The manifold roles of protein S-nitrosylation in the life of insulin (original) (raw)
. Author manuscript; available in PMC: 2022 Aug 1.
Published in final edited form as: Nat Rev Endocrinol. 2021 Nov 17;18(2):111–128. doi: 10.1038/s41574-021-00583-1
Abstract
Insulin, released by pancreatic islet β cells in response to elevated levels of glucose in the blood, is a critical regulator of metabolism. Insulin triggers the uptake of glucose and fatty acids into liver, adipose tissue and muscle, and promotes the storage of these nutrients in the form of glycogen and lipids. Dysregulation of insulin synthesis, secretion, transport, degradation or signal transduction all cause failure to take up and store nutrients, resulting in diabetes and metabolic dysfunction. Here we make the case that insulin signaling is intimately coupled to protein S-nitrosylation, in which nitric oxide groups are conjugated to cysteine thiols to form S-nitrosothiols (SNOs), within effectors of insulin action. We discuss the role of S-nitrosylation in the life cycle of insulin, from its synthesis and secretion in pancreatic β cells, to its signaling and degradation in target tissues. Finally, we consider how aberrant S-nitrosylation contributes to metabolic disease, including the roles of human genetic mutations and cellular events that alter S-nitrosylation of insulin-regulating proteins. Given the growing influence of S-nitrosylation in cellular metabolism, the field of metabolic signaling may benefit from renewed focus on S-nitrosylation in type 2 diabetes and insulin-related disorders.
Introduction
Insulin serves as an endocrine hormone that facilitates the uptake of glucose from blood and its conversion into energy storage macromolecules (glycogen and lipids) in liver, adipose tissue and skeletal muscle[1]. Insulin is a heterodimer consisting of an A-chain and B-chain linked together by two intermolecular disulfide bonds, and acts via the cell surface insulin receptor (INSR), a receptor tyrosine kinase. Pancreatic islets in mammals contain β-cells that are the primary source of circulating insulin[2, 3]. Synthesis and release of insulin in islets, transport to target tissue, signal transduction, physiological effects and degradation of insulin are all highly regulated processes that are essential for proper insulin action. Dysregulation of any step may cause diabetes mellitus (DM). Given the high (and growing) incidence of diabetes and other metabolic diseases globally, the insulin pathway is among the most intensively studied in medicine and biology[4].
Nitric oxide (NO) is a gaseous signaling molecule that has been implicated in the life cycle of insulin, from synthesis and secretion in the β-cells to physiological effects in skeletal muscle, adipose tissue and liver. NO is synthesized from L-arginine by a family of NO synthases (NOS 1–3). NO in β-cells, adipocytes, hepatocytes and skeletal myocytes is produced in large part by NOS3 (eNOS) and NOS1 (nNOS), and is generally considered to be required for normal insulin action. By contrast, NO produced by NOS2 (iNOS) is deleterious to insulin signaling, contributing to DM[5, 6]. For better or worse, NO acts either indirectly through activation of the soluble guanylyl cyclase (sGC)–cGMP–protein kinase G pathway or through direct modification of protein thiols by S-nitrosylation. The consequences of the NO–cGMP pathway on insulin signaling have been reviewed[7, 8]. Here we focus on the much larger but less-appreciated role of protein S-nitrosylation.
The oxidative modification of a Cys thiol by NO forms an S-nitrosothiol (SNO). Inasmuch as most proteins have free thiols and most cells generate NO, the post-translational modification of proteins by SNO provides a general means to control cellular function. Over 20,000 SNO sites have been identified in the published literature (ref), and predictions hold that ~70% of the proteome is modified in this way (ref). SNO groups in proteins have been shown to regulate proteins in all functional classes, across phylogeny, and this regulation includes altering activity, interactions, stability and subcellular localization[9]. Accumulating new evidence indicates that protein S-nitrosylation is essentially enzymatic both under physiological conditions and in disease states (Fig.1)[10, 11]. Enzymatic S-nitrosylation requires the coordinated operation of three classes of enzymes: NO synthases to generate NO, SNO synthases that convert NO into SNO, and transnitrosylases to transfer NO to target proteins. SNO synthases and transnitrosylases can be further grouped under the heading of protein S-nitrosylases i.e., enzymes that generate SNO-proteins (Fig.1A)[10]. By contrast, denitrosylases remove NO groups from SNO-proteins and come in two main classes: low molecular weight (LMW) thiol-cofactor dependent denitrosylases and thioredoxin-related denitrosylases. LMW denitrosylases act on LMW SNOs formed from cellular thiols, including SNO-glutathione and SNO-Co-enzyme A, and thereby effectively regulate an equilibrium between LMW-SNOs and protein SNOs[11]. By contrast, the thioredoxin-related proteins act directly on SNO-proteins. Denitrosyases by definition interact directly with their protein or LMW substrates (Fig.1B). Overall, cellular SNO levels reflect a balance of de novo SNO production and conjugation to proteins, and SNO removal.
Figure 1. Working model of protein S-nitrosylation and denitrosylation.
