Therapeutic Strategies to Attenuate Hemorrhagic Transformation After Tissue Plasminogen Activator Treatment for Acute Ischemic Stroke (original) (raw)
Abstract
This review focuses on the mechanisms and emerging concepts of stroke and therapeutic strategies for attenuating hemorrhagic transformation (HT) after tissue plasminogen activator (tPA) treatment for acute ischemic stroke (AIS). The therapeutic time window for tPA treatment has been extended. However, the patients who are eligible for tPA treatment are still <5% of all patients with AIS. The risk of serious or fatal symptomatic hemorrhage increases with delayed initiation of treatment. HT is thought to be caused by 1) ischemia/reperfusion injury; 2) the toxicity of tPA itself; 3) inflammation; and/or 4) remodeling factor-mediated effects. Modulation of these pathophysiologies is the basis of direct therapeutic strategies to attenuate HT after tPA treatment. Several studies have revealed that matrix metalloproteinases and free radicals are potential therapeutic targets. In addition, we have demonstrated that the inhibition of the vascular endothelial growth factor-signaling pathway and supplemental treatment with a recombinant angiopoietin-1 protein might be a promising therapeutic strategy for attenuating HT after tPA treatment through vascular protection. Moreover, single-target therapies could be insufficient for attenuating HT after tPA treatment and improving the therapeutic outcome of patients with AIS. We recently identified progranulin, which is a growth factor and a novel target molecule with multiple therapeutic effects. Progranulin might be a therapeutic target that protects the brain through suppression of vascular remodeling (vascular protection), neuroinflammation, and/or neuronal death (neuroprotection). Clinical trials which evaluate the effects of anti-VEGF drugs or PGRN-based treatment with tPA will be might worthwhile.
Keywords: tPA, Therapeutic time window, Hemorrhagic transformation, Vascular protection, Brain protection
Introduction
Tissue plasminogen activator (tPA) is the only thrombolytic drug approved to treat acute ischemic stroke (AIS) and is a class-I recommendation in the American Heart Association/American Stroke Association guidelines. The guidelines for the administration of tPA were revised to extend the therapeutic time window (within 4.5 h after the onset of symptoms) in 2012. However, the patients who are eligible for tPA treatment are still between 3.4% and 5.2% of all patients with AIS because of the very narrow therapeutic time window1). In the pooled analysis, the risks of serious or fatal symptomatic hemorrhage increased with later initiation of treatment2). The third International Stroke Trial sought to determine whether a wider range of patients undergoing tPA treatment up to 6 h from stroke onset would benefit3). At 6 months, the patients in the tPA group scored better on the Oxford handicap scale than those in the control group. However, fatal or nonfatal symptomatic intracranial hemorrhages occurred within 7 days in 7% of the patients in the tPA group versus 1% of the patients in the control group. Additionally, a very recent study has demonstrated that early treatment is very important for patients with severe strokes because of the increasing risk of symptomatic hemorrhagic transformation (HT), even within 4.5 h of onset4). Therefore, attenuation of the incidence of HTs after tPA treatment is an important therapeutic strategy against AIS, and it will enable extension of the therapeutic time window and increase the number of patients who are eligible for tPA treatment and the probability of achieving excellent outcomes.
A retrospective clinical study has shown that early disruption of the blood-brain barrier (BBB) after tPA administration, which was indicated by early gadolinium enhancement, predicted a higher risk for symptomatic HT5). Moreover, experimental animal models have revealed that the incorrect timing of reperfusion by thrombolysis increases the incidence of intracerebral HT6). tPA itself is neurotoxic, and it aggravates the neurodamage caused by glutamic acid release after ischemia if it leaks into the brain parenchyma. In addition, tPA promotes the infiltration of leukocytes and activated microglia and the production of free radicals in ischemic lesions7). Furthermore, vascular remodeling factors are upregulated, and microvascular structures are destabilized after cerebral ischemia. These factors also play roles in BBB disruption. Thus, these observations suggest that these factors play dual roles, simultaneously harmful and beneficial, and have diverse heterogeneity, which results in a biphasic clinical course8). Multiple cells, such as neurons, astrocytes, microglia, pericytes, and endothelial cells, make up the neurovascular unit (NVU) and are involved in neurovascular dysfunction in the acute phase of ischemic stroke9). Alterations in the remodeling factors in multiple target cells might be a therapeutic strategy for attenuating HT after tPA treatment in patients with AIS. Eventually, single-target therapies might be insufficient for attenuating HT after tPA treatment in patients with AIS.
In this Review, we describe the mechanisms underlying HT that occurs after tPA treatment in patients with AIS. In addition, we briefly outline the therapeutic vascular and brain protective strategies for attenuating HT after tPA treatment and improving the therapeutic outcomes of patients with AIS.
Mechanisms of Intracerebral HT After tPA Treatment
In order to suppress tPA-induced HT by BBB disruption, an understanding of the underlying mechanisms is essential. The major causes of disruption of the BBB, which is involved in the intracerebral HTs that occur after tPA treatment, are the following: (1) cerebral ischemia/reperfusion injury, (2) the direct toxicity of tPA, (3) inflammation, and (4) remodeling factor-mediated effects. It is important to understand the pathophysiologies underlying the disruption of the BBB in order to attenuate intracerebral HT after tPA treatment (Fig. 1). Modulating these pathophysiologies are direct therapeutic strategies.
Fig. 1.
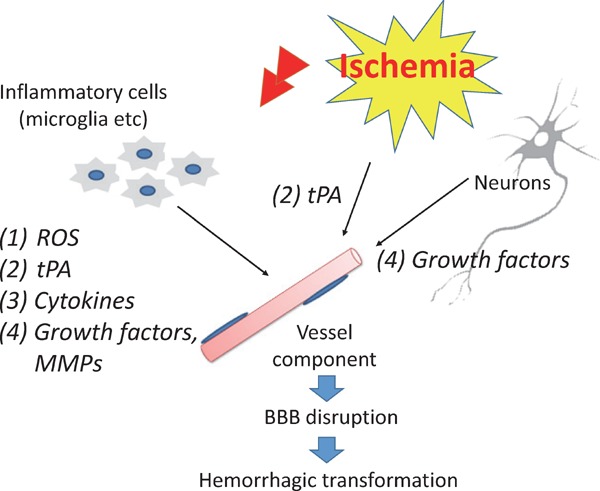
Mechanisms of intracerebral hemorrhagic transformation after tPA treatment and therapeutic targets
BBB, blood-brain barrier; MMP, matrix metalloproteinase; ROS, reactive oxygen species; tPA, tissue plasminogen activator
Reactive Oxygen Species Produced by Cerebral Ischemia/Reperfusion
Cerebral ischemia/reperfusion results in the activation of several reactive oxygen species (ROS)-generating enzymatic systems. In ischemia, the resulting increase in cytosolic Ca2+ activates the superoxide-producing enzyme nicotinamide adenine dinucleotide phosphate-oxidase through protein kinase C and nitric oxide (NO) that are derived from neuronal nitric oxide synthase (NOS)10, 11). The increased ROS that are produced by ischemia-reperfusion can disrupt the NVU through damage to endothelial cells, pericytes, smooth muscle cells, and astrocytes. This increases the likelihood of HT through increased BBB permeability. Damage of the NVU at the capillary level by ROS species might predispose to petechial hemorrhage, whereas ROS injury to both endothelial cells and pericytes at the small arteriolar level could produce larger parenchymal hemorrhages.
A number of experimental models have shown that ROS are involved in early HT. After 2 h of focal cerebral ischemia and 3 h of reperfusion, ROS levels were increased in microvessels and astrocytic endfeet12). The ischemia-induced generation of ROS occurs prior to the upregulation of matrix metalloproteinases (MMPs)13). Therefore, ROS play important roles in very early HT.
