Quantification of Intermediates Formed during the Reduction of Nitrite by Deoxyhemoglobin (original) (raw)
Abstract
Nitric oxide (NO) plays a crucial role in human physiology by regulating vascular tone and blood flow. The short life-span of NO in blood requires a mechanism to retain NO bioactivity in the circulation. Recent studies have suggested a mechanism involving the reduction of nitrite back to NO by deoxyhemoglobin in RBCs. A role for RBCs in transporting NO must, however, bypass the scavenging of NO in RBCs by hemoglobin. To understand how the nitrite reaction can deliver bioactive NO to the vasculature, we have studied the intermediates formed during the reaction. A reliable measure of the total concentration of heme-associated nitrite/NO intermediates formed was provided by combining filtration to measure free nitrite by chemiluminescence and electron paramagnetic resonance to measure the final product Hb(II)NO. By modifying the chemiluminescence method used to detect NO, we have been able to identify two intermediates: 1) a heme-associated nitrite complex that is released as NO in acid solution in the presence of ascorbate and 2) an intermediate that releases NO at neutral pH in the presence of ferricyanide when reacted with an Fe(III) ligand like azide. This species designated as “Hb(II)NO+ ⇆ Hb(III)NO” has properties of both isomeric forms resulting in a slower NO dissociation rate and much higher stability than Hb(III)NO, but provides a potential source for bioactive NO, which can be released from the RBC. This detailed analysis of the nitrite reaction with deoxyHb provides important insights into the mechanism for nitrite induced vasodilation by RBCs.
Nitric oxide (NO), also known as the endothelium-derived relaxing factor, is an important messenger molecule involved in the regulation of vascular tone and blood flow (1). The primary source for the synthesis of NO in the circulatory system involves endothelial nitric-oxide synthase (2). This enzyme requires oxygen for the synthesis of NO and is, therefore, less effective in the microcirculation where hypoxic vasodilation regulates the delivery of oxygen. Because nitric oxide has a life-time in blood of <2 ms (3), a mechanism is required to allow for more distal and sustained effects of NO at the reduced oxygen pressures found in the microcirculation. Recent studies have suggested that the bioactivity of NO can be conserved in the blood by the uptake of NO and/or nitrite by red blood cells (RBCs)2 and its interaction with hemoglobin (4–7). However, any role for the red cell in transporting nitric oxide must be able to avoid the very efficient scavenging of nitric oxide by both oxyhemoglobin (oxyHb) and deoxyhemoglobin (deoxyHb) that destroy and trap NO, respectively, preventing a physiological role for RBC NO.
In a series of studies, Stamler and co-workers (7–10) have hypothesized that NO can bypass this difficulty by being transferred to the β-93 thiol group of hemoglobin (Hb) forming _S_-nitrosylated hemoglobin (SNO-Hb) when partially heme nitrosylated hemoglobin (Hb(II)NO) is oxygenated. The allosteric quaternary conformational change of hemoglobin at low oxygen pressure destabilizes the β-93 nitrosylated thiol and results in the transfer of NO to membrane thiol groups facilitating the release of the NO to the plasma and the vasculature. However, the extremely low levels of SNO-Hb (11) found in human blood and its instability (12) as a result of intracellular reducing conditions within the RBCs do not support the SNO-Hb hypothesis as the major mechanism for NO transport (11–13).
The 2003 studies by Rifkind and Gladwin and their collaborators (4,5,14,15) proposed an alternative mechanism that involved the reduction of nitrite, formed by the oxidation of NO, back to NO by a reaction with deoxyHb. Nitrite is present in the blood at fairly high levels (0.1–0.5 μmol/liter) (4,16–18), and it is much more stable than NO or _S_-nitrosothiols (6), making nitrite an ideal storage pool that can be converted to NO. However, the mechanism by which the NO produced in the red cell by nitrite reduction is exported without being trapped or destroyed is still unclear. Recent studies by Rifkind and co-workers (5,13,19) have suggested that the trapping of NO by deoxyHb and/or oxyHb can be bypassed by the formation of a metastable intermediate(s) that retains the NO in a state that is not quenched by reacting with oxyHb or deoxyHb.
In this report, we quantitate the two intermediate species that are formed during the reduction of nitrite by deoxyHb when an excess of hemoglobin is present. We also demonstrate that one of the intermediate species designated as “Hb(II)NO+ ⇆ Hb(III)NO” has properties of Hb(II)NO+ and Hb(III)NO, respectively. This species has a slower NO dissociation rate and a much higher stability than Hb(III)NO. This intermediate is a potential source for bioactive NO that can be released from RBCs.
EXPERIMENTAL PROCEDURES
_Reagents_—All reagents were obtained from Sigma-Aldrich unless mentioned.
_Solution Preparations_—All hemoglobin solutions were prepared in 50 mm NaCl and 4 mm phosphate buffer (PBS), pH 7.4.
_DeoxyHb Preparation_—Human hemoglobin was purified from fresh hemolysate by gel filtration using a Sephadex G-100 column equilibrated with PBS, pH 7.4 at 4 °C. A 1 mm hemoglobin solution was deoxygenated in an anaerobic Coy glove box until the visible spectrum of hemoglobin corresponded to that of deoxyHb. A 0.1-cm path length cuvette was used for spectroscopic measurements of the 1 mm hemoglobin solutions.
_Hb(II)NO Preparation_—NO gas (2.5%) was purified, by passing through concentrated NaOH (5 m). Small amounts of this purified gas were transferred to a septum-sealed 100 μm deoxyHb solution to produce Hb(II)NO as confirmed by visible spectroscopy. The Hb(II)NO was then quickly passed down a Sephadex G-25 column anaerobically to remove unreacted NO. The final concentration of Hb(II)NO was determined by visible spectroscopy using the millimolar extinction coefficient for Hb(II)NO of 11.4 at 544 nm. The Hb(II)NO standard solution (∼44 μm) was flushed with inert gas, and stored in the freezer at –150 °C.
_MetHb Preparation_—Purified human hemoglobin (∼1.5 mm) was oxidized with a 50 mm solution of K3Fe(CN)6 (50 μl/ml) for 5 mins. K3Fe(CN)6 was removed from the metHb solution by gel filtration using a Sephadex G-25 column equilibrated with PBS, pH 7.4. The final concentration of metHb was determined by visible spectroscopy using the millimolar extinction coefficient for metHb of 4.4 at 631 nm. The metHb standard solution (∼0.75 mm) was stored in the freezer at –150 °C.
