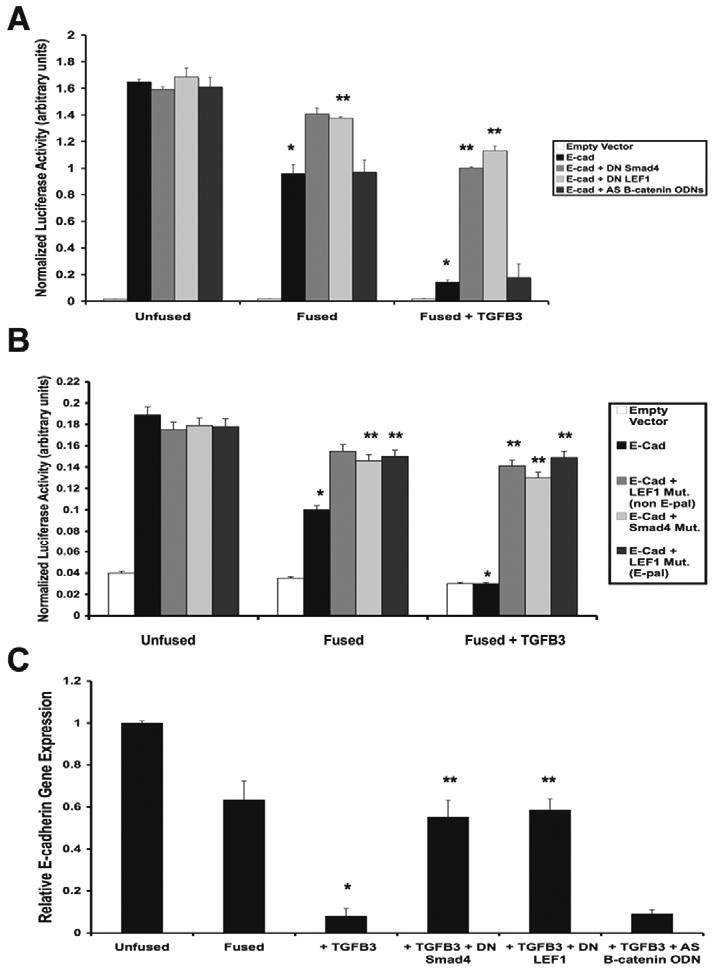
Smad2-_P_-Smad4-LEF1 directly inhibits E-cadherin gene expression. (A) Reporter gene analysis of E-cadherin promoter activity (pGL3-E-cad-Lux) demonstrated decreased expression under the influence of TGFβ3. Treatment of cells with dominant-negative Smad4 or LEF1 (DN Smad4 or DN LEF1, respectively) prevented the repression of E-cadherin, but antisense β-catenin/γ-catenin oligodeoxynucleotide (AS B-catenin ODN) did not (mean ± s.d.; _n_=3; *P<0.001 compared with Unfused; **P<0.05 compared with E-cad). (B) Site directed mutagenesis of the Smad4-binding (SBE) and LEF1-binding (E-pal and non E-pal) regions also prevented loss of E-cadherin promoter activity (mean ± s.d.; _n_=3; *P<0.001 compared with Unfused; **P<0.01 compared with E-cad). (C) Real-time quantitative PCR for E-cadherin gene expression relative to the unfused MEE negative control showed a steady decrease in fused MEE cells and fused cells treated with TGFβ3. DN-LEF1 and DN Smad4 prevented this suppression, whereas AS B-catenin ODN had no effect (mean ± s.d.; _n_=3; *P<0.001 compared with Unfused; **P<0.01 compared with + TGFβ3).
E-cadherin protein expression was assessed by immunocytochemistry (Fig. 6A) and immunoblotting (Fig. 6B), showing moderate loss upon palatal fusion, and a complete loss when exposed to exogenous TGFβ3. Furthermore, addition of dominant-negative Smad4 or dominant-negative LEF1 prevented E-cadherin loss, whereas treatment with antisense oligodeoxynucleotide against β-catenin/γ-catenin transcripts did not. To confirm EMT, we also assessed expression of mesenchymal markers vimentin and fibronectin by immunoblotting. We found that, inversely to E-cadherin, expression of these mesenchymal markers was steadily increased upon palatal fusion and stimulation with TGFβ3. Addition of dominant-negative Smad4 and dominant-negative LEF1 prevented these increases, but antisense β-catenin/γ-catenin oligodeoxynucleotide (AS β-catenin ODN) had no effect (Fig. 6B). Cell migration was determined under the same conditions by scratch wound (Fig. 7A) and transwell (Fig. 7B) assays. We found that no migration occurred in unfused or fused cells, whereas fused cells treated with TGFβ3 had high levels of migration. Dominant-negative LEF1 and dominant-negative Smad4 prevented TGFβ3 induced migration, but AS β-catenin ODN did not. These results provide a new model for E-cadherin repression during EMT of palate MEE cells (Fig. 8).