The experimental evidence implicating oxidative stress in stroke suggests the use of a combination treatment of tPA and radical-trapping agents (free radical scavengers). However, the translation of these fundamental concepts into clinical applications has proven challenging. In animal models of AIS, the free radical-trapping agent NXY-059 had shown promise as a neuroprotectant. SAINT I and II, which were randomized, placebo-controlled, double-blind trials, were then conducted to investigate the efficacy of NXY-059 in patients with AIS. However, these trials failed to reduce HT and improve the outcomes of the patients with AIS14). Edaravone is another free radical scavenger that can reduce HT in rat stroke15). Notably, the postmarketing registry of the PROTECT4.5 trial on edaravone treatment in acute cerebral infarction in the 4.5-h time window has shown that the frequency of intracerebral HT is lower with the use of edaravone in combination with tPA than with tPA alone16). In addition, edaravone might be a good partner to use in combination therapy with tPA to enhance recanalization and reduce HT17). The results of a tPA and edaravone combination therapy (YAMATO) study are not yet available. However, a worldwide clinical trial to assess the efficacy of edaravone has not been conducted. Therefore, strong evidence for the efficacy of edaravone is needed. Isoflurane may increase ROS by inhibiting superoxide dismutase and catalase. It increases HT in rats with focal ischemia18). Hydrogen gas, which might reduce oxidative stress in the brain, reduces hyperglycemia-enhanced HT in a rat stroke model19). However, small animal studies cannot delineate the evidence for suppressing HT and are less likely to translate to the clinic.
Direct Endothelial Injuries by tPA
tPA is thought to cause neuronal damage and be directly involved in BBB disruption (Fig. 2). The results of animal studies have indicated that tPA increases neuronal damage after focal cerebral ischemia that is mediated by glutamatergic receptors by modifying the properties of the N-methyl-D-aspartate (NMDA) receptor20). In addition, tPA potentiates apoptosis in ischemic endothelium by shifting the apoptotic pathways from caspase-9 to caspase-8, which directly activates caspase-321). Activated protein C, which is a serine protease with anticoagulant activity, inhibits tPA-induced caspase-8 induction and caspase-3 activation in endothelium and hemorrhage21, 22). tPA cleaves the low-density lipoprotein receptor-related protein (LRP) in the plasma membrane of astrocytes, which are located around blood vessels, and the cleaved extracellular fragments induce MMP-9 through nuclear factor-κB pathway activation23). In addition, tPA promotes neutrophil degranulation and MMP-9 release24). The administration of tPA results in the degradation of the protein components of the basal lamina and extracellular matrixes by plasmin and MMP-925–27) (Fig. 3). MMP-9 might directly degrade tight junction proteins27, 28). Several mechanisms of tPA-induced BBB disruption have been described. However, no evidence currently exists for direct injury effects of tPA in the degradation of tight junction proteins of the BBB in the acute time frame of the use of tPA because the administration of tPA within a few hours after onset is not clinical evidence of the induction of HT27).
Fig. 2.
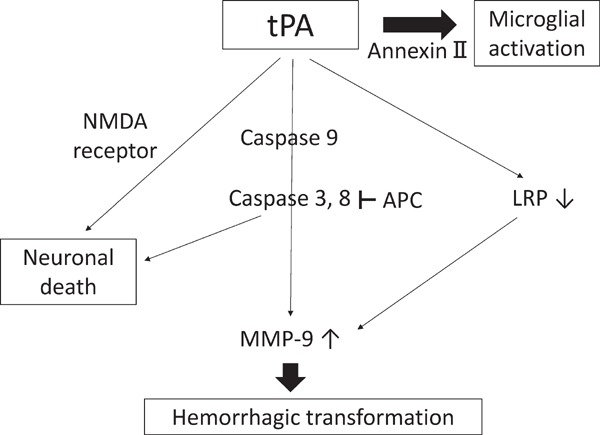
The cascade of tPA-induced adverse effects
APC, activated protein C; LRP, Low-density lipoprotein receptor-related protein; MMP, matrix metalloproteinase; NMDA, N-methyl-D-aspartate; tPA, tissue plasminogen activator
Fig. 3.
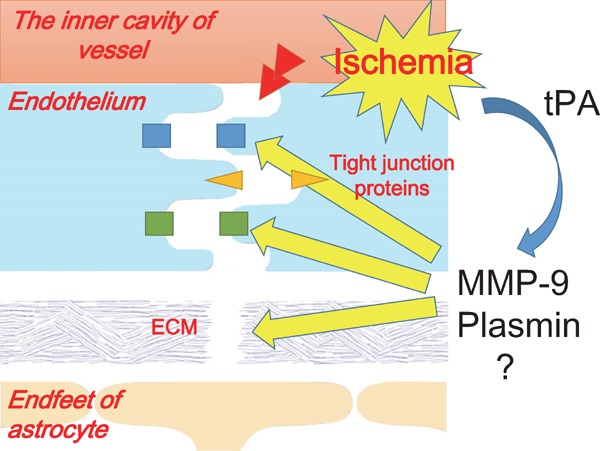
The schema of protease-mediated BBB disruption
BBB, blood-brain barrier; ECM, extracellular matrix; MMP, matrix metalloproteinase; tPA, tissue plasminogen activator
BBB Disruption by Inflammation
The inflammatory response to ischemic stroke expands cerebral infarct volume and induces BBB disruption. Leucocytes infiltrate in increasing numbers within several hours. Macrophages (both microglia-derived and blood-derived) are visible within 24 h, with decreasing numbers of neutrophils seen within 48 h29). Activated microglia and infiltrating inflammatory cells secrete proinflammatory mediators that amplify the inflammatory response, as well as various effector molecules, including proteases, prostaglandins, and ROS, such as NO, through inducible NOS (iNOS), which can directly damage cells or the extracellular matrix30). Cytokines might also directly lead to cell death31). Damage to the endothelium and other components of the BBB can lead to uncontrolled vasogenic edema, microvascular ischemia, or HT.
Microglia and neutrophils are sources of MMP-932). MMP-9 induction also results from tPA/LRP interactions in microglia33). MMPs directly degrade tight junction proteins, including occludin and claudin-5, and components of the extracellular matrix of the basement membrane, such as fibronectin, laminin, collagen, and proteoglycans, and MMPs thereby produce NVU impairments, leukocyte infiltration, brain edema, and HT25, 26, 34) (Fig. 3). Again, infiltrated leukocytes might, in fact, aggravate BBB leakage as tPA has been shown to promote the degranulation of neutrophils, which results in the massive release of MMPs24).
In addition, tPA itself might mediate neuroinflammatory processes. Briefly, tPA promotes microglial chemotaxis by the processing of monocyte chemotactic protein-1 (MCP-1)35), which is a chemokine, and activating microglia following excitotoxic injury and expanding inflammation36). Moreover, tPA activates microglia by binding to LRP-133) or annexin-II37) (Fig. 2). However, microglia are a source of tPA, and tPA deficiency reduces microglia activation by bacterial lipopolysaccharide stimuli, which suggests that tPA acts on microglia activation in an autocrine fashion38). Inflammatory cells are strongly associated with HT through several mechanisms (Fig. 1). Thus, the suppression of inflammation might be an important strategy to attenuate HT.
Minocycline and a pan-MMP inhibitor might also be therapeutic candidates. Minocycline, which is a tetracycline derivative, reduces inflammation and MMP-9 activation and protects against focal cerebral ischemia39, 40). Furthermore, minocycline is clinically safe and well tolerated in combination with tPA41). However, a multinational clinical trial of minocycline has not been conducted. In contrast, the focal ischemic model of experimental stroke proposes that the administration of the monoclonal antibody against the programmed death-1 receptor (PD-L1), which is expressed on T cells, has been reported to reduce infarct volumes and improve neurological outcomes after 96 h of reperfusion42). Thus, fingolimod, which essentially traps lymphocytes in lymph nodes, attenuates the neurological deficits and reduces infarct volume after in situ thromboembolic occlusion of the middle cerebral artery43). The combination of fingolimod and tPA improves the neurological outcomes of the thrombolytic therapy and reduces the risk of HT that is associated with the delayed administration of tPA. These results support the use of the available humanized anti-PD-L1 antibody and fingolimod in the treatment of human stroke subjects. In fact, a clinical trial of fingolimod has started.
