Essential Role of Hemoglobin βCys93 in Cardiovascular Physiology (original) (raw)
Abstract
The supply of oxygen to tissues is controlled by microcirculatory blood flow. One of the more surprising discoveries in cardiovascular physiology is the critical dependence of microcirculatory blood flow on a single conserved cysteine within the β-subunit (βCys93) of hemoglobin (Hb). βCys93 is the primary site of Hb _S_-nitrosylation [i.e., _S_-nitrosothiol (SNO) formation to produce _S_-nitrosohemoglobin (SNO-Hb)]. Notably, _S_-nitrosylation of βCys93 by NO is favored in the oxygenated conformation of Hb, and deoxygenated Hb releases SNO from βCys93. Since SNOs are vasodilatory, this mechanism provides a physiological basis for how tissue hypoxia increases microcirculatory blood flow (hypoxic autoregulation of blood flow). Mice expressing βCys93A mutant Hb (C93A) have been applied to understand the role of βCys93, and RBCs more generally, in cardiovascular physiology. Notably, C93A mice are unable to effect hypoxic autoregulation of blood flow and exhibit widespread tissue hypoxia. Moreover, reactive hyperemia (augmentation of blood flow following transient ischemia) is markedly impaired. C93A mice display multiple compensations to preserve RBC vasodilation and overcome tissue hypoxia, including shifting SNOs to other thiols on adult and fetal Hbs and elsewhere in RBCs, and growing new blood vessels. However, compensatory vasodilation in C93A mice is uncoupled from hypoxic control, both peripherally (e.g., predisposing to ischemic injury) and centrally (e.g., impairing hypoxic drive to breathe). Altogether, physiological studies utilizing C93A mice are confirming the allosterically controlled role of SNO-Hb in microvascular blood flow, uncovering essential roles for RBC-mediated vasodilation in cardiovascular physiology and revealing new roles for RBCs in cardiovascular disease.
Keywords: hemoglobin, microcirculation, _S_-nitrosothiol, _S_-nitrosylation, vasodilation
Introduction
Cardiovascular physiology encompasses the circulation of blood to deliver oxygen and nutrients and remove carbon dioxide and metabolic waste from tissues. The ability to improve oxygen delivery to hypoxic tissues would be a great benefit to medicine, since poor oxygen delivery is a component of a multitude of diseases (24, 37). However, standard treatments, including breathing oxygen or increasing circulating red blood cells (RBCs) by transfusion, are generally ineffective at relieving tissue hypoxia (40, 50, 59). These treatments increase the oxygen content of blood but do not affect the primary driver of tissue oxygenation, namely, microcirculatory blood flow. Currently, no treatment is available in medicine to improve microcirculatory blood flow directly, since all vasodilators address large vessels preferentially, predisposing to shunting of blood. Elucidation of the mechanism of blood flow coupling to tissue oxygenation is the last major frontier in cardiovascular physiology, and drugs to exploit this microcirculatory mechanism remain a holy grail in cardiovascular medicine.
In this review, we show how studies using genetically modified mice unable to be _S_-nitrosylated at one specific residue in hemoglobin, β-globin Cys93, have illuminated the role of red blood cells, hemoglobin, and _S_-nitrosylation in multiple aspects of cardiovascular physiology, including specifically hypoxic vasodilation in the microvasculature.
Classic View of Oxygen Delivery: A Physiological Paradox
Oxygen delivery is generally considered a relatively passive process, where oxygen-carrying hemoglobin (Hb) within RBCs transiting the tissue microcirculation encounters relative hypoxia and releases oxygen. Briefly, four subunits (two α-type and two β-type globin proteins) comprise Hb, each of which contains a heme that coordinates a single iron atom that can bind to a single O2 molecule (or other diatomic gas ligand, including CO or NO). Due to allosteric linkage among the four Hb subunits, Hb is able to bind and release oxygen in a highly cooperative manner: oxygen binding to each heme increases the affinity of unliganded hemes for oxygen, and likewise oxygen release promotes further release. Thus individual Hb molecules are essentially either fully unoccupied or bound to four oxygens. Structural studies by Perutz of oxygenated and deoxygenated Hb provided a molecular basis for allosteric cooperativity, whereby Hb transitions between two structural states: relaxed (R), which is bound to four oxygen molecules, and tense/taut (T) with unliganded hemes (38). T-state deoxy-Hb binds lung oxygen to transition to the fully liganded R-state, whereas, in hypoxic tissues, release of oxygen from R-state oxy-Hb promotes the T-state conformation that offloads the remaining oxygen.
This view of oxygen delivery, whereby four molecules of oxygen are off-loaded in tissues, is hard to reconcile with basic fundamentals of cardiovascular physiology. Blood is ~70% oxygenated in the right heart and ~100% in the left (25). Thus mammals utilize only one of four molecules of oxygen off-loaded in the respiratory cycle (the other three are picked up again in the venous circulation through counter-currents and the like). Furthermore, in adults, blood in the left heart that is only 50–70% oxygenated is, for all intents and purposes, unable to support life, yet plenty of oxygen is seemingly available. The solution to this paradox can be best understood in terms of the effect of oxygen utilization on microvascular blood flow, as discussed herein. In short, microvascular blood flow is the primary determinant of oxygen delivery, and it is impaired greatly by Hb oxygen saturation of <50% through loss of an allosterically controlled RBC-derived vasodilator.
Properties and Principles of Microvascular Blood Flow
Metabolically active tissues rely on increased tissue perfusion by blood to increase oxygen delivery. Classic blood flow experiments by Roy and Brown (46), Krogh (20, 21), and many others established general principles of the microcirculation. 1) Capillaries are often smaller than RBCs, so that both the capillary and RBCs must deform as RBCs pass through in single file, yet RBCs can still move rapidly through these too-small capillaries while in direct contact with the surrounding endothelial cell. 2) Only a small fraction of capillaries in a tissue are open to RBC or even plasma flow at one time. 3) The set of active capillaries changes constantly so that all parts of a tissue are perfused, albeit intermittently. 4) In an inactive tissue, oxygen must diffuse further to reach all regions of that tissue due to larger average distance between active capillaries. 5) Upon increased metabolic oxygen demand (as when a muscle becomes active), capillaries are recruited to the active state to reduce the distance between active capillaries (decreasing diffusion distance) and to increase total perfusion of the tissue by oxygenated blood to greatly enhance effective oxygen delivery. In Nobel Prize-winning experiments by Krogh a century ago, muscle activity was shown to increase the density of active capillaries by over 100-fold and correspondingly to decrease the distance oxygen must diffuse by over 20-fold (20).
One common explanation for these results is that smooth muscle cells at terminal arterioles act as sphincters to control blood flow through the individual capillaries they feed, and one prominent proposed mechanism is that metabolically active tissues release vasomediators such as adenosine that enter the bloodstream to increase blood flow (18). However, such vasomediators are released into the capillaries of oxygen-demanding tissues, downstream of arteriolar muscle regulating blood flow through that region of tissue, necessitating a mechanism to signal to upstream arterioles (or, conversely, to signal vasoconstrictive signals upstream in lungs to drive ventilation-perfusion matching). Electrical coupling of capillary endothelial cells via gap junctions has been proposed as one means by which such signals could reach upstream arterioles to alter capillary flow (49). However, such mechanisms do not account fully for the temporal and spatial aspects of tissue hypoxia-driven vasodilation to increase oxygen delivery.
