Pericyte Requirement for Anti-Leak Action of Angiopoietin-1 and Vascular Remodeling in Sustained Inflammation (original) (raw)
Abstract
Blood vessel leakiness is an early, transient event in acute inflammation but can also persist as vessels undergo remodeling in sustained inflammation. Angiopoietin/Tie2 signaling can reduce the leakiness through changes in endothelial cells. The role of pericytes in this action has been unknown. We used the selective PDGF-B-blocking oligonucleotide aptamer AX102 to determine whether disruption of pericyte-endothelial crosstalk alters vascular leakiness or remodeling in the airways of mice under four different conditions: i) baseline, ii) acute inflammation induced by bradykinin, iii) sustained inflammation after 7-day infection by the respiratory pathogen Mycoplasma pulmonis, or iv) leakage after bradykinin challenge in the presence of vascular stabilization by the angiopoietin-1 (Ang1) mimic COMP-Ang1 for 7 days. AX102 reduced pericyte coverage but did not alter the leakage of microspheres from tracheal blood vessels at baseline or after bradykinin; however, AX102 exaggerated leakage at 7 days after M. pulmonis infection and increased vascular remodeling and disease severity at 14 days. AX102 also abolished the antileakage effect of COMP-Ang1 at 7 days. Together, these findings show that pericyte contributions to endothelial stability have greater dependence on PDGF-B during the development of sustained inflammation, when pericyte dynamics accompany vascular remodeling, than under baseline conditions or in acute inflammation. The findings also show that the antileakage action of Ang1 requires PDGF-dependent actions of pericytes in maintaining endothelial stability.
Stability of the microvasculature in health and disease is governed by bidirectional signaling between endothelial cells and pericytes (mural cells).1–3 The interaction of these cells changes under conditions that increase vascular permeability.4–6 Platelet-derived growth factor subunit B (PDGF-B) is an important vascular stabilizing factor that acts through PDGF receptor-β (PDGFR-β) signaling in pericytes.7–9 PDGF-B from endothelial cells promotes proliferation, migration, attraction, and survival of pericytes.7,10–12 Angiopoietins (Ang1, Ang2, and Ang3 in mice; ANG1, ANG2, and ANG4 in humans), which act through Tie2 receptor signaling in endothelial cells and possibly in pericytes, are other key regulators of vascular plasticity and stability.13–15 Ang1 from pericytes and other sources and Ang2 primarily from endothelial cells balance one another in the processes of endothelial cell growth, remodeling, and maturation.15–18 Together, PDGF-B and Ang1 have reciprocal interactions, regulating expression of one another19 and controlling vascular stability through their respective signaling pathways.2,20
The vascular stabilizing actions of Ang1 include protection against leakage by preventing the formation of intercellular gaps in the endothelium after exposure to inflammatory mediators.21–24 In contrast, overexpression of Ang2 tends to weaken pericyte-endothelial interactions and can result in pericyte loss and lead to vascular regression or proliferation.14,25,26 The vascular stabilizing action of Ang1 is more potent when Ang2 is blocked, and the reverse occurs when Ang1 is inhibited.14,27 It is not certain whether PDGF-B and pericytes contribute to these actions of Ang1 and Ang2.
Vascular remodeling is a conspicuous and functionally important component of the pathophysiology of chronic inflammatory conditions of the airways, skin, gastrointestinal tract, and other organs.28 Persistent inflammation is accompanied by changes in the structure and function of the vasculature, with capillaries acquiring the phenotype of venules that support leukocyte influx.29–31 In this remodeling, endothelial cells undergo distinctive changes in molecular phenotype that support innate and adaptive immune responses.32–34 Angiopoietin signaling through Tie2 receptors can drive vascular remodeling and participates in endothelial cell changes that occur in inflammation.31,32,35,36
The important role of pericytes in vascular stability is well documented by genetic and pharmacological studies that interfere with PDGF-B signaling,3,7,10,11 but less is known about how pericytes influence the vasculature in inflammation, in which leakage and vascular remodeling are prominent features. Pericytes respond to lipopolysaccharide and produce multiple cytokines.4,6 Pericytes may limit leakage by covering endothelial gaps.37 Pericytes have protective functions in models of diabetic retinopathy,14,38 and pericyte loss has the opposite effect.26,39 Pericyte loss is reduced by deletion of Ang2 and is promoted by Ang2 overexpression.26,39 Pericyte associations with endothelial cells change as capillaries enlarge in response to angiopoietins or undergo remodeling into venules in sustained inflammation,35,36 but the mechanisms and consequences are unknown.
In the present study, we examined the role of pericytes in maintenance of vascular stability as blood vessels undergo remodeling in the airways of mice. In particular, we determined the effects of PDGF-B-dependent interactions of endothelial cells and pericytes on vessel pericyte coverage, leakiness, and extent of remodeling. PDGF-B was selectively inhibited by administering the DNA aptamer AX102, which is known to affect pericytes in normal airways and tumors.12 Normal blood vessels were compared to an early stage of vascular remodeling in inflamed airways after Mycoplasma pulmonis infection23,29,31 or to the corresponding stage of angiopoietin-mediated remodeling driven by the Ang1 mimic COMP-Ang1.35,40
The experiments revealed that PDGF-B signaling in pericytes played a more important role in maintaining vascular stability while blood vessels were undergoing remodeling after M. pulmonis infection or after COMP-Ang1 than under baseline conditions or in the acute response of otherwise normal blood vessels to bradykinin.
Materials and Methods
Experimental Design
C57BL/6 mice (Charles River, Hollister, CA) were housed under barrier conditions to maintain their pathogen-free state and were studied at 8 to 10 weeks of age. Each group consisted of five mice. All experiments were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of California, San Francisco.
Normal blood vessels at baseline or after bradykinin challenge were compared with an early stage (7 days) of sustained inflammation after M. pulmonis infection or with the same stage of angiopoietin-mediated remodeling. Most treatments lasted 7 days, to enable comparison of different experimental conditions. PDGF-B was inhibited for 7 days by administration of AX102 daily in studies of Ang1 overexpression by adenoviral COMP-Ang1, and every other day in studies of M. pulmonis infection. AX102 was generously provided by Archemix Corporation (Cambridge, MA). AX102 treatment began the day before adenoviral injection or infection and covered the duration of the 7-day action of COMP-Ang1 and 7-day infection. Mice were also treated with AX102 for 7 days in studies of 14-day infections, in which the inhibitor was injected from day 7 to 14. Studies of 14-day infection showed that AX102 had the same effect on pericyte coverage when administered during the second week as when given for both weeks. Mice injected with adenoviral β-galactosidase (LacZ) were used as controls in COMP-Ang1 studies, and pathogen-free mice were used as controls in infection studies.
Data were collected on changes in vascular caliber, pericyte number, coverage, and phenotype, and PDGF-B expression, which were examined after AX102, COMP-Ang1 alone or with AX102, or infection for 7 days. Changes in pericytes and PDGF-B expression were also examined after infection for 14 days.
Blood vessel leakiness was used as a functional indicator of vascular instability in pathogen-free mice that were treated with COMP-Ang1 alone or together with AX102 for 7 days and then anesthetized and challenged by intravenous injection of bradykinin, followed by fluorescent microspheres. Similarly, mice infected with M. pulmonis for 7 days and treated concurrently with vehicle or AX102 were anesthetized and injected intravenously with microspheres. Controls included untreated pathogen-free mice and mice treated with saline or AX102 for 7 days and then challenged with bradykinin.
M. pulmonis Infection
Infected mice were studied at 7 days or 14 days after inoculation with 25 μL of broth containing 1 × 107 colony-forming units per milliliter of M. pulmonis strain UAB CT841,42 into each nostril after anesthesia induced by intramuscular injection of ketamine (87 mg/kg) and xylazine (13 mg/kg).29,34
Inhibition of PDGF-B by AX102
PDGF-B was selectively inhibited by intraperitoneal injection of AX102 at a dose of 50 mg/kg for 7 days.12 AX102 is a 34-nucleotide DNA aptamer that has high selectivity for PDGF-B and is modified on the 5′ terminus with a 40-kDa polyethylene glycol moiety to reduce renal filtration and to prolong plasma half-life.12
Adenoviral Overexpression of Ang1
Ang1 was overexpressed systemically in pathogen-free mice by intravenous injection of an adenoviral vector expressing the stable and potent Ang1 mimic COMP-Ang1 at a dose of 1 × 109 plaque-forming units.35,40 Pathogen-free mice were anesthetized and then received a single injection of adenoviral COMP-Ang1, or the same dose of adenoviral LacZ as a control, into the left jugular vein; the mice were studied 7 days later.