_Nitrite Preparation_—A 10 mm nitrite solution was prepared by dissolving NaNO–2 in 10 ml of an argon-saturated PBS solution, pH 7.4.
_Nitrite Reduction by DeoxyHb_—All experiments were carried out inside an anaerobic Coy glove box. 1 mm deoxyHb was reacted with 0.25 mm nitrite in PBS, pH 7.4 for 60 min at room temperature (22 °C). The free (non-reacted nitrite) was determined as a function of time by filtering aliquots of the reaction mixture at different time intervals using 10,000 mol. wt. cutoff microfilterfuge tubes (Rainin Instrument Co. Inc. filters) inside the anaerobic glove box. The amount of nitrite in the filtrate was determined by chemiluminescence (see below). The total nitrite reacted with hemoglobin was calculated by subtracting the free nitrite from the initial nitrite added. Aliquots of the same reaction mixture were used to determine Hb(II)NO by electron paramagnetic resonance (EPR).
_NOA Chemiluminescence Assays_—A Model-280 Nitric Oxide Analyzer (NOA) from Sievers Instruments was used to determine free nitrite and the various heme-associated nitrite/NO species. The NOA purge vessel was also kept inside the glove box and gas lines were connected through the gas ports of the glove box to the argon gas tank and the instrument. Argon was bubbled through the purge vessel to carry any released NO to the detector, which quantitates NO by measuring the gas-phase chemiluminescence NO reaction with ozone. The glove box temperature was maintained at 30 °C. The computer was operated using a wireless mouse from inside the glove box.
_Determination of Hb(II)NO and Nitrite Standards by Chemiluminescence_—5-μl injections of 5.5, 11, 22, 33, and 44 μm solutions of Hb(II)NO prepared by appropriate dilutions of the stock Hb(II)NO standard solution (∼44 μm) previously described, were injected into the purge vessel that contained 1.5 ml of 0.8m K3Fe(CN)6 plus 6.5 ml of 85% glacial acetic acid with 100 mm sulfanilamide to release the NO. The concentration dependent chemiluminescence signals for Hb(II)NO were used to prepare a calibration curve. Similarly, 5 μl of 5, 10, 20, 40, and 80 μm nitrite standard solutions prepared by appropriate dilutions of the stock 10 mm nitrite standard were injected into the purge vessel that contained 7 ml of glacial acetic acid and 1 ml of 0.5 m ascorbic acid to obtain a calibration curve for nitrite. Four injections were made for each sample using a Hamilton gas-tight syringe. The data were transferred to Origin 6.1 software program and the area of under the peak of each signal was integrated. The Hb(II)NO and nitrite standard curves were prepared by plotting the area of each standard against its concentration. The nitric oxide concentration of unknown samples was determined using the Hb(II)NO or nitrite standard curve prepared under the same conditions.
_Determination of Free Nitrite by Chemiluminescence_—5 ml of a 1 mm deoxyHb solution in a mini Petri dish was reacted with 0.25 mm nitrite inside the anaerobic glove box for 60 min at room temperature. At specific time intervals a 0.5 ml aliquot of the reaction mixture was placed into a microfilterfuge tube (Rainin filter with a 10,000 MW cutoff) and centrifuged for 2 min at 6,500 rpm using a microfuge (LK Scientific). After centrifugation, the filtration solutions were diluted accordingly, and 5 μl of the filtrate obtained at various time intervals was injected into the NOA purge vessel containing 7 ml of glacial acetic acid and 1 ml of 0.5 m ascorbic acid in order to determine the concentration of nitrite as previously described (16). Simultaneously, at each time interval, 0.4 ml of the remaining reaction mixture were placed anaerobically into 4-mm clear fused quartz EPR tubes (707 SQ250M-WILMAND) and frozen by submerging into liquid nitrogen for Hb(II)NO analysis by EPR.
_Determination of Heme-NO Species by Chemiluminescence_—1 mm deoxyHb solution contained in a septum-sealed cuvette, was reacted with 0.25 mm nitrite. 5 μl of the reaction mixture were withdrawn at varying time points and injected into the NOA purge vessel containing various reagents (see below) to determine the amount of different NO-related species (products) formed (released) from the reaction mixture.
_Reaction System for Determination of Heme-NO Species under Acidic Conditions_—The purge vessel contained 1.5 ml of 0.8 m K3Fe(CN)6 plus 6.5 ml of 85% glacial acetic acid with 100 mm sulfanilamide for determination of exclusively heme-NO species. For determination of heme-NO species and part of nitrite, sulfanilamide was not added to the purge vessel. For determination of heme-associated nitrite/NO species and any free nitrite, no sulfanilamide was present in the purge vessel but 0.5 m ascorbate was added, so that the purge vessel contained 1.5 ml of 0.8 m K3Fe(CN)6 plus 1.0 ml of 0.5 m ascorbic acid and 5.5 ml of glacial acetic acid. For determination of heme-associated nitrite/NO species without free nitrite the purge vessel contained 1.5 ml of 0.8 m K3Fe(CN)6 plus 1.0 ml of 0.5m ascorbic acid and 5.5 ml of glacial acetic that contained 100 mm sulfanilamide. The total volume of each reaction system was 8 ml.
Potassium ferricyanide was used to help release the NO from Hb(II)NO by oxidizing the hemoglobin. Sulfanilamide was used to react with nitrite under acidic conditions to eliminate the NO signal coming from nitrite. Acetic acid enhances the release of NO from Hb(II)NO and prevents protein foam formation. Ascorbic acid is a strong reducing agent that will reduce any nitrite to NO under acidic conditions. In the presence of sulfanilamide it provides a measure of the total nitrite associated or reacted with hemoglobin except for nitrosated (thiol nitrosylated) species that are stable in acid.
_Determination of Heme-NO species from the Residue_—1 mm deoxyHb and 0.25 mm nitrite were reacted for 60 min, the reaction mixture was then filtered to completion in a microfilter (Rainin filter with 10,000 MW cutoff) using a microfuge (LK Scientific) at 6,500 rpm. The residue on the filter was resuspended in PBS buffer pH 7.4 to an initial volume. The residue was analyzed by chemiluminescence at acidic pH with added ascorbate but without sulfanilamide (see above).