Fig. 6.
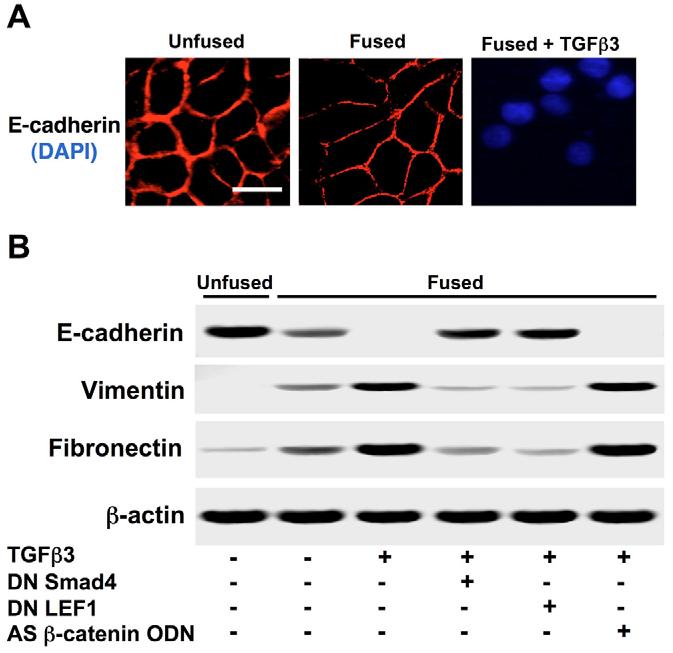
Loss of E-cadherin is associated with increased expression of mesenchymal markers. (A) Immunocytochemistry demonstrated progressive repression of E-cadherin from fused MEE cells to those exposed to exogenous TGFβ3. Bar, 10 μm. (B) E-cadherin expression was also assessed by immunoblotting, showing a moderate loss in fused cells, followed by a complete loss in fused cells treated with TGFβ3. Dominant-negative Smad4 or LEF1 (DN Smad4 or DN LEF1, respectively) prevented this repression, whereas antisense β-catenin/γ-catenin oligodeoxynucleotide (AS β-catenin ODN) had no effect. Contrarily, we found that expression of the mesenchymal markers vimentin and Fibronectin was steadily increased; DN Smad4 and DN LEF1 prevented these increases. AS β-catenin ODN had no effect on these proteins.
Fig. 7.
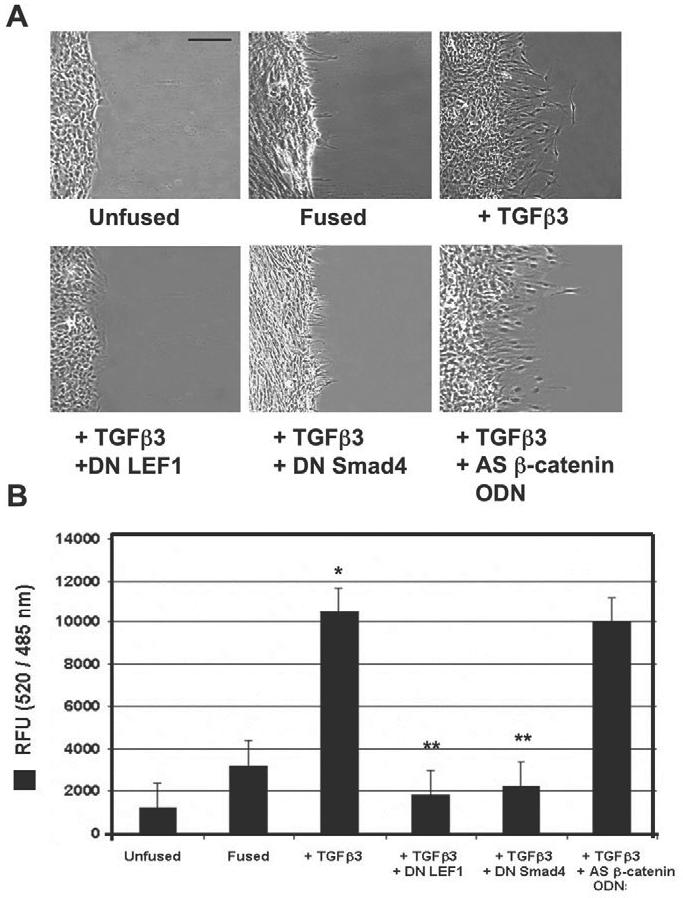
TGFβ3 signaling promotes post-EMT cell migration of MEE cells. (A) Scratch wound assays demonstrated lack of cell migration in unfused or fused MEE cells. Addition of exogenous TGFβ3 stimulated cell migration of fused cells, whereas addition of dominant-negative LEF1or Smad4 (DN LEF-1 or DN Smad4, respectively) prevented migration under these conditions. Antisense β-catenin/γ-catenin oligodeoxynucleotide (AS β-catenin ODN) did not prevent TGFβ3-induced migration. Bar, 60 μm. (B) Transwell cell migration assays showed little migration of unfused or fused MEE cells, whereas fused cells exposed to TGFβ3 were highly migratory. DN Smad4 and DN LEF1 prevented migration, but AS β-catenin ODN did not (mean ± s.d.; _n_=3; *P<0.001 compared with Unfused; **P<0.05 compared with + TGFβ3).