Vascular Leakage after Bradykinin Challenge or M. pulmonis Infection
Leakage was assessed by determining the amount and distribution of extravasated fluorescent microspheres. 500-nm diameter microspheres were used after bradykinin challenge and 50-nm microspheres were used after M. pulmonis infection, because the leakage rate after bradykinin is greater, albeit much more transient, than after infection.23,43 Pilot studies revealed abundant extravasated 500-nm microspheres after bradykinin but not after infection; however, abundant 50-nm microspheres were extravasated after infection. Microspheres of both sizes are sold as 1% solids: the injected volume of 100 μL of aqueous suspension contains 1.4 × 1010 500-nm microspheres, or 1000 times as many 50-nm microspheres (1.4 × 1013) are present in the same volume (Thermo Fisher–Duke Scientific, Palo Alto, CA).
For studies of bradykinin-induced leakage, pathogen-free mice were anesthetized and challenged by intravenous injection of bradykinin (Sigma-Aldrich, St. Louis, MO) at a dose of 1 mg/kg diluted in 100 μL of 0.9% NaCl (saline). Control mice received an injection of saline. One minute later, 100 μL of 500-nm diameter fluorescent polymer microspheres was injected into a femoral vein. At 2 minutes after the microsphere injection, blood and intravascular microspheres were washed from the circulation by systemic vascular perfusion of fixative, leaving only the extravasated microspheres.
For studies of leakage after M. pulmonis infection, mice were anesthetized at 7 days after infection and received an intravenous injection of 50 μL of 50-nm microspheres. Ten minutes later, blood and intravascular microspheres were washed from the circulation by perfusion of fixative.
Measurement of Microsphere Leakage
The fluorescence of extravasated microspheres that marked sites of leakage was measured in digital fluorescent images of regions of tracheal mucosa over five to eight cartilage rings in each trachea with a Zeiss Axiophot fluorescence microscope (10× objective, 1× Optovar) equipped with single and dual fluorescence filters and a low-light, three-chip, charge-coupled device (CCD) camera (480 × 640 pixel RGB-color images). Camera settings were constant for images from all groups in each experiment. The RGB color images were converted to 8-bit grayscale, and then the fractional area (area density) of extravasated microspheres was measured with ImageJ software version 1.44 (NIH, Bethesda, MD). Pixels included in the measurement had a fluorescence intensity equal to or greater than the threshold value of 50 (range, 0 to <255) that distinguished the microsphere signal from tissue autofluorescence. The mean area density for each group of mice was calculated from the median value of all images of the trachea of each mouse (5 mice/group).
Immunohistochemistry
Anesthetized mice were perfused through the left ventricle for 2 minutes with 1% paraformaldehyde in PBS, pH 7.4, at room temperature. Tracheas were removed, immersed in fixative for 1 hour, pinned flat on Sylgard 184 silicone elastomer (Dow Corning, Midland, MI), washed for 4 hours and stained as whole mounts for 16 hours at room temperature using primary antibodies to PECAM-1 (CD31) (hamster anti-mouse PECAM-1 antibody, clone 2H8, 1:500; Thermo Fisher Scientific–Lab Vision, Fremont, CA), desmin (rabbit monoclonal anti-mouse desmin antibody, clone Y66, 1:1000; Millipore, Billerica, MA), PDGFR-β (rat anti-mouse PDGFR-β, clone APB5, 1:500; eBioscience, San Diego, CA), α-SMA (mouse FITC-labeled anti-chicken α-SMA, clone 1A4, 1:1000; Sigma-Aldrich), activated caspase-3 (rabbit anti-caspase-3 antibody, 1:1000; R&D Systems, Minneapolis, MN), or phosphohistone H3 (rabbit anti-phosphohistone H3 antibody, 1:500, Millipore). Primary antibodies were followed by staining for 4 hours at room temperature with anti-hamster, anti-rabbit, or anti-rat secondary antibodies conjugated to FITC, Cy3 or Cy5 (Jackson ImmunoResearch, West Grove, PA). Fluorescence microscopic images were acquired as for microsphere measurements. Other images were obtained with a Zeiss LSM-510 confocal microscope with argon, helium-neon, and UV lasers and Zeiss AIM 3.2.2 software (512 × 512 or 1024 × 1024 pixel RGB-color images).
Real-Time Quantitative PCR
For real-time quantitative polymerase chain reaction (qPCR), RNA was isolated from tracheas using an RNeasy Plus mini isolation kit (Qiagen, Valencia, CA). RNA yield and purity were determined by spectrophotometry. cDNAs were synthesized with 1 μg of total RNA using the cDNA synthesis kit (Roche Applied Science, Indianapolis, IN). The qPCR was performed with Kapa SYBR Fast qRT-PCR master mix (Kapa Biosystems, Woburn, MA) using a MyiQ detection system (Bio-Rad Laboratories, Hercules, CA). The relative expression of each target gene was normalized to the expression of β-actin by calculation of the difference between the Ct target and the Ct reference gene, where Ct is the fractional PCR cycle number at which the reporter fluorescence was greater than the threshold. The following primers were used: M. pulmonis 16S RNA forward 5′-CCCTAAGTATGACGGTACCTTGTCA-3′, reverse 5′-GCGGCTGCTGGCACAT-3′; mouse PDGF-B forward 5′-GATCTCTCGGAACCTCATCG-3′, reverse 5′-GGCTTCTTTCGCACAATCTC-3′; and mouse P-selectin forward 5′-GCATACTCATGGAATAACTCACG-3′, reverse 5′-GACGTCATTGAGGTGAGCG-3′
Measurement of Vascular Remodeling and Pericyte Coverage
The average diameter of capillaries was measured in tracheal whole mounts stained for PECAM-1 and desmin immunoreactivities at the rostro-caudal midpoint of cartilage rings, roughly half way between the arteriole and venule.36,44 At least five vessels were measured over each of 10 cartilage rings in each trachea. Measurements of pericytes were made in three steps: i) the length of 25 capillaries was measured in each trachea; ii) pericyte bodies were counted on these vessels; and iii) the length of vessel segments devoid of pericyte cell bodies or processes was measured. Measurements were made on real-time fluorescent images from the CCD camera on the fluorescence microscope (10× objective for capillaries, 20× objective for pericytes, 1× Optovar for both) linked to a digitizing tablet (Digi-Pad; GTCO CalComp, Columbia, MD).33,44,45 Percentages of vessel length with or without pericyte coverage were calculated from these values.
Statistical Analysis
Data are reported as means ± SE for groups of five mice, unless otherwise indicated. The significance of differences among groups was assessed by analysis of variance followed by the Bonferroni-Dunn test for multiple comparisons. Differences between groups were considered significant at P < 0.05.
Results
Normal Tracheal Vasculature
Pericyte Distribution
The tracheal vasculature of pathogen-free mice had a distinctive segmental organization in which most arterioles and venules were located between cartilage rings, and most capillaries were arranged in a ladder-like pattern in the mucosa overlying cartilage rings (Figure 1A). Smooth muscle cells on arterioles had a circumferential orientation and covered most of the vascular surface (Figure 1B). Pericytes on capillaries had a longitudinal orientation, with long processes that ran the entire vessel length but covered only a small proportion of the surface (Figure 1C). Pericytes on venules were irregularly shaped, had multiple short processes, and covered much but not all of the endothelium (Figure 1D).
Figure 1.
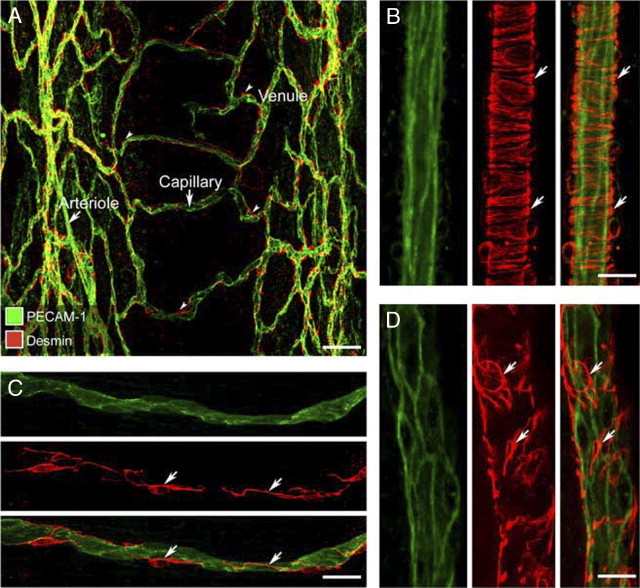
Distribution of pericytes on vasculature of normal tracheal mucosa. A: Confocal microscopic image showing endothelial cells (green, PECAM-1) and pericytes (red, desmin) of the mucosal vasculature in a whole mount of normal mouse trachea. B–D: Higher magnification images with arrows marking desmin-positive smooth muscle cells in the wall of an arteriole (B) and pericytes in the wall of a capillary (C) and a venule (D). Scale bars: 100 μm (A); 20 μm (B–D).