_Reaction System for Determination of Heme-NO Species under Neutral Conditions_—The purge vessel contained 0.2 m K3Fe(CN)6 in 50 mm phosphate buffer, pH 7.4, 0.1 ml of anti-foam reagent with or without 1 mm sodium azide for a total volume of 8 ml.
Azide binds to Fe(III) and was used to release NO from the intermediate species “Hb(II)NO+ ⇆ Hb(III)NO”. Although Hb(III)NO releases NO readily even at neutral pH, the different isoforms present in this species stabilizes the bound NO preventing the release of this NO at neutral pH. However, the interaction of azide with the Fe(III) isoform of this complex, is nevertheless, able to release NO.
_Determination of Heme-NO Species from the Reaction of metHb with Nitrite and Nitrate_—250 μm metHb solution in PBS buffer, pH 7.4 was mixed with a nitrite solution (at 1:1, 1:2, and 1:5, metHb:nitrite ratios) or a nitrate solution (at 1:1, metHb:nitrate ratio) in a septum-sealed cuvette. The reaction mixture was then analyzed by chemiluminescence at neutral pH in the presence of 1 mm sodium azide (see above).
_Determination of Heme-NO Species from the Reaction of oxyHb with Nitrite and Nitrate_—250 μm oxyHb solution in PBS buffer, pH 7.4 was mixed with a nitrite solution (at 1:1, oxyHb: nitrite ratio) or a nitrate solution (at 1:1, oxyHb:nitrate ratio). The reaction mixture was then analyzed by for the formation of heme-NO species by chemiluminescence at neutral pH in the presence of 1 mm sodium azide (see above).
_Determination of Heme-NO Species from the Reaction of metHb and NO_—100 μm metHb was reacted with 100 μm NO obtained from a NO-saturated PBS buffer, pH 7.4 solution in a septum-sealed cuvette for 60 min. The reaction mixture was then analyzed by chemiluminescence at neutral pH in the absence and presence of 1 mm sodium azide (see above).
_EPR Measurements_—EPR spectra were measured using a Bruker EMX-61A spectrometer with an Oxford continuous flow ESR-900 cryogenic unit. An Oxford ITC 502 temperature controller was used to maintain the sample temperature in the cavity at 10 K with an Oxford VC41 gas flow controller for liquid helium flow through the cryostat. EPR spectra were recorded in the region of 500–4500 G with a sweep time of 335.54 s, a time constant of 80.42 ms, 2 milliwatt power, and 100 kHz modulation frequency.
_Determination of Hb(II)NO Standards by EPR_—The Hb(II)NO standard solution (see above) was diluted accordingly with anaerobic 3× diluted PBS, pH 7.4 to make 0.4-ml Hb(II)NO standards of 44, 33, 22, 11, and 5.5 μm. The 0.4-ml aliquots were transferred anaerobically into 4-mm clear fused quartz EPR tubes (707 SQ250M-WILMAD), and frozen by submerging the tubes into liquid nitrogen. The samples were stored in the freezer at –150 °C until recorded. The EPR spectrum of each Hb(II)NO standard was recorded, and the region from 3150–3550 gauss (g = 2 signal) was integrated using the Bruker WINEPR software version 2.22 revision 10. A standard calibration curve for Hb(II)NO was produced which compared the concentration of Hb(II)NO versus the double integration obtained for each standard.
_Determination of metHb Spectrum by EPR_—The EPR spectrum of the metHb standard solution (see above) was recorded in the region of 900–3550 gauss.
_Determination of Heme-NO Species Formed during the Reaction of deoxyHb with Nitrite by EPR_—0.25 mm nitrite was reacted with a 1 mm deoxyHb in an anaerobic glove box. At various time intervals, 0.4-ml aliquots were transferred into 4-mm EPR tubes and immediately frozen by submerging into liquid nitrogen. The EPR signal was measured from 900 to 3550 gauss. The reaction of nitrite with deoxyHb produces metHb as well as Hb(II)NO, both of which have EPR signals in the g = 2 region. To quantitate the formation of Hb(II)NO it was, therefore, necessary to correct the Hb(II)NO spectrum in the g = 2 region from the contribution of metHb. For this purpose, the spectrum of a metHb standard was recorded, which resulted in signals in the g = 6 (900–1300 gauss) and g = 2 (3150–3550 gauss) regions. Because the g = 6 region observed in the spectrum of the reaction mixture contains the dominant contribution from metHb, but no contribution from Hb(II)NO, it was possible to use the g = 6 amplitude in the region of 900–1300 gauss to quantitate the formation of metHb. Furthermore, the amplitude contribution from metHb in the g = 2 region, overlapping with the Hb(II)NO contribution was determined by subtracting the metHb standard from the sample spectrum using the WINEPR algebra/subtraction function. The resulting spectrum corresponded to a spectrum with a corrected g = 2 amplitude of the Hb(II)NO signal in the region from 3150–3550 gauss. From the corrected spectra, the concentration of Hb(II)NO was determined during nitrite reduction using the Hb(II)NO standard curve.
RESULTS
_Filtration Studies to Determine the Total Nitrite Reacted with deoxyHb_—Nitrite reduction by deoxyHb involves the proton-assisted reduction of nitrite to NO (Reaction 1).
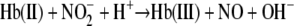 |
REACTION 1 |
|---|
The released NO will rapidly bind to deoxygenated hemoglobin chains producing nitrosylhemoglobin (Hb(II)NO) (Reaction 2) (20),
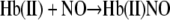 |
REACTION 2 |
|---|
or react with any oxygenated hemoglobin chains producing metHb and nitrate (Reaction 3) (21).