Fig. 8.
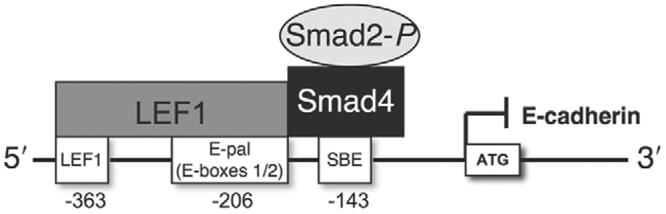
Diagram of the proposed interaction between the Smad2-_P_-Smad4-LEF1 transcription complex and the E-cadherin gene promoter is shown to demonstrate our hypothesis of transcriptional repression.
Discussion
Repression of E-cadherin gene transcription is perhaps the most significant molecular change within cells that undergo EMT. Loss of this epithelial marker has been proven to stimulate (or be a contributing factor of) the transformation of epithelium to the mesenchymal morphology (Hay, 1995; Peinado et al., 2004). During craniofacial development, loss of E-cadherin in palate MEE cells is essential for correct formation of palatal tissue. Failure of this mechanism within MEE cells can lead to palatal clefting (Nawshad et al., 2004).
Our results prove that LEF1, as part of an activated transcription complex with Smad2-P and Smad4, acts as a transcriptional repressor of the E-cadherin gene during EMT of palate MEE cells. When stimulated by TGFβ3, these Smad proteins bind and activate LEF1 to form a complex that can either induce or repress transcription. This portrays a major discovery in this system, because LEF1 has most commonly been described as being activated by β-catenin, as demonstrated in traditional Wnt signaling. These data are justified by previous findings that Wnt-knockout mice show no evidence of palatal clefting, whereas LEF1- (Galceran et al., 1999) and TGFβ3- (Taya et al., 1999) knockout mice have severe craniofacial deformities, including cleft palate. Recent findings of Smad2 knockdown by small interfering RNA (siRNA) in preventing palatal confluence (Shiomi et al., 2006) further support these data. Why β-catenin remains in the cytoplasm during palatal EMT remains to be determined. Also, whereas E-cadherin may be suppressed in other systems by β-catenin and/or LEF1 at a single binding site (Jamora et al., 2003), Smad2-_P_-Smad4-LEF1 appears to require two LEF1-binding regions for E-cadherin repression in palate MEE cells.
Our data also show that TGFβ3 mediates EMT in a Smad and/or LEF1 dependent manner by promoting expression of the mesenchymal markers vimentin and fibronectin. We observed a clear change in cell morphology from epithelial cobblestone to mesenchymal spindle shaped cells with filopodia, thus providing anatomic proof of MEE transdifferentiation. Increased levels of cell migration associated with EMT were also observed. Interestingly, although fused cells showed moderate levels of TGFβ3 signaling, no EMT was observed. Only when exogenous TGFβ3 was added to cultures did we observe this transition. This is consistent with the in vivo model of palatal EMT. Levels of endogenous TGFβ3 are known to continually increase in the MEE after adherence as palate development progresses, and remains until EMT is complete (Nawshad et al., 2004). After isolation of primary MEE cells from adhered palate tissue (12 hours post initial contact) this continuous upregulation of TGFβ3 was lost, producing only moderate levels that appear insufficient for EMT induction. Since we were unable to isolate cells from the peak stage of EMT (36 hours post initial contact), we added excess TGFβ3 (for 24 hours) to fused cells to mimic the higher levels observed in vivo. Whereas it is not known why higher amounts of nuclear Smad2-_P_-Smad4-LEF1 are necessary for EMT, it is clear that the mechanism is TGFβ3 dose-dependent.