Effects of PDGF-B Blockade
Inhibition of PDGF-B in pathogen-free mice by administration of aptamer AX102 daily for 7 days was accompanied by a significant reduction in pericyte coverage and enlargement of some capillaries over cartilage rings in the trachea (Figure 2, A and B). After AX102, the proportion of capillary length without pericyte coverage assessed by desmin staining increased from 7% to 37%, and the number of pericyte cell bodies decreased by 37%, from 19 to 12 per millimeter of vessel length (Figure 2, C and D).
Figure 2.
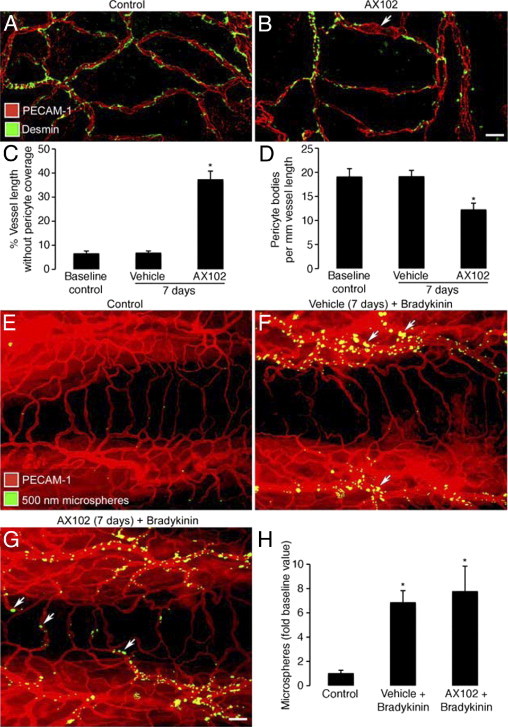
Effect of PDGF-B blockade by AX102 on pericyte coverage and bradykinin-induced leakage. A and B: Confocal microscopic images of the tracheal vasculature (endothelial cells, PECAM-1, red); compare the continuous row of pericytes (green, desmin) after treatment with vehicle (A) with incomplete pericyte coverage after AX102 for 7 days (arrow, B). C and D: Pericyte coverage of tracheal vasculature of baseline control mice versus mice treated with vehicle or AX102 for 7 days. The AX102 group had significantly less pericyte coverage (C) and fewer desmin-positive pericyte cell bodies (D). E–G: Fluorescence micrographs showing extravasated 500-nm microspheres (blue-white) at sites of leakage from vessels stained for PECAM-1 immunoreactivity (red). Bradykinin (1 mg/kg) was injected 1 minute before microspheres that circulated for 2 minutes before fixation by vascular perfusion. Microspheres are sparse in trachea of baseline control mouse after saline (E) but are abundant after bradykinin injected after 7-day treatment with vehicle (F). Leakage was not further increased by 7-day treatment with AX102 (G). Most extravasated microspheres are near postcapillary venules after vehicle (F, arrows), but after AX102 some microspheres extravasated from capillaries that cross cartilage rings (G, arrows). H: Abundance of extravasated microspheres (mean area density scaled to baseline control value = 1) in tracheas of the three groups. *P < 0.05 versus control. Scale bars: 20 μm (A and B); 50 μm (E–G).
Endothelial barrier function in pathogen-free mice was assessed by measuring leakage of 500-nm fluorescent microspheres after the microspheres remaining in the bloodstream were washed away by fixative perfusion.23 Few or no extravasated microspheres were found near tracheal blood vessels of control mice (Figure 2E) or in mice pretreated with AX102 (data not shown) under baseline conditions. However, after bradykinin, extravasated microspheres were abundant around venules, but not capillaries, of control mice (Figure 2F). When preceded by AX102 for 7 days, bradykinin induced leakage from venules and some capillaries (Figure 2G), and the overall amount of leakage tended to be greater than without AX102, but the difference was not significant (Figure 2H).
Pericyte Plasticity after M. pulmonis Infection
Change in Shape and Number
As in previous studies by our research group,30–32 we found that the tracheal vasculature underwent conspicuous remodeling after M. pulmonis infection (Figure 3A–C). At 7 days after infection, blood vessels in the location of capillaries over cartilage rings were larger than normal capillaries, without evidence of sprouting angiogenesis, and had a venular phenotype, as reflected by expression of P-selectin, ICAM-1, and Eph-B4.31
Figure 3.
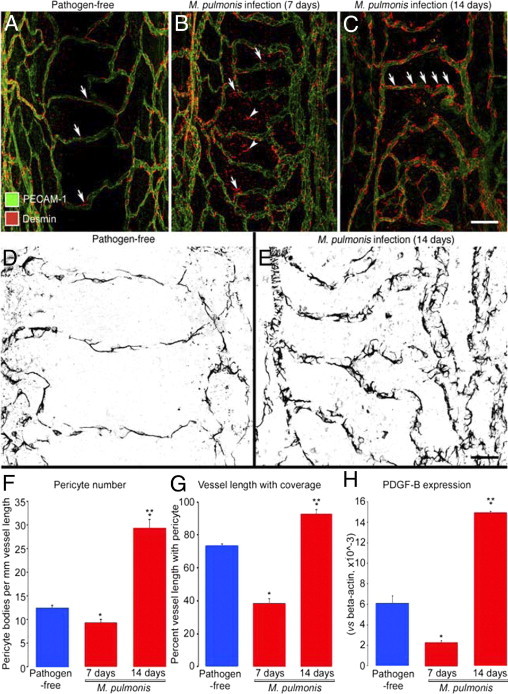
Pericytes on tracheal vasculature after M. pulmonis infection. A–C: Confocal micrographs showing pericytes (red, desmin, arrows) and endothelial cells (PECAM-1, green) of normal capillaries (A) and remodeled vessels in tracheas at 7 days (B) or 14 days (C) after infection with M. pulmonis. Arrowheads in (B) mark desmin-positive cells not in contact with vasculature. Arrows in C mark abundance of pericytes on remodeled vessels after 14 days of infection. D and E: Higher magnification images; compare pericytes (desmin, black) on tracheal capillaries of pathogen-free mouse (D) with those on remodeled tracheal vessels after infection for 14 days (E). F–H: Number of pericyte cell bodies (F), percentage of vessel length with pericyte coverage (G), and PDGF-B mRNA expression normalized to β-actin × 10−3 (H) in tracheas of pathogen-free mice and mice infected for 7 or 14 days. *P < 0.05 versus pathogen-free; **P < 0.05 versus M. pulmonis infection for 7 days. Scale bars: 100 μm (A–C); 20 μm (D and E).
Pericytes on tracheal blood vessels underwent distinctive changes after M. pulmonis infection. Unlike normal tracheal capillaries, in which pericytes had a longitudinal orientation, long processes, and tight attachment to the endothelium (Figure 3, A and D), the remodeled vessels at 7 days after infection had pericytes that were more rounded, had short processes, and were loosely attached to the endothelium (Figure 3B). Counts of pericyte cell bodies revealed that pericyte number was approximately 30% less than the pathogen-free baseline, and measurements of the proportion of vessel length with pericyte coverage showed a reduction to half the baseline value (Figure 3, F and G). The larger decrease in pericyte coverage than in number of pericytes indicated that the shape change (from elongated to rounded) made a greater contribution to the reduced coverage than did loss of pericytes. In addition, round desmin-positive cells (Figure 3B) were scattered in the mucosa with no apparent association with blood vessels. No evidence of activated caspase-3 immunoreactivity indicative of apoptosis was found in pericytes at 7 days.
At 14 days of infection, pericytes had multiple short processes, like those at 7 days but much more abundant than at 7 days or in the pathogen-free state (Figure 3, C and E). At 14 days, pericyte number and coverage were more than twice baseline (Figure 3, F and G). Phosphohistone H3 immunoreactivity indicative of dividing nuclei was not detected in pericytes but was widespread in the trachea and particularly strong in the epithelium and in cells scattered in the mucosa. A low level of staining of pericytes could not be excluded, however, because of the abundance of proliferating cells.
Expression of PDGF-B mRNA measured in the trachea by qPCR fit the observed changes in pericyte coverage: PDGF-B mRNA at 7 days was less than half the pathogen-free value but at 14 days was more than double this baseline value (Figure 3H).
Changes in Desmin, PDGFR-β, and α-SMA
Pericyte phenotype was assessed by comparing the staining for three pericyte markers: desmin, which marks most if not all pericytes, PDGFR-β, which marks more immature pericytes, and α-SMA, which marks more mature pericytes. Pericytes on normal capillaries had strong desmin immunoreactivity (Figure 3A), but did not stain for PDGFR-β or α-SMA (Figure 4, A and D). Staining for α-SMA was restricted to arterioles and large venules. At 7 days after infection, many pericytes on remodeled vessels had weak desmin immunoreactivity (Figure 3B), but also stained for PDGFR-β (Figure 4B); α-SMA immunoreactivity was still limited to smooth muscle cells on arterioles (Figure 4E) and large venules.