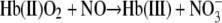 |
REACTION 3 |
|---|
In the absence of oxygen all of the reduced nitrite should react with deoxyHb to produce Hb(II)NO. In our original study on nitrite reduction, we compared the level of Hb(II)NO determined by EPR with a chemiluminescence method designed to measure total heme-associated NO (5). The finding that the chemiluminescence signal accounted for appreciably more NO than EPR indicated the presence of a significant pool of NO associated with hemoglobin that was not Hb(II)NO. This finding indicated that there are intermediate species formed in Reaction 1 before NO is released. Other investigators (14) have, however, challenged the implied stability of any hemoglobin associated intermediates. While EPR as a measure of Hb(II)NO is accepted, the validity of low pH chemiluminescence to measure heme-associated NO species can be open to uncertainties (the chemiluminescence method used in that study involved ferricyanide in acetic acid to release all the heme-associated NO and sulfanilamide to react with any free nitrite present). In a subsequent study (22), we used visible spectroscopy to analyze the species formed when nitrite reacts with deoxyHb at neutral pH. The analysis of this data supported the formation of a metastable intermediate, but was unable to delineate the actual visible spectrum of the intermediate leaving open uncertainties about the intermediates formed.
A determination of free nitrite as a function of time and a comparison of the nitrite consumed with the EPR determined Hb(II)NO is the most direct way to determine whether metastable intermediates are formed. For this purpose we designed a rapid filtration method that makes it possible to collect the unreacted nitrite from the reaction between deoxyHb and nitrite as a function of time. Fig. 1_A_ shows that the concentration of unreacted nitrite (free nitrite) removed from the reaction mixture (1 mm deoxyHb and 0.25 mm nitrite) at various time intervals up to 60 min exponentially decreases as a function of time. The amount of nitrite consumed (decrease in the free nitrite concentration) as the reaction proceeds is a direct measure of the heme-associated nitrite/NO species that are produced. After 60 min of reaction time only 29 μm of the original 250 μm is free, indicating that > 88% of the nitrite has reacted with hemoglobin. However,Fig. 1_B_ indicates that the nitrite consumed is much greater than the Hb(II)NO determined by EPR, which is 43 μm instead of the 221 μm calculated from the filtration experiment.
FIGURE 1.

Formation of heme-NO species during the reduction of nitrite (0.25 mm) by deoxyHb (1 mm) at 22 °C. A, consumption of nitrite as a function of reaction time. The amount of reacted nitrite was determined by the chemiluminescence method for measuring free nitrite (un-reacted nitrite) from the filtrate of the reaction mixture (see “Experimental Procedures” for details). B, the total amount of heme-NO species formed after 60 min calculated and observed by chemiluminescence and EPR. Calculated (initial minus free): these data correspond to the total amount of nitrite consumed during the reaction that is expected to have been converted to heme-NO species. This value was obtained by subtracting the amount of un-reacted nitrite at 60 min (A) from the initial amount of 250 μm. EPR: these data correspond to the total amount of Hb(II)NO detected by EPR.Observed (residue with ascorbate): these data correspond to the total amount of heme-NO species detected in the residue by the chemiluminescence assay under acidic pH in the absence of sulfanilamide and the presence of ascorbate (see “Experimental Procedures” for details). Four injections were made for each sample. The average concentration from the integrated signals is shown. Values represent the mean ± S.D. of three independent experiments for each sample.
To determine whether the nitrite consumed reflects the nitrite associated with the hemoglobin, we collected the hemoglobin residue after all the low molecular weight material was removed at the end of a 60-min reaction time. We then determined the total chemiluminescence signal of the residue in acidic pH (acetic acid with K3Fe(CN)6 and ascorbate) in order to determine all possible heme-associated NO species. As shown inFig. 1_B_ the observed chemiluminescence of the residue corresponds to 201 μm of the original nitrite added or 91% of the nitrite consumed. The EPR, therefore, does not detect 158 μm or 78.6% of the nitrite that has reacted with hemoglobin.
The comparison of the total consumed nitrite (221 μm) with the total nitrite reacted products detected in the hemoglobin residue (201 μm) results in only 20 μm (8%) not accounted for. This discrepancy can be attributed to other low molecular weight species not detected by this chemiluminescence method and/or to the formation of SNO-Hb, which would also not be detected by low pH chemiluminescence (13).
_Chemiluminescence Determination of an Intermediate Released from Hemoglobin as NO by Ascorbate_—The filtration experiment (Fig. 1) in comparison to the EPR data indicates that other heme-NO species in addition to Hb(II)NO are formed during the nitrite reaction. Our earlier studies used ferricyanide in acetic acid with a large excess of sulfanilamide to measure what we assumed to be the total heme-associated nitrite/NO species. By this method the non-reacted (free) nitrite was removed by sulfanilamide and the ferricyanide in acid was assumed to release NO from all heme-associated NO species except SNO-Hb, which is stabilized in acid solution. As shown inFig. 2_A_, the chemiluminescence signal under these conditions after 60 min of reaction time accounted for only 100 μm (45.2% of the nitrite consumed or 49.8% of the total heme-associated NO species as indicated by the chemiluminescence of the residue). The finding that the signal intensity is nearly the same without the addition of sulfanilamide is consistent with the low levels of free nitrite after 60 min (Fig. 1_A_).
FIGURE 2.

The total amount of NO species determined by the chemiluminescence method at acidic pH under various experimental conditions when 0.25 mm nitrite is reacted with 1 mm deoxyHb up to 60 min at pH 7.4. A, total heme-NO species measured after 60 min of reaction time. The purge vessel contained 85% glacial acetic acid with 0.15m ferricyanide with or without sulfanilamide (100 mm) and/or ascorbate (62.5 mm). residue: sample with all free nitrite removed by filtration and ascorbate added to the purge vessel without sulfanilamide; the other samples involved the total reaction mixture with the additives indicated in the figure (see “Experimental Procedures” for additional details). B, total NO species measured after 1 min of reaction time. The purge vessel contained 85% glacial acetic acid with 0.15m ferricyanide with or without sulfanilamide (100 mm) and/or ascorbate (62.5 mm) as indicated in the figure. Results are shown for nitrite reacted with deoxyHb and for nitrite without hemoglobin under similar experimental conditions. C, comparison of the time course for the formation of the expected (•) and the observed (▪) total amount of heme-NO species. The expected total heme-NO species (•) was obtained by subtracting at each time point the free nitrite from the chemiluminescence obtained at acidic pH when ascorbate was present in the purge vessel without sulfanilamide. The observed total heme-NO species (▪) was obtained by the chemiluminescence assay at acidic pH with sulfanilamide and no ascorbate in the purge vessel. D, the time course for the formation of the nitrite-associated intermediate species. This time course is the difference spectrum between the expected and the observed time courses in C. Values represent the mean ± S.D. of three independent experiments for each sample at each time point.