Disappearance of palate medial-edge epithelium during craniofacial development is a topic of much controversy. Whereas most of the early work in this field has provided evidence for EMT (Fitchett and Hay, 1989; Griffith and Hay, 1992; Shuler et al., 1992; Sun et al., 1998; Nawshad and Hay, 2003) others have suggested that apoptosis (Martinez-Alvarez et al., 2000; Cuervo and Covarrubias, 2004) or cell migration (Carette and Ferguson, 1992) may be the major fate of the MEE. Conflicting reports of cell tracing experiments have been shown to either support (Griffith and Hay, 1992; Shuler et al., 1992; Sun et al., 1998) or rule out (Vaziri Sani et al., 2005; Xu et al., 2006) EMT, adding more confusion to this area of research. The purpose of our work was not to provide further support for in vivo EMT but, rather, to establish the existence of Smad2-_P_-Smad4-LEF1 complexes that inhibit gene expression. However, our primary MEE cells did undergo EMT in vitro as a result of the TGFβ3 and Smad2-_P_-Smad4-LEF1-dependent suppression of E-cadherin transcription.
Whereas others have previously described Smad3-Smad4 complexes being able to bind and functionally activate LEF1 (Labbe et al., 2000; Nishita et al., 2000), we provide the first evidence of Smad2-Smad4-LEF1 complexes. Also, whereas Smad-LEF1 complexes have been described to promote transcription of reporter genes such as Xtwn (Labbe et al., 2000), our work presents the first evidence that these complexes can repress gene transcription. This work provides a foundation to isolate differences between Smad-mediated and β-catenin-mediated LEF1 activation and EMT. The unique mechanism of cell adhesion loss described here will provide new insight into EMT in other systems of embryonic development and pathology.
Materials and Methods
Cell Culture
Palate medial edge epithelial cells from CF-1 mouse (Charles River Laboratories) embryos were separated from underlying mesenchyme [using dispase II (Roche)], from unfused single palatal shelves and fused palatal shelves adhered for 12 hours in organ culture, as previously described (LaGamba et al., 2005). Cells were then cultured into primary cell lines in F12 media (Gibco) + 10% FBS + 1% penicillin-streptomycin. Fetal bovine serum was removed for all experimental conditions. Isolation of MEE cells during the peak of in vivo LEF1 expression (36 hours post initial contact; 24 hours post established adherence) (Nawshad and Hay, 2003) could not be achieved because of dissociation of the MEE seam. When fused cells were removed from primary tissue, increases in TGFβ3 halt to only a moderate expression that is not sufficient to induce EMT. Since TGFβ3 expression continually increases during MEE dissociation (LaGamba et al., 2005), we added exogenous TGFβ3 to fused cells to mimic the in vivo results. Cells were treated with recombinant TGFβ3 (R&D Systems) at a concentration of 10 ng/ml for 24 hours for all relative experiments. The dominant-negative LEF1 and dominant-negative Smad4 plasmids (which produce proteins that lack the ability to bind DNA) were kind gifts from M. Waterman (University of California, Irvine, CA) and D. M. Simeone (University of Michigan, Ann Arbor, MI), respectively. All adenoviral constructs were made using the AdEasy vector system (Adenovirus Technologies), a gift from B. Vogelstein, (The Johns Hopkins School of Medicine, Baltimore, MD). The above constructs have been used in our laboratory previously (Nawshad and Hay, 2003). All viruses were used at a concentration of 1:100 with treatments for 24 hours. Antisense oligodeoxynucleotide against β-catenin/γ-catenin (AS β-catenin ODN) with the sequence 5′-GTGGTCCACAGAACTTCTC-3′ was used as previously described (Kim et al., 1998). All experiments were performed in triplicate.
Immunocytochemistry, immunoblotting, and coimmunoprecipitation (CoIP)
The antibodies against the following proteins were used for our immunoassays: E-cadherin (Zymed), Smad2, LEF1 (Santa Cruz Biotechnology), Smad2-P (generously provided by P. ten Dijke, The Netherlands Cancer Institute, Amsterdam, The Netherlands), Smad3-P (BioSource), Smad4 (Cell Signaling Technology), fibronectin, γ-catenin (Transduction Laboratories), GAPDH, histone H3 (Chemicon), β-catenin, vimentin, β-actin (Sigma). Dilutions were used according to the recommendation of the respective manufacturers. Fluorescein- and Rhodamine-conjugated secondary antibodies (Pierce) were used at a concentration of 1:250. Anti-mouse HRP-conjugated secondary antibodies (Chemicon) were used at a 1:1000 concentration. Immunocytochemistry (Nawshad and Hay, 2003) and immunoblotting (Li et al., 2002) experiments were conducted as described elsewhere. Isolation of cytoplasmic and/or membrane and nuclear protein extracts were achieved using the Compartment Protein Extraction Kit (Chemicon) and protocol. For immunoprecipitation we used the IP50 Protein G Immunoprecipitation Kit (Sigma) and followed the protocol as suggested by the manufacturer. Immunocytochemistry images were acquired using a Nikon 80i fluorescence microscope. Adjustments of image size, brightness and contrast were made using Adobe Photoshop CS.