Figure 4.
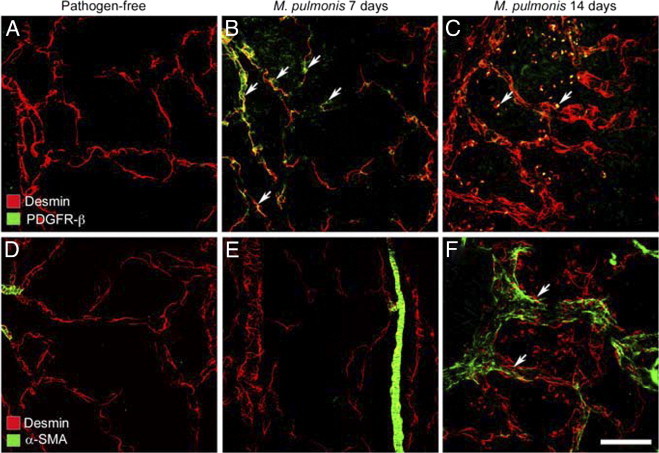
Pericyte changes after M. pulmonis infection. A–F: Confocal micrographs showing pericytes (desmin, red) on normal capillaries (A and D) and remodeled vessels in tracheas after M. pulmonis infection for 7 days (B and E) or 14 days (C and F). A–C: Pericytes with PDGFR-β immunoreactivity (green) are absent in normal capillaries (A) but are present after infection for 7 days (B, arrows). After 14 days of infection, round desmin-positive cells (C, arrows), some with faint PDGFR-β immunoreactivity, are scattered among blood vessels but do not resemble pericytes. D and E: Mural cells with α-SMA immunoreactivity (green) not present on capillaries of normal tracheas (D) or after 7 days of infection, except on arterioles (E), but are present on remodeled vessels (previously capillaries) after 14 days of infection (F, arrows). Scale bar = 50 μm (A–F).
The staining pattern was different at 14 days after infection, when pericytes on remodeled vessels again had strong desmin immunoreactivity (Figure 3C), and some stained for α-SMA but not for PDGFR-β (Figure 4, C and F). Small, round desmin-positive cells scattered around some blood vessels after 14 days of infection had faint PDGFR-β immunoreactivity, lacked α-SMA, and did not resemble normal pericytes (Figure 4, C and F).
PDGF-B Dependence
In mice infected with M. pulmonis for 14 days, tracheal capillaries were enlarged and had heavy pericyte coverage (Figure 5, A and B). To test the involvement of PDGF-B in the recruitment of pericytes, as suggested by mRNA expression (Figure 3H), we examined the effect of AX102 on the tracheal vasculature. When AX102 was administered for the final 7 days of infection (or throughout the infection), pericyte coverage was conspicuously less, and vascular enlargement was exaggerated (Figure 5, C–E).
Figure 5.
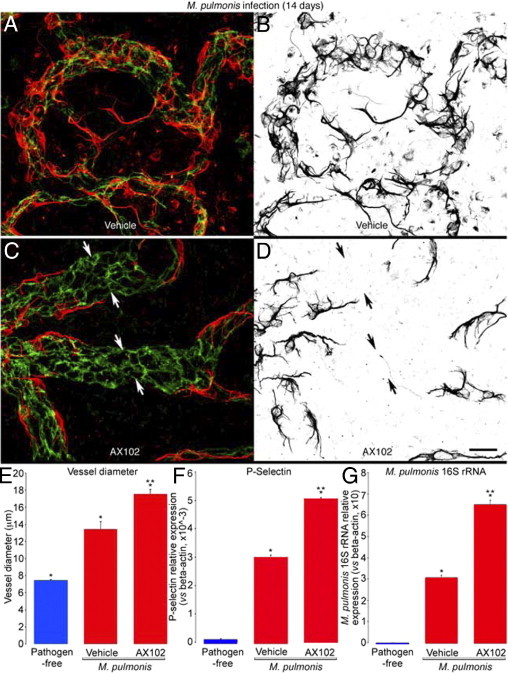
PDGF-B-dependent changes in pericytes after M. pulmonis infection. A–D: Confocal micrographs showing pericytes (desmin, red) and endothelial cells (PECAM-1, green) of remodeled tracheal blood vessels after M. pulmonis infection for 14 days with daily treatment with vehicle (A and B) or AX102 (C and D) during the final 7 days. The distribution of pericytes in A and C is highlighted in B and D, where desmin immunoreactivity is shown in black. After infection, pericytes are very numerous and densely packed (A and B), but with AX102 treatment the pericytes are more sparse, and pericyte-free regions are present on some vessels (C and D, arrows). E–G: Diameter of capillaries/remodeled vessels measured at the center of cartilage rings (E) and measurements by qPCR of expression of P-selectin mRNA (F) and of M. pulmonis 16S rRNA (G) in tracheas of pathogen-free mice or infected mice treated daily with vehicle or AX102 during the final 7 days of the 14-day infection. Expression values for P-selectin are normalized to β-actin × 10−3; values for M. pulmonis 16S rRNA are normalized to β-actin × 10. *P < 0.05 versus pathogen-free; **P < 0.05 versus vehicle treatment after infection. Scale bars = 15 μm (A–D).
P-selectin expression, which is absent in normal capillaries of pathogen-free mice but is strong in remodeled capillaries after M. pulmonis infection,31 was increased by 50% at 14 days (50-fold increase from pathogen-free) by AX102 given during the infection (Figure 5F).
The functional consequence of PDGF-B blockade on the severity of the infection at 14 days was examined by measuring the expression of M. pulmonis 16S rRNA in the trachea by qPCR.46 The expression of M. pulmonis 16S rRNA was significantly greater in infected mice treated with AX102, consistent with PDGF-B inhibition leading to more severe infection (Figure 5G).
Vessel Leakiness
Tracheal blood vessels that undergo remodeling after M. pulmonis infection become leaky during the first week, when endothelial cell proliferation peaks,43,47 but the role of pericytes in this leakiness is unclear. To assess the effect of pericyte dynamics on leakage, we examined the extravasation of 50-nm fluorescent microspheres from the tracheal vasculature at 7 days after infection, when pericyte coverage was reduced. Extravasation was much greater at 7 days than in pathogen-free mice (Figure 6, A and B), and extravasated microspheres were most abundant near remodeled capillaries over cartilage rings (Figure 6B). This distribution differed from bradykinin-induced leakage, in which extravasated microspheres were absent in capillaries and were most numerous around venules between cartilage rings (Figure 2F).
Figure 6.
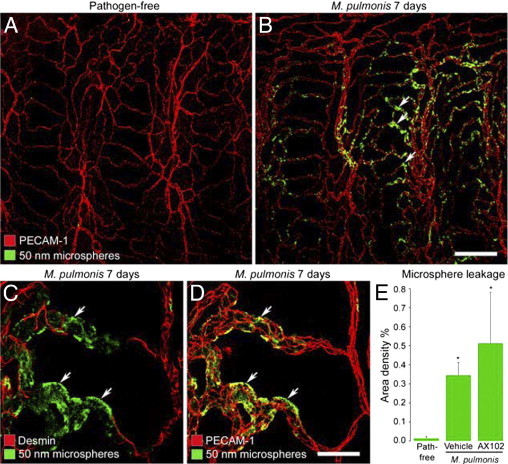
Microsphere leakage during M. pulmonis infection. A and B: Confocal microscopic images showing extravasated 50-nm microspheres (green) in the tracheal vasculature (PECAM-1, red) of a pathogen-free mouse (A) and a mouse infected with M. pulmonis for 7 days (B). C and D: Higher magnification images showing extravasated microspheres (green) in remodeled vessels with pericytes stained for desmin (C, red) or endothelial cells stained for PECAM-1 (D, red) after infection for 7 days. Arrows in C and D mark extravasated microspheres in pericyte-free regions of remodeled vessels. E: Abundance of extravasated microspheres, reflected by area density of microsphere fluorescence (threshold ≥50), in trachea of pathogen-free mice or mice infected with M. pulmonis for 7 days accompanied by treatment with vehicle or AX102. *P < 0.05 versus pathogen-free mice. Scale bars: 100 μm (A and B); 20 μm (C and D).
After 7 days of infection, extravasated microspheres were most abundant in regions of remodeled capillaries that had little or no pericyte coverage (Figure 6, C and D). Leakage of microspheres after infection was 24 times the pathogen-free value (Figure 6E). In mice treated with AX102 during the 7-day infection, leakage averaged 50% greater, but there was considerable variability, and the difference from vehicle-treated infected mice was not significant (Figure 6E).
