Epithelial–mesenchymal transition is regulated at post-transcriptional levels by transforming growth factor-β signaling during tumor progression (original) (raw)
Abstract
Transforming growth factor (TGF)-β acts as a tumor suppressor during cancer initiation, but as a tumor promoter during tumor progression. It has become increasingly clear that TGF-β plays fundamental roles in multiple steps of tumor progression, including epithelial-mesenchymal transition (EMT). The EMT, first described by developmental biologists at the beginning of the 1980s, plays crucial roles in appropriate embryonic development, but also functions in adults during wound healing, organ fibrosis, and tumor progression. During EMT, epithelial cells lose their epithelial polarity and acquire mesenchymal phenotypes, endowing them with migratory and invasive properties. Many secreted polypeptides are implicated in this process, and act in a sequential or cooperative manner. TGF-β induces EMT by propagating intracellular signaling pathways and activating transcriptional factors. Here, I discuss new insights into the molecular mechanisms underlying induction of EMT by TGF-β in cooperation with Ras or growth factors, along with the signals that induce EMT through transcriptional and post-transcriptional regulation.
Keywords: Cancer biology, EMT, signal transduction, Smad, TGF-β
Transforming Growth Factor-β-Smad Signaling
Transforming growth factor (TGF)-β is a prototypic member of a large superfamily of more than 30 secreted cytokines, including bone morphogenetic proteins, growth differentiation factors, activins, myostatin, and TGF-β.1 These pleotropic cytokines regulate numerous biological functions, such as proliferation, apoptosis, differentiation, and migration, in various types of cells, thereby regulating embryonic patterning, stem cell maintenance, and the immune system.2 Transforming growth factor-β and related factors bind to serine/threonine kinase receptors and transduce signals principally through Smad proteins. The Smad family comprises of eight structurally related proteins, categorized into three distinct groups: R-Smads, common mediator Smad (Smad4), and inhibitory Smads. R-Smads, including Smad2 and Smad3, are involved in TGF-β signaling. R-Smads possess an N-terminal MH1 domain and a C-terminal MH2 domain, which are connected by a proline-rich acidic linker region. The MH1 domain is bound to DNA, whereas the MH2 domain is involved in complex formation and transcriptional regulation.3 The linker region contains several serine and threonine residues that are phosphorylated by several kinases downstream of growth factor receptors. Phosphorylation of the SXS motif in the MH2 domain of R-Smads by receptor kinase leads to their activation and translocation (along with Smad4) into the nucleus, where transcription of target genes are regulated in cooperation with various transcription factors and transcriptional coactivators or co-repressors. On the other hand, phosphorylation of the linker region induces retention of R-Smad proteins in the cytoplasm and promotes their ubiquitin-dependent degradation, resulting in inhibition of TGF-β signaling.4 However, phosphorylation of the Smad3 linker region is required for maximal transcriptional activation by TGF-β,5,6 and to transduce signals independent of TGF-β.7 Hence, the physiological significance of the phosphorylation of the linker region is still controversial.
Opposing Roles of TGF-β in Tumorigenesis and Cancer Progression
TGF-β was initially identified as a factor that induces proliferation and transformation of fibroblasts.8 So far, it is the most widely studied member of its superfamily in multiple types of cells. In normal epithelial cells, TGF-β suppresses proliferation of the cells. Indeed, the growth inhibitory effects of TGF-β in epithelial cells have also been established by gain- or loss-of-function experiments in mice. Overexpression of TGF-β in epidermis decreases proliferation of keratinocytes and protects mice from tumorigenesis and hyperplasia after treatment with carcinogens. By contrast, ectopic expression of dominant-negative receptors in the epidermis promotes hyperplasia or malignant conversion of epithelial cells. Moreover, aberrations in TGF-β signaling by mutations in signaling components have been observed in various types of tumors. In particular, loss of chromosome 18q21, which contains the Smad4 gene, is observed in 60% of pancreatic and 30% of colorectal cancers. These genetic alterations are not as frequent in other cancers, although inhibitory molecules of TGF-β signaling, such as Smad7 and c-Ski, are also upregulated in certain cancers. Based on these observations, TGF-β is considered to act as a tumor suppressor.9
These genetic alterations and insensitivity to TGF-β are not detected in all types of tumors. In the advanced stages of cancer, TGF-β is often overexpressed in tumor tissues, where it induces migration and invasion of cancer cells. Because TGF-β regulates cell viability and cellular function of immune cells, endothelial cells, and fibroblasts in the tumor microenvironment, it facilitates proliferation and motile properties of tumor cells by helping them evade immune surveillance, promoting angiogenesis and lymphangiogenesis, and depositing ECM proteins. Consequently, TGF-β promotes tumor progression by modifying tumor microenvironments.10 However, blockade of TGF-β signaling in malignant tumor cells suppresses cell survival, intravasation, and motile properties of the cells. In addition, chronic exposure of tumor cells to TGF-β results in loss of TGF-β-mediated growth inhibition, increased cell motility and invasion, and marked changes in cell morphology, leading to EMT.11,12 Therefore, TGF-β in tumor tissues promotes tumor progression by modulating tumor cells themselves, as well as normal cells present in tumor microenvironments.