The Remodeling Factor Mechanisms of HT
The classical definition of ischemic penumbra is the region of salvage of peri-infarct lesions by any treatment44). Thus, one of the definitions of an ischemic penumbra is a region consisting of multiple molecules45). The penumbra consists of stratified layers, such as the selective cell death zone, heat shock protein 70-inducible zone, hypoxia inducible factor (HIF) zone, and spreading depression zone. In the HIF zone, HIF induces vascular endothelial growth factor (VEGF), iNOS, and erythropoietin. VEGF promotes vascular remodeling, and iNOS increases blood flow by the production of NO. These phenomena cause vascular remodeling in the ischemic penumbra, which might cause HT through BBB disruption.
Another new definition of an ischemic penumbra is the region of transition from an injury to repair by various mediators8) (Fig. 4). Interestingly, the factors that are associated with cell death and tissue damage during the acute period might also play roles in tissue recovery in the chronic period. In other words, these mediators have biphasic roles as a harmful and beneficial target in stroke pathophysiology. During the acute phase, most of these targets mediate injury. In contrast, during the recovery phase, the same mediators induce vascular remodeling/angiogenesis and neurogenesis after stroke. New vessels would not be fully matured. Therefore, during this vascular remodeling, vessels are leakier and prone to HT because of vascular unsteadiness46). The modulation of remodeling factors after stroke with tPA treatment might be one of the ideal therapeutic strategies to attenuate HT.
Fig. 4.
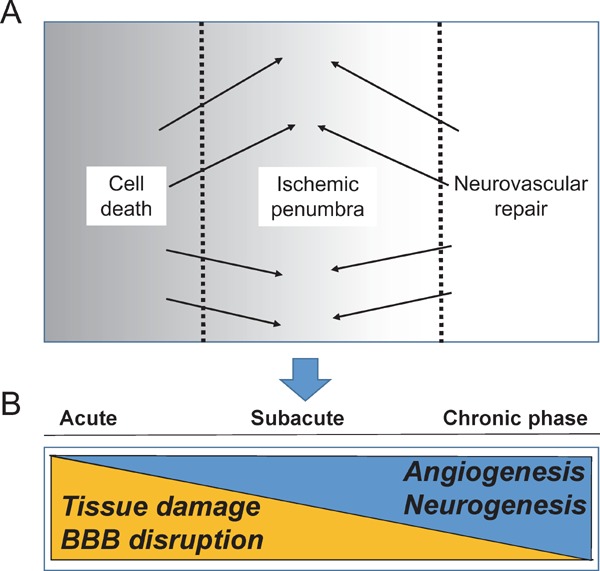
Ischemic penumbra (modified by reference 8)
The new definition of ischemic penumbra is the transition region from injury to repair (A) and with time course after injury (B).
The Biphasic Nature of Molecular Signals in the Ischemic Penumbra
Various drugs that attenuate intracerebral HT after tPA treatment have been investigated in experimental animal models (Table 1). Interestingly, these therapeutic target molecules, including the NMDA-type glutamate receptor, tumor necrosis factor-α, MMPs, and VEGF, have a biphasic nature8). The NMDA-type glutamate receptor is involved not only in the neuronal damage that is mediated by excitotoxicity in the early acute phase but also in neuronal regeneration in the recovery phase47). MMPs cause BBB disruption in the acute phase but are essential for angiogenesis/remodeling in the recovery phase. The knockout of genes encoding MMPs and the suppression of selective MMP inhibitors have all proven considerably protective in animal models of stroke26). The degradation of BBB components by MMP causes edema, HT, and neuronal death. In addition, the importance of MMPs is underscored by the fact that they are upregulated by tPA. Thus, these findings suggest that blocking MMPs might attenuate the HTs that currently limit the widespread application of tPA treatment48). However, as usual, things are never as simple as we wish them to be. Although MMPs disrupt the neurovascular matrix and cause injury during acute stroke, they can promote neurovascular remodeling in the peri-infarct cortex during the delayed stages of stroke recovery49). In addition, MMPs mediate the movement of neuroblasts during the endogenous neurogenic response that is triggered after brain injury50). Whereas the use of MMP inhibitors during the first few hours after stroke reduces infarction, the same inhibitors could worsen the outcomes when they are applied several days later51).
Table 1. Drug candidates to attenuate intracerebral HT after tPA treatment in animal models (modified by reference 58).
| Drug candidates | Reference | Animal | Model |
|---|---|---|---|
| MMP inhibitor | |||
| BB-94 (pan-MMP inhibitor) | Sumii et al. Stroke 2002 | SHR | eMCAO |
| Activated protein | Cheng et al. Nat Med 2006 | rat | eMCAO |
| Anti-TNF-α antibody | Lapchak. Brain Res 2007 | rabbit | eMCAO |
| Minocycline | Murata et al. Stroke 2008 | SHR | eMCAO |
| Cilostazol | Ishiguro et al. Plos One 2010 | mouse | tMCAO |
| Anti-VEGF antibody/receptor inhibitor | Kanazawa et al. JCBFM 2011 | rat | eMCAO |
| Free radical scavenger | |||
| NXY-059 | Lapchak et al. Stroke 2002 | rabbit | eMCAO |
| Edaravone | Yamashita et al. JCBFM 2009 | SHR | tMCAO |
| Immunosuppressant | |||
| FK506 | Okubo et al. Brain Res 2007 | rat | eMCAO |
| Fingolimod | Campos et al. Stroke 2013 | mouse | eMCAO |
| Statin | |||
| Atorvastatin | Zhang et al. JCBFM 2009 | rat | eMCAO |
| Simvastatin | Lapchak et al. Brain Res 2009 | rabbit | eMCAO |
| Others | |||
| Caffeinol | Aronowski et al. Stroke 2003 | rat | tMCAO |
| Imatinib (PDGFR-α antagonist) | Su et al. Nat Med 2008 | mouse | eMCAO |
| High density lipoprotein | Lapergue et al. Stroke 2013 | rat | eMCAO |
| Insulin | Fan et al. Stroke 2013 | rat | eMCAO |
| Angiopoietin 1 | Kawamura et al. PLoS One. 2014 | rat | eMCAO |
| Annexin A2 | Jiang et al. Neurosci lett 2015 | rat | eMCAO |
| Bryostatin | Tan et al. Eur J Pharmacol 2015 | rat | eMCAO |
| Gas | |||
| Hyperbaric oxygen therapy | Qin et al. Stroke 2007 | rat | tMCAO |
| Normobaric hyperoxia therapy (100% O2) | Liang et al. Stroke 20015 | rat | tMCAO |
| Xenon | David et al. JCBFM 2010 | rat | eMCAO |
VEGF also plays a role in the MMP-9-mediated disruption of the BBB in the early acute phase52) and in angiogenesis/remodeling in the recovery phase53). Remodeling factors play dual roles in the acute and recovery period. From this point of view, drugs that inhibit cell death in the acute phase and that do not adversely affect the subsequent repair of neuronal cells and blood vessels are preferred.
Vascular Remodeling Factors, Vascular Endothelial Growth Factor, and Angiopoietin-1, as Novel Therapeutic Target Molecules
We identified the remodeling factors, VEGF and anigiopoietin-1 (Ang1), as therapeutic target molecules for the prevention of intracerebral HT after tPA treatment52, 54). VEGF induces the proliferation, migration, and enhanced permeability of vascular endothelial cells55). The administration of VEGF to animal models in the early phase of acute cerebral ischemia enhances vascular permeability, while the administration in the recovery phase promotes angiogenesis56). Employing embolic middle cerebral artery occlusion models, we demonstrated that the VEGF signal cascade is activated at the BBB in the ischemic penumbra, thereby activating MMP-9 and degrading protein components of the basal lamina, which in turn results in HT52). These changes were evident when tPA was administered after the therapeutic time window. Moreover, any of the changes, including MMP-9 activation and the degradation of the BBB components, were inhibited by an anti-VEGF neutralizing antibody and receptor antagonist. Thus, we demonstrated that the VEGF signal cascade that is related to tPA treatment is located upstream of MMP-9 and that VEGF is a promising therapeutic target molecule that is involved in intracerebral HT after tPA treatment (Fig. 5)52, 57).
Fig. 5.