Our studies on the method to detect nitrite (16) show that nitrite is not efficiently detected at low pH unless a reducing agent such as ascorbate is added. This observation suggested that nitrite associated hemoglobin may not be detected even at low pH without adding a reducing agent like ascorbate. The presence of a complex involving nitrite associated with the hemoglobin that has not yet been reduced was confirmed by the increase in the total amount of heme-associated nitrite/NO species from 100 μm (no ascorbate) to 154 μm (added ascorbate) even when sulfanilamide is also added to react with any free nitrite (Fig. 2_A_). This increase, however, does not account for all of the heme-associated nitrite/NO species found in the residue (201 μm) (Fig. 2_A_). With ascorbate and without sulfanilamide the chemiluminescence signal corresponds to 227 μm, which is consistent with the total heme-associated nitrite/NO species from the residue (201 μm) plus the free nitrite (29 μm). This indicates that all of the undetected heme-associated species at acidic pH (without ascorbate) can be attributed to nitrite bound to hemoglobin that is not yet reduced. In the presence of sulfanilamide some of the heme-associated nitrite that is reduced by ascorbate reacts with sulfanilamide in the purge vessel and is not detected.
In the presence of a large pool of free nitrite after about 1 min of reaction time, only a small amount of heme-associated NO is detected by low pH chemiluminescence in the presence of sulfanilamide (Fig. 2_B_). Even without sulfanilamide only part of the added nitrite is recovered, however, with added ascorbate and without sulfanilamide, we are able to detect all the added nitrite including free nitrite plus any heme-associated nitrite/NO species made (Fig. 2_B_). Also shown inFig. 2_B_ are the results obtained with a 250 μm nitrite solution without added hemoglobin. Without ascorbate no signal is obtained with sulfanilamide and only part of the nitrite is detected without sulfanilamide. Only with ascorbate and without sulfanilamide is the total 250 μm nitrite detected. These results indicate that low pH chemiluminescence will only quantitatively detect heme-associated nitrite in the presence of ascorbate. Furthermore, in the presence of sulfanilamide without ascorbate the heme-associated nitrite is not detected.
Fig. 2_C_ plots the total amount of heme-associated NO species expected and the total amount detected by low pH chemiluminescence at various time intervals in the reaction up to 60 min. The expected curve for the total amount of heme-associated NO species generated by subtracting the amount of free nitrite from the heme-associated NO species detected in the presence of ascorbate shows that the heme-associated NO species form slowly with a half-time of 25 min reaching a level of ∼220 μm after 60 min. The curve generated by plotting the total amount of heme-associated NO species detected in the presence of sulfanilamide without added ascorbate also shows the slow formation of reaction products with a half-time of 25 min but only reaching a level of 100 μm after 60 min. The unaccounted fraction of ∼120 μm corresponds to the heme-associated nitrite species that cannot be detected by low pH chemiluminescence without added ascorbate.Fig. 2_D_ plots the total amount of heme-associated nitrite species formed as a function of time by subtracting the actual curve from the expected curve inFig. 2_C_. This curve represents nitrite that is associated with hemoglobin that has not yet been reduced. The kinetic curve is best fitted with a double exponential function, indicating that the initial more rapid reaction involves the initial association of nitrite with the deoxyHb followed by a slower reaction that suggests a conformational change in the heme pocket that stabilizes the binding of nitrite to deoxyHb and may be required to facilitate the reduction of nitrite.
_Chemiluminescence Determination of an Intermediate Released from Hemoglobin as NO by Azide at Neutral pH_—The acidic pH chemiluminescence assay without added ascorbate has been shown (Fig. 2) to measure the heme-associated NO species where the nitrite has already reacted with the hemoglobin and not the nitrite associated with hemoglobin that has not yet reacted. A comparison of the heme-associated NO species detected by acidic pH chemiluminescence and by EPR (method specific for Hb(II)NO), however, indicates that other species in addition to Hb(II)NO are included in this chemiluminescence signal. To further investigate the species involved, we performed the chemiluminescence assay at neutral pH with ferricyanide in the purge vessel. Under these conditions the Hb(II)NO is oxidized to Hb(III)NO. Because of the rapid off-rate for NO from Hb(III)NO (NO off-rate ∼1 s–1) (23), this NO is released by the argon flushing through the purge vessel, and is included in the chemiluminescence signal detected under these conditions.
As shown in Fig. 3_A_, the chemiluminescence signal obtained at neutral pH (∼47 μm) corresponds to the amount of Hb(II)NO detected by EPR (∼43 μm). The comparison with the acidic pH chemiluminescence results without ascorbate (Fig. 2_A_), however, indicates that there is a heme-associated intermediate NO species that is not released by ferricyanide at neutral pH.
FIGURE 3.

The total amount of heme-NO species during the reaction of 0.25 mm nitrite with 1 mm deoxyHb up to 75 min at pH 7.4 that are detected without ascorbate reduction. A, the total amount of heme-NO species detected after 60 min reaction time by EPR and by chemiluminescence at acidic pH and neutral pH in the absence and presence of 1 mm sodium azide. Acidic pH: the purge vessel contained 100 mm sulfanilamide in 85% glacial acetic acid with 0.15 m ferricyanide. Neutral pH: the purge vessel contained 0.2 m ferricyanide in 50 mm phosphate buffer, pH 7.4 with or without 1 mm azide for a total volume of 8 ml. The 60-min reaction mixture was pretreated with 1 mm azide (s) or 1 mm azide was added in the purge vessel (v) (see “Experimental Procedures” for details). B, the time course for the formation of heme-NO species obtained by EPR (○); neutral pH without azide (•); neutral pH with azide (□); acidic pH (▪). C, time course for the formation of the azide released isomeric intermediate. The data were generated by subtracting the time course of the reaction mixture analyzed by the chemiluminescence assay under neutral pH conditions from the time course of the reaction analyzed under neutral pH conditions but in the presence of azide. Values represent the mean ± S.D. of three independent experiments for each sample at each time point.