Regulation of EMT by Transcriptional Factors and miRNAs
Epithelial–mesenchymal transition (EMT) is a phenotypic conversion that facilitates embryonic development and wound healing in physiological processes; the process was initially described by developmental biologists.13,14 However, it became apparent that EMT is also associated with fibrotic diseases and tumor progression in adult tissues. EMT involves dramatic cellular changes, including a decrease in intercellular adhesion and cell polarity, as well as an increase in matrix remodeling and motility. The EMT is characterized by the loss of epithelial marker proteins, such as cytokeratin. The process is also accompanied by dissolution of adherens junction proteins, such as E-cadherin, β-catenin, γ-catenin, and p120 catenin, and by disruption of tight junctions through downregulation or delocalization of tight junction proteins such as Zo-1, occludin, and claudins, resulting in dissociation of epithelial cells and loss of apical-basal polarity. Concomitantly, EMT induces mesenchymal features, like spindle-shaped morphology, reorganization of actin stress fibers, and expression of mesenchymal marker proteins such as N-cadherin, vimentin, and fibronectin.15
Occurs in epithelial carcinoma cells in primary nodules, which differentiate into metastatic tumor cells. The process of tumor cell invasion involves the loss of cell-cell interactions together with acquisition of migratory properties, both of which are also often associated with EMT. As well as acquiring mesenchymal behaviors, cancer cells undergoing EMT exhibit more aggressive phenotypes, including resistance to drugs and stresses, inhibition of senescence and anoikis, and acquisition of immunosuppression and stem cell-like features (Fig.1). These phenotypic changes are regulated by crucial roles of ECM components and soluble factors. Recent studies have revealed the involvement of several transcription factors, known as key EMT regulators, in this process; these include the Snai family of zinc-finger transcription factors (Snail, Slug, and Smuc), the δEF1 family of two-handed zinc-finger factors (δEF1/ZEB1 and SIP1/ZEB2), and the basic helix–loop–helix factors Twist and E12/E47. Except for Twist, these transcription factors repress expression of E-cadherin by direct binding to the E-box sites in its promoter, resulting in transcriptional repression of E-cadherin.16,17
Fig 1.
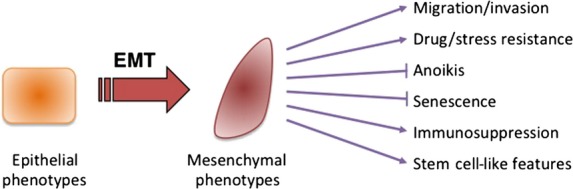
Features of epithelial–mesenchymal transition (EMT).
Several miRNAs involved in specifying epithelial phenotypes have been identified. The miRNAs of the miR-200 family target and repress δEF1 and SIP1 (Fig.2), which in turn inhibit the transcription of two loci that encode five members of the miR-200 family (miR-200b/200a/429 and miR-200c/141), resulting in a double negative feedback loop (Fig.3).18,19 In various types of cancer cells, expression profiles of δEF1 are similar to those of SIP1, both of which are functionally redundant due to the same binding sites in promoter regions of target genes.20 Recently, it was reported that miR-200s also represses Snai family proteins,21 suggesting that the negative feedback loop is a hierarchical machinery for repressing several key EMT regulators during EMT. Indeed, reduced expression of δEF1 results in increased expression of miR-200s and subsequently represses SIP1 expression. In vivo studies show that overexpression of miR-200s inhibits EMT and metastasis through regulating δEF1/SIP1 expression, and that a correlation between miR-200s and liver metastasis is clearly observed in colorectal cancers.22
Fig 2.
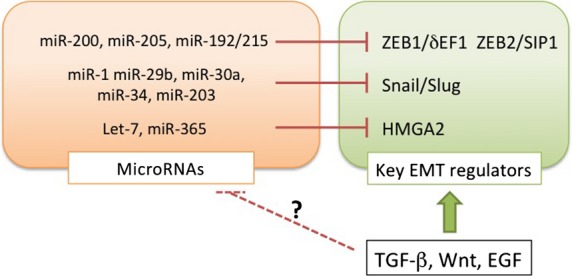
Induction of the epithelial–mesenchymal transition (EMT) by transforming growth factor (TGF)-β through transcriptional regulation of miRNAs and transcription factors. TGF-β directly or indirectly upregulates the key EMT regulators at the transcriptional level, and appears to modulate expression of miRNAs that target the key EMT regulators. δEF1, δ-crystallin/E2-box factor 1; EGF, epidermal growth factor; HMGA2, high mobility group AT-hook 2; ZEB1, zinc finger E-box binding homeobox 1.
Fig 3.

Schematic illustrations of a double-negative feedback loop and the ceRNA pathway. Expression levels of the key EMT regulators and miRNAs suppress each other. Some of the key EMT regulators function as ceRNAs. δEF1, δ-crystallin/E2-box factor 1; HMGA2, high mobility group AT-hook 2; PTEN, phosphatase and tensin homolog; SIP1, Smad-interacting protein 1; TGF-β, transforming growth factor-β; ZEB1, zinc finger E-box binding homeobox 1.