Indeed, vasoactive responses of capillaries and arterioles can readily be demonstrated to be independent. For example, in the skin, arteriole plus capillary vasodilation leads to increased skin temperature with blushing color due to increased blood flow near the skin surface; arteriole vasodilation coupled with capillary vasoconstriction leads to increased skin temperature without reddening due to increased blood flow through shunts but not through capillaries at the skin surface (as in a hot environment); arteriole vasoconstriction coupled with capillary vasodilation (as in a cold environment) leads to blue-tinged cold skin due to increased deoxygenation of abundant (but essentially immobile) RBCs near the skin surface (21). These distinct responses of arterioles, shunt vessels, and capillaries must be mediated by independent mechanisms.
In landmark experiments assessing hypoxia-induced blood flow, Arthur Guyton demonstrated in the 1960s that blood flow through muscle increased proportionately as blood oxygen-saturation levels were decreased (45) (FIGURE 1). That is, the amount of oxygen in the blood, not the level of oxygen in the tissue, determines blood flow. This finding demonstrates that blood flow through tissues can be regulated by oxygenation at the level of the perfused tissue itself, not just by tissue metabolic activity. This he termed autoregulation of blood flow by oxygen lack (i.e., hypoxia) (45). This should not be confused with the similarly named autoregulation of blood flow by blood pressure, where organs (most notably brain, kidney, or gut) react to the pressure of their incoming blood supply to maintain blood flow through an organ, such as in hemorrhage or in shunting after a meal or during a fight-or-flight response (30). These mechanisms together regulate blood flow reaching a tissue or organ, but only hypoxic autoregulation of blood flow directly regulates the perfusion within the tissue to regulate oxygen delivery by increasing utilization of arterioles and capillaries, whereas autoregulation via blood pressure maintains overall flow through the tissue (adjusting the pressure gradient) without maximizing local perfusion and oxygen delivery (30). Conflation of these distinct responses has led to much misunderstanding and confusion (34, 53, 55). Guyton’s data, and subsequent work by others (reviewed in Ref. 51), point to the fact that blood flow autoregulation is linked to Hb oxygen saturation, not Po2, implying a central role for Hb allostery in control of blood flow (39). That is, Hb itself regulates microcirculatory blood flow.
FIGURE 1.
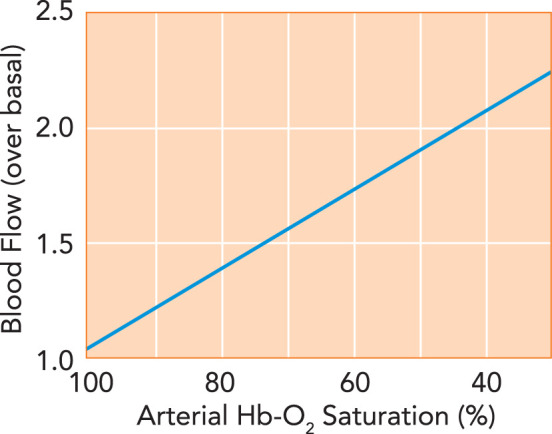
Hypoxic autoregulation of blood flow
In the classic demonstration of hypoxic autoregulation of blood flow (45), blood flow through muscle increases linearly as the oxygen saturation of the Hb entering the muscle decreases, maintaining relatively constant oxygen delivery.
βCYS93, _S_-NITROSYLATION, AND NITROSOTHIOL-MEDIATED VASODILATION
NO and S-Nitrosothiol
Nitric oxide bioactivity is a global regulator of the cardiovascular system. In the form of NO gas derived from endothelial cell nitric oxide synthase (eNOS), it regulates blood vessel tone to control blood pressure through smooth muscle relaxation mediated primarily by cGMP (31). NO gas acts locally, because the heme content of blood Hb is extraordinarily high, and heme in Hb inactivates NO (39, 51). Indeed, NO gas was initially identified as endothelial-derived relaxation factor (EDRF) in part due to its inactivation by Hb (15). Furthermore, capillaries lack smooth muscle (25), so this NO-cGMP pathway cannot operate there.
However, NO gas is not the only form of NO bioactivity that exhibits vasorelaxant or EDRF effect. When NO is redox-activated to NO+, it is able to react with free thiol (i.e., SH group in cysteine) to form an _S_-nitrosothiol (SNO). These free thiols could be in proteins such as Hb or in thiol-containing molecules such as glutathione or coenzyme A, or even free cysteine (13, 14, 54). Protein modification by SNO is a general NO- and redox-dependent protein posttranslational modification that is able to regulate the activity of proteins through multiple means, akin to other posttranslational modifications like phosphorylation (56). These include direct modification of active-site cysteine or altering protein allostery, turnover, localization, or partner binding. Once formed, SNO can be transferred from one thiol to another, allowing dynamic equilibria among SNO species (13, 14). Exogenous SNO molecules can act as donors to transfer SNO to target proteins (13, 14). Within cells, SNO formation, transfer, and degradation is enzyme mediated via SNO synthases that generate SNO from NO (to create a SNO-signaling messenger), SNO transnitrosylases that move SNO from one specific thiol to another to transmit signals, and SNO reductases that eliminate SNO from thiols to terminate signaling (56). Hb is a prototypic protein that is modified by _S_-nitrosylation. Hb exhibits activity both as a SNO synthase and as an allosterically regulated SNO transferase, creating SNO from NO and distributing SNO bioactivity around the body. Details of this activity and of how _S_-nitrosylation functions in general are beyond the scope of this discussion, but one principle helpful to understand is that SNOs typically elicit _S_-nitrosylation more readily than NO itself; interested readers are referred to comprehensive reviews (13, 39, 51, 56).
The vasorelaxant activity of SNO in general is well recognized and contributes broadly to bioactivity of “NO” in vivo (13, 14). Nevertheless, the precise targets for SNO-mediated vasodilation in the microcirculation remain unknown overall, and this is an area that needs additional work to understand detailed mechanisms. However, it is clear that SNO can stimulate generation of cGMP to induce vasorelaxation in a manner equivalent to NO gas (6), providing one such mechanism, and that SNOs are more effective vasodilators than NO in microcirculatory vessels (22).
Hemoglobin βCys93 as a Purveyor of SNO
One physiological system where SNO regulation has been shown to be essential is autoregulation of blood flow. As described below, Hb acts as an allosterically controlled purveyor of SNO, exchanging SNO in Hb onto other erythrocyte thiols under hypoxia that then transfer SNO to blood vessels to elicit hypoxic relaxation (7, 52). One way to think about this is in terms of a “SNO bucket brigade” between erythrocyte and blood vessel, where SNO only need transfer from hemoglobin onto other erythrocyte thiols for another SNO at the end of the line to elicit relaxation (39).