Our earlier studies (22) suggested the formation of an intermediate of the form “Hb(II)NO+ ⇆ Hb(III)NO”, with properties of Hb(III)NO and of Hb(II)NO+ with a possible contribution from the β-93 sulfhydryl (13). We have shown (22) that the electronic structure of this intermediate, which favors the Fe(III)NO state, inhibits CO binding and results in a very slow reaction with CO, indicative of its stability. At the same time the electronic structure that favors the Hb(II)NO+ state is responsible for the slow off-rate of NO. It is the presence of both of these isomers that result in the observation that unlike Hb(II)NO, which is oxidized to Hb(III)NO and releases its NO in the presence of ferricyanide, the intermediate does not release NO in the presence of ferricyanide at neutral pH. However, it may release its NO under acidic pH.
To determine whether it is this species that is responsible for the difference in the chemiluminescence signal detected at neutral pH and at acidic pH we sought a reagent which would release NO from this intermediate species at neutral pH. For this purpose we used azide (N–3) due to its high affinity to Hb(III) (24). The reaction of azide with this putative intermediate species is expected to shift the distribution of isomeric forms toward Hb(III)NO, which would react with azide releasing NO (Reaction 4).
 |
REACTION 4 |
|---|
As shown in Fig. 3_A_, both pretreatment of the reaction mixture with azide at 60 min or addition of azide in the purge vessel at neutral pH resulted in an increase in the neutral pH release of NO. In both cases the chemiluminescence signal was similar to that obtained in acidic pH with ferricyanide (without ascorbate), and 100 μm (45.2%) of the nitrite consumed is detected, suggesting the release of NO from a stable intermediate with Hb(III) character, such as, “Hb(II)NO+ ⇆ Hb(III)NO”.
The reaction mixture studied by EPR, acidic pH and neutral pH chemiluminescence in the absence and presence of azide was monitored at various time intervals. The kinetics inFig. 3_B_ show that EPR and neutral pH chemiluminescence monitor the formation of the same reaction product, Hb(II)NO, which results in a final average concentration of ∼45 μm after 60 min. Meanwhile, the same reaction products (Hb(II)NO and “Hb(II)NO+ ⇆ Hb(III)NO”) can be monitored by chemiluminescence at acidic pH and at neutral pH in the presence of azide. The formation of the intermediate with different isomeric forms (“Hb(II)NO+ ⇆ Hb(III)NO”) as a function of time was determined by subtracting the time course of the reaction monitored at neutral pH from that at neutral pH in the presence of azide (Fig. 3_C_).Fig. 3_C_ shows that this intermediate forms slowly, and reaches a concentration of 55 μm at 60 min.
Therefore, out of the total amount of heme-associated NO species (100 μm) detected by the chemiluminescence method at acidic pH and at neutral pH in the presence of azide, ∼45 μm correspond to the formation of Hb(II)NO and ∼55 μm correspond to the intermediate “Hb(II)NO+ ⇆ Hb(III)NO”.
To confirm that the chemiluminescence signal obtained with azide is due to the reaction of azide with the intermediate and not with any of the other species present, we tested the intensity of the chemiluminescence signal obtained when azide was added to metHb or oxyHb in the presence of nitrite or nitrate (supplemental Fig. S1). In all cases, the signal observed corresponded to <1 μm NO, thereby eliminating the contribution of other species to the signal detected in the presence of azide.
_Azide Reaction with the Intermediate Formed When NO Reacts with MetHb_—Hb(III)NO formed when NO binds to metHb undergoes reductive nitrosylation forming Hb(II)NO (13,25). This reaction is known to involve the formation of the same intermediate formed during nitrite reduction, “Hb(II)NO+ ⇆ Hb(III)NO” (13,19). To confirm that azide can release NO from this intermediate, the affect of azide on the chemiluminescent signal obtained when NO reacts with metHb was investigated.
A solution of metHb mixed with NO saturated buffer was analyzed by chemiluminescence after 60 min at neutral pH in the absence and presence of azide. Fig. 4 shows that when a 1:1 solution of metHb and NO are mixed, in addition to the NO released from the Hb(II)NO present after 60 min an additional pool of NO is released from the reaction mixture at neutral pH when azide is added.
FIGURE 4.

The total amount of heme-NO species detected by the chemiluminescence assay when 100 μm metHb is reacted with 100 μm NO for 60 min. The purge vessel contained 0.2 m ferricyanide in 50 mm phosphate buffer, pH 7.4 with or without 1 mm azide for a total volume of 8 ml (see “Experimental Procedures” for details). Four injections were made for each sample, the average concentration from the integrated signals is shown. Values represent the mean ± S.D. of three independent experiments for each sample.
The initial product formed during this reaction is Hb(III)NO, which readily releases NO. However, Hb(III)NO is no longer detected after 10 min (22). The Hb(III)NO is converted to the same intermediate with multiple forms that we see during the reaction of nitrite with deoxyHb, which is slowly converted to Hb(II)NO (26). Studies involving the reaction of CO with metHb that has reacted with NO indicate that this intermediate is present even after 60 min (22).
The intermediate known to be produced during the reaction of NO with metHb, thus, releases NO in the presence of added azide in the same way as the intermediate formed by the reaction of nitrite with deoxyHb.
DISCUSSION
Intermediates some of which have properties of NO+ are known to form during the reduction of nitrite by deoxyHb. It has however, been assumed by most investigators (14,15,27,28) that these intermediates are only transients with negligible concentrations present at anytime. We have now unambiguously demonstrated that this is not the case and that there are appreciable concentrations of two intermediates formed during the reduction of nitrite by deoxyHb.
The final release of NO during the reduction of nitrite by deoxyHb can be monitored by the formation of Hb(II)NO in the presence of an excess of deoxyHb. However, even after 60 min of reaction time the EPR data accounts for only 43 μm or 21.4% of the nitrite shown to be associated with hemoglobin detected by chemiluminescence of the residue (Fig. 1). The filtration and chemiluminescence data, thus, indicates that there are appreciable concentrations of heme-associated nitrite/NO species other than Hb(II)NO that are not detected by EPR.
Chemiluminescence studies on the nitrite reduction by deoxyHb at acidic pH and neutral pH confirmed that most of the consumed nitrite is associated with two heme-associated intermediates (Scheme 1). The initial intermediate formed during the reaction involves nitrite bound to hemoglobin that is not yet reduced (Reaction 5).