Recently, an idea called the “ceRNA theory” has emerged.23 In this model, miRNAs are sequestered by ceRNAs to regulate mRNA transcripts containing common miRNA recognition elements; for example, miR-25 and miR-200 target both the SIP1 and PTEN mRNAs, both of which possess the common miRNA recognition elements in their 3′-UTRs (Fig.3). Attenuation of the SIP1 mRNA liberates the miRNAs to bind and silence the PTEN mRNA, resulting in a reduction in PTEN protein levels. Importantly, this process is dependent on miRNA binding but independent of protein coding. Thus, suppression of SIP1 mRNA leads to activation of the phosphatidylinositol 3-kinase/AKT pathway in human cancer cells. In this case, multiple putative miRNAs targeting PTEN validate the SIP1 mRNA as a PTEN ceRNA.24,25 The transcription factor HMGA2 is also involved in EMT through Snail induction in mouse epithelial cells.26 Expression of HMGA2 mRNA is regulated by the let-7 miRNA, which also binds the mRNA of TGF-β receptor, suggesting that HMGA2 mRNA acts as a ceRNA for let-7 miRNA (Fig.3).27 Therefore, several types of machinery mediated by ceRNAs might be involved in EMT induction through multiple gene expression networks in cancer cells.
Regulation of EMT through Post-translational Regulation of Snail and δEF-1
Snail, one of the most critical factors involved in EMT induction, undergoes post-transcriptional modifications through phosphorylation by GSK-3β. Because Snail possesses a typical GSK-3β phosphorylation motif (DSGxxS) within its nuclear export signal, GSK-3β-mediated phosphorylation provokes its cytoplasmic export and subsequent ubiquitin-mediated proteasome degradation by β-TrCP, as in the case of β-catenin. Thus, GSK-3β regulates tumorigenesis and tumor progression by controlling protein levels of β-catenin and Snail, respectively.16,17 By contrast, the small C-terminal domain phosphatase (SCP) dephosphorylates these phosphorylation sites and inhibits degradation of Snail. Snail is also degraded by an E3 ligase, F-box protein 11, which is dependent on phosphorylation of Snail at Ser-11 by protein kinase D1.28 Although these phosphorylation sites are not conserved in other members of the Snai family, post-translational regulation of Snail is deeply associated with EMT induction and cancer metastasis.
Two well-characterized members of the δEF1 family, δEF1 (also known as ZEB1) and SIP1 (ZEB2), are involved in TGF-β-induced EMT in mouse epithelial cells.20 The amino acid similarity between δEF1 and SIP1 is approximately 60%.29,30 The former was initially identified as a molecule that regulates embryonic development in mice,31 and it activates TGF-β signaling. The latter was identified by a two-hybrid screen using Smad as the bait, and it inhibits TGF-β signaling by interaction with R-Smads.29 The basis of the functional difference between δEF1 and SIP1 has been elucidated: δEF1 forms a complex with the transcriptional activators p300 and PCAF through its N-terminal region, whereas SIP1 does not bind these factors. Moreover, both δEF1 and SIP1 directly bind the promoter region of E-cadherin and suppress its transcription. Interaction between SIP1 and R-Smads takes place through the SBD at its N-terminus, whereas δEF1 only interacts weakly with R-Smads, possibly due to the low degree of sequence similarity in the two proteins' SBDs. A SIP1 deletion mutant lacking the SBD still represses the transcription of E-cadherin,20 suggesting that binding to Smads is dispensable for E-cadherin repression by both δEF1 and SIP1. In addition, δEF1 regulates the TGF-β-mediated epithelial-myofibroblastic transition,32,33 and positively regulates the transcription of smooth muscle α-actin, a representative myofibroblast marker, by direct binding to its promoter region in vascular smooth muscle cells, whereas SIP1 does not. Therefore, although SIP1 functions as a transcriptional repressor, δEF1 acts both as a transcriptional repressor and as a transcriptional activator (or de-repressor).
Induction of EMT by Synergism between TGF-β and other Signals
Nearly all cases of EMT described to date are regulated by ECM components and soluble growth factors or cytokines, including Wnt, FGFs, hepatocyte growth factor, EGF, and TGF-βs.15,34 Of these, TGF-β was first described as an inducer of EMT during development, and is now thought to promote metastasis through induction of EMT on the invasive fronts of metastatic cancers. TGF-β upregulates expression of key EMT regulators, including Snail and δEF1/SIP1, in epithelial and cancer cells. Several intracellular signals enhance TGF-β signaling to promote tumor invasion/metastasis and EMT. For example, tumor necrosis factor-α, which is secreted from macrophages, promotes TGF-β-induced EMT in lung cancer A549 cells.35 FGF-2 and FGF-4, as well as EGF and hepatocyte growth factor, also enhance TGF-β-induced EMT in epithelial and cancer cells (Fig.4a).33,36 Thus, it is possible that EGF-induced EMT, for example, is facilitated by autocrine TGF-β secreted from the cells. Indeed, cancer cells autonomously produce large quantities of TGF-β, and TGF-β inhibitors affect the phenotypes of cancer cells.37 Taken together, TGF-β secreted from cancer cells could prime the EMT in these cells, in cooperation with other growth factors from cells in the cancer microenvironment. EMT resulting from synergism in cancer cells surrounding the tumor nest may be associated with invasion into the stroma; these phenomena are consistent with previous pathological findings that expression of E-cadherin is preferentially repressed at the invasive edge of a metastatic tumor, but not in the central region of the tumor nest.
Fig 4.