Hb was among the first proteins demonstrated to be modified endogenously by SNO, predominantly at a single conserved cysteine within the β-subunit (βCys93) (17). A cognate Cys residue is found in all mammalian β-type globins (β, γ, δ, ε) expressed throughout development and adulthood, but not in α-globin. Thus each Hb tetramer contains two Cys93-equivalent sites. βCys93 is one of only three residues that have remained invariant in mammalian Hb over the course of evolution (and the other two, His that coordinates heme iron and Phe that keeps the heme in place, are required for oxygen binding) (32, 33, 38).
βCys93 is the most reactive cysteine in Hb (16), and its reaction with NO is linked to the allosteric transition in Hb. Specifically, although modifications at βCys93 have little effect on oxygen affinity of Hb (16), oxygen binding to Hb (or, more specifically, R-state conformation) greatly increases reactivity of βCys93 (17, 28): βCys93 reactivity is high in oxygenated R-state Hb compared with deoxygenated T-state Hb (51, 52). Accordingly, SNO-βCys93 is stabilized in oxygenated Hb. By contrast, liberation of oxygen in tissues causes SNO to be transferred from deoxygenated T-state Hb to other erythrocytic thiols (7, 52), ultimately shuttling SNO out of RBCs (39). Specifically, SNO derived from Hb can be exported from RBCs through _S_-nitrosylation of RBC transmembrane proteins, including AE1/Band 3, leading to coating of the outer RBC membrane with SNO that can transfer to proteins on adjacent endothelial cells in microvessels (39), and potentially to low-molecular weight thiol SNOs such as SNO-glutathione or SNO-cysteine (6, 36, 51). In addition, some SNO-proteins derived from SNO-Hb may be exported from RBCs directly (19). Because SNOs in RBCs and SNOs in blood vessels are likely in equilibrium, deoxygenation of Hb will shift the equilibrium toward SNO-blood vessels, increasing vasodilation (see “SNO bucket brigade” mentioned above). This allosteric linkage of oxygen and SNO delivery is key to the function of SNO-Hb as a hypoxia-dependent vasodilator (FIGURE 2). The role of Hb as a SNO transferase and mechanisms for SNO export from RBCs leading to microvascular dilation are more fully described in Refs. 39, 51.
FIGURE 2.

Allosteric linkage of Hb conformation to O2 and SNO release
Left: due to cooperativity, Hb in the presence of air in the lung will bind fully to four oxygen molecules (oxygenated, R-state Hb). A fraction of oxygenated Hb will also bind and stabilize SNO at Hb βCys93. Right: on reaching hypoxic tissues, Hb will cooperatively give off all four oxygen molecules during transition to the deoxygenated T-state (improving tissue oxygenation). Liberation of oxygen will cause T-state Hb βCys93-SNO to liberate SNO, which can now transfer to other cellular thiols (R-S-H), leading to vasorelaxation (SNO-mediated vasodilation).
βCys93, a Solution to the Oxygen Delivery Paradox
The shift in Hb conformation between T (deoxy)- and R (oxy)-state takes place on binding the third (of four) oxygen molecules. SNO loading of Hb is therefore dependent on blood oxygenation, and blood that is less than 75% oxygenated will be deficient in vasodilator SNO-Hb (29). It has long been a mystery as to why blood that is less than 75% oxygenated threatens mammalian life, since plenty of oxygen is seemingly available. However, it should now be clear that hypoxic autoregulation of blood flow, which couples blood flow with tissue demand, will be impaired with SNO-Hb deficiency, creating tissue hypoxia (39, 51). That is, paradoxically, hypoxemia (low oxygen content in blood) will lead to tissue hypoxia through impairment of blood flow that is caused by loss of a RBC-derived vasodilator, not by the deficiency of oxygen itself.
The C93A Mouse Model and Caveats
To address the physiological significance of Hb βCys93 as a reactive site able to carry SNO, a mouse model that expresses in its RBCs human adult HbA in place of mouse HbA (60) was modified to mutant human β-globin Cys93Ala (C93A) (16). The human and mouse α-globin and β-globin loci are complex, with multiple coding regions in each locus that undergo sequential expression through development (2). These humanized mice contain human adult α-globin in place of mouse α-globin, and one of the two human fetal γ-globins (γA-globin) and human adult β-globin in place of the two mouse β-globins (βmaj-globin and βmin-globin) (60). The adult minor globins (human δ-globin and mouse βmin-globin) are both missing (60). These mice express human HbA, with minor expression of both human HbF (with human γΑ-globin) and mouse HbF (with mouse βH1-globin, the fetal γ-globin equivalent).
As with all animal models, there are caveats involved in interpreting physiological data in C93A mice and in extrapolating this to humans. First, all β-type globin isoforms contain a residue cognate to Cys93 (including fetal γ-globin) that is able to be _S_-nitrosylated (42). Thus these humanized mice are expected to develop relatively normally, since they do not rely on β-globin itself until birth when they transition to HbA. Second, although human HbA makes up the majority of Hb in C93 RBCs, a significant amount of fetal HbF (containing γ-globin rather than adult β-globin) continues to be expressed throughout adult life (43). Thus, even in adult C93A mice, residual Cys93 function remains in HbF: ~1% of Hb in the C93A mouse (8) or ~100 μM Cys93 within an adult C93A RBC (i.e., a large excess over NO). Third, Hb contains additional Cys residues that can carry SNO (61), albeit lacking the allosteric oxygen-dependent reactivity of βCys93, and RBCs also contain other proteins and low-molecular weight thiols (e.g., glutathione) that can carry SNO (6, 16), so C93A does not eliminate SNO carriage or release by Hb/RBCs (see below). Fourth, mouse and human RBCs differ substantially [e.g., mouse RBCs are far smaller than human (11), mouse RBCs exhibit higher ATP-mediated vasodilation activity than human (16), and human Hb does not bind to mouse RBC membrane AE1 proteins as tightly as it does human (required for SNO export) (4); also mouse and human RBCs express completely different amounts of met Hb and SNO-Hb when treated with NO donors], so care must be taken when comparing experiments performed using human RBCs, mouse RBCs, and these humanized mouse RBCs.
Physiological Evidence for βCys93 Function in the Cardiovascular System
Multiple studies since have identified specific aspects of cardiovascular function, including hypoxic vasodilation, that are indeed critically impaired in this mouse model (8, 61, 62) (FIGURE 3). Altogether, these studies support the hypothesis that Hb βCys93 is an important regulator of the cardiovascular system through its carriage and dissemination of SNO.
FIGURE 3.