Promoter analysis and chromatin immunoprecipitation (ChIP)
Analysis of the murine E-cadherin gene promoter was performed using MAT Inspector (Genomatix) software. Single-stranded oligonucleotide primers representing forward and reverse sequences of the wild-type and mutant LEF1-and Smad4-binding sites were commercially synthesized (IDT, Coralville, IA). The ChIP assay was performed using ChIP-ITTM (ActiveMotif, Carlsbad CA) and its protocol as described by the manufacturer. PCR (and real-time quantitative PCR) analysis was performed on DNA isolated through ChIP using an ABI 7500 cycler, with 40 cylces per sample. The following primers were used: LEF1-binding site (non E-pal): forward, 5′-CATGCCACCAACTACAGACAG-3′; reverse, 5′-CTAGCAGAAGTTCTTGGGAAC-3′; LEF1-binding site (E-pal): forward, 5′-TAGGAAGCTGGGAAG-3′; reverse, 5′-TGCGGTCGGGCAGGG-3′; Smad4-binding site (SBE): forward, 5′-CCCTCTTGGTGGAAGAAGAG-3′; reverse, 5′-CATCATCTAGGTTTCCG-3′.
Luciferase reporter gene assays
Luciferase reporter assays were conducted using the Luciferase Assay System (Promega) and the corresponding protocol. Light units were measured with a Luminometer TD-20/20 (Turner Designs). Assays were normalized for transfection efficiency by co-transfection with a β-gal control plasmid and detected with the Luminescent β-gal control assay kit (Clontech). Experimental (luciferase) results were divided by the β-gal results to provide normalized values of arbitrary units. All plasmids (500 ng) were transfected into cells using Lipofectamine and LipofectaminePlus reagents (Invitrogen) according to the manufacturer's guidelines. The p3TP-Lux reporter plasmid was provided by J. Massague (Memorial Sloan-Kettering Cancer Center, New York, NY). The pTOPFLASH-Lux and pFOPFLASH-Lux reporter constructs were kindly provided by H. Clevers (Netherlands Institute for Developmental Biology, Utrecht, The Netherlands). The pGL3-E-cad-Lux luciferase construct was generously provided by S. Dedhar (University of British Columbia, Vancouver, Canada).
Site-directed mutagenesis
A -800 bp E-cadherin gene promoter construct (generously provided by J. Behrens, University Erlangen-Nürnberg, Erlangen, Germany) was cloned into the pGL3-Lux reporter plasmid. Site-directed mutagenesis (Mutant-Max) was used to create mutant -800 bp promoter constructs. One construct was mutated at the LEF1-binding site located outside the traditional E-pal site, the second was mutated at the Smad4-binding site, the third (kindly provided by A. Cano, Instituto de Investigaciones Biomedicas, Madrid, Spain) was mutated at the E-pal site where LEF1 commonly binds. The mutant sites were all confirmed by sequencing. These constructs were then transfected into cells for luciferase assays as described above. The following regions were mutated (mutated base are underlined). Construct 1 (LEF1, non E-pal) wild-type LEF1: 5′-CTTTGTAACTCC A-3′; mutant LEF1: 5′-CTTGTCGACTCCA-3′; construct 2 (Smad4) wild-type Smad4: 5′-GGCCGCAGCCT-3′; mutant Smad4: 5′-GGCTTGAGCCT-3′; construct 3 (LEF1: E-pal) wild-type E-pal: 5′-CACCTAAAGGTG-3′; mutant E-pal: 5′-CACCTTTAGGTG-3′.
Real-time quantitative PCR
RNA samples were extracted using the RNeasy Mini Kit (Qiagen) and according to the manufacturer's protocol. Samples were submitted to a core facility (Biopolymers Facility, Harvard Medical School, Department of Genetics) where real-time PCR experiments were conducted using the Syber Green PCR system (ABI) on an ABI 7500 cycler, with 40 cycles per sample. Cycling temperatures were as follows: denaturing 95°C; annealing and extension, 60°C. The following primers were used. E-cadherin forward 5′- CGTGATGAAGGTCTCAGCC-3′, reverse 5′-ATGGGGGCTTCATTCAC-3′; GAPDH forward 5′-TGAAGGTCGGTGTGAACGGATTTGGC-3′, reverse 5′-CATGTAGGCCATGAGGTCCACCAC-3′.