EMT by TGF-β in cooperation with FGF-2 or oncogenic K-Ras. (a) Treatment with both TGF-β and FGF-2 dramatically enhanced epithelial–mesenchymal transition, as determined by phalloidin staining. (b) When K-Ras was knocked down with the siRNA in Panc-1 cells harboring an endogenous oncogenic K-Ras mutation, TGF-β-induced Snail expression was reduced. (c) When RasG12V was transfected into HeLa cells harboring wild-type Ras, Snail (shown in blue) was synergistically upregulated by TGF-β, whereas Smad7 (shown in red), a representative target gene of TGF-β, was slightly inhibited.
Regarding the synergism mentioned above, TGF-β induces Snail expression in cooperation with oncogenic (constitutively active) Ras in cancer cells. When epithelial cells are transformed by oncogenic Ras, they not only become resistant to growth inhibition by TGF-β, but also undergo EMT with invasive and metastatic phenotypes. In MDCK cells and human pancreatic cancer Panc-1 cells, induction of Snail by TGF-β occurs in cooperation with active Ras signals.36,38 Oncogenic Ras dramatically enhances TGF-β-induced expression of Snail, whereas representative direct targets of TGF-β, including Smad7 and plasminogen activator inhibitor 1, are either unaffected or slightly inhibited by Ras signaling (Fig.4b,c).36 Consistent with these observations, cancer cells harboring activated alleles of Ras revealed remarkable induction of Snail after exposure to TGF-β alone.36 This finding is intriguing, because phosphorylation at the linker regions of R-Smads by kinases downstream of Ras prevents nuclear translocation of the R-Smads and stimulates their degradation.10 In addition, Smad3, but not Smad2, has the ability to enhance Snail induction in collaboration with Ras (unpublished data). Mutations in Smad3 in which either four putative phosphorylation sites in the linker region or all serine–proline sites were ablated enhance the responsiveness of representative TGF-β target genes,39 but do not affect the induction of Snail by a combination of TGF-β with Ras.36 Thus, this synergism between TGF-β and Ras signaling leads selectively to induction of Snail, which is dependent on Smad3 but independent of phosphorylation at the serine–proline sites of Smad3. The underlying mechanism, which is not well understood, seems to switch the TGF-β response from antitumor effects to tumor-promoting effects, and is thus sometimes called the “TGF-β switch”.40 Elucidation of the molecular mechanism by which oncogenic Ras regulates the opposing roles of TGF-β might lead to understanding of the TGF-β switch.
Post-transcriptional Regulation of EMT by TGF-β
As described above, FGF-2 and FGF-4 enhance the TGF-β-induced EMT. FGF-2 and FGF-4 bind preferentially to the mesenchymal IIIc isoforms of FGF receptors, whereas FGF-7 (also known as keratinocyte growth factor) and FGF-10 bind exclusively to the epithelial IIIb isoforms.33 These isoforms are regulated by the alternative splicing machinery through the ESRPs.41 Epithelial splicing regulatory proteins 1 (ESRP1) and 2 (ESRP2) were previously known as RNA-recognition motif-containing proteins Rbm35a and Rbm35b, respectively. They bind directly to hexamers containing repeats of UGG or GGU motifs, which are enriched in alternatively spliced regions. As expected, TGF-β downregulates the expression of ESRPs, and subsequently switches the IIIb isoforms of FGFRs to IIIc isoforms, which become sensitized to FGF-2/-4 ligands during EMT. Addition of FGF-2 to TGF-β-treated cells dramatically enhanced EMT (Fig.4a). Importantly, aggressive breast cancer cells express only IIIc isoforms of FGFRs, whereas non-aggressive cells express IIIb isoforms.42 In addition, silencing ESRPs induces switching of alternative splicing of FGFRs from the IIIb isoform to the IIIc isoform, whereas overexpression of ESRPs induces a switch from IIIc to IIIb. Besides FGFs, ESRPs changed the splicing profiles of CD44, Ste 20-like kinase, p120 catenin, and Mena (a member of the Enabled (Ena)/vasodilator-stimulated phosphoprotein family of proteins).43 Together with previous findings that a large number of mRNAs was altered by alternative splicing in TGF-β-induced EMT,42 the splicing program seems to be an essential feature of the epithelial phenotype, and many changes in cell behavior associated with EMT are due to alterations in functions of proteins that undergo isoform switching during this process (Fig.5).
Fig 5.
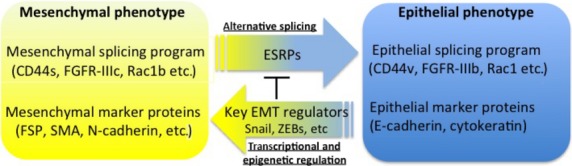
Schematic illustrations of epithelial–mesenchymal transition (EMT) regulated by transcriptional and post-transcriptional mechanisms. The key EMT regulators decrease expression of epithelial splicing regulatory proteins (ESRPs), leading to changes in alternative splicing events. FGFR, fibroblast growth factor receptor; FSP, fibroblast-specific protein; SMA, smooth muscle α-actin; ZEB, zinc finger E-box binding homeobox.
Recently, we found functional differences between ESRP1 and ESRP2.44 When ESRP1 was knocked down in cancer cells, CD44 variants (CD44v) were switched to the standard form (CD44s). Moreover, ESRP1 siRNA altered the splicing isoform of Rac1, generating another alternative splicing variant (Rac1b). Rac1b has 19 additional amino acid residues that correspond to the exon 3b (57 nucleotides) included into the Rac1 mRNA. This insertion accelerates GDP/GTP exchange and impairs GTP hydrolysis. Thus, Rac1b is preferentially in a GTP-bound active form and involved in motility of tumor cells.45–47 However, when ESRP2 was knocked down in these cells, CD44v was not switched to CD44s, but E-cadherin expression was transcriptionally repressed.44 Therefore, ESRPs play crucial roles in alternative splicing and transcriptional regulation during EMT in cancer cells.