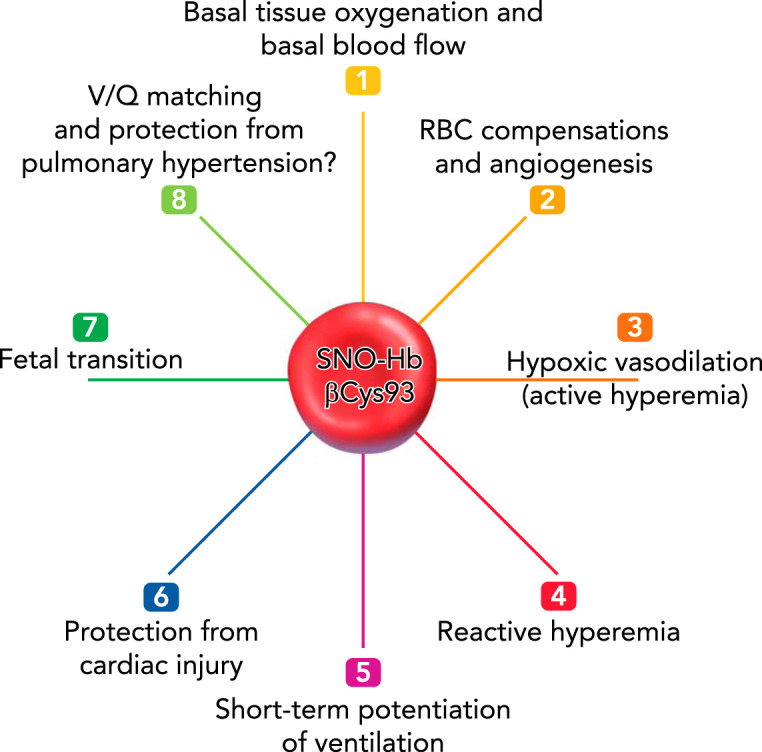
Cardiovascular physiology regulated by βCys93
Mice lacking βCys93 have altered physiological responses due to the inability to carry and deliver SNO from Hb βCys93. These include reduced basal tissue oxygenation and blood flow (1), RBC adaptation to loss of function (2), reduced hypoxic vasodilation (active hyperemia) (3), reduced reactive hyperemia (4), absent short-term potentiation of ventilation (5), reduced resilience to cardiac injury (ischemia- and pressure overload-induced) (6), and reduced fetal transition to free-living adulthood (7). Hypothesized effects also include altered ventilation-perfusion (V/Q) matching in the lung and reduced resilience to hypoxia-driven pulmonary hypertension (8).
Reduced Basal Tissue Oxygenation and Blood Flow
Direct measurement of tissue oxygenation and perfusion, and indirect assessments of tissue oxygen sufficiency indicated that C93A mice exhibit chronic tissue hypoxia and reduced perfusion while breathing room air. Specifically, blood perfusion of skeletal muscle at baseline (room air, resting muscle) in C93A mice is decreased by 20% compared with C93 controls, accompanied by a 30% decrease in muscle Po2 (61). Similarly, electrocardiographic traces revealed that C93A mice displayed significantly reduced T-wave amplitude compared with C93 controls (61), a sign of myocardial ischemia and reduced coronary blood flow (5). (Note: Cardiac function at baseline remains entirely normal.) We are unaware of another point mutation in Hb or another protein that causes myocardial ischemia at baseline. Thus C93A mice live with chronic underperfusion and hypoxia, reflecting the invariant role of βCys93.
Adaptation to Loss of βCys93 Function
Indirect physiological evidence of the importance of Hb βCys93 in RBC function is that loss of βCys93 results in dramatic compensatory changes to RBCs and blood circulation (see Reduced Resilience to Cardiac Injury and Heart Failure below) that help mitigate against loss of SNO function (FIGURE 4). If βCys93 had no functional role in RBC SNO functions, as some have contended (16, 27, 35, 57), there would be no driving force for such compensations, nor would Cys93 be invariant in mammals. As expected from studies indicating the βCys93 is the most reactive cysteine in Hb (28), Hb from C93A mice contained far less reactive cysteine than C93 controls (16). However, total RBC SNO levels appeared unchanged in C93A mice compared with C93 controls, reflecting a dramatic shift in SNO distribution from the normal >10 kDa fraction (containing Hb) into a low molecular mass (<10 kDa) fraction (16); the identity of this low molecular weight SNO species remains to be determined. Furthermore, βC93A-Hb carried SNO at secondary sites on both α-globin and β-globin (61) [human α-globin has only one Cys residue (Cys104), and human β-globin has one additional residue (Cys112)], and in fetal Hb (which retains γCys93). Thus C93A RBCs undergo dramatic compensation to maintain SNO levels by enlisting additional sites on Hb itself as well as utilizing fetal Hb and low molecular weight thiol(s) as SNO carriers. Given this degree of compensation, it is not surprising that some studies of C93A mice or RBCs (16, 27, 57) have concluded that these appear relatively normal in assays that assessed primarily non-specific SNO donation rather than specific hypoxia-driven, allosterically coupled SNO release, as exploited physiologically (41, 55). Angiogenic effects of βCys93 mutation, indicative of hypoxia, are discussed in detail below.
FIGURE 4.

_S_-nitrosothiol compensation by humanized βCys93Ala red blood cells
Left: in normal RBCs and humanized Hb RBCs, SNO is carried on Hb primarily on βCys93, with minor contributions by the other Cys residues on Hb, αCys104, and βCys112. Additionally, other low molecular weight thiols and proteins, including fetal Hb γCys93, can carry SNO within RBCs (LMW-SNO; γCys93-SNO). Right: in C93A RBCs, the βCys93Ala residue (Ala93) is unable to carry SNO, but the other Hb cysteine residues show elevated SNO levels, and fetal Hb retains γCys93-SNO, which provides essential residual function (since Cys93 is invariant and βC93A mice show reduced survival). Furthermore, LMW-SNO levels are also increased. Although total RBC SNO level is essentially unchanged by C93A mutation, the distribution within RBCs and on Hb is greatly altered. Most importantly, these other Hb Cys residues carrying SNO and the LMW-SNO species do not show allosteric coupling of SNO release to hypoxia and Hb R-to-T-state transition, but instead will release SNO independent of oxygen level.
Microcirculatory Blood Flow and Tissue Oxygenation Under Hypoxia (Active Hyperemia)
If SNO-Hb βCys93 regulates blood flow to counteract tissue oxygen deficiency, C93A mice should exhibit reduced blood flow under tissue hypoxia. Blood flow through gastrocnemius muscle was measured as global hypoxia was induced by progressively reducing the amount of oxygen in inspired air, from the normal 21% down to 5% (61). Although control C93 mice exhibited stable to increased flow down to 10% inspired O2, C93A mice were unable to sustain muscle blood flow in reduced oxygen (61). Analysis of the rate of change in blood flow during the transitions to the next lower O2 level showed that flow rate remained stable in control mice, until finally dropping during the transition to 5% O2 (likely due to redistribution to critical tissues), whereas the C93A mice showed large reductions in flow rate on transition to 10% O2 (61). Direct measurement of oxygen within muscle during decreases in inspired O2 showed drops in muscle oxygen levels in both control and C93A mice, but the C93A mice were less able to maintain oxygenation than C93 mice at all inspired O2 levels (61). Reduced muscle blood flow and muscle oxygenation were also observed in mutant mice breathing room air (61). Reducing inspired oxygen also led to elevated hyperacute T, ST area in C93A mice (61), an electrocardiographic sign of markedly reduced cardiac oxygenation.