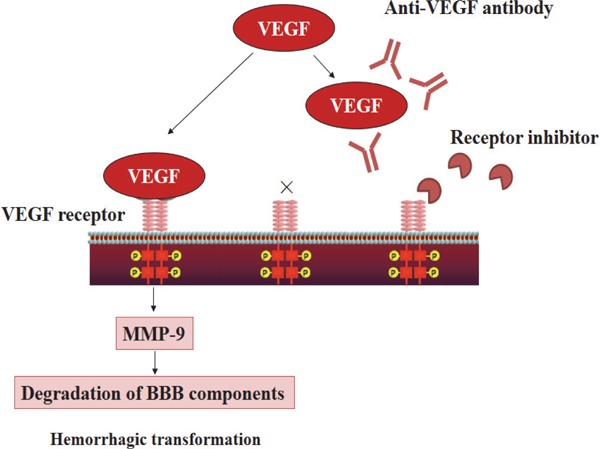
VEGF signaling cascade and anti-VEGF therapy (quoted from reference 57)
After cerebral ischemia, vascular endothelial growth factor (VEGF) is expressed in the microvascular wall, and receptors that are conjugated to VEGF as a ligand are phosphorylated and activated. The subsequent activation of matrix metalloproteinase-9 (MMP-9) and degradation of protein components of the basal lamina cause intracerebral hemorrhage. The VEGF signaling cascade is inhibited by the anti-VEGF antibody that neutralizes VEGF and VEGF receptor inhibitors that inhibit VEGF receptors from being phosphorylated. BBB, blood-brain barrier.
Another endothelial cell-specific growth factor, Ang158), binds to its receptor Tie-2, which is expressed in various types of cells, such as endothelial cells, pericytes, and neuronal cells59). Ang1 participates in the survival of endothelial cells, vascular remodeling, and vascular maturation and stability60). Ang1 has been reported to reduce the postischemic vascular hyperpermeability that is triggered by VEGF61). We confirmed that reduced endogenous Ang1 expression was also involved in HT after tPA treatment54). We demonstrated that the administration of a recombinant Ang1 protein suppressed HT, as well as cerebral edema, after tPA treatment. Several studies have reported that Ang1 suppresses vascular hyperpermeability by increasing glycocalyx in endothelial cells62), acting on tight junction proteins63), and working through the signaling of the platelet-derived growth factor-B in pericytes64). Future studies are needed to confirm whether Ang1 prevents HT and cerebral edema after tPA treatment by suppressing the permeability that is mediated by platelet-derived growth factor-B signaling in pericytes.
Pleiotropic Mechanisms by Progranulin in Ischemic Stroke
As described above, we have demonstrated that the inhibition of the VEGF signaling pathway and the administration of Ang1 attenuates HT after tPA treatment of ischemic stroke. Although this treatment can enable vascular protection, it cannot reduce the cerebral infarct volume52, 54) because it does not have neuroprotective or anti-inflammatory effects. We suggest that brain protection, which includes vascular protection, neuroprotection, and anti-inflammation, is an ideal therapeutic strategy for ischemic stroke. We identified a target molecule, progranulin (PGRN) (Fig. 6)65). In the central nervous system, PGRN is a growth factor that is thought to play crucial roles in maintaining physiological functions66) because mutations of the PGRN gene cause the familial dementia, TAR DNA binding protein-43 (TDP-43)-positive frontotemporal lobar degeneration67–69). We have reported nuclear the TDP-43 might be involved in neuronal cell death prior to cell death after cerebral ischemia65, 70). We have demonstrated dynamic changes in PGRN expression, including increased levels of PGRN expression in microglia within the ischemic core and in surviving neurons, as well as the induction of PGRN expression in endothelial cells within the ischemic penumbra, in ischemic rats. We have observed that PGRN protects against acute focal cerebral ischemia through brain protection, including neuroprotection that occurs in part by the inhibition of the cytoplasmic redistribution of nuclear TDP-43, suppression of neuroinflammation through anti-inflammatory interleukin-10 in microglia, and attenuation of BBB disruption through the inhibition of VEGF. Finally, intravenously administered recombinant PGRN significantly reduces the volumes of cerebral infarcts and edema, suppresses HT, and improves motor outcomes in thromboembolic rats with delayed tPA treatment65). Several other researchers have also shown the pleiotropic protective effects of PGRN71–73). PGRN might be a novel therapeutic target that provides brain protection.
Fig. 6.
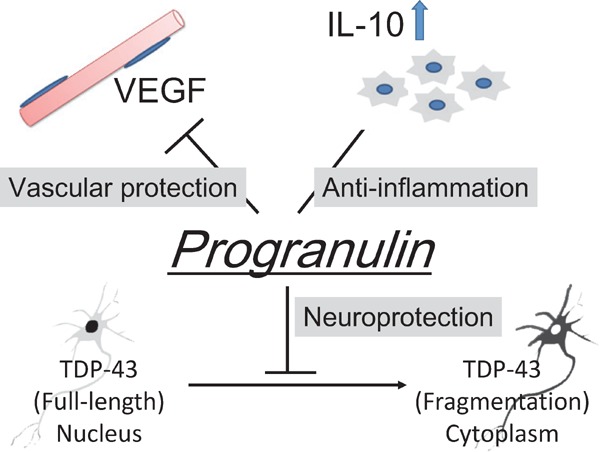
The pleiotropic effects of the brain protective target progranulin (PGRN)
The growth factor PGRN could protect against acute focal cerebral ischemia through a variety of mechanisms, which we call brain protection. The nuclear protein TAR DNA binding protein-43 (TDP-43) is localized in the nucleus. However, cytoplasmic redistribution of TDP-43 occurs after ischemia. Intravenously administered recombinant PGRN significantly reduced the cerebral infarct and edema volumes, suppressed hemorrhagic transformation, and improved motor outcome in thromboembolic rats with delayed tissue plasminogen activator (tPA) treatment because of neuroprotection that occurred in part through the inhibition of the cytoplasmic redistribution of TDP-43, suppression of neuroinflammation through anti-inflammatory interleukin-10 (IL-10) in microglia, and attenuation of BBB disruption through the vascular endothelial growth factor (VEGF). PGRN might be a novel therapeutic target that provides brain protection through processes, such as vascular protection, anti-neuroinflammation, and neuroprotection.
Next-Generation Therapeutic Strategies
A very recent meta-analysis of randomized trials of endovascular treatment with alteplase showed that the therapeutic time window was 6 h after the onset of stroke74). According to the guidelines, patients who are eligible for intravenous tPA should receive intravenous tPA, even if endovascular treatments are being considered (Class I; Level of Evidence A). Moreover, patients should receive endovascular treatment with a stent retriever if they meet the criterion of being within 6 h after onset of stroke (Class I; Level of Evidence A). These suggestions consider the therapeutic limitations of only alteplase and alteplase with endovascular treatments within 6 h after the onset of stroke. Indeed, the need for the development of protective agents for ischemic stroke was discussed at the 2015 International Stroke Conference in Nashville, TN. The next-generation therapeutic strategies are the following: 1) the selection of eligible patients, 2) development of new thrombolytic agents, and 3) combination treatments with protective agents and only alteplase and alteplase with endovascular treatments.
To extend the therapeutic time window of tPA for the selection of eligible patients, a multicenter, randomized, double-blinded, and placebo-controlled Phase III study to investigate EXtending the time for Thrombolysis in Emergency Neurological Deficits (EXTEND) study75) and the European Cooperative Acute Stroke Study-4: Extending the time for thrombolysis in emergency neurological deficits (ECASS-4:ExTEND) are ongoing. These studies deal with ischemic stroke patients with diffusion-weighted image and perfusion-weighted image mismatch in patients 4.5 to 9 h after stroke onset. The final results are not yet available.
Some Recent New Thrombolytic Agents
Desmoteplase (Desmodus rotundus salivary plasminogen activator) is a desirable and attractive alternative to alteplase, and it has several theoretical advantages. It has demonstrated minimal neurotoxicity, high selectivity, specificity for fibrin, and a long half-life. However, a randomized, placebo-controlled, phase-III clinical trial (DIAS-3) that enrolled AIS patients presenting within 3–9 h of onset showed that the frequency of symptomatic intracranial hemorrhage was the same between the desmoteplase group and placebo groups76). The treatment with desmoteplase did not cause safety concerns and did not improve functional outcome when given to patients with ischemic stroke beyond 3 h of onset.