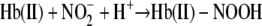 |
REACTION 5 |
|---|
This complex accounts for ∼50% of the nitrite shown to be associated with hemoglobin after a reaction time of 60 min. The quantitative conversion of this species to NO requires low pH ferricyanide and ascorbate (Fig. 2 andReaction 6).
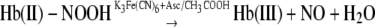 |
REACTION 6 |
|---|
Included in this species are not only the initial nitrite complex that rapidly binds to deoxyHb, but also additional species that may be stabilized by conformational changes in the ligand pocket that occur more slowly, but do not yet involve the cleavage of OH– from the nitrite forming NO+. Because these complexes involve nitrite and not NO+ or NO, they do not generate NO unless they are reduced by ascorbate. It is presumably this complex which is responsible for the reported higher nitrite levels in red cells than in plasma (18). While this complex is associated with deoxyHb it is presumably displaced by ligands that bind to Fe(II) like oxygen or CO and explains the reported decrease in the chemiluminescence signal in the presence of oxygen (5). This pool of hemoglobin associated nitrite provides an intracellular pool of nitrite that can be reduced to NO.
SCHEME 1.

The reaction scheme for the reduction of nitrite by deoxyhemoglobin.
The heme-associated nitrite subsequently has the OH– cleaved forming “Hb(II)NO+” inReaction 7.
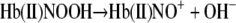 |
REACTION 7 |
|---|
The reaction with CO (22) is indicative of the Fe(II) properties of this species. However, because of the appreciable electrophilic properties of NO+, this species (the second intermediate) also has properties of Hb(III)NO, where the NO+ takes an electron from the Fe(II) forming NO and Fe(III) inReaction 8.
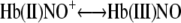 |
REACTION 8 |
|---|
The reaction with azide is indicative of the Fe(III) properties of this species. We also have evidence for a contribution from the β-93 thiol that can transfer an electron to NO+ forming a thiyl radical “·SHb(II)NO” (13).
While contributions from all three of these components are clear, the actual extent of the electronic delocalization is not known. A unique electronic state with complete delocalization between Hb(II)NO+ and Hb(III)NO need not be present. Instead the intermediate can be considered a resonance structure, and the species may actually reflect the presence of different isomers involving different protonation states or amino acid conformations in the heme pocket, which alter the tilt angle of the ligand bound to iron. These changes will result in different ligand affinities and different electronic distributions.
The structure of this intermediate must, however, explain the stability of this intermediate and the finding that it does not release NO when flushed with argon at neutral pH even with ferricyanide added to the purge vessel. It is, therefore, only detected at neutral pH in the presence of a strong Fe(III) ligand like azide (Fig. 3 andReaction 4).
The nature of this intermediate was confirmed by showing that azide also releases NO from the same intermediate formed when NO reacts with metHb (Fig. 4) and is not released from metHb or oxyHb in the presence of nitrite or nitrate (supplemental Fig. S1). This intermediate is analogous to the intermediate formed during nitrite reduction by heme cd1 nitrite reductase (29). The stability of this same species formed during the reaction of NO with metHb has been demonstrated by the slow reaction with CO (22). This intermediate “Hb(II)NO+ ⇆ Hb(III)NO” accounts for ∼27% of the nitrite associated with hemoglobin after a reaction time of 60 min. The stability of this complex explains its accumulation in the red cell even in the presence of oxyHb and deoxyHb (5). This pool of NO thus provides a pool of potentially bioactive NO in the red cell.
This intermediate does however slowly release NO producing Hb(III) and NO. However, with excess deoxyHb present in solution, this NO becomes rapidly inaccessible by binding to the heme forming the final reaction product of Hb(II)NO (Reaction 2), which accounts for the remaining ∼23% of the nitrite associated with hemoglobin at 60 min.
As the reaction proceeds and conformational changes take place in the ligand pocket which inhibit the stability of the Hb(II)NO+ isomer of the intermediate, the isomer with properties of Hb(III)NO predominates as a transient species, which rapidly releases its NO.
_The Nitrite Reductase Reaction by deoxyHb in the RBC and Its Putative Effect on Vascular Activity_—We have demonstrated that the nitrite reductase activity of deoxyHb and presumably the deoxygenated hemoglobin in the RBC is not a simple reaction where nitrite is converted to NO. The reaction of nitrite with deoxyHb is instead demonstrated to involve two intermediate species (Scheme 1). The initial intermediate retains nitrite associated hemoglobin providing a ready source of nitrite that can be reduced to NO. This intermediate is converted to an intermediate with properties of Hb(II)NO+ and Hb(III)NO that, particularly at low nitrite/hemoglobin ratios (22) found in vivo, is stable even in the presence of a large excess of oxyHb and deoxyHb. This intermediate provides a pool of potentially bioactive NO. Our earlier studies indicate that the release of NO from this intermediate is facilitated by excess nitrite and is conformationally regulated (22). More recently we have shown (30,31) that the R quaternary conformation favors formation of this intermediate, but that the T-quaternary conformation favors the release of NO from the intermediate. The release of NO from hemoglobin in the T-state that also displays an increase in the affinity for the RBC membrane can, thus, explain conformationally induced transfer of NO from the RBC to the vasculature. This regulated release of NO from the RBC provides a potential mechanism for RBC-induced vasodilation.
Supplementary Material
[Supplemental Data]
*
This work was supported, in whole or in part, by the National Institutes of Health NIA Intramural Research Program.
S⃞
The on-line version of this article (available athttp://www.jbc.org) contains supplemental Fig. S1.
Footnotes
2
The abbreviations used are: RBC, red blood cell; PBS, phosphate-buffered saline; EPR, electron paramagnetic resonance; SNO-Hb, _S_-nitrosylated hemoglobin; oxyHb, oxyhemoglobin; deoxyHb, deoxyhemoglobin.