Thus βCys93 and Hb itself play essential roles in maintaining tissue oxygenation through blood perfusion under hypoxia, indicating a role for Hb-derived SNO in this process. Although the initial report on C93A mice concluded that SNO-Hb was not essential for RBC-mediated hypoxic vasodilation (16), this conclusion was based on misunderstanding of physiological principles: experiments did not measure hypoxic vasodilation, blood flow, or tissue oxygenation, but rather assessed blood pressure and run time to exhaustion. Neither blood pressure nor time to exhaustion is linked to hypoxic autoregulation of blood flow or predicted to be allosterically regulated by Hb or SNO-Hb. The same authors have subsequently contended that the decline in blood flow in C93A mice is caused by impaired cardiac function (57), but cardiac function was not at all impaired under basal conditions or in mice breathing 10% O2 (61). [And even if cardiac output was reduced in C93A mice, this is not how the heart works: cardiac output is only the cause of inadequate tissue blood flow in frank heart failure, when function is greatly compromised and pressure cannot be maintained (3).] The point is that impaired blood flow was the cause of (and not caused by) impaired heart function: at extreme hypoxia (5%) when heart function actually dropped, C93A mice could not compensate.
Reactive Hyperemia
Reactive hyperemia is the response of a tissue to temporary circulatory interruption by augmenting flow above normal for a time after flow is restored (e.g., the transient flushing after release in a limb “fallen asleep”). Reactive hyperemia is traditionally conceived as being mediated by stimulation of eNOS by shear stress when flow is resumed (48) or by the build-up in non-perfused tissue of metabolites that induce vasodilation (e.g., adenosine from depleted ATP) (12, 23), but hypoxia induces release of SNO from Hb in proportion to hypoxia (39), so that resumption of flow should induce release of SNO from fresh RBCs entering into hypoxic tissue to cause highly elevated blood perfusion until tissue oxygenation normalizes. Indeed, dramatic differences in hyperemic responses were seen in C93 versus C93A mice when limb blood flow was restored after 5 min of blockage; muscle hyperemia in the C93A mice was only half that in the C93 control mice (61). Thus βCys93 and Hb have a major, newly discovered role in reactive hyperemia.
Central Ventilatory Drive
Central ventilatory drive is regulated by detection of Pco2/pH of cerebral spinal fluid by chemoreceptors in the brain stem and secondarily by detection of Pco2/pH of blood by chemoreceptors in the carotid body to regulate the endogenous pacemaker function of the brain stem (25). So it is notable that a role for hypoxia sensing via SNO-Hb has been revealed by studies of C93A mice (8). When the oxygen content of inspired air is reduced, the rate and depth of breathing is increased for a brief time; this inability to sustain high ventilation is termed ventilatory roll-off or hypoxic ventilatory suppression. When the oxygen content of inspired air is increased to normal after such an episode of hypoxia, the rate and depth of breathing similarly increases for a short period; this temporary overcompensation of breathing is called short-term potentiation of ventilation (9). When C93A mice breathing 10% O2 were returned to 21% O2, they completely failed to exhibit short-term potentiation of ventilation, although ventilatory roll-off appeared normal (8). This reveals a physiological role for SNO-Hb βCys93 in oxygen/hypoxia sensing to regulate ventilatory drive. Although the molecular target of this response remains unknown, the mechanistic basis of ventilation impairment likely reflects allosteric uncoupling of blood flow from tissue oxygenation in mutant mice. Future studies are also needed to discern the role of CO2/pH versus Po2, since each parameter can induce the T state in Hb that liberates SNO.
Reduced Resilience to Cardiac Injury and Heart Failure
C93A mouse hearts show electrophysiological evidence for chronic hypoxia even in the absence of hypoxic stress or vessel blockage (61). In an animal model of acute myocardial infarction (ischemia-reperfusion injury), C93A mice were less resilient than C93 controls (62). Although only 15% of control C93 mice failed to survive this procedure, 32% of C93A mice failed to survive (62). C93A mice much more frequently exhibited collateral arterial vessels that supplied the left ventricle than did C93 mice (60% vs 15%) (62). Mice of either genotype with such collaterals survived ischemia-reperfusion, whereas mice lacking such collaterals preferentially died [and at a far higher rate for C93A (~60%) compared with C93 (~20%)] (62). Indexes of cardiac damage were all worse in C93A mice lacking collaterals compared with C93A mice with collaterals or to C93 mice (62). Measurement of cardiac function by echocardiography in surviving mice indicated that C93A mice were worse than C93 controls (62). That C93A mice much more frequently develop coronary collateral arteries indicates compensation for poor cardiac muscle perfusion and oxygenation during development and chronic hypoxia throughout life.
C93A mice were also subjected to transverse-aortic constriction (TAC) pressure overload model of heart failure (62). During TAC, C93A mice died more frequently, with only 5% of control mice dying compared with over 60% of C93A mice (which mainly died within the first 5 days) (62). Short-term (2 days) and long-term (28 days) TAC resulted in worse cardiac performance in C93A mice compared with controls, measured by echocardiographic parameters including ejection fraction, fractional shortening, end-systolic diameter, and cardiac output (62). After 28 days, C93A mice also exhibited increased cardiac hypertrophy and lung edema compared with C93 controls (62).
Overall, SNO-Hb βCys93 provides cardioprotective activity that is lost in C93A mice (62). This was observed when mice were subjected to both ischemic injury and pressure overload-induced heart failure. This protective function is attributed to SNO-mediated hypoxic vasodilation of the coronary microcirculation to support cardiomyocyte health, contractility, and remodeling (62), confirming the importance of microvascular blood flow in the origins of ischemic heart disease and heart failure. Compensatory development of coronary collaterals can partially offset cardiac resilience deficits in C93A mice.
Fetal Transition
Mice (and humans) utilize distinct Hb subunits during embryonic and fetal development (HbE before the appearance of RBCs, and HbF when erythropoiesis is primarily in the developing liver) compared with free-living individuals breathing air and making RBCs in bone marrow (47). Transition from fetal to adult Hb does not occur in humans at birth or upon exposure of lungs to air but shifts over the first year of life (1, 58), and both SNO-HbF and SNO-HbA can be detected simultaneously in blood (42). Failure to transition from HbF to HbA is generally an adaptive response to Hb diseases such as sickle cell disease or β-thalassemia, and is not overtly pathological (43). However, during transition from HbF to HbA in the C93A mouse, levels of Cys93 and SNO-Cys93 will decline precipitously, which might lead to chronic hypoxia and death of the animals. Indeed, C93A pups exhibit reduced viability (61), suggesting that uncompensated loss of Hb βCys93 leads to pups dying, although it is unclear whether this is an in utero developmental problem or a perinatal lethality problem.