Tenecteplase is a genetically engineered variant of tPA that has a longer half-life and is more fibrin-specific than tPA. These properties give tenecteplase more complete clot lysis with less bleeding complications. In the tenecteplase versus alteplase treatment for patients with AIS who were within 6 h after onset of the ischemic stroke, the tenecteplase group had greater reperfusion rates and better clinical improvement at 24 h compared with the tPA group77). Evaluations of tenecteplase in larger trials of patients with acute stroke are warranted because randomized controlled phase-III trials are lacking. The Norwegian Tenecteplase Stroke Trial (NOR-TEST) is ongoing to compare the efficacy and safety of tenecteplase vs. alteplase in larger groups of patients78).
Another next-generation thrombolytic agent, Stachybotrys microspora triphenyl phenol-7 (SMTP-7), might be another ideal candidate agent. SMTP-7 was discovered from the fungus Stachybotrys microspore. SMTP-7 promotes the urokinase-catalyzed conversion of plasminogen to plasmin, fibrin binding to plasminogen, and the enhancement of thrombolysis in focal ischemic models of rodents and primates. Notably, SMTP-7 suppressed neuroinflammation after reperfusion through the suppression of proinflammatory cytokines79, 80). The results indicated that SMTP-7 decreases infarct volume, HT, mortality, and neurological deficits, and it may be a safe thrombolytic agent to use following cerebral ischemia under warfarin-treated conditions81).
Combination Treatments with Protective Agents
Clinical trials should be performed on thrombolytic therapies and/or endovascular treatments plus concomitant drugs. The Albumin in Acute Ischemic Stroke (ALIAS) parts 1 and 2 trials evaluated whether 25% human serum albumin improved clinical outcomes after acute ischemic stroke82). During the trial, there was a rising use of both intravenous thrombolysis and endovascular stroke treatment. Albumin increased the risk of symptomatic intracerebral hemorrhage in combination with thrombolysis (intravenous and endovascular), although the absolute risk increase was too small to account for the difference between the treatment groups. The ALIAS trials are the latest in a string of clinical trials of putative neuroprotection that have failed to demonstrate clinical efficacy, despite strong preclinical evidence. The vast majority of preclinical models of so-called neuroprotective agents have shown efficacy in models of ischemia-reperfusion. Yet, in humans, on average, early reperfusion was achieved much less than 50% of the time. However, reperfusion was not commonly evaluated. We look forward to new studies that will reexamine the neuroprotection hypothesis in an era of proven early reperfusion83). The glycoprotein IIb/IIIa receptor antagonists (GPIs) in terms of platelet inhibition eptifibatide have demonstrated that AIS patients with tPA plus eptifibatide showed lower incident rates of symptomatic HT than those with tPA alone. The comparison outcomes in patients with tPA and eptifibatide were better than tPA only subjects in ALIAS Part 2 and Interventional Management of Stroke III84). A phase III trial to establish the efficacy of tPA plus eptifibatide for improving AIS outcomes is warranted.
In addition, clinical trials with tPA have been conducted with substances, such as atorvastatin, and edaravone, although a very recent study did not show that simvastatin plus tPA combination treatment suppress HT and improve outcome85). The trials with interventions need a large number of patients and adequate timing for the intervention not to prevent recovery from the point of view of the new ischemic penumbra. Therapeutic agents with pleiotropic protective mechanism are ideal. Clinical trials which evaluate the effects of anti-VEGF drugs or PGRN-based treatment with tPA will be might worthwhile.
Acknowledgments
None.
Conflict of Interest
TS is an academic adviser of the ShimoJani LLC biotech company.
Funding
This work was supported by a Grant-in-Aid for Scientific Research (Research Project Number: 15K19478), a grant from SENSHIN Medical Research Foundation, Uehara Memorial Foundation, and Takeda Science Foundation, and Young Investigator Okamoto Award (MK), a Grant-in-Aid for Scientific Research (Research Project Number: 26430066), and a grant from The Daiichi Sankyo TaNeDS (Take a New Challenge for Drug diScovery) Global Program (TS).
References
- 1).Writing Group Members. Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de Ferranti S, Després JP, Fullerton HJ, Howard VJ, Huffman MD, Isasi CR, Jiménez MC, Judd SE, Kissela BM, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Magid DJ, McGuire DK, Mohler ER, 3rd, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Rosamond W, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Woo D, Yeh RW, Turner MB, American Heart Association Statistics Committee; Stroke Statistics Subcommittee : Executive Summary: Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association: Circulation, 2016; 133: e38-360 [DOI] [PubMed] [Google Scholar]
- 2).Lees KR, Bluhmki E, von Kummer R, Brott TG, Toni D, Grotta JC, Albers GW, Kaste M, Marler JR, Hamilton SA, Tilley BC, Davis SM, Donnan GA, Hacke W, ECASS, ATLANTIS, NINDS and EPITHET rt-PA Study Group. Allen K, Mau J, Meier D, del Zoppo G, De Silva DA, Butcher KS, Parsons MW, Barber PA, Levi C, Bladin C, Byrnes G. Time to treatment with intravenous alteplase and outcome in stroke: an updated pooled analysis of ECASS, ATLANTIS, NINDS, and EPITHET trials. Lancet, 2010; 375: 1695-1703 [DOI] [PubMed] [Google Scholar]
- 3).IST-3 collaborative group. Sandercock P, Wardlaw JM, Lindley RI, Dennis M, Cohen G, Murray G, Innes K, Venables G, Czlonkowska A, Kobayashi A, Ricci S, Murray V, Berge E, Slot KB, Hankey GJ, Correia M, Peeters A, Matz K, Lyrer P, Gubitz G, Phillips SJ, Arauz A: The benefits and harms of intravenous thrombolysis with recombinant tissue plasminogen activator within 6 h of acute ischaemic stroke (the third international stroke trial [IST-3]): a randomised controlled trial. Lancet, 2012; 379: 2352-2363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4).Whiteley WN, Emberson J, Lees KR, Blackwell L, Albers G, Bluhmki E, Brott T, Cohen G, Davis S, Donnan G, Grotta J, Howard G, Kaste M, Koga M, von Kummer R, Lansberg MG, Lindley RI, Lyden P, Olivot JM, Parsons M, Toni D, Toyoda K, Wahlgren N, Wardlaw J, Del Zoppo GJ, Sandercock P, Hacke W, Baigent C, Stroke Thrombolysis Trialists' Collaboration : Risk of intracerebral haemorrhage with alteplase after acute ischaemic stroke: a secondary analysis of an individual patient data meta-analysis. Lancet Neurol, 2016; 15: 925-933 [DOI] [PubMed] [Google Scholar]
- 5).Kastrup A, Groschel K, Ringer TM, Redcker C, Cordesmeyer R, Witte OW, Terborg C: Early disruption of the blood-brain barrier after thrombolytic therapy predicts hemorrhage in patients with acute stroke. Stroke, 2008; 39: 2385-2387 [DOI] [PubMed] [Google Scholar]
- 6).Schaller B, Graf R: Cerebral ischemia and reperfusion: the pathophysiologic concept as a basis for clinical therapy. J Cereb Blood Flow Metab, 2004; 24: 351-371 [DOI] [PubMed] [Google Scholar]