Epithelial–mesenchymal Transition in Cancer
Metastasis occurs through several steps: local invasion, intravasation, transport (circulating tumor cells), extravasation, and colonization. EMT has been shown to play pivotal roles in these steps to promote metastasis. For examples, EMT is observed in only a limited area of tumor sections, such as a leading edge. However, cancer cells that are morphologically well differentiated take part in the metastasis process, known as “collective migration”, in which cells do not disseminate and invade as individuals.48 The migrating cancer cells with epithelial characteristics appear to follow fibroblasts/cancer-associated fibroblasts or motile invasive cells derived by EMT at the invasive front of the tumor. It has been reported that EMT cells at the invasive front degrade the ECM and lead non-EMT cells that can attach to the vessel wall, resulting in extravasation.49 In addition, EMT, which occurs in circulating tumor cells, causes acquisition of resistance to anoikis and stress from blood pressure. Once tumor cells have extravasated into distant organs, the EMT cells regain their epithelial phenotypes (mesenchymal–epithelial transition) to form a solid tumor. The phenomenon of switching between EMT and mesenchymal–epithelial transition is evident in bladder, colorectal, and ovarian cancers. Otherwise, the EMT cells may continue to further invade into the secondary site where only non-EMT cells can colonize and initiate growth to form metastasized tumor tissues.
Breast cancer is well characterized in EMT studies and is classified into two subtypes, luminal and basal-like, corresponding to two distinct types of epithelial cells found in the normal mammary gland.50 The luminal subtype is correlated with a good prognosis, whereas the basal-like subtype is associated with aggressive behavior and poor prognosis. The expression of E-cadherin was inversely correlated with both expression of δEF1/SIP1 and progression of breast cancer. Most cell lines with high levels of E-cadherin and low levels of δEF1/SIP1 are categorized into the luminal subtype. By contrast, most of the cell lines with low E-cadherin levels and high δEF1/SIP1 levels are categorized into the basal-like subtype.42 ESRPs are also expressed at low levels in the basal-like subtype, and repressed by direct binding of δEF1/SIP1 to their promoter regions, similar to the repression of E-cadherin by δEF1/SIP1. Recently, we found that ITGA3 was highly expressed in the basal-like subtype, and that ERK inhibitor downregulated expression of ITGA3.51 When ITGA3 is functionally blocked by neutralizing antibodies or its expression is reduced by ERK inhibitor or its specific siRNA, invasive properties are significantly repressed in breast cancer cells and glioma cells.51,52 Interestingly, ERK inhibitor also suppressed the expression of δEF1.51 Thus, the ERK pathway, which is constitutively activated in the basal-like subtype, upregulates expression of ITGA3 as well as δEF1. In the basal-like breast cancer cells, δEF1 interacted with DNMT1 through the SBD domain. Sustained knockdown of both δEF1 and SIP1 reduces the number of DNA methylations at the C-5 position of cytosine sites in the E-cadherin promoter region, suggesting that these proteins maintain the C-5 position of cytosine through interaction with DNMT1. Moreover, δEF1 also interacts with some components of the nucleosome remodeling and deacetylase complex, including the metastasis-associated proteins and the methyl-CpG binding domain family of proteins.53 Previous studies reported that Snail also interacts with DNMT1, as well as multiple chromatin-modifying proteins including histone lysine-specific demethylase, Polycomb repressive complex 2, and histone methyltransferase responsible for the trimethylation of H3K9.54,55 Thus, it seems that δEF1 and Snail regulate both methylation status and chromatin modification of the E-cadherin gene, acting as transcriptional repressors both directly at the transcriptional level and indirectly at the epigenetic level during tumor progression.
Analyses using more than 1000 samples of breast cancer show that recurrence-free survival of the tumor is reduced with increase expression of the EMT markers, including Snail, Twist, and vimentin.56 In colon cancer, the upregulation of genes involved in EMT defines a distinct subtype with very unfavorable prognosis. Moreover, immunohistochemical analyses in prostate cancer suggest that expression levels of the EMT markers are useful predictors for recurrence following surgery and associated with increased Gleason score, advanced clinical stage, and poor prognosis.57 The EMT status (E-cadherin-negative/Snail-positive) in endometrial cancer significantly correlates with histopathological type, myometrial invasion and positive peritoneal cytology, but reversely correlates with overall survival.58 Taken together, studies on EMT in a variety of tumors show statistical correlations between alternations of various EMT markers and patient prognosis.
Conclusions and Perspectives
The key EMT regulators modify expressions of EMT marker proteins at the post-transcriptional, transcriptional, and epigenetic levels. TGF-β, probably secreted from cancer cells, regulates expression of the key EMT regulators and plays a role in tumor progression in cooperation with additional signals from other factors secreted from cells in the tumor microenvironment.