Future Directions and Achieving Consensus
Despite the large body of accumulated physiological evidence outlined above and elsewhere (39, 51) for the importance of SNO-Hb βCys93 function as a purveyor of vasodilatory activity and oxygen delivery in response to hypoxic conditions, some controversy persists about this function. Nonetheless, areas of agreement and consensus continue to expand. Broadly accepted concepts introduced by this laboratory that had once been disputed (39, 57) now include: SNOs retain vasodilatory activity in the presence of excess Hb, whereas NO does not (17, 51); RBCs and Hb carry SNO (17, 29); RBCs and Hb can release SNO, particularly under hypoxic conditions (17, 29, 51); native RBCs (replete in SNO) and SNO-Hb itself can be vasodilatory (17, 29, 36, 52); and SNO-βCys93-Hb and RBCs can act as allosterically controlled vasodilators, exchanging SNO with other thiols (7, 29, 39, 52) as a function of Hb deoxygenation. The core of remaining contention has centered on physiology: whether Hb βCys93 in particular accounts for hypoxic vasodilation subserving tissue oxygenation (autoregulation of blood flow) (57); but this would seem resolved based on results described here. Remaining disagreements stem in large part from reliance on non-physiological systems and misunderstanding of physiology, especially where non-SNO or non-βCys93 mechanisms are conflated with SNO-βCys93-mediated ones.
Others have proposed alternate mechanisms for the phenomenon of RBC-mediated vasodilation that we originally discovered (17, 36), including reduction of nitrite by Hb to form bioactive NO (10, 26) or the release of vasoactive substances such as ATP that signal via endothelial NO synthase (44). Note that these mechanisms are not incompatible with βCys93-mediated vasodilation, and each could generate NO bioactivity and vasorelaxation under distinct circumstances. However, hypoxic vasodilation by nitrite is a pharmacological response (physiological context is still lacking for nitrite), and the ability of nitrite at endogenous blood levels to effect hypoxia-mediated microvascular perfusion is unclear [lowering nitrite does not impair RBC vasodilation or autoregulation (39)]. Similarly, ATP released from RBCs does not play a discernable role in autoregulation (39). A broader discussion of this topic has been recently published (39). Our experimental focus (and this review) is strictly on hypoxic autoregulation responses, with a strong emphasis on physiological responses in vivo over more-contrived in vitro measures of function.
C93A mice are likely to have additional hypoxia-driven phenotypes to divulge, consistent with the essential role of βCys93-SNO in the integrated response to hypoxia. These may include ventilation-perfusion matching, where blood seeking oxygenation is diverted away from sections of lung that lack full access to fresh oxygenated air, and an elevated susceptibility to pulmonary hypertension in response to hypoxia, reflecting impaired vasodilation by RBCs (FIGURE 3). Clinical correlates of SNO-Hb βCys93 function include loss of SNO from RBCs at high altitude (and possibly in altitude sicknesses), in sickle cell disease, in pulmonary hypertension, in peripheral arterial disease/critical limb ischemia, and in stored RBCs used for transfusion (39). Clinical trials are ongoing to assess the effectiveness of RBC renitrosylation to increase oxygen delivery (39). Other diseases likely to involve SNO-Hb βCys93 function include myocardial infarction, stroke, anemias, lung diseases, and kidney injury. Lessons learned from βCys93 physiology extend understanding of microcirculatory blood flow—the last remaining frontier in cardiovascular physiology—and may promise new treatments for millions of patients (39).
Acknowledgments
The authors are supported by National Institutes of Health Grants P01 HL-075443, P01 HL-126900, R01 HL-128192, and R01 DK-119506, and American Heart Association-Allen Brain Health Initiative Grant 19PABHI34580006 (to J.S.S.).
J.S.S. has patents related to re-nitrosylation of hemoglobin and to the use of _S_-nitrosothiol donors as therapeutics. CWRU and UHCMC are aware of these conflicts, and management plans are in place.
R.T.P. prepared figures; R.T.P. drafted manuscript; R.T.P. and J.S.S. approved final version of manuscript; J.S.S. edited and revised manuscript.
References
- 1.Bard H. Postnatal fetal and adult hemoglobin synthesis in early preterm newborn infants. J Clin Invest 52: 1789–1795, 1973. doi: 10.1172/JCI107360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bulger M, van Doorninck JH, Saitoh N, Telling A, Farrell C, Bender MA, Felsenfeld G, Axel R, Groudine M. Conservation of sequence and structure flanking the mouse and human beta-globin loci: the beta-globin genes are embedded within an array of odorant receptor genes. Proc Natl Acad Sci USA 96: 5129–5134, 1999. doi: 10.1073/pnas.96.9.5129. A correction for this article is available at . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Camm AJ, Lüscher TF, Maurer G, Serruys PW (Editors). ESC CardioMed. Oxford, UK: Oxford University Press, 2018. [DOI] [PubMed] [Google Scholar]
- 4.Chen Q, Balazs TC, Nagel RL, Hirsch RE. Human and mouse hemoglobin association with the transgenic mouse erythrocyte membrane. FEBS Lett 580: 4485–4490, 2006. doi: 10.1016/j.febslet.2006.07.021. [DOI] [PubMed] [Google Scholar]
- 5.de Luna AB, Zareba W, Fiol M, Nikus K, Birnbaum Y, Baranowski R, Goldwasser D, Kligfield P, Piotrowicz R, Breithardt G, Wellens H. Negative T wave in ischemic heart disease: a consensus article. Ann Noninvasive Electrocardiol 19: 426–441, 2014. doi: 10.1111/anec.12193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Diesen DL, Hess DT, Stamler JS. Hypoxic vasodilation by red blood cells: evidence for an_S_-nitrosothiol-based signal. Circ Res 103: 545–553, 2008. doi: 10.1161/CIRCRESAHA.108.176867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Doctor A, Platt R, Sheram ML, Eischeid A, McMahon T, Maxey T, Doherty J, Axelrod M, Kline J, Gurka M, Gow A, Gaston B. Hemoglobin conformation couples erythrocyte_S_-nitrosothiol content to O2 gradients. Proc Natl Acad Sci USA 102: 5709–5714, 2005. doi: 10.1073/pnas.0407490102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gaston B, May WJ, Sullivan S, Yemen S, Marozkina NV, Palmer LA, Bates JN, Lewis SJ. Essential role of hemoglobin beta-93-cysteine in posthypoxia facilitation of breathing in conscious mice. J Appl Physiol (1985) 116: 1290–1299, 2014. doi: 10.1152/japplphysiol.01050.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gauda ES, Martin RJ. Control of breathing. In: Avery’s Diseases of the Newborn, edited by Gleason C, Devaskar S. Philadelphia: Saunders, 2012, p. 584–597. [Google Scholar]