- 7).Flavin MP, Zhao G: Tissue plasminogen activator protects hippocampal neurons from oxygen-glucose deprivation injury. J Neurosci Res, 2001; 63: 388-394 [DOI] [PubMed] [Google Scholar]
- 8).Lo EH: A new penumbra: transitioning from injury into repair after stroke. Nat Med, 2008; 14: 497-500 [DOI] [PubMed] [Google Scholar]
- 9).del Zoppo GJ: Stroke and neurovascular protection. N Engl J Med, 2006; 354: 553-555 [DOI] [PubMed] [Google Scholar]
- 10).Brennan AM, Suh SW, Won SJ, Narasimhan P, Kauppinen TM, Lee H, Edling Y, Chan PH, Swanson RA: NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nat Neurosci, 2009; 12: 857-863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11).Girouard H, Wang G, Gallo EF, Anrather J, Zhou P, Pickel VM, Iadecola C: NMDA receptor activation increases free radical production through nitric oxide and NOX2. J Neurosci, 2009; 29: 2545-2552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12).Gürsoy-Ozdemir Y, Can A, Dalkara T: Reperfusion-induced oxidative/nitrative injury to neurovascular unit after focal cerebral ischemia. Stroke, 2004; 35: 1449-1453 [DOI] [PubMed] [Google Scholar]
- 13).Jickling GC, Liu D, Stamova B, Ander BP, Zhan X, Lu A, Sharp FR: Hemorrhagic transformation after ischemic stroke in animals and humans. J Cereb Blood Flow Metab, 2014; 34: 185-199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14).Shuaib A, Lees KR, Lyden P, Grotta J, Davalos A, Davis SM, Diener HC, Ashwood T, Wasiewski WW, Emeribe U, SAINT II Trial Investigators : NXY-059 for the treatment of acute ischemic stroke. N Engl J Med, 2007; 357: 562-571 [DOI] [PubMed] [Google Scholar]
- 15).Yamashita T, Kamiya T, Deguchi K, Inaba T, Zhang H, Shang J, Miyazaki K, Ohtsuka A, Katayama Y, Abe K: Dissociation and protection of the neurovascular unit after thrombolysis and reperfusion in ischemic rat brain. J Cereb Blood Flow Metab, 2009; 29: 715-725 [DOI] [PubMed] [Google Scholar]
- 16).Yamaguchi T, Yamada T, Hirota T, Tanabashi N: Reducing frequency of symptomatic intracranial hemorrhage in patients with acute ischemic stroke treated by recombinant tissue-plasminogen activator; Interim result of a prospective observational cohort study. Stroke, 2013; 44: A195 [Google Scholar]
- 17).Kimura K, Aoki J, Sakamoto Y, Kobayashi K, Sakai K, Inoue T, Iguchi Y, Shibazaki K: Administration of edaravone, a free radical scavenger, during t-PA infusion can enhance early recanalization in acute stroke patients--a preliminary study. J Neurol Sci, 2012; 313: 132-136 [DOI] [PubMed] [Google Scholar]
- 18).Hu Q, Ma Q, Zhan Y, He Z, Tang J, Zhou C, Zhang J: Isoflurane enhanced hemorrhagic transformation by impairing antioxidant enzymes in hyperglycemic rats with middle cerebral artery occlusion. Stroke, 2011; 42: 1750-1756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19).Chen CH, Manaenko A, Zhan Y, Liu WW, Ostrowki RP, Tang J, Zhang JH: Hydrogen gas reduced acute hyperglycemia-enhanced hemorrhagic transformation in a focal ischemia rat model. Neuroscience, 2010; 169: 402-414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20).Tsirka SE, Gualandris A, Amaral DG, Strickland S: Excitotoxin-induced neuronal degeneration and seizure are mediated by tissue plasminogen activator. Nature, 1995; 377: 340-344 [DOI] [PubMed] [Google Scholar]
- 21).Liu D, Cheng T, Guo H, Fernández JA, Griffin JH, Song X, Zlokovic BV: tPA neurovascular toxicity is controlled by activated protein C. Nat Med, 2004; 10: 1379-1383 [DOI] [PubMed] [Google Scholar]
- 22).Cheng T, Petraglia AL, Li Z, Thiyagarajan M, Zhong Z, Wu Z, Liu D, Maggirwar SB, Deane R, Fernández JA, LaRue B, Griffin JH, Chopp M, Zlokovic BV: Activated protein C inhibits tissue plasminogen activator-induced brain hemorrhage. Nat Med, 2006; 12: 1278-1285 [DOI] [PubMed] [Google Scholar]
- 23).Zhang X, Polavarapu R, She H, Mao Z, Yepes M: Tissue-type plasminogen activator and the low-density lipoprotein receptor-related protein mediate cerebral ischemia-induced nuclear factor-kappaB pathway activation. Am J Pathol, 2007; 171: 1281-1290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24).Cuadrado E, Ortega L, Hernández-Guillamon M, Penalba A, Fernández-Cadenas I, Rosell A, Montaner J: Tissue plasminogen activator (t-PA) promotes neutrophil degranulation and MMP-9 release. J Leukoc Biol, 2008; 84: 207-214 [DOI] [PubMed] [Google Scholar]
- 25).Rosell A, Cuadrado E, Ortega-Aznar A, Hernández-Guillamon M, Lo EH, Montaner J: MMP-9-positive neutrophil infiltration is associated to blood-brain barrier breakdown and basal lamina type IV collagen degradation during hemorrhagic transformation after human ischemic stroke. Stroke, 2008; 39: 1121-1126 [DOI] [PubMed] [Google Scholar]
- 26).Cunningham LA, Wetzel M, Rosenberg GA: Multiple roles for MMPs and TIMPs in cerebral ischemia. Glai 2005; 50: 329-339 [DOI] [PubMed] [Google Scholar]
- 27).del Zoppo GJ, Izawa Y, Hawkins BT: Hemostasis and alterations of the central nervous system. Semin Thromb Hemost, 2013; 39: 856-875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28).Yang Y, Estrada EY, Thompson JF, Liu W, Rosenberg GA: Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J Cereb Blood Flow Metab, 2007; 27: 697-709 [DOI] [PubMed] [Google Scholar]
- 29).Mabuchi T, Kitagawa K, Ohtsuki T, Kuwabara K, Yagita Y, Yanagihara T, Hori M, Matsumoto M: Contribution of microglia/macrophages to expansion of infarction and response of oligodendrocytes after focal cerebral ischemia in rats. Stroke, 2000; 31: 1735-1743 [DOI] [PubMed] [Google Scholar]
- 30).Iadecola C, Anrather J: The immunology of stroke: from mechanisms to translation. Nat Med, 2011; 17: 796-808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31).Kleinig TJ, Vink R: Suppression of inflammation in ischemic and hemorrhagic stroke: therapeutic options. Curr Opin Neurol, 2009; 22: 294-301 [DOI] [PubMed] [Google Scholar]
- 32).del Zoppo GJ, Frankowski H, Gu YH, Osada T, Kanazawa M, Milner R, Wang X, Hosomi N, Mabuchi T, Koziol JA: Microglial cell activation is a source of metalloproteinase generation during hemorrhagic transformation. J Cereb Blood Flow Metab, 2012; 32: 919-932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33).Zhang C, An J, Haile WB, Echeverry R, Strickland DK, Yepes M: Microglial low-density lipoprotein receptor-related protein 1 mediates the effect of tissue-type plasminogen activator on matrix metalloproteinase-9 activity in the ischemic brain. J Cereb Blood Flow Metab, 2009; 29: 1946-1954 [DOI] [PubMed] [Google Scholar]
- 34).Adibhatla RM, Hatcher JF: Tissue plasminogen activator (tPA) and matrix metalloproteinases in the pathogenesis of stroke: therapeutic strategies. CNS Neurol Disord Drug Targets, 2008; 7: 243-253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35).Sheehan JJ, Zhou C, Gravanis I, Rogove AD, Wu YP, Bogenhagen DF, Tsirka SE: Proteolytic activation of monocyte chemoattractant protein-1 by plasmin underlies excitotoxic neurodegeneration in mice. J Neurosci, 2007; 27: 1738-1745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36).Siao CJ, Fernandez SR, Tsirka SE: Cell type-specific roles for tissue plasminogen activator released by neurons or microglia after excitotoxic injury. J Neurosci, 2003; 23: 3234-3242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37).Siao CJ, Tsirka SE: Tissue plasminogen activator mediates microglial activation via its finger domain through annexin II. J Neurosci, 2002; 22: 3352-3358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38).Rogove AD, Siao C, Keyt B, Strickland S, Tsirka SE: Activation of microglia reveals a non-proteolytic cytokine function for tissue plasminogen activator in the central nervous system. J Cell Sci, 1999; 112: 4007-4016 [DOI] [PubMed] [Google Scholar]