So far, it is still unclear to what extent cells undergo a complete conversion of cell type/morphology by EMT in vivo. However, recent studies prove that an EMT-like morphology or expression of the key EMT regulators is associated with cancer stem cell and tumor aggressivity in a variety of malignant tumors. Thus, the molecules underlying EMT would be promising targets for novel antitumor drugs. Indeed, current treatment modalities remain limited in their efficacy in targeting cells with EMT, due in part to potential drug resistance in the cells. Therefore, appropriate pharmacological targets for the EMT program remains to be elucidated in future studies.
Acknowledgments
I thank Dr. K. Miyazawa for critical reading of the manuscript. I also thank Dr. K. Horiguchi, Dr. K. Miyazono, and Dr. T. Shirakihara for collaboration. This work was supported by JSPS KAKENHI Grant Number 24390419. This work was also supported by the JSPS Core-to-Core Program Cooperative International Framework in TGF-β Family Signaling, the Astellas Foundation for Research on Metabolic Disorders, and the Kobayashi Foundation for Cancer Research.
Glossary
Abbreviations
ceRNA
competing (competitive) endogenous RNA
CtBP
C-terminal binding protein
δEF1
δ-crystallin/E2-box factor 1
DNMT1
DNA methyltransferase 1
EGF
epidermal growth factor
EMT
epithelial–mesenchymal transition
ESRP
epithelial splicing regulatory protein
FGF
fibroblast growth factor
FGFR
fibroblast growth factor receptor
GSK-3β
glycogen synthase kinase 3β
HMGA2
high mobility group AT-hook 2
ITGA3
integrin α3
MH
Mad homology
PTEN
phosphatase and tensin homolog
R-Smad
receptor-regulated Smad
SBD
Smad-binding domain
SIP
Smad-interacting protein
SRF
serum response factor
TGF-β
transforming growth factor-β
ZEB
zinc finger E-box binding homeobox
Disclosure Statement
The author has no conflict of interest.
References
- Derynck R, Miyazono K. The TGF-ß Family. Long Island, New York: Cold Spring Harbor Laboratory Press; 2008. [Google Scholar]
- Massague J, Blain SW, Lo RS. TGFβ signaling in growth control, cancer, and heritable disorders. Cell. 2000;103:295–309. doi: 10.1016/s0092-8674(00)00121-5. [DOI] [PubMed] [Google Scholar]
- Wrana JL, Attisano L. The Smad pathway. Cytokine Growth Factor Rev. 2000;11:5–13. doi: 10.1016/s1359-6101(99)00024-6. [DOI] [PubMed] [Google Scholar]
- Massague J. Integration of Smad and MAPK pathways: a link and a linker revisited. Genes Dev. 2003;17:2993–7. doi: 10.1101/gad.1167003. [DOI] [PubMed] [Google Scholar]
- Kamaraju AK, Roberts AB. Role of Rho/ROCK and p38 MAP kinase pathways in transforming growth factor-β-mediated Smad-dependent growth inhibition of human breast carcinoma cells in vivo. J Biol Chem. 2005;280:1024–36. doi: 10.1074/jbc.M403960200. [DOI] [PubMed] [Google Scholar]
- Motizuki M, Isogaya K, Miyake K, et al. Oligodendrocyte transcription factor 1 (Olig1) is a Smad cofactor involved in cell motility induced by transforming growth factor-β. J Biol Chem. 2013;288:18911–22. doi: 10.1074/jbc.M113.480996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki K. Smad phospho-isoforms direct context-dependent TGF-β signaling. Cytokine Growth Factor Rev. 2013;24:385–99. doi: 10.1016/j.cytogfr.2013.06.002. [DOI] [PubMed] [Google Scholar]
- Sporn MB, Roberts AB. Transforming growth factor-β: recent progress and new challenges. J Cell Biol. 1992;119:1017–21. doi: 10.1083/jcb.119.5.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierie B, Moses HL. TGF-β and cancer. Cytokine Growth Factor Rev. 2006;17:29–40. doi: 10.1016/j.cytogfr.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Bierie B, Moses HL. Tumour microenvironment: TGFβ: the molecular Jekyll and Hyde of cancer. Nat Rev Cancer. 2006;6:506–20. doi: 10.1038/nrc1926. [DOI] [PubMed] [Google Scholar]
- Miyazono K. Transforming growth factor-β signaling in epithelial-mesenchymal transition and progression of cancer. Proc Jpn Acad Ser B Phys Biol Sci. 2009;85:314–23. doi: 10.2183/pjab.85.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh M, Miyazawa K. Transcriptional and post-transcriptional regulation in TGF-β -mediated epithelial-mesenchymal transition. J Biochem. 2012;151:563–71. doi: 10.1093/jb/mvs040. [DOI] [PubMed] [Google Scholar]
- Greenburg G, Hay ED. Epithelia suspended in collagen gels can lose polarity and express characteristics of migrating mesenchymal cells. J Cell Biol. 1982;95:333–9. doi: 10.1083/jcb.95.1.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay ED. An overview of epithelio-mesenchymal transformation. Acta Anat (Basel) 1995;154:8–20. doi: 10.1159/000147748. [DOI] [PubMed] [Google Scholar]
- Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–90. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- Nieto MA. The snail superfamily of zinc-finger transcription factors. Nat Rev Mol Cell Biol. 2002;3:155–66. doi: 10.1038/nrm757. [DOI] [PubMed] [Google Scholar]
- Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7:415–28. doi: 10.1038/nrc2131. [DOI] [PubMed] [Google Scholar]
- Bracken CP, Gregory PA, Kolesnikoff N, et al. A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer Res. 2008;68:7846–54. doi: 10.1158/0008-5472.CAN-08-1942. [DOI] [PubMed] [Google Scholar]
- Lamouille S, Subramanyam D, Blelloch R, Derynck R. Regulation of epithelial-mesenchymal and mesenchymal-epithelial transitions by microRNAs. Curr Opin Cell Biol. 2013;25:200–7. doi: 10.1016/j.ceb.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirakihara T, Saitoh M, Miyazono K. Differential regulation of epithelial and mesenchymal markers by δEF1 proteins in epithelial mesenchymal transition induced by TGF-β. Mol Biol Cell. 2007;18:3533–44. doi: 10.1091/mbc.E07-03-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HF, Xu LY, Li EM. A family of pleiotropically acting microRNAs in cancer progression, miR-200: potential cancer therapeutic targets. Curr Pharm Des. 2014;20:1896–903. doi: 10.2174/13816128113199990519. [DOI] [PubMed] [Google Scholar]
- Hur K, Toiyama Y, Takahashi M, et al. MicroRNA-200c modulates epithelial-to-mesenchymal transition (EMT) in human colorectal cancer metastasis. Gut. 2013;62:1315–26. doi: 10.1136/gutjnl-2011-301846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay Y, Rinn J, Pandolfi PP. The multilayered complexity of ceRNA crosstalk and competition. Nature. 2014;505:344–52. doi: 10.1038/nature12986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karreth FA, Tay Y, Perna D, et al. In vivo identification of tumor- suppressive PTEN ceRNAs in an oncogenic BRAF-induced mouse model of melanoma. Cell. 2011;147:382–95. doi: 10.1016/j.cell.2011.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay Y, Kats L, Salmena L, et al. Coding-independent regulation of the tumor suppressor PTEN by competing endogenous mRNAs. Cell. 2011;147:344–57. doi: 10.1016/j.cell.2011.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thuault S, Valcourt U, Petersen M, Manfioletti G, Heldin CH, Moustakas A. Transforming growth factor-beta employs HMGA2 to elicit epithelial-mesenchymal transition. J Cell Biol. 2006;174:175–83. doi: 10.1083/jcb.200512110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar MS, Armenteros-Monterroso E, East P, et al. HMGA2 functions as a competing endogenous RNA to promote lung cancer progression. Nature. 2014;505:212–7. doi: 10.1038/nature12785. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Zheng H, Shen M, Zha YL, et al. PKD1 phosphorylation-dependent degradation of SNAIL by SCF-FBXO11 regulates epithelial-mesenchymal transition and metastasis. Cancer Cell. 2014;26:358–733. doi: 10.1016/j.ccr.2014.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postigo AA. Opposing functions of ZEB proteins in the regulation of the TGFβ/BMP signaling pathway. EMBO J. 2003;22:2443–52. doi: 10.1093/emboj/cdg225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postigo AA, Depp JL, Taylor JJ, Kroll KL. Regulation of Smad signaling through a differential recruitment of coactivators and corepressors by ZEB proteins. EMBO J. 2003;22:2453–62. doi: 10.1093/emboj/cdg226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funahashi J, Sekido R, Murai K, Kamachi Y, Kondoh H. δ-crystallin enhancer binding protein δEF1 is a zinc finger-homeodomain protein implicated in postgastrulation embryogenesis. Development. 1993;119:433–46. doi: 10.1242/dev.119.2.433. [DOI] [PubMed] [Google Scholar]
- Nishimura G, Manabe I, Tsushima K, et al. δEF1 mediates TGF-β signaling in vascular smooth muscle cell differentiation. Dev Cell. 2006;11:93–104. doi: 10.1016/j.devcel.2006.05.011. [DOI] [PubMed] [Google Scholar]
- Shirakihara T, Horiguchi T, Miyazawa M, et al. TGF-β regulates isoform switching of FGF receptors and epithelial-mesenchymal transition. EMBO J. 2011;30:783–7895. doi: 10.1038/emboj.2010.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moustakas A, Heldin CH. Signaling networks guiding epithelial-mesenchymal transitions during embryogenesis and cancer progression. Cancer Sci. 2007;98:1512–20. doi: 10.1111/j.1349-7006.2007.00550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawata M, Koinuma D, Ogami T, et al. TGF-β-induced epithelial-mesenchymal transition of A549 lung adenocarcinoma cells is enhanced by pro-inflammatory cytokines derived from RAW 264.7 macrophage cells. J Biochem. 2012;151:205–16. doi: 10.1093/jb/mvr136. [DOI] [PubMed] [Google Scholar]
- Horiguchi K, Shirakihara T, Nakano A, Imamura T, Miyazono K, Saitoh M. Role of Ras signaling in the induction of snail by transforming growth factor-β. J Biol Chem. 2009;284:245–53. doi: 10.1074/jbc.M804777200. [DOI] [PubMed] [Google Scholar]
- Ehata S, Hanyu A, Hayashi M, et al. Transforming growth factor-β promotes survival of mammary carcinoma cells through induction of antiapoptotic transcription factor DEC1. Cancer Res. 2007;67:9694–703. doi: 10.1158/0008-5472.CAN-07-1522. [DOI] [PubMed] [Google Scholar]