Transwell migration assays
To assess post-EMT activities we used the InnocyteTM Cell Migration Assay (EMD Biosciences). Transwell migration chambers (8 μm pore size) in 96-well plates were used for migration analyses. Unfused and untreated fused MEE cells were used as negative controls. Fused MEE cells were treated with either exogenous recombinant TGFβ3 alone or in combination with dominant-negative LEF1, dominant-negative Smad4, AS β-catenin ODN as described above. MEE cells were allowed to migrate across the membrane insert towards medium in the presence of serum (10%) for 24 hours at 37°C (chemotactic migration). Cells that migrated through the membrane attached to the lower side of the cell culture insert and were subsequently detached using cell detachment buffer containing Calcein-AM fluorescent dye (excitation maximum 485 nm, emission maximum 520 nm).. The data were obtained using a standard fluorescent plate reader (BD FACSArrayTM bioanalyzer).
Scratch wound assays
Unfused (control) and fused MEE cells were grown to 100% confluence in six-well culture plates. A straight-line-shaped wound (no cell zone) was made by scraping the MEE cell monolayer with a sterile pipette tip to a uniformly placed scratch among the cells. Wounded cultures were incubated for 24 hours with or without TGFβ3 or in combination with dominant-negative LEF1, dominant-negative Smad4 and AS β-catenin ODN as described above. Migration of cells (or gap filling) was observed 24 hours post treatment through a phase-contrast microscope where cells were morphologically assessed for the migratory mesenchymal morphology. The center of the scratch line was used for positioning.
Acknowledgments
We thank Hector Peinado and Amparo Cano (Instituto de Investigaciones Biomedicas, Madrid) for their helpful advice regarding this study. This work was supported by R01-DE11142 (to E.D.H.) and RR018759 NIH-CoBRE for the Nebraska Center for Cellular Signaling (to M. J. Wheelock) from the National Institutes of Health.
Footnotes
References
- Behrens J, von Kries JP, Kuhl M, Bruhn L, Wedlich D, Grosschedl R, Birchmeier W. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature. 1996;382:638–642. doi: 10.1038/382638a0. [DOI] [PubMed] [Google Scholar]
- Birchmeier W, Behrens J, Weidner KM, Hulsken J, Birchmeier C. Epithelial differentiation and the control of metastasis in carcinomas. Curr. Top. Microbiol. Immunol. 1996;213:117–135. doi: 10.1007/978-3-642-61109-4_6. [DOI] [PubMed] [Google Scholar]
- Brunet CL, Sharpe PM, Ferguson MW. Inhibition of TGF-beta 3 (but not TGF-beta 1 or TGF-beta 2) activity prevents normal mouse embryonic palate fusion. Int. J. Dev. Biol. 1995;39:345–355. [PubMed] [Google Scholar]
- Carette MJ, Ferguson MW. The fate of medial edge epithelial cells during palatal fusion in vitro: an analysis by DiI labelling and confocal microscopy. Development. 1992;114:379–388. doi: 10.1242/dev.114.2.379. [DOI] [PubMed] [Google Scholar]
- Chen D, Xu W, Bales E, Colmenares C, Conacci-Sorrell M, Ishii S, Stavnezer E, Campisi J, Fisher DE, Ben-Ze′ev A, et al. SKI activates Wnt/beta-catenin signaling in human melanoma. Cancer Res. 2003;63:6626–6634. [PubMed] [Google Scholar]
- Cuervo R, Covarrubias L. Death is the major fate of medial edge epithelial cells and the cause of basal lamina degradation during palatogenesis. Development. 2004;131:15–24. doi: 10.1242/dev.00907. [DOI] [PubMed] [Google Scholar]
- Derynck R, Zhang Y, Feng XH. Smads: transcriptional activators of TGF-beta responses. Cell. 1998;95:737–740. doi: 10.1016/s0092-8674(00)81696-7. [DOI] [PubMed] [Google Scholar]
- Eastman Q, Grosschedl R. Regulation of LEF-1/TCF transcription factors by Wnt and other signals. Curr. Opin. Cell Biol. 1999;11:233–240. doi: 10.1016/s0955-0674(99)80031-3. [DOI] [PubMed] [Google Scholar]