COMP-Ang1–Mediated Vascular Enlargement
Reduction in Pericyte Coverage
Angiopoietin/Tie2 signaling has been implicated in vascular remodeling after M. pulmonis infection.31,48 Involvement of angiopoietins in vascular remodeling is in accord with multiple studies showing that overexpression of Ang1 or its mimic COMP-Ang1 promotes the enlargement of blood vessels in the airways and other organs.35,36,49,50 Vascular enlargement after Ang1 has similarities to vascular remodeling after M. pulmonis infection.31 It is unclear, however, whether Ang1-induced vascular enlargement is accompanied by the distinctive pericyte changes found after M. pulmonis infection. Ang1 is thought to promote pericyte recruitment during development,13,49 but COMP-Ang1–mediated vascular enlargement is apparently accompanied by pericyte loss rather than pericyte recruitment.35
To better understand the pericyte changes after Ang1 in relation to corresponding changes after M. pulmonis infection for 7 days, we determined whether the pericyte coverage decreased as vessels enlarged after COMP-Ang1 and whether PDGF-B blockade by AX102 amplified the reduction. The 7-day treatment with AX102 matched that used during M. pulmonis infection.
Expression of COMP-Ang1 for 7 days after systemic delivery of adenoviral COMP-Ang1 resulted in enlargement of tracheal capillaries into funnel-shaped venules (Figure 7, A and B), as previously described.35,50 This was not found in control mice that received the adenoviral LacZ vector. The remodeled capillaries had conspicuously less pericyte coverage than normal capillaries (Figure 7, A and B). This reduction in pericytes was restricted to the parts of the vasculature that were enlarged by COMP-Ang1 treatment. Daily administration of AX102 to block PDGF-B during the 7-day period of COMP-Ang1 exposure resulted in significantly less pericyte coverage and fewer pericyte cell bodies (Figure 7, C–E).
Figure 7.
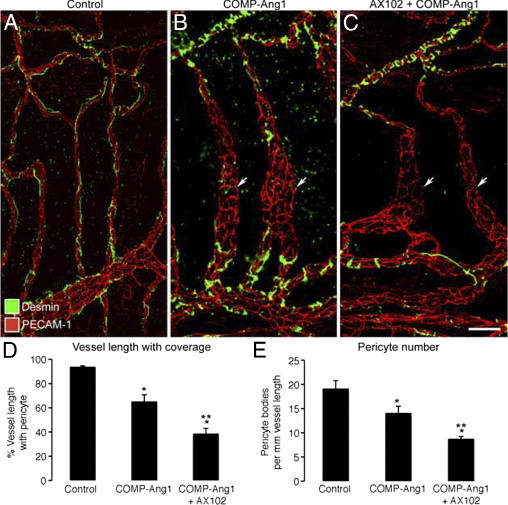
PDGF-dependent changes in pericytes after COMP-Ang1. A–C: Confocal micrographs of blood vessels (PECAM-1, red) in tracheal whole mounts showing a continuous, narrow row of pericytes (desmin, green) on capillaries in a control mouse treated with adenoviral LacZ (A) but pericyte-free regions of enlarged, remodeled vessels (arrows) in tracheas of mice treated with adenoviral COMP-Ang1 (B) or COMP-Ang1 with concurrent AX102 (C) for 7 days. Fewer pericytes are present on remodeled vessels in mice treated with AX102. D and E: Pericyte coverage, expressed as percentage of vessel length with pericytes (D) and number of pericyte cell bodies per millimeter (E), on tracheal vessels from pathogen-free mice treated for 7 days with adenoviral LacZ (control), COMP-Ang1, or COMP-Ang1 accompanied by AX102. *P < 0.05 versus vehicle; **P < 0.05 versus COMP-Ang1.
COMP-Ang1–Mediated Antileakage Action
Dependence on PDGF-B
Overexpression of Ang1 protects the vasculature from leakage produced by inflammatory mediators.21–23 We used AX102 as a tool to determine whether PDGF-B-dependent actions of pericytes play a role in the antileakage effect of COMP-Ang1. In agreement with previous work by our research group,23 500-nm extravasated microspheres were sparse or absent in tracheas examined under baseline conditions (Figure 2E), but microspheres were abundant around tracheal venules challenged by intravenous injection of bradykinin into adenoviral LacZ-injected control mice (Figure 8A). By comparison, bradykinin-induced leakage of microspheres was appreciably less at 7 days after intravenous injection of adenoviral COMP-Ang1 (Figure 8B). However, when COMP-Ang1 was accompanied by daily doses of AX102 for 7 days, COMP-Ang1 had no antileakage effect, and extravasated microspheres after bradykinin were even more numerous than in the absence of COMP-Ang1 (Figure 8, C and D). Measurements showed that AX102 completely prevented the antileakage effect of COMP-Ang1 (Figure 8D), consistent with a significant contribution of PDGF-B-dependent actions of pericytes to Ang1-mediated vascular stabilization.
Figure 8.
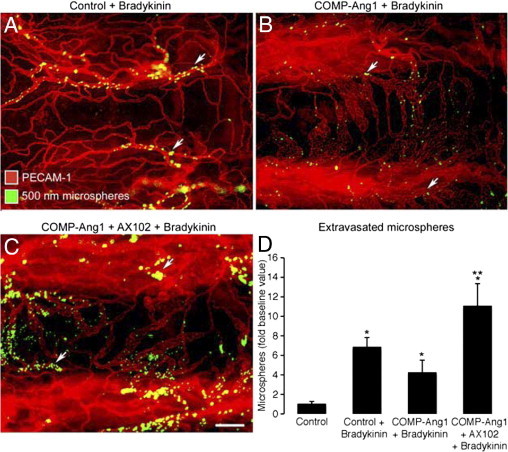
PDGF-B-dependence of antileakage effect of Ang1. A–C: Fluorescence micrographs of blood vessels (PECAM-1, red) in tracheal whole mounts from mice pretreated with adenoviral LacZ (control) (A) or COMP-Ang1 (B) or COMP-Ang1 accompanied by AX102 (C). After 7 days, all mice received an injection of bradykinin, followed 1 minute later by fluorescent 500-nm microspheres (blue-white, arrows), which circulated for 2 minutes before vascular perfusion of fixative. Many extravasated microspheres are visible in trachea prepared after bradykinin was preceded by adenoviral LacZ (A). Although few microspheres are visible when bradykinin was preceded by COMP-Ang1 (B), much leakage is evident when exposure to COMP-Ang1 was accompanied by PDGF-B blockade by daily doses of AX102 (C). D: Measurements of area density of extravasated microspheres in tracheas showed that AX102 treatment during COMP-Ang1 exposure increased the leakage effect of bradykinin by 61% over LacZ controls and by 162% over COMP-Ang1 without AX102. Values are scaled to the control (no bradykinin) group = 1. *P < 0.05 versus bradykinin group; **P < 0.05 versus COMP-Ang1 + bradykinin group. Scale bar = 30 μm (A–C).
Discussion
The purpose of this study was to determine the importance of PDGF-B signaling in pericytes for blood vessel stability under four conditions: i) baseline; ii) leakiness in acute inflammation induced by bradykinin; iii) leakiness in sustained inflammation after M. pulmonis infection for 7 days when pericyte coverage was reduced during vascular remodeling; and iv) the antileakage effect of COMP-Ang1 for 7 days.
Inhibition of PDGF-B by AX102 in otherwise normal mice led to a reduction in pericyte coverage of tracheal capillaries. This reduction in coverage was greater than could be explained by the decrease in number of pericytes, indicating contributions of both change in pericyte shape and loss of cells. Despite the decrease in pericyte coverage, baseline leakage from venules was not greater, nor was leakage after bradykinin increased, but minor leakage did occur in capillaries, where bradykinin normally had no effect.
Other other studies using bradykinin in pathogen-free mice revealed that the antileakage action of COMP-Ang1 was abolished by AX102 inactivation of PDGF-B. This change was accompanied by more extreme reduction in pericyte coverage and remodeling of the leaky vessels at 7 days. The findings are consistent with the involvement of PDGF-B-dependent effects of pericytes in the antileakage action and provide evidence that Ang1 regulates vascular stability on two levels: through endothelial-endothelial interactions and through endothelial-pericyte interactions.
Infection with M. pulmonis was accompanied by previously unrecognized biphasic changes in pericytes. Infection for 7 days was accompanied by reduced pericyte coverage and number of cells, decreased PDGF-B expression, and vessel leakiness. Pericyte phenotype changed, as reflected by reduced desmin and increased PDGFR-β immunoreactivities. In contrast, after 14 days of infection, the number of pericytes and PDGF-B expression were more than double the values for pathogen-free controls, desmin staining increased, PDGFR-β staining decreased, and some pericytes had α-SMA immunoreactivity. PDGF-B blockade by treatment with AX102 during the second week reduced the expansion of pericyte coverage, exaggerated the vascular remodeling, and increased the severity of the infection. These effects of AX102 indicate that PDGF-B played an essential role in pericyte dynamics after infection.