- Peinado H, Quintanilla M, Cano A. Transforming growth factor β-1 induces snail transcription factor in epithelial cell lines: mechanisms for epithelial mesenchymal transitions. J Biol Chem. 2003;278:21113–23. doi: 10.1074/jbc.M211304200. [DOI] [PubMed] [Google Scholar]
- Nakano A, Koinuma D, Miyazawa K, et al. Pin1 down-regulates transforming growth factor-β signaling by inducing degradation of Smad proteins. J Biol Chem. 2009;284:6109–15. doi: 10.1074/jbc.M804659200. [DOI] [PubMed] [Google Scholar]
- Inman GJ. Switching TGFβ from a tumor suppressor to a tumor promoter. Curr Opin Genet Dev. 2011;21:93–9. doi: 10.1016/j.gde.2010.12.004. [DOI] [PubMed] [Google Scholar]
- Warzecha CC, Sato TK, Nabet B, Hogenesch JB, Carstens RP. ESRP1 and ESRP2 are epithelial cell-type-specific regulators of FGFR2 splicing. Mol Cell. 2009;33:591–601. doi: 10.1016/j.molcel.2009.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiguchi K, Sakamoto K, Koinuma D, et al. TGF-β drives epithelial-mesenchymal transition through δEF1-mediated downregulation of ESRP. Oncogene. 2012;31:3190–201. doi: 10.1038/onc.2011.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warzecha CC, Jiang P, Amirikian K, et al. An ESRP-regulated splicing programme is abrogated during the epithelial-mesenchymal transition. EMBO J. 2010;29:3286–300. doi: 10.1038/emboj.2010.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii H, Saitoh M, Sakamoto K, et al. Epithelial Splicing Regulatory Proteins 1 (ESRP1) and 2 (ESRP2) suppress cancer cell motility via different mechanisms. J Biol Chem. 2014;289:27386–99. doi: 10.1074/jbc.M114.589432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncalves V, Matos P, Jordan P. Antagonistic SR proteins regulate alternative splicing of tumor-related Rac1b downstream of the PI3-kinase and Wnt pathways. Hum Mol Genet. 2009;18:3696–707. doi: 10.1093/hmg/ddp317. [DOI] [PubMed] [Google Scholar]
- Radisky DC, Levy DD, Littlepage LE, et al. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature. 2005;436:123–7. doi: 10.1038/nature03688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiegen D, Haeusler LC, Blumenstein L, et al. Alternative splicing of Rac1 generates Rac1b, a self-activating GTPase. J Biol Chem. 2004;279:4743–9. doi: 10.1074/jbc.M310281200. [DOI] [PubMed] [Google Scholar]
- Gaggioli C, Hooper S, Hidalgo-Carcedo C, et al. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat Cell Biol. 2007;9:1392–400. doi: 10.1038/ncb1658. [DOI] [PubMed] [Google Scholar]
- Tsuji T, Ibaragi S, Hu GF. Epithelial-mesenchymal transition and cell cooperativity in metastasis. Cancer Res. 2009;69:7135–9. doi: 10.1158/0008-5472.CAN-09-1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neve RM, Chin K, Fridlyand J, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10:515–27. doi: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirakihara T, Kawasaki T, Fukagawa A, et al. Identification of integrin α3 as a molecular marker of cells undergoing epithelial-mesenchymal transition and of cancer cells with aggressive phenotypes. Cancer Sci. 2013;104:1189–97. doi: 10.1111/cas.12220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawataki T, Yamane T, Naganuma H, et al. Laminin isoforms and their integrin receptors in glioma cell migration and invasiveness: evidence for a role of α5-laminin(s) and α3β1 integrin. Exp Cell Res. 2007;313:3819–31. doi: 10.1016/j.yexcr.2007.07.038. [DOI] [PubMed] [Google Scholar]
- Fukagawa A, Ishii H, Miyazawa K, Saitoh M. δEF1 associates with DNMT1 and maintains DNA methylation of the E-cadherin promoter in breast cancer cells. Cancer Med. 2015;4:125–35. doi: 10.1002/cam4.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espada J, Peinado H, Lopez-Serra L, et al. Regulation of SNAIL1 and E-cadherin function by DNMT1 in a DNA methylation-independent context. Nucleic Acids Res. 2011;39:9194–205. doi: 10.1093/nar/gkr658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Dong C, Zhou BP. Epigenetic regulation of EMT: the Snail story. Curr Pharm Des. 2014;20:1698–705. doi: 10.2174/13816128113199990512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobin NP, Sims AH, Lundgren KL, Lehn S, Landberg G. Cyclin D1, Id1 and EMT in breast cancer. BMC Cancer. 2011;11:417. doi: 10.1186/1471-2407-11-417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behnsawy HM, Miyake H, Harada K, Fujisawa M. Expression patterns of epithelial-mesenchymal transition markers in localized prostate cancer: significance in clinicopathological outcomes following radical prostatectomy. BJU Int. 2013;111:30–7. doi: 10.1111/j.1464-410X.2012.11551.x. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Terai Y, Kawaguchi H, et al. Prognostic impact of EMT (epithelial-mesenchymal-transition)-related protein expression in endometrial cancer. Cancer Biol Ther. 2013;14:13–9. doi: 10.4161/cbt.22625. [DOI] [PMC free article] [PubMed] [Google Scholar]