- Fitchett JE, Hay ED. Medial edge epithelium transforms to mesenchyme after embryonic palatal shelves fuse. Dev. Biol. 1989;131:455–474. doi: 10.1016/s0012-1606(89)80017-x. [DOI] [PubMed] [Google Scholar]
- Galceran J, Farinas I, Depew MJ, Clevers H, Grosschedl R. Wnt3a-/--like phenotype and limb deficiency in Lef1(-/-)Tcf1(-/-) mice. Genes Dev. 1999;13:709–717. doi: 10.1101/gad.13.6.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith CM, Hay ED. Epithelial-mesenchymal transformation during palatal fusion: carboxyfluorescein traces cells at light and electron microscopic levels. Development. 1992;116:1087–1099. doi: 10.1242/dev.116.4.1087. [DOI] [PubMed] [Google Scholar]
- Hay ED. An overview of epithelio-mesenchymal transformation. Acta Anat. 1995;154:8–20. doi: 10.1159/000147748. [DOI] [PubMed] [Google Scholar]
- Jamora C, DasGupta R, Kocieniewski P, Fuchs E. Links between signal transduction, transcription and adhesion in epithelial bud development. Nature. 2003;422:317–322. doi: 10.1038/nature01458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaartinen V, Cui XM, Heisterkamp N, Groffen J, Shuler CF. Transforming growth factor-beta3 regulates transdifferentiation of medial edge epithelium during palatal fusion and associated degradation of the basement membrane. Dev. Dyn. 1997;209:255–260. doi: 10.1002/(SICI)1097-0177(199707)209:3<255::AID-AJA1>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Kim K, Daniels KJ, Hay ED. Tissue-specific expression of beta-catenin in normal mesenchyme and uveal melanomas and its effect on invasiveness. Exp. Cell Res. 1998;245:79–90. doi: 10.1006/excr.1998.4238. [DOI] [PubMed] [Google Scholar]
- Kim K, Lu Z, Hay ED. Direct evidence for a role of beta-catenin/LEF-1 signalling pathway in the induction of EMT. Cell Biol. Int. 2002;26:463–476. doi: 10.1006/cbir.2002.0901. [DOI] [PubMed] [Google Scholar]
- Labbe E, Letamendia A, Attisano L. Association of Smads with lymphoid enhancer binding factor 1/T cell-specific factor mediates cooperative signaling by the transforming growth factor-beta and wnt pathways. Proc. Natl. Acad. Sci. USA. 2000;97:8358–8363. doi: 10.1073/pnas.150152697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaGamba D, Nawshad A, Hay ED. Microarray analysis of gene expression during epithelial-mesenchymal transformation. Dev. Dyn. 2005;234:132–142. doi: 10.1002/dvdy.20489. [DOI] [PubMed] [Google Scholar]
- Li W, Murai Y, Okada E, Matsui K, Hayashi S, Horie M, Takano Y. Modified and simplified western blotting protocol: use of intermittent microwave irradiation (IMWI) and 5% skim milk to improve binding specificity. Pathol. Int. 2002;52:234–238. doi: 10.1046/j.1440-1827.2002.01342.x. [DOI] [PubMed] [Google Scholar]
- Martinez-Alvarez C, Tudela C, Perez-Miguelsanz J, O′Kane S, Puerta J, Ferguson MW. Medial edge epithelial cell fate during palatal fusion. Dev. Biol. 2000;220:343–357. doi: 10.1006/dbio.2000.9644. [DOI] [PubMed] [Google Scholar]
- Medici D, Hay ED, Goodenough DA. Cooperation between Snail and LEF-1 transcription factors is essential for TGF-beta1-induced epithelial-mesenchymal transition. Mol. Biol. Cell. 2006;17:1871–1879. doi: 10.1091/mbc.E05-08-0767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamed OA, Clarke HJ, Dufort D. Beta-catenin signaling marks the prospective site of primitive streak formation in the mouse embryo. Dev. Dyn. 2004;231:416–424. doi: 10.1002/dvdy.20135. [DOI] [PubMed] [Google Scholar]
- Murakami T, Toda S, Fujimoto M, Ohtsuki M, Byers HR, Etoh T, Nakagawa H. Constitutive activation of Wnt/beta-catenin signaling pathway in migration-active melanoma cells: role of LEF-1 in melanoma with increased metastatic potential. Biochem. Biophys. Res. Commun. 2001;288:8–15. doi: 10.1006/bbrc.2001.5719. [DOI] [PubMed] [Google Scholar]