- 10.Gladwin MT, Grubina R, Doyle MP. The new chemical biology of nitrite reactions with hemoglobin: R-state catalysis, oxidative denitrosylation, and nitrite reductase/anhydrase. Acc Chem Res 42: 157–167, 2009. doi: 10.1021/ar800089j. [DOI] [PubMed] [Google Scholar]
- 11.Gregory TR. Nucleotypic effects without nuclei: genome size and erythrocyte size in mammals. Genome 43: 895–901, 2000. doi: 10.1139/g00-069. [DOI] [PubMed] [Google Scholar]
- 12.Hellsten Y, Nyberg M, Jensen LG, Mortensen SP. Vasodilator interactions in skeletal muscle blood flow regulation. J Physiol 590: 6297–6305, 2012. doi: 10.1113/jphysiol.2012.240762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol 6: 150–166, 2005. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- 14.Hess DT, Matsumoto A, Nudelman R, Stamler JS. S-nitrosylation: spectrum and specificity. Nat Cell Biol 3: E46–E49, 2001. doi: 10.1038/35055152. [DOI] [PubMed] [Google Scholar]
- 15.Ignarro LJ, Buga GM, Wood KS, Byrns RE, Chaudhuri G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc Natl Acad Sci USA 84: 9265–9269, 1987. doi: 10.1073/pnas.84.24.9265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Isbell TS, Sun CW, Wu LC, Teng X, Vitturi DA, Branch BG, Kevil CG, Peng N, Wyss JM, Ambalavanan N, Schwiebert L, Ren J, Pawlik KM, Renfrow MB, Patel RP, Townes TM. SNO-hemoglobin is not essential for red blood cell-dependent hypoxic vasodilation. Nat Med 14: 773–777, 2008. doi: 10.1038/nm1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jia L, Bonaventura C, Bonaventura J, Stamler JS. S-nitrosohaemoglobin: a dynamic activity of blood involved in vascular control. Nature 380: 221–226, 1996. doi: 10.1038/380221a0. [DOI] [PubMed] [Google Scholar]
- 18.Johnson PC. Autoregulation of blood flow. Circ Res 59: 483–495, 1986. doi: 10.1161/01.RES.59.5.483. [DOI] [PubMed] [Google Scholar]
- 19.Kallakunta VM, Slama-Schwok A, Mutus B. Protein disulfide isomerase may facilitate the efflux of nitrite derived_S_-nitrosothiols from red blood cells. Redox Biol 1: 373–380, 2013. doi: 10.1016/j.redox.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krogh A. The supply of oxygen to the tissues and the regulation of the capillary circulation. J Physiol 52: 457–474, 1919. doi: 10.1113/jphysiol.1919.sp001844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krogh A. The Anatomy and Physiology of Capillaries. New Haven, CT: Yale University Press, 1922. [Google Scholar]
- 22.Kurz MA, Lamping KG, Bates JN, Eastham CL, Marcus ML, Harrison DG. Mechanisms responsible for the heterogeneous coronary microvascular response to nitroglycerin. Circ Res 68: 847–855, 1991. doi: 10.1161/01.RES.68.3.847. [DOI] [PubMed] [Google Scholar]
- 23.Layland J, Carrick D, Lee M, Oldroyd K, Berry C. Adenosine: physiology, pharmacology, and clinical applications. JACC Cardiovasc Interv 7: 581–591, 2014. doi: 10.1016/j.jcin.2014.02.009. [DOI] [PubMed] [Google Scholar]
- 24.Leach RM, Treacher DF. Oxygen transport-2. Tissue hypoxia. BMJ 317: 1370–1373, 1998. doi: 10.1136/bmj.317.7169.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Levitsky MG. Pulmonary Physiology. New York: McGraw-Hill Education, 2018. [Google Scholar]
- 26.Liu C, Wajih N, Liu X, Basu S, Janes J, Marvel M, Keggi C, Helms CC, Lee AN, Belanger AM, Diz DI, Laurienti PJ, Caudell DL, Wang J, Gladwin MT, Kim-Shapiro DB. Mechanisms of human erythrocytic bioactivation of nitrite. J Biol Chem 290: 1281–1294, 2015. doi: 10.1074/jbc.M114.609222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu Y, Sun CW, Honavar J, Townes T, Patel RP. Role of the b93cys, ATP and adenosine in red cell dependent hypoxic vasorelaxation. Int J Physiol Pathophysiol Pharmacol 5: 21–31, 2013. [PMC free article] [PubMed] [Google Scholar]
- 28.Luchsinger BP, Rich EN, Gow AJ, Williams EM, Stamler JS, Singel DJ. Routes to S-nitroso-hemoglobin formation with heme redox and preferential reactivity in the beta subunits. Proc Natl Acad Sci USA 100: 461–466, 2003. doi: 10.1073/pnas.0233287100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McMahon TJ, Moon RE, Luschinger BP, Carraway MS, Stone AE, Stolp BW, Gow AJ, Pawloski JR, Watke P, Singel DJ, Piantadosi CA, Stamler JS. Nitric oxide in the human respiratory cycle. Nat Med 8: 711–717, 2002. doi: 10.1038/nm718. [DOI] [PubMed] [Google Scholar]
- 30.Meng L, Wang Y, Zhang L, McDonagh DL. Heterogeneity and variability in pressure autoregulation of organ blood flow: lessons learned over 100+ years. Crit Care Med 47: 436–448, 2019. doi: 10.1097/CCM.0000000000003569. [DOI] [PubMed] [Google Scholar]
- 31.Murad F. Cyclic guanosine monophosphate as a mediator of vasodilation. J Clin Invest 78: 1–5, 1986. doi: 10.1172/JCI112536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nagai K, Perutz MF, Poyart C. Oxygen binding properties of human mutant hemoglobins synthesized in Escherichia coli. Proc Natl Acad Sci USA 82: 7252–7255, 1985. doi: 10.1073/pnas.82.21.7252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Olson JS, Mathews AJ, Rohlfs RJ, Springer BA, Egeberg KD, Sligar SG, Tame J, Renaud JP, Nagai K. The role of the distal histidine in myoglobin and haemoglobin. Nature 336: 265–266, 1988. doi: 10.1038/336265a0. [DOI] [PubMed] [Google Scholar]
- 34.Palmer LA, Doctor A, Gaston B. SNO-hemoglobin and hypoxic vasodilation. Nat Med 14: 1009, 2008. doi: 10.1038/nm1008-1009a. [DOI] [PubMed] [Google Scholar]
- 35.Patel R, Townes T. Reply to SNO-hemoglobin and hypoxic vasodilation. Nat Med 14: 1009–1010, 2008. doi: 10.1038/nm1008-1009b. [DOI] [PubMed] [Google Scholar]
- 36.Pawloski JR, Hess DT, Stamler JS. Export by red blood cells of nitric oxide bioactivity. Nature 409: 622–626, 2001. doi: 10.1038/35054560. [DOI] [PubMed] [Google Scholar]
- 37.Perdrizet GA. Chronic diseases as barriers to oxygen delivery: a unifying hypothesis of tissue reoxygenation therapy. In: Oxygen Transport to Tissue XXXIX, edited by Halpern HJ, LaManna JC, Harrison DK, Epel B. Cham, Switzerland: Springer International Publishing, 2017, p. 15–20. [DOI] [PubMed] [Google Scholar]