Together, these results are consistent with two phases of vascular remodeling during the first 2 weeks after M. pulmonis infection. Airway blood vessels were abnormal in both phases, but had contrasting features. The first phase, evident at 7 days, was characterized by reductions in PDGF-B expression and pericyte coverage and transformation of pericytes into a more immature phenotype. The second phase, evident at 14 days, was manifested by increases in PDGF-B expression and pericyte coverage and conversion of pericytes into a more mature phenotype.
The findings at 14 days after M. pulmonis infection suggest that pericyte stabilization of blood vessels limits vascular remodeling in sustained inflammation as part of the host response. The findings also raise the question of whether pericyte recruitment and stabilization dictate the evolution and reversibility of vascular remodeling in inflammation. Nonreversible vascular remodeling could affect the resolution of chronic inflammation.46
The abundance of pericytes at 14 days raises the question of the source of pericytes at this stage of M. pulmonis infection. Pericytes are thought to arise from other pericytes, local mesenchymal precursors, or bone marrow-derived precursors that form a pool of stem cells.51–55 Expansion of the pericyte precursor niche may be a tissue repair mechanism that contributes to pericyte recruitment and vessel stabilization and limits tissue injury in sustained inflammation. Fate-mapping studies should be useful for testing this possibility.
Further studies are needed to determine more precisely the onset and duration of the changes in pericytes and leakage after M. pulmonis infection. Additional studies should determine the dose-response and time-course of AX102 effects on the vasculature and infection severity, to learn whether the observed variability in responses reflects less than maximal inhibition of PDGF-B or changes that occur before 7 days or after 14 days. Also to be determined is whether the reduction in PDGF-B expression at 7 days after infection limits the effect of AX102 at that stage of the inflammatory response.
The essential role of PDGF-B in vascular remodeling during normal development has been demonstrated by targeted deletion of PDGF-B or its receptor PDGFR-β.7,56–58 Loss of PDGF-B or the receptor in mice results in embryonic death during late gestation due to edema, hemorrhage, and cardiovascular failure. Vascular defects in PDGF-B-deficient mice include endothelial hyperplasia, vascular enlargement, and abnormal cell shape.8,57,58
Pericyte changes are also evident in tumors and other conditions associated with vascular remodeling and angiogenesis. Pericytes are loosely attached to the abnormal, disorganized blood vessels in tumors.59 Defective pericyte coverage contributes to the structural and functional abnormalities of tumor vessels and supports the use of pericytes as targets for antiangiogenic therapy in tumors.10,60 Furthermore, one of the earliest indications of diabetic retinopathy is loss of pericytes from retinal capillaries.38 This phenomenon is associated with capillary occlusion, dilatation, and leakiness and is thought to contribute to disease progression.
An important role of Ang1/Tie2 signaling in vascular remodeling and stability is evident from the phenotype of mice deficient in Ang1 or Tie2, which die at approximately embryonic day E9.5 to day E11.5 because of cardiac defects, edema, and hemorrhage.13,16 In these mice, the vasculature does not become a hierarchical network of vessels and lacks mural cell coverage, suggesting that Ang1/Tie2 signaling is essential for pericyte recruitment. This interpretation has been challenged, however, by data showing that pericyte coverage is normal in Tie2 chimeras containing a large proportion of Tie2-deficient cells, suggesting that pericyte deficiency in Tie2-deficient mice may involve other mechanisms, such as disturbed blood flow.61,62 Overexpression of Ang1 during development, or in the adult, leads to vascular remodeling and resistance to leakage induced by inflammatory mediators.21–24 The antileakage effect of Ang1 is associated with the inhibition of gap formation at endothelial cell junctions.23 Ang1 also protects blood vessels in the neonatal mouse retina from the destabilizing effects of inhibition of PDGF-B/PDGFR-β signaling by a function-blocking antibody.63
In conclusion, responses of endothelial cells and pericytes influence one another through crosstalk between angiopoietin and PDGF-B signaling. Under baseline conditions, vascular stability is maintained by Ang1-mediated activation of Tie2 in endothelial cells, which promotes junctional integrity, and by PDGF-B activation of PDGFR-β receptors in pericytes, which promotes pericyte attachment and vascular stability. Vascular remodeling after M. pulmonis infection during the first week involves vessel destabilization, mediated via inactivation of Tie2 by Ang2 and inactivation of PDGFR-β due to decreased PDGF-B expression. Ang2 blockade prevents this step.48 During the second week, PDGF-B signaling increases, pericytes proliferate, and the remodeled vessels stabilize in an abnormal phenotype that sustains leakage and leukocyte traffic. Inhibition of PDGF-B prevents this step and intensifies the infection.
Our experiments revealed that pericytes located on functionally strategic blood vessels in the airway mucosa undergo rapid changes after infection by M. pulmonis. Under normal conditions, these vessels have the phenotype of capillaries, but they transform into venules in the first week after infection. During this period, pericytes loosen their attachment to endothelial cells, and some may round up, migrate away, or die. As capillaries remodel into venules during the first week, the vessels enlarge and become leaky. During the second week, pericytes become much more numerous on remodeled vessels through proliferation and/or recruitment of precursors. Expansion and stabilization of the pericyte population is dependent on PDGF-B signaling. This process is key to the regulation of the severity of the infection. PDGF-B-dependent pericyte actions are also essential for the antileakage action of Ang1 at 7 days, when the same vessels affected by M. pulmonis infection are enlarged and resistant to inflammatory mediators. Overall, our findings indicate that PDGF-B-mediated actions of pericytes are particularly important for vascular stability when blood vessels undergo remodeling.
Acknowledgments
We thank Mary B. Brown, Ph.D., (University of Florida, Gainesville, FL) for help with M. pulmonis production and Karen A. Olson, Ph.D. (Archemix Corporation, Cambridge, MA), for supplying the PDGF-B binding aptamer AX102.
Footnotes
Supported in part by the National Institutes of Health (NIH HL-24136, HL-59157, and HL-096511 to D.M.M.); by a fellowship from the Belgian American Educational Foundation, a fellowship of the Anticancer Center (“Centre Anticancéreux”) of the University of Liège, Belgium, and funding from the Neoangio Program of the Walloon Region (S.T.); by funding from the Swedish Wenner-Gren Foundation and a UICC American Cancer Society International Fellowship for Beginning Investigators (J.F.); and by funding from the Dean's Research Fund at the University of California, San Francisco, and the American Heart Association (H.Z.). AX102 was generously provided by Archemix Corporation (Cambridge, MA).
J.F. and S.T. contributed equally to the present work and each is considered first author; K.C. and H.Z. contributed equally to the present work and each is considered second author.