- Nawshad A, Hay ED. TGF[beta]3 signaling activates transcription of the LEF1 gene to induce epithelial-mesenchymal transformation during mouse palate development. J. Cell Biol. 2003;163:1291–1301. doi: 10.1083/jcb.200306024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawshad A, LaGamba D, Hay ED. Transforming growth factor beta (TGFbeta) signalling in palatal growth, apoptosis and epithelial mesenchymal transformation (EMT) Arch. Oral Biol. 2004;49:675–689. doi: 10.1016/j.archoralbio.2004.05.007. [DOI] [PubMed] [Google Scholar]
- Nishita M, Hashimoto MK, Ogata S, Laurent MN, Ueno N, Shibuya H, Cho KW. Interaction between Wnt and TGF-beta signaling pathways during formation of Spemann′s organizer. Nature. 2000;403:781–785. doi: 10.1038/35001602. [DOI] [PubMed] [Google Scholar]
- Peinado H, Portillo F, Cano A. Transcriptional regulation of cadherins during development and carcinogenesis. Int. J. Dev. Biol. 2004;48:365–375. doi: 10.1387/ijdb.041794hp. [DOI] [PubMed] [Google Scholar]
- Proetzel G, Pawlowski SA, Wiles MV, Yin M, Boivin GP, Howles PN, Ding J, Ferguson MW, Doetschman T. Transforming growth factor-beta 3 is required for secondary palate fusion. Nat. Genet. 1995;11:409–414. doi: 10.1038/ng1295-409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiomi N, Cui XM, Yamamoto T, Saito T, Shuler CF. Inhibition of SMAD2 expression prevents murine palatal fusion. Dev. Dyn. 2006;235:1785–1793. doi: 10.1002/dvdy.20819. [DOI] [PubMed] [Google Scholar]
- Shuler CF, Halpern DE, Guo Y, Sank AC. Medial edge epithelium fate traced by cell lineage analysis during epithelial-mesenchymal transformation in vivo. Dev. Biol. 1992;154:318–330. doi: 10.1016/0012-1606(92)90071-n. [DOI] [PubMed] [Google Scholar]
- Sun D, Vanderburg CR, Odierna GS, Hay ED. TGFbeta3 promotes transformation of chicken palate medial edge epithelium to mesenchyme in vitro. Development. 1998;125:95–105. doi: 10.1242/dev.125.1.95. [DOI] [PubMed] [Google Scholar]
- Takeichi M. Cadherins: key molecules for selective cell-cell adhesion. IARC Sci. Publ. 1988;92:76–79. [PubMed] [Google Scholar]
- Taya Y, O′Kane S, Ferguson MW. Pathogenesis of cleft palate in TGF-beta3 knockout mice. Development. 1999;126:3869–3879. doi: 10.1242/dev.126.17.3869. [DOI] [PubMed] [Google Scholar]
- Ten Dijke P, Hill CS. New insights into TGF-beta-Smad signaling. Trends Biochem. Sci. 2004;29:265–273. doi: 10.1016/j.tibs.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Thiery JP. Epithelial-mesenchymal transitions in tumor progression. Nat. Rev. Cancer. 2002;2:442–454. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- Thiery JP. Epithelial-mesenchymal transitions in development and pathologies. Curr. Opin. Cell Biol. 2003;15:740–746. doi: 10.1016/j.ceb.2003.10.006. [DOI] [PubMed] [Google Scholar]
- Tudela C, Formoso MA, Martinez T, Perez R, Aparicio M, Maestro C, Del Rio A, Martinez E, Ferguson M, Martinez-Alvarez C. TGF-beta3 is required for the adhesion and intercalation of medial edge epithelial cells during palate fusion. Int. J. Dev. Biol. 2002;46:333–336. [PubMed] [Google Scholar]
- Vaziri Sani F, Hallberg K, Harfe BD, McMahon AP, Linde A, Gritli-Linde A. Fate-mapping of the epithelial seam during palatal fusion rules out epithelial-mesenchymal transformation. Dev. Biol. 2005;285:490–495. doi: 10.1016/j.ydbio.2005.07.027. [DOI] [PubMed] [Google Scholar]
- Xu L, Chen YG, Massague J. The nuclear import function of Smad2 is masked by SARA and unmasked by TGFbeta-dependent phosphorylation. Nat. Cell Biol. 2000;2:559–562. doi: 10.1038/35019649. [DOI] [PubMed] [Google Scholar]
- Xu X, Han J, Ito Y, Bringas P, Jr, Urata MM, Chai Y. Cell autonomous requirement for Tgfbr2 in the disappearance of medial edge epithelium during palatal fusion. Dev. Biol. 2006;297:238–248. doi: 10.1016/j.ydbio.2006.05.014. [DOI] [PubMed] [Google Scholar]