- 39).Yrjänheikki J, Tikka T, Keinänen R, Goldsteins G, Chan PH, Koistinaho J: A tetracycline derivative, minocycline, reduces inflammation and protects against focal cerebral ischemia with a wide therapeutic window. Proc Natl Acad Sci USA, 1999; 96: 13496-13500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40).Murata Y, Rosell A, Scannevin RH, Rhodes KJ, Wang X, Lo EH: Extension of the thrombolytic time window with minocycline in experimental stroke. Stroke, 2008; 39: 3372-3277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41).Fagan SC, Waller JL, Nichols FT, Edwards DJ, Pettigrew LC, Clark WM, Hall CE, Switzer JA, Ergul A, Hess DC: Minocycline to improve neurologic outcome in stroke (MINOS): a dose-finding study. Stroke, 2010; 41: 2283-2287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42).Bodhankar S, Chen Y, Lapato A, Dotson AL, Wang J, Vandenbark AA, Saugstad JA, Offner H: PD-L1 monoclonal antibody treats ischemic stroke by controlling central nervous system inflammation. Stroke, 2015; 46: 2926-2934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43).Campos F, Qin T, Castillo J, Seo JH, Arai K, Lo EH, Waeber C: Fingolimod reduces hemorrhagic transformation associated with delayed tissue plasminogen activator treatment in a mouse thromboembolic model. Stroke, 2013; 44: 505-511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44).Astrup J, Siesjö BK, Symon L: Thresholds in cerebral ischemia - the ischemic penumbra. Stroke, 1981; 12: 723-725 [DOI] [PubMed] [Google Scholar]
- 45).Sharp FR, Lu A, Tang Y, Millhorn DE: Multiple molecular penumbras after focal cerebral ischemia. J Cereb Blood Flow Metab, 2000; 20: 1011-1032 [DOI] [PubMed] [Google Scholar]
- 46).Durukan A, Marinkovic I, Strbian D, Pitkonen M, Pedrono E, Soinne L, Abo-Ramadan U, Tatlisumak T: Post-ischemic blood-brain barrier leakage in rats: one-week follow-up by MRI. Brain Res, 2009; 1280: 158-165 [DOI] [PubMed] [Google Scholar]
- 47).Platel JC, Dave KA, Gordon V, Lacar B, Rubio ME, Bordey A: NMDA receptors activated by subventricular zone astrocytic glutamate are critical for neuroblast survival prior to entering a synaptic network. Neuron, 2010; 65: 859-872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48).Wang X, Tsuji K, Lee SR, Ning M, Furie KL, Buchan AM, Lo EH: Mechanisms of hemorrhagic transformation after tissue plasminogen activator reperfusion therapy for ischemic stroke. Stroke, 2004; 35: 2726-2730 [DOI] [PubMed] [Google Scholar]
- 49).Zhao BQ, Wang S, Kim HY, Storrie H, Rosen BR, Mooney DJ, Wang X, Lo EH: Role of matrix metalloproteinases in delayed cortical responses after stroke. Nat Med 2006; 12: 441-445 [DOI] [PubMed] [Google Scholar]
- 50).Lee SR, Kim HY, Rogowska J, Zhao BQ, Bhide P, Parent JM, Lo EH: Involvement of matrix metalloproteinase in neuroblast cell migration from the subventricular zone after stroke. J Neurosci, 2006; 26: 3491-3495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51).Zhao BQ, Tejima E, Lo EH: Neurovascular proteases in brain injury, hemorrhage and remodeling after stroke. Stroke, 2007; 38: 748-752 [DOI] [PubMed] [Google Scholar]
- 52).Kanazawa M, Igarashi H, Kawamura K, Takahashi T, Kakita A, Takahashi H, Nakada T, Nishizawa M, Shimohata T: Inhibition of VEGF signaling pathway attenuates hemorrhage after tPA treatment. J Cereb Blood Flow Metab, 2011; 31: 1461-1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53).Sun Y, Jin K, Xie L, Childs J, Mao XO, Logvinova A, Greenberg DA: VEGF-induced neuroprotection, neurogenesis, and angiogenesis after focal cerebral ischemia. J Clin Invest, 2003; 111: 1843-1851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54).Kawamura K, Takahashi T, Kanazawa M, Igarashi H, Nakada T, Nishizawa M, Shimohata T: Effects of angiopoietin-1 on hemorrhagic transformation and cerebral edema after tissue plasminogen activator treatment for ischemic stroke in rats. PLoS One, 2014; 9: e98639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55).Senger DR, Ledbetter SR, Claffey KP, Papadopoulos-Sergiou A, Peruzzi CA, Detmar M: Stimulation of endothelial cell migration by vascular permeability factor/vascular endothelial growth factor through cooperative mechanisms involving the alphavbeta3 integrin, osteopontin, and thrombin. Am J Pathol, 1996; 149: 293-305 [PMC free article] [PubMed] [Google Scholar]
- 56).Zhang ZG, Zhang L, Jiang Q, Zhang R, Davies K, Powers C, Bruggen N, Chopp M: VEGF enhances angiogenesis and promotes blood-brain barrier leakage in the ischemic brain. J Clin Invest, 2000; 106: 829-838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57).Shimohata T, Kanazawa M, Kawamura K, Takahashi T, Nishizawa M: Therapeutic strategies to attenuate hemorrhagic transformation after tissue plasminogen activator treatment for acute ischemic stroke. Neurology and Clinical Neuroscience 2013; 1: 201-208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58).Suri C, Jones PF, Patan S, Bartunkova S, Maisonpierre PC, Davis S, Sato TN, Yancopoulos GD: Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell, 1996; 87: 1171-1780 [DOI] [PubMed] [Google Scholar]
- 59).Davis S, Aldrich TH, Jones PF, Acheson A, Compton DL, Jain V, Ryan TE, Bruno J, Radziejewski C, Maisonpierre PC, Yancopoulos GD: Isolation of angiopoietin-1, a ligand for the TIE2 receptor, by secretion-trap expression cloning. Cell 1996; 87: 1161-1169 [DOI] [PubMed] [Google Scholar]
- 60).Kim I, Kim HG, So JN, Kim JH, Kwak HJ, Koh GY: Angiopoietin-1 regulates endothelial cell survival through the phosphatidylinositol 3'-Kinase/Akt signal transduction pathway. Circ Res 2000; 86: 24-29 [DOI] [PubMed] [Google Scholar]
- 61).Zhang ZG, Zhang L, Croll SD, Chopp M: Angiopoi etin-1 reduces cerebral blood vessel leakage and ischemic lesion volume after focal cerebral embolic ischemia in mice. Neuroscience, 2002; 113: 683-687 [DOI] [PubMed] [Google Scholar]
- 62).Salmon AH, Neal CR, Sage LM, Glass CA, Harper SJ, Bates DO: Angiopoietin-1 alters microvascular permeability coefficients in vivo via modification of endothelial glycocalyx. Cardiovasc Res, 2009; 83: 24-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63).Yu H, Wang P, An P, Xue Y, Yixue X: Recombinant human angiopoietin-1 ameliorates the expressions of ZO-1, occludin, VE-cadherin, and PKCα signaling after focal cerebral ischemia/reperfusion in rats. J Mol Neurosci, 2012; 46: 236-427 [DOI] [PubMed] [Google Scholar]
- 64).Fuxe J, Tabruyn S, Colton K, Zaid H, Adams A, Baluk P, Lashnits E, Morisada T, Le T, O'Brien S, Epstein DM, Koh GY, McDonald DM: Pericyte requirement for antileak action of angiopoietin-1 and vascular remodeling in sustained inflammation. Am J Pathol, 2011; 178: 2897-2909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65).Kanazawa M, Kawamura K, Takahashi T, Miura M, Tanaka Y, Koyama M, Toriyabe M, Igarashi H, Nakada T, Nishihara M, Nishizawa M, Shimohata T: Multiple therapeutic effects of progranulin on experimental acute ischaemic stroke. Brain, 2015; 138: 1932-1948 [DOI] [PubMed] [Google Scholar]