References
- 1.Armulik A., Abramsson A., Betsholtz C. Endothelial/pericyte interactions. Circ Res. 2005;97:512–523. doi: 10.1161/01.RES.0000182903.16652.d7. [DOI] [PubMed] [Google Scholar]
- 2.Gaengel K., Genove G., Armulik A., Betsholtz C. Endothelial-mural cell signaling in vascular development and angiogenesis. Arterioscler Thromb Vasc Biol. 2009;29:630–638. doi: 10.1161/ATVBAHA.107.161521. [DOI] [PubMed] [Google Scholar]
- 3.von Tell D., Armulik A., Betsholtz C. Pericytes and vascular stability. Exp Cell Res. 2006;312:623–629. doi: 10.1016/j.yexcr.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 4.Speyer C.L., Steffes C.P., Ram J.L. Effects of vasoactive mediators on the rat lung pericyte: quantitative analysis of contraction on collagen lattice matrices. Microvasc Res. 1999;57:134–143. doi: 10.1006/mvre.1998.2134. [DOI] [PubMed] [Google Scholar]
- 5.Sims D.E. Diversity within pericytes. Clin Exp Pharmacol Physiol. 2000;27:842–846. doi: 10.1046/j.1440-1681.2000.03343.x. [DOI] [PubMed] [Google Scholar]
- 6.Edelman D.A., Jiang Y., Tyburski J.G., Wilson R.F., Steffes C.P. Cytokine production in lipopolysaccharide-exposed rat lung pericytes. J Trauma. 2007;62:89–93. doi: 10.1097/TA.0b013e31802dd712. [DOI] [PubMed] [Google Scholar]
- 7.Lindahl P., Johansson B.R., Levéen P., Betsholtz C. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science. 1997;277:242–245. doi: 10.1126/science.277.5323.242. [DOI] [PubMed] [Google Scholar]
- 8.Hellström M., Gerhardt H., Kalen M., Li X., Eriksson U., Wolburg H., Betsholtz C. Lack of pericytes leads to endothelial hyperplasia and abnormal vascular morphogenesis. J Cell Biol. 2001;153:543–554. doi: 10.1083/jcb.153.3.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lindblom P., Gerhardt H., Liebner S., Abramsson A., Enge M., Hellstrom M., Backstrom G., Fredriksson S., Landegren U., Nystrom H.C., Bergstrom G., Dejana E., Ostman A., Lindahl P., Betsholtz C. Endothelial PDGF-B retention is required for proper investment of pericytes in the microvessel wall. Genes Dev. 2003;17:1835–1840. doi: 10.1101/gad.266803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bergers G., Song S., Meyer-Morse N., Bergsland E., Hanahan D. Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. J Clin Invest. 2003;111:1287–1295. doi: 10.1172/JCI17929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wilkinson-Berka J.L., Babic S., De Gooyer T., Stitt A.W., Jaworski K., Ong L.G., Kelly D.J., Gilbert R.E. Inhibition of platelet-derived growth factor promotes pericyte loss and angiogenesis in ischemic retinopathy. Am J Pathol. 2004;164:1263–1273. doi: 10.1016/s0002-9440(10)63214-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sennino B., Falcón B.L., McCauley D., Le T., McCauley T., Kurz J.C., Haskell A., Epstein D.M., McDonald D.M. Sequential loss of tumor vessel pericytes and endothelial cells after inhibition of platelet-derived growth factor B by selective aptamer AX102. Cancer Res. 2007;67:7358–7367. doi: 10.1158/0008-5472.CAN-07-0293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Suri C., Jones P.F., Patan S., Bartunkova S., Maisonpierre P.C., Davis S., Sato T.N., Yancopoulos G.D. Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell. 1996;87:1171–1180. doi: 10.1016/s0092-8674(00)81813-9. [DOI] [PubMed] [Google Scholar]
- 14.Cai J., Kehoe O., Smith G.M., Hykin P., Boulton M.E. The angiopoietin/Tie-2 system regulates pericyte survival and recruitment in diabetic retinopathy. Invest Ophthalmol Vis Sci. 2008;49:2163–2171. doi: 10.1167/iovs.07-1206. [DOI] [PubMed] [Google Scholar]
- 15.Augustin H.G., Koh G.Y., Thurston G., Alitalo K. Control of vascular morphogenesis and homeostasis through the angiopoietin-Tie system. Nat Rev Mol Cell Biol. 2009;10:165–177. doi: 10.1038/nrm2639. [DOI] [PubMed] [Google Scholar]
- 16.Sato T.N., Tozawa Y., Deutsch U., Wolburg-Buchholz K., Fujiwara Y., Gendron-Maguire M., Gridley T., Wolburg H., Risau W., Qin Y. Distinct roles of the receptor tyrosine kinases Tie-1 and Tie-2 in blood vessel formation. Nature. 1995;376:70–74. doi: 10.1038/376070a0. [DOI] [PubMed] [Google Scholar]
- 17.Morisada T., Kubota Y., Urano T., Suda T., Oike Y. Angiopoietins and angiopoietin-like proteins in angiogenesis. Endothelium. 2006;13:71–79. doi: 10.1080/10623320600697989. [DOI] [PubMed] [Google Scholar]
- 18.Fiedler U., Augustin H.G. Angiopoietins: a link between angiogenesis and inflammation. Trends Immunol. 2006;27:552–558. doi: 10.1016/j.it.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 19.Nishishita T., Lin P.C. Angiopoietin 1, PDGF-B, and TGF-beta gene regulation in endothelial cell and smooth muscle cell interaction. J Cell Biochem. 2004;91:584–593. doi: 10.1002/jcb.10718. [DOI] [PubMed] [Google Scholar]
- 20.Saharinen P., Eklund L., Miettinen J., Wirkkala R., Anisimov A., Winderlich M., Nottebaum A., Vestweber D., Deutsch U., Koh G.Y., Olsen B.R., Alitalo K. Angiopoietins assemble distinct Tie2 signalling complexes in endothelial cell-cell and cell-matrix contacts. Nat Cell Biol. 2008;10:527–537. doi: 10.1038/ncb1715. [DOI] [PubMed] [Google Scholar]
- 21.Thurston G., Suri C., Smith K., McClain J., Sato T.N., Yancopoulos G.D., McDonald D.M. Leakage-resistant blood vessels in mice transgenically overexpressing angiopoietin-1. Science. 1999;286:2511–2514. doi: 10.1126/science.286.5449.2511. [DOI] [PubMed] [Google Scholar]
- 22.Thurston G., Rudge J.S., Ioffe E., Zhou H., Ross L., Croll S.D., Glazer N., Holash J., McDonald D.M., Yancopoulos G.D. Angiopoietin-1 protects the adult vasculature against plasma leakage. Nat Med. 2000;6:460–463. doi: 10.1038/74725. [DOI] [PubMed] [Google Scholar]
- 23.Baffert F., Le T., Thurston G., McDonald D.M. Angiopoietin-1 decreases plasma leakage by reducing number and size of endothelial gaps in venules. Am J Physiol Heart Circ Physiol. 2006;290:H107–H118. doi: 10.1152/ajpheart.00542.2005. [DOI] [PubMed] [Google Scholar]
- 24.Mammoto T., Parikh S.M., Mammoto A., Gallagher D., Chan B., Mostoslavsky G., Ingber D.E., Sukhatme V.P. Angiopoietin-1 requires p190 RhoGAP to protect against vascular leakage in vivo. J Biol Chem. 2007;282:23910–23918. doi: 10.1074/jbc.M702169200. [DOI] [PubMed] [Google Scholar]
- 25.Maisonpierre P.C., Suri C., Jones P.F., Bartunkova S., Wiegand S.J., Radziejewski C., Compton D., McClain J., Aldrich T.H., Papadopoulos N., Daly T.J., Davis S., Sato T.N., Yancopoulos G.D. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science. 1997;277:55–60. doi: 10.1126/science.277.5322.55. [DOI] [PubMed] [Google Scholar]
- 26.Pfister F., Wang Y., Schreiter K., vom Hagen F., Altvater K., Hoffmann S., Deutsch U., Hammes H.P., Feng Y. Retinal overexpression of angiopoietin-2 mimics diabetic retinopathy and enhances vascular damages in hyperglycemia. Acta Diabetol. 2010;47:59–64. doi: 10.1007/s00592-009-0099-2. [DOI] [PubMed] [Google Scholar]
- 27.Falcón B.L., Hashizume H., Koumoutsakos P., Chou J., Bready J.V., Coxon A., Oliner J.D., McDonald D.M. Contrasting actions of selective inhibitors of angiopoietin-1 and angiopoietin-2 on the normalization of tumor blood vessels. Am J Pathol. 2009;175:2159–2170. doi: 10.2353/ajpath.2009.090391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McDonald D.M. Angiogenesis and vascular remodeling in inflammation and cancer: biology and architecture of the vasculature. In: Figg W.D., Folkman J., editors. Angiogenesis: An Integrative Approach from Science to Medicine. Springer; New York: 2008. pp. 15–31. [Google Scholar]
- 29.Thurston G., Murphy T.J., Baluk P., Lindsey J.R., McDonald D.M. Angiogenesis in mice with chronic airway inflammation: strain-dependent differences. Am J Pathol. 1998;153:1099–1112. doi: 10.1016/S0002-9440(10)65654-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thurston G., Maas K., Labarbara A., McLean J.W., McDonald D.M. Microvascular remodelling in chronic airway inflammation in mice. Clin Exp Pharmacol Physiol. 2000;27:836–841. doi: 10.1046/j.1440-1681.2000.03342.x. [DOI] [PubMed] [Google Scholar]
- 31.Fuxe J., Lashnits E., O'Brien S., Baluk P., Tabruyn S.P., Kuhnert F., Kuo C., Thurston G., McDonald D.M. Angiopoietin/Tie2 signaling transforms capillaries into venules primed for leukocyte trafficking in airway inflammation. Am J Pathol. 2010;176:2009–2018. doi: 10.2353/ajpath.2010.090976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McDonald D.M. Angiogenesis and remodeling of airway vasculature in chronic inflammation. Am J Respir Crit Care Med. 2001;164:S39–S45. doi: 10.1164/ajrccm.164.supplement_2.2106065. [DOI] [PubMed] [Google Scholar]