References
- 1.Ignarro, L. J., Cirino, G., Casini, A., and Napoli, C. (1999) J. Cardiovasc. Pharmacol. 34 879–886 [DOI] [PubMed] [Google Scholar]
- 2.Gautier, C., van, F. E., Mikula, I., Martasek, P., and Slama-Schwok, A. (2006) Biochem. Biophys. Res. Commun. 341 816–821 [DOI] [PubMed] [Google Scholar]
- 3.Liu, X., Miller, M. J., Joshi, M. S., Sadowska-Krowicka, H., Clark, D. A., and Lancaster, J. R., Jr. (1998) J. Biol. Chem. 273 18709–18713 [DOI] [PubMed] [Google Scholar]
- 4.Cosby, K., Partovi, K. S., Crawford, J. H., Patel, R. P., Reiter, C. D., Martyr, S., Yang, B. K., Waclawiw, M. A., Zalos, G., Xu, X., Huang, K. T., Shields, H., Kim-Shapiro, D. B., Schechter, A. N., Cannon, R. O., III, and Gladwin, M. T. (2003) Nat. Med. 9 1498–1505 [DOI] [PubMed] [Google Scholar]
- 5.Nagababu, E., Ramasamy, S., Abernethy, D. R., and Rifkind, J. M. (2003) J. Biol. Chem. 278 46349–46356 [DOI] [PubMed] [Google Scholar]
- 6.Lundberg, J. O., and Weitzberg, E. (2005) Arterioscler. Thromb. Vasc. Biol. 25 915–922 [DOI] [PubMed] [Google Scholar]
- 7.Jia, L., Bonaventura, C., Bonaventura, J., and Stamler, J. S. (1996) Nature 380 221–226 [DOI] [PubMed] [Google Scholar]
- 8.McMahon, T. J., Moon, R. E., Luschinger, B. P., Carraway, M. S., Stone, A. E., Stolp, B. W., Gow, A. J., Pawloski, J. R., Watke, P., Singel, D. J., Piantadosi, C. A., and Stamler, J. S. (2002) Nat. Med. 8 711–717 [DOI] [PubMed] [Google Scholar]
- 9.Luchsinger, B. P., Rich, E. N., Gow, A. J., Williams, E. M., Stamler, J. S., and Singel, D. J. (2003) Proc. Natl. Acad. Sci. U. S. A. 100 461–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pawloski, J. R., Hess, D. T., and Stamler, J. S. (2001) Nature 409 622–626 [DOI] [PubMed] [Google Scholar]
- 11.Gladwin, M. T., Ognibene, F. P., Pannell, L. K., Nichols, J. S., Pease-Fye, M. E., Shelhamer, J. H., and Schechter, A. N. (2000) Proc. Natl. Acad. Sci. U. S. A. 97 9943–9948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gladwin, M. T., Wang, X., Reiter, C. D., Yang, B. K., Vivas, E. X., Bonaventura, C., and Schechter, A. N. (2002) J. Biol. Chem. 277 27818–27828 [DOI] [PubMed] [Google Scholar]
- 13.Nagababu, E., Ramasamy, S., and Rifkind, J. M. (2006) Nitric. Oxide 15 20–29 [DOI] [PubMed] [Google Scholar]
- 14.Kim-Shapiro, D. B., Gladwin, M. T., Patel, R. P., and Hogg, N. (2005) J. Inorg. Biochem. 99 237–246 [DOI] [PubMed] [Google Scholar]
- 15.Huang, K. T., Keszler, A., Patel, N., Patel, R. P., Gladwin, M. T., Kim-Shapiro, D. B., and Hogg, N. (2005) J. Biol. Chem. 280 31126–31131 [DOI] [PubMed] [Google Scholar]
- 16.Nagababu, E., and Rifkind, J. M. (2007) Free Radic. Biol. Med. 42 1146–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kehmeier, E. S., Kropp, M., Kleinbongard, P., Lauer, T., Balzer, J., Merx, M. W., Heusch, G., Kelm, M., Lepper, W., and Rassaf, T. (2008) Free Radic. Biol. Med. 44 1945–1950 [DOI] [PubMed] [Google Scholar]
- 18.Dejam, A., Hunter, C. J., Pelletier, M. M., Hsu, L. L., Machado, R. F., Shiva, S., Power, G. G., Kelm, M., Gladwin, M. T., and Schechter, A. N. (2005) Blood 106 734–739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rifkind, J. M., Nagababu, E., and Ramasamy, S. (2006) Antioxid. Redox. Signal. 8 1193–1203 [DOI] [PubMed] [Google Scholar]
- 20.Doyle, M. P., Pickering, R. A., DeWeert, T. M., Hoekstra, J. W., and Pater, D. (1981) J. Biol. Chem. 256 12393–12398 [PubMed] [Google Scholar]
- 21.Lissi, E. (1998) Free Radic. Biol. Med. 24 1535–1536 [DOI] [PubMed] [Google Scholar]
- 22.Nagababu, E., Ramasamy, S., and Rifkind, J. M. (2007) Biochemistry 46 11650–11659 [DOI] [PubMed] [Google Scholar]
- 23.Sharma, V. S., Traylor, T. G., Gardiner, R., and Mizukami, H. (1987) Biochemistry 26 3837–3843 [DOI] [PubMed] [Google Scholar]
- 24.Brittain, T. (2000) J. Inorg. Biochem. 81 99–103 [DOI] [PubMed] [Google Scholar]
- 25.Addison, A. W., and Stephanos, J. J. (1986) Biochemistry 25 4104–4113 [DOI] [PubMed] [Google Scholar]
- 26.Hoshino, M., Maeda, M., Konishi, R., Seki, H., and Ford, P. C. (1996) J. Am. Chem. Soc. 118 5702–5707 [Google Scholar]
- 27.Luchsinger, B. P., Rich, E. N., Yan, Y., Williams, E. M., Stamler, J. S., and Singel, D. J. (2005) J. Inorg. Biochem. 99 912–921 [DOI] [PubMed] [Google Scholar]
- 28.Angelo, M., Singel, D. J., and Stamler, J. S. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 8366–8371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang, Y., and Averill, B. A. (1960) J. Am. Chem. Soc. 118 3972–3973 [Google Scholar]
- 30.Rifkind, J. M., Ramasamy, S., and Nagababu, E. (2007) Biophys. J. 383A–384A
- 31.Nagababu, E., Cao, Z., Ramasamy, S., and Rifkind, J. M. (2008) XVth International Conference on Oxygen Binding and Sensing Proteins P27
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
[Supplemental Data]