- 66).He Z, Bateman A: Progranulin (granulin-epithelin precursor, PC-cell-derived growth factor, acrogranin) mediates tissue repair and tumorigenesis. J Mol Med (Berl), 2003; 81: 600-612 [DOI] [PubMed] [Google Scholar]
- 67).Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S, Cannon A, Dwosh E, Neary D, Melquist S, Richardson A, Dickson D, Berger Z, Eriksen J, Robinso T, Zehr C, Dickey CA, Crook R, McGowan E, Mann D, Boeve B, Feldman H, Hutton M: Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature, 2006; 442: 916-919 [DOI] [PubMed] [Google Scholar]
- 68).Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, Rademakers R, Vandenberghe R, Dermaut B, Martin JJ, van Duijn C, Peeters K, Sciot R, Santens P, De Pooter T, Mattheijssens M, Van den Broeck M, Cuijt I, Vennekens K, De Deyn PP, Kumar-Singh S, Van Broeckhoven C: Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature, 2006; 442: 920-924 [DOI] [PubMed] [Google Scholar]
- 69).Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM: Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science, 2006; 314: 130-133 [DOI] [PubMed] [Google Scholar]
- 70).Kanazawa M, Kakita A, Igarashi H, Takahashi T, Kawamura K, Takahashi H, Nakada T, Nishizawa M, Shimohata T: Biochemical and histopathological alterations in TAR DNA binding protein-43 after acute ischemic stroke in rats. J Neurochem, 2011; 116: 957-965 [DOI] [PubMed] [Google Scholar]
- 71).Tao J, Ji F, Wang F, Liu B, Zhu Y: Neuroprotective effects of progranulin in ischemic mice. Brain Res, 2012; 1436: 130-136 [DOI] [PubMed] [Google Scholar]
- 72).Egashira Y, Suzuki Y, Azuma Y, Takagi T, Mishiro K, Sugitani S, Tsuruma K, Shimazawa M, Yoshimura S, Kashimata M, Iwama T, Hara H: The growth factor progranulin attenuates neuronal injury induced by cerebral ischemia-reperfusion through the suppression of neutrophil recruitment. J Neuroinflammation, 2013; 10: 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73).Jackman K, Kahles T, Lane D, Garcia-Bonilla L, Abe T, Capone C, Hochrainer K, Voss H, Zhou P, Ding A, Anrather J, Iadecola C: Progranulin Deficiency Promotes Post-Ischemic Blood-Brain Barrier Disruption. J Neurosci, 2013; 33: 19579-19589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74).Goyal M, Menon BK, van Zwam WH, Dippel DW, Mitchell PJ, Demchuk AM, Dávalos A, Majoie CB, van der Lugt A, de Miquel MA, Donnan GA, Roos YB, Bonafe A, Jahan R, Diener HC, van den Berg LA, Levy EI, Berkhemer OA, Pereira VM, Rempel J, Millán M, Davis SM, Roy D, Thornton J, Román LS, Ribó M, Beumer D, Stouch B, Brown S, Campbell BC, van Oostenbrugge RJ, Saver JL, Hill MD, Jovin TG, HERMES collaborators : Endovascular thrombectomy after large-vessel ischaemic stroke: a meta-analysis of individual patient data from five randomised trials. Lancet, 2016; 387: 1723-1731 [DOI] [PubMed] [Google Scholar]
- 75).Ma H, Parsons MW, Christensen S, Campbell BC, Churilov L, Connelly A, Yan B, Bladin C, Phan T, Barber AP, Read S, Hankey GJ, Markus R, Wijeratne T, Grimley R, Mahant N, Kleinig T, Sturm J, Lee A, Blacker D, Gerraty R, Krause M, Desmond PM, McBride SJ, Carey L, Howells DW, Hsu CY, Davis SM, Donnan GA, EXTEND investigators : A multicentre, randomized, double-blinded, placebo-controlled Phase III study to investigate EXtending the time for Thrombolysis in Emergency Neurological Deficits (EXTEND). Int J Stroke, 2012; 7: 74-80 [DOI] [PubMed] [Google Scholar]
- 76).Albers GW, von Kummer R, Truelsen T, Jensen JK, Ravn GM, Grønning BA, Chabriat H, Chang KC, Davalos AE, Ford GA, Grotta J, Kaste M, Schwamm LH, Shuaib A, DIAS-3 Investigators : Safety and efficacy of desmoteplase given 3–9 h after ischaemic stroke in patients with occlusion or high-grade stenosis in major cerebral arteries (DIAS-3): a double-blind, randomised, placebo-controlled phase 3 trial. Lancet Neurol, 2015; 14: 575-584 [DOI] [PubMed] [Google Scholar]
- 77).Parsons M, Spratt N, Bivard A, Campbell B, Chung K, Miteff F, O'Brien B, Bladin C, McElduff P, Allen C, Bateman G, Donnan G, Davis S, Levi C: A randomized trial of tenecteplase versus alteplase for acute ischemic stroke. N Engl J Med, 2012; 366: 1099-1107 [DOI] [PubMed] [Google Scholar]
- 78).Logallo N, Kvistad CE, Nacu A, Naess H, Waje-Andreassen U, Asmuss J, Aamodt AH, Lund C, Kurz MW, Rønning OM, Salvesen R, Idicula TT, Thomassen L: The Norwegian tenecteplase stroke trial (NOR-TEST): randomised controlled trial of tenecteplase vs. alteplase in acute ischaemic stroke. BMC Neurol, 2014; 14: 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79).Miyazaki T, Kimura Y, Ohata H, Hashimoto T, Shibata K, Hasumi K, Honda K: Distinct effects of tissue-type plasminogen activator and SMTP-7 on cerebrovascular inflammation following thrombolytic reperfusion. Stroke, 2011; 42: 1097-1104 [DOI] [PubMed] [Google Scholar]
- 80).Sawada H, Nishimura N, Suzuki E, Zhuang J, Hasegawa K, Takamatsu H, Honda K, Hasumi K: SMTP-7, a novel small-molecule thrombolytic for ischemic stroke: a study in rodents and primates. J Cereb Blood Flow Metab, 2014; 34: 235-241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81).Ito A, Niizuma K, Shimizu H, Fujimura M, Hasumi K, Tominaga T: SMTP-7, a new thrombolytic agent, decreases hemorrhagic transformation after transient middle cerebral artery occlusion under warfarin anticoagulation in mice. Brain Res, 2014; 1578: 38-48 [DOI] [PubMed] [Google Scholar]
- 82).Martin RH, Yeatts SD, Hill MD, Moy CS, Ginsberg MD, Palesch YY, for the ALIAS Parts 1 and 2 and NETT Investigators : ALIAS (Albumin in Acute Ischemic Stroke) Trials: Analysis of the Combined Data From Parts 1 and 2. Stroke, 2016. July 26 pii: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83).Adeoye O, Sucharew H, Khoury J, Vagal A, Schmit PA, Ewing I, Levine SR, Demel S, Eckerle B, Katz B, Kleindorfer D, Stettler B, Woo D, Khatri P, Broderick JP, Pancioli AM: Combined approach to lysis utilizing eptifibatide and recombinant tissue-type plasminogen activator in acute ischemic stroke-full dose regimen stroke trial. Stroke, 2015; 46: 2529-2533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84).Adeoye O, Sucharew H, Khoury J, Tomsick T, Khatri P, Palesch Y, Schmit PA, Pancioli AM, Broderick JP, CLEAR-ER, IMS III, and ALIAS Part 2 Investigators Recombinant tissue-type plasminogen activator plus eptifibatide versus recombinant tissue-type plasminogen activator alone in acute ischemic stroke: propensity score-matched post hoc analysis. Stroke, 2015; 46: 461-464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85).Montaner J, Bustamante A, García-Matas S, Martínez-Zabaleta M, Jiménez C, de la Torre J, Rubio F, Segura T, Masjuán J, Cánovas D, Freijo M, Delgado-Mederos R, Tejada J, Lago A, Bravo Y, Corbeto N, Giralt D, Vives-Pastor B, de Arce A, Moniche F, Delgado P, Ribó M, on Behalf of the STARS Investigators Combination of thrombolysis and statins in acute stroke is safe: results of the STARS randomized trial (stroke treatment with acute reperfusion and simvastatin). Stroke, 2016. published October 6, 2016. 10.1161/STROKEAHA.116.014600 [DOI] [PubMed] [Google Scholar]