- 33.Baluk P., Tammela T., Ator E., Lyubynska N., Achen M.G., Hicklin D.J., Jeltsch M., Petrova T.V., Pytowski B., Stacker S.A., Ylä-Herttuala S., Jackson D.G., Alitalo K., McDonald D.M. Pathogenesis of persistent lymphatic vessel hyperplasia in chronic airway inflammation. J Clin Invest. 2005;115:247–257. doi: 10.1172/JCI22037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aurora A.B., Baluk P., Zhang D., Sidhu S.S., Dolganov G.M., Basbaum C., McDonald D.M., Killeen N. Immune complex-dependent remodeling of the airway vasculature in response to a chronic bacterial infection. J Immunol. 2005;175:6319–6326. doi: 10.4049/jimmunol.175.10.6319. [DOI] [PubMed] [Google Scholar]
- 35.Cho C.H., Kim K.E., Byun J., Jang H.S., Kim D.K., Baluk P., Baffert F., Lee G.M., Mochizuki N., Kim J., Jeon B.H., McDonald D.M., Koh G.Y. Long-term and sustained COMP-Ang1 induces long-lasting vascular enlargement and enhanced blood flow [Erratum appeared in Circ Res 2006;98: e69] Circ Res. 2005;97:86–94. doi: 10.1161/01.RES.0000174093.64855.a6. [DOI] [PubMed] [Google Scholar]
- 36.Thurston G., Wang Q., Baffert F., Rudge J., Papadopoulos N., Jean-Guillaume D., Wiegand S., Yancopoulos G.D., McDonald D.M. Angiopoietin 1 causes vessel enlargement, without angiogenic sprouting, during a critical developmental period. Development. 2005;132:3317–3326. doi: 10.1242/dev.01888. [DOI] [PubMed] [Google Scholar]
- 37.Sims D.E. Recent advances in pericyte biology–implications for health and disease. Can J Cardiol. 1991;7:431–443. [PubMed] [Google Scholar]
- 38.Hammes H.P. Pericytes and the pathogenesis of diabetic retinopathy. Horm Metab Res. 2005;37(Suppl 1):39–43. doi: 10.1055/s-2005-861361. [DOI] [PubMed] [Google Scholar]
- 39.Pfister F., Feng Y., vom Hagen F., Hoffmann S., Molema G., Hillebrands J.L., Shani M., Deutsch U., Hammes H.P. Pericyte migration: a novel mechanism of pericyte loss in experimental diabetic retinopathy. Diabetes. 2008;57:2495–2502. doi: 10.2337/db08-0325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hwang S.J., Kim S.H., Kim H.Z., Steinmetz M.O., Koh G.Y., Lee G.M. High-level expression and purification of a designed angiopoietin-1 chimeric protein, COMP-Ang1, produced in Chinese hamster ovary cells. Protein J. 2008;27:319–326. doi: 10.1007/s10930-008-9140-5. [DOI] [PubMed] [Google Scholar]
- 41.Davidson M.K., Davis J.K., Lindsey J.R., Cassell G.H. Clearance of different strains of Mycoplasma pulmonis from the respiratory tract of C3H/HeN mice. Infect Immun. 1988;56:2163–2168. doi: 10.1128/iai.56.8.2163-2168.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Davidson M.K., Lindsey J.R., Parker R.F., Tully J.G., Cassell G.H. Differences in virulence for mice among strains of Mycoplasma pulmonis. Infect Immun. 1988;56:2156–2162. doi: 10.1128/iai.56.8.2156-2162.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ezaki T., Baluk P., Thurston G., La Barbara A., Woo C., McDonald D.M. Time course of endothelial cell proliferation and microvascular remodeling in chronic inflammation. Am J Pathol. 2001;158:2043–2055. doi: 10.1016/S0002-9440(10)64676-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baffert F., Thurston G., Rochon-Duck M., Le T., Brekken R., McDonald D.M. Age-related changes in vascular endothelial growth factor dependency and angiopoietin-1-induced plasticity of adult blood vessels. Circ Res. 2004;94:984–992. doi: 10.1161/01.RES.0000125295.43813.1F. [DOI] [PubMed] [Google Scholar]
- 45.Baffert F., Le T., Sennino B., Thurston G., Kuo C.J., Hu-Lowe D., McDonald D.M. Cellular changes in normal blood capillaries undergoing regression after inhibition of VEGF signaling. Am J Physiol Heart Circ Physiol. 2006;290:H547–H559. doi: 10.1152/ajpheart.00616.2005. [DOI] [PubMed] [Google Scholar]
- 46.Yao L.C., Baluk P., Feng J., McDonald D.M. Steroid-resistant lymphatic remodeling in chronically inflamed mouse airways. Am J Pathol. 2010;176:1525–1541. doi: 10.2353/ajpath.2010.090909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kwan M.L., Gómez A.D., Baluk P., Hashizume H., McDonald D.M. Airway vasculature after mycoplasma infection: chronic leakiness and selective hypersensitivity to substance P. Am J Physiol Lung Cell Mol Physiol. 2001;280:L286–L297. doi: 10.1152/ajplung.2001.280.2.L286. [DOI] [PubMed] [Google Scholar]
- 48.Tabruyn S.P., Colton K., Morisada T., Fuxe J., Wiegand S.J., Thurston G., Coyle A.J., Connor J., McDonald D.M. Angiopoietin-2-driven vascular remodeling in airway inflammation. Am J Pathol. 2010;177:3233–3243. doi: 10.2353/ajpath.2010.100059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Suri C., McClain J., Thurston G., McDonald D.M., Zhou H., Oldmixon E.H., Sato T.N., Yancopoulos G.D. Increased vascularization in mice overexpressing angiopoietin-1. Science. 1998;282:468–471. doi: 10.1126/science.282.5388.468. [DOI] [PubMed] [Google Scholar]
- 50.Kim K.E., Cho C.H., Kim H.Z., Baluk P., McDonald D.M., Koh G.Y. In vivo actions of angiopoietins on quiescent and remodeling blood and lymphatic vessels in mouse airways and skin. Arterioscler Thromb Vasc Biol. 2007;27:564–570. doi: 10.1161/01.ATV.0000256458.82320.be. [DOI] [PubMed] [Google Scholar]
- 51.Ding R., Darland D.C., Parmacek M.S., D'Amore P.A. Endothelial-mesenchymal interactions in vitro reveal molecular mechanisms of smooth muscle/pericyte differentiation. Stem Cells Dev. 2004;13:509–520. doi: 10.1089/scd.2004.13.509. [DOI] [PubMed] [Google Scholar]
- 52.De Palma M., Venneri M.A., Galli R., Sergi Sergi L., Politi L.S., Sampaolesi M., Naldini L. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell. 2005;8:211–226. doi: 10.1016/j.ccr.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 53.Ozerdem U., Alitalo K., Salven P., Li A. Contribution of bone marrow-derived pericyte precursor cells to corneal vasculogenesis. Invest Ophthalmol Vis Sci. 2005;46:3502–3506. doi: 10.1167/iovs.05-0309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Song S., Ewald A.J., Stallcup W., Werb Z., Bergers G. PDGFRbeta(+) perivascular progenitor cells in tumours regulate pericyte differentiation and vascular survival. Nat Cell Biol. 2005;7:870–879. doi: 10.1038/ncb1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lamagna C., Bergers G. The bone marrow constitutes a reservoir of pericyte progenitors. J Leukoc Biol. 2006;80:677–681. doi: 10.1189/jlb.0506309. [DOI] [PubMed] [Google Scholar]
- 56.Hellström M., Kalén M., Lindahl P., Abramsson A., Betsholtz C. Role of PDGF-B and PDGFR-beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development. 1999;126:3047–3055. doi: 10.1242/dev.126.14.3047. [DOI] [PubMed] [Google Scholar]
- 57.Levéen P., Pekny M., Gebre-Medhin S., Swolin B., Larsson E., Betsholtz C. Mice deficient for PDGF B show renal, cardiovascular, and hematological abnormalities. Genes Dev. 1994;8:1875–1887. doi: 10.1101/gad.8.16.1875. [DOI] [PubMed] [Google Scholar]
- 58.Soriano P. Abnormal kidney development and hematological disorders in PDGF beta-receptor mutant mice. Genes Dev. 1994;8:1888–1896. doi: 10.1101/gad.8.16.1888. [DOI] [PubMed] [Google Scholar]
- 59.Morikawa S., Baluk P., Kaidoh T., Haskell A., Jain R.K., McDonald D.M. Abnormalities in pericytes on blood vessels and endothelial sprouts in tumors. Am J Pathol. 2002;160:985–1000. doi: 10.1016/S0002-9440(10)64920-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gerhardt H., Semb H. Pericytes: gatekeepers in tumour cell metastasis? J Mol Med. 2008;86:135–144. doi: 10.1007/s00109-007-0258-2. [DOI] [PubMed] [Google Scholar]
- 61.Puri M.C., Partanen J., Rossant J., Bernstein A. Interaction of the TEK and TIE receptor tyrosine kinases during cardiovascular development. Development. 1999;126:4569–4580. doi: 10.1242/dev.126.20.4569. [DOI] [PubMed] [Google Scholar]
- 62.Jones N., Voskas D., Master Z., Sarao R., Jones J., Dumont D.J. Rescue of the early vascular defects in Tek/Tie2 null mice reveals an essential survival function. EMBO Rep. 2001;2:438–445. doi: 10.1093/embo-reports/kve093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Uemura A., Ogawa M., Hirashima M., Fujiwara T., Koyama S., Takagi H., Honda Y., Wiegand S.J., Yancopoulos G.D., Nishikawa S. Recombinant angiopoietin-1 restores higher-order architecture of growing blood vessels in mice in the absence of mural cells. J Clin Invest. 2002;110:1619–1628. doi: 10.1172/JCI15621. [DOI] [PMC free article] [PubMed] [Google Scholar]