- 38.Perutz MF, Kendrew JC, Watson HC. Structure and function of haemoglobin: II. Some relations between polypeptide chain configuration and amino acid sequence. J Mol Biol 13: 669–678, 1965. doi: 10.1016/S0022-2836(65)80134-6. [DOI] [Google Scholar]
- 39.Premont RT, Reynolds JD, Zhang R, Stamler JS. Role of nitric oxide carried by hemoglobin in cardiovascular physiology: developments on a three-gas respiratory cycle. Circ Res 126: 129–158, 2020. doi: 10.1161/CIRCRESAHA.119.315626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rees PJ, Dudley F. Oxygen therapy in chronic lung disease. BMJ 317: 871–874, 1998. doi: 10.1136/bmj.317.7162.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reynolds JD, Premont RT, Stamler JS. Letter by Reynolds et al. regarding article, “Hemoglobin β93 cysteine is not required for export of nitric oxide bioactivity from the red blood cell”. Circulation 140: e758–e759, 2019. doi: 10.1161/CIRCULATIONAHA.119.041389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Riccio DA, Malowitz JR, Cotten CM, Murtha AP, McMahon TJ. S-Nitrosylated fetal hemoglobin in neonatal human blood. Biochem Biophys Res Commun 473: 1084–1089, 2016. doi: 10.1016/j.bbrc.2016.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rochette J, Craig JE, Thein SL. Fetal hemoglobin levels in adults. Blood Rev 8: 213–224, 1994. doi: 10.1016/0268-960X(94)90109-0. [DOI] [PubMed] [Google Scholar]
- 44.Rosenblum WI. Endothelium-dependent responses in the microcirculation observed in vivo. Acta Physiol (Oxf) 224: e13111, 2018. doi: 10.1111/apha.13111. [DOI] [PubMed] [Google Scholar]
- 45.Ross JM, Fairchild HM, Weldy J, Guyton AC. Autoregulation of blood flow by oxygen lack. Am J Physiol 202: 21–24, 1962. doi: 10.1152/ajplegacy.1962.202.1.21. [DOI] [PubMed] [Google Scholar]
- 46.Roy CS, Brown JG. The blood-pressure and its variations in the arterioles, capillaries and smaller veins. J Physiol 2: 323–446.1, 1880. doi: 10.1113/jphysiol.1880.sp000068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sankaran VG, Orkin SH. The switch from fetal to adult hemoglobin. Cold Spring Harb Perspect Med 3: a011643, 2013. doi: 10.1101/cshperspect.a011643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schuler D, Sansone R, Freudenberger T, Rodriguez-Mateos A, Weber G, Momma TY, Goy C, Altschmied J, Haendeler J, Fischer JW, Kelm M, Heiss C. Measurement of endothelium-dependent vasodilation in mice–brief report. Arterioscler Thromb Vasc Biol 34: 2651–2657, 2014. doi: 10.1161/ATVBAHA.114.304699. [DOI] [PubMed] [Google Scholar]
- 49.Segal SS. Integration and modulation of intercellular signaling underlying blood flow control. J Vasc Res 52: 136–157, 2015. doi: 10.1159/000439112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shah DM, Gottlieb ME, Rahm RL, Stratton HH, Barie PS, Paloski WH, Newell JC. Failure of red blood cell transfusion to increase oxygen transport or mixed venous PO2 in injured patients. J Trauma 22: 741–746, 1982. doi: 10.1097/00005373-198209000-00004. [DOI] [PubMed] [Google Scholar]
- 51.Singel DJ, Stamler JS. Chemical physiology of blood flow regulation by red blood cells: the role of nitric oxide and S-nitrosohemoglobin. Annu Rev Physiol 67: 99–145, 2005. doi: 10.1146/annurev.physiol.67.060603.090918. [DOI] [PubMed] [Google Scholar]
- 52.Stamler JS, Jia L, Eu JP, McMahon TJ, Demchenko IT, Bonaventura J, Gernert K, Piantadosi CA. Blood flow regulation by S-nitrosohemoglobin in the physiological oxygen gradient. Science 276: 2034–2037, 1997. doi: 10.1126/science.276.5321.2034. [DOI] [PubMed] [Google Scholar]
- 53.Stamler JS, Reynolds JD, Hess DT. Letter by Stamler et al. regarding article, “Nitrite and S-nitrosohemoglobin exchange across the human cerebral and femoral circulation: relationship to basal and exercise blood flow responses to hypoxia”. Circulation 135: e1135–e1136, 2017. doi: 10.1161/CIRCULATIONAHA.117.027071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stamler JS, Simon DI, Osborne JA, Mullins ME, Jaraki O, Michel T, Singel DJ, Loscalzo J. S-nitrosylation of proteins with nitric oxide: synthesis and characterization of biologically active compounds. Proc Natl Acad Sci USA 89: 444–448, 1992. doi: 10.1073/pnas.89.1.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stamler JS, Singel DJ, Piantadosi CA. SNO-hemoglobin and hypoxic vasodilation. Nat Med 14: 1008–1009, 2008. doi: 10.1038/nm1008-1008. [DOI] [PubMed] [Google Scholar]
- 56.Stomberski CT, Hess DT, Stamler JS. Protein S-nitrosylation: determinants of specificity and enzymatic regulation of S-nitrosothiol-based signaling. Antioxid Redox Signal 30: 1331–1351, 2019. doi: 10.1089/ars.2017.7403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sun CW, Yang J, Kleschyov AL, Zhuge Z, Carlström M, Pernow J, Wajih N, Isbell TS, Oh JY, Cabrales P, Tsai AG, Townes T, Kim-Shapiro DB, Patel RP, Lundberg JO. Hemoglobin β93 cysteine is not required for export of nitric oxide bioactivity from the red blood cell. Circulation 139: 2654–2663, 2019. doi: 10.1161/CIRCULATIONAHA.118.039284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Terrenato L, Bertilaccio C, Spinelli P, Colombo B. The switch from haemoglobin F to A: the time course of qualitative and quantitative variations of haemoglobins after birth. Br J Haematol 47: 31–41, 1981. doi: 10.1111/j.1365-2141.1981.tb02759.x. [DOI] [PubMed] [Google Scholar]
- 59.Tsai AG, Hofmann A, Cabrales P, Intaglietta M. Perfusion vs. oxygen delivery in transfusion with “fresh” and “old” red blood cells: the experimental evidence. Transfus Apheresis Sci 43: 69–78, 2010. doi: 10.1016/j.transci.2010.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wu LC, Sun CW, Ryan TM, Pawlik KM, Ren J, Townes TM. Correction of sickle cell disease by homologous recombination in embryonic stem cells. Blood 108: 1183–1188, 2006. doi: 10.1182/blood-2006-02-004812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang R, Hess DT, Qian Z, Hausladen A, Fonseca F, Chaube R, Reynolds JD, Stamler JS. Hemoglobin βCys93 is essential for cardiovascular function and integrated response to hypoxia. Proc Natl Acad Sci USA 112: 6425–6430, 2015. doi: 10.1073/pnas.1502285112. A correction for this article is available at https://doi.org/10.1073/pnas.1506997112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang R, Hess DT, Reynolds JD, Stamler JS. Hemoglobin S-nitrosylation plays an essential role in cardioprotection. J Clin Invest 126: 4654–4658, 2016. doi: 10.1172/JCI90425. [DOI] [PMC free article] [PubMed] [Google Scholar]