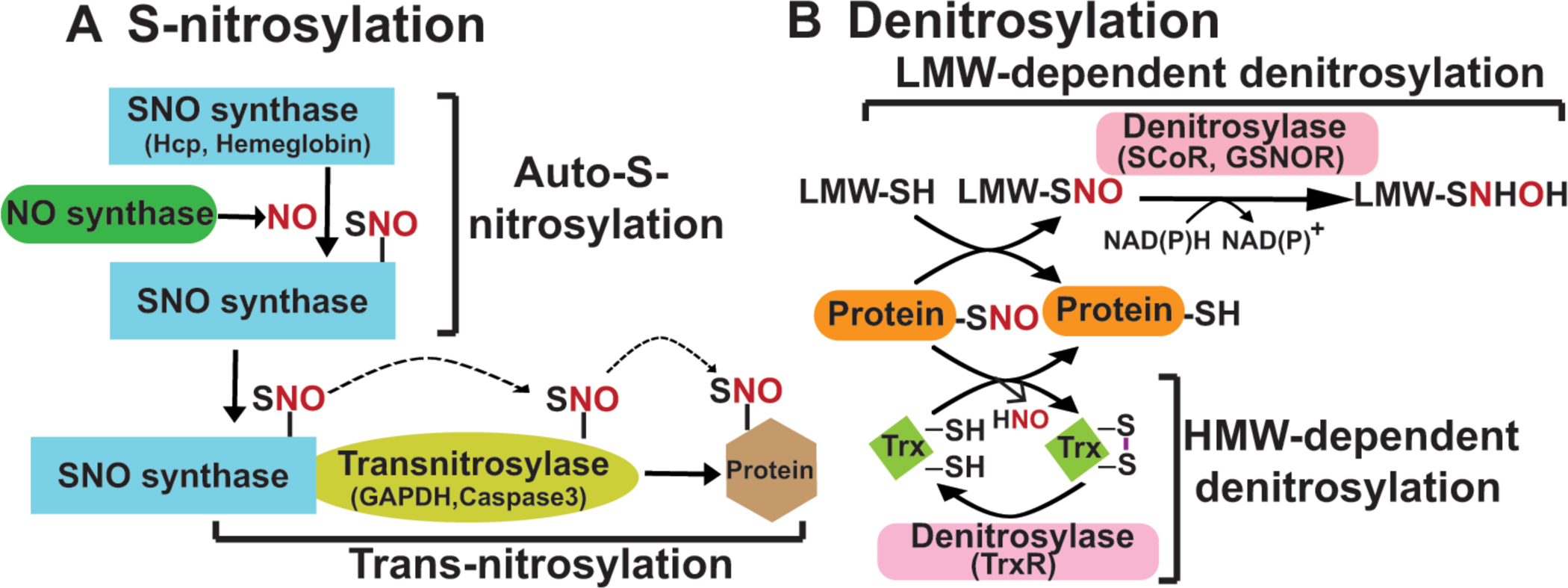
(A) The complete machinery for enzymatic S-nitrosylation includes three classes of enzymes: NO synthases, SNO synthases and transnitrosylases. NO synthases catalyze the production of NO from L-arginine and include three members in mammals: eNOS, iNOS and nNOS. SNO synthases convert NO to NO+ through one-electron oxidation, and conjugate it to thiol to generate protein S-nitrosothiol. There are two identified SNO synthases: Hybrid cluster protein (HCP) in bacteria and hemoglobin in mammals (others are anticipated). Transnitrosylases propagate SNO-based signaling through transferring the SNO group from themselves to Cys residues on substrates. Multiple transnitrosylases have been identified. GAPDH and S100A9 are well-known examples. (B) There are two main classes of S-nitrosothiol denitrosylases: low molecular weight (LMW) thiol-co-factor dependent denitrosylases and thioredoxin (Trx)-related denitrosylases. Low molecular weight thiol (LMW)-SNOs, including SNO-glutathione (GSNO) and SNO-coenzyme A (SNO-CoA), are in equilibrium with SNO-proteins through transnitrosylation. GSNO reductase (GSNOR) and SNO-CoA reductase (SCoR) are enzymes that denitrosylate SNO-substrates by reducing GSNO and SNO-CoA, respectively. Trx/TrxR (thioredoxin reductase) systems denitrosylate SNO-proteins directly using vicinal thiols.
Accumulating evidence suggests that the insulin signaling pathway is regulated at multiple steps by S-nitrosylation. This review summarizes this new understanding, and suggests how S-nitrosylation is integrated with more commonly-known modes of regulation in both health and disease.
S-nitrosylation in insulin secretion
Production and secretion of insulin are largely independent. Prepro-insulin synthesis and insulin maturation by proteolytic processing in β-cells occur inside the ER/Golgi. Mature insulin is stored in insulin-secretion granules (ISG) within β-cells, awaiting signals for secretion. The glucose-stimulated insulin secretion (GSIS) pathway increases intracellular calcium to trigger ISG fusion and insulin secretion in response to elevated blood glucose. GSIS has two phases, a transient initial phase followed by a sustained phase, each mediated by distinct exocytotic machinery[12–14]. In islets, NO is mainly generated by nNOS, and can act as either a mediator or negative feedback inhibitor of GSIS[7, 15]. The complex physiologic roles of NO on GSIS can be explained by S-nitrosylation of different target proteins over time, leading to activation vs inhibition of insulin release. Protein S-nitrosylation is known to be involved in both the GSIS pathway and exocytosis of ISG, as summarized in Fig.2.
Figure 2. Role of S-nitrosylation in insulin secretion from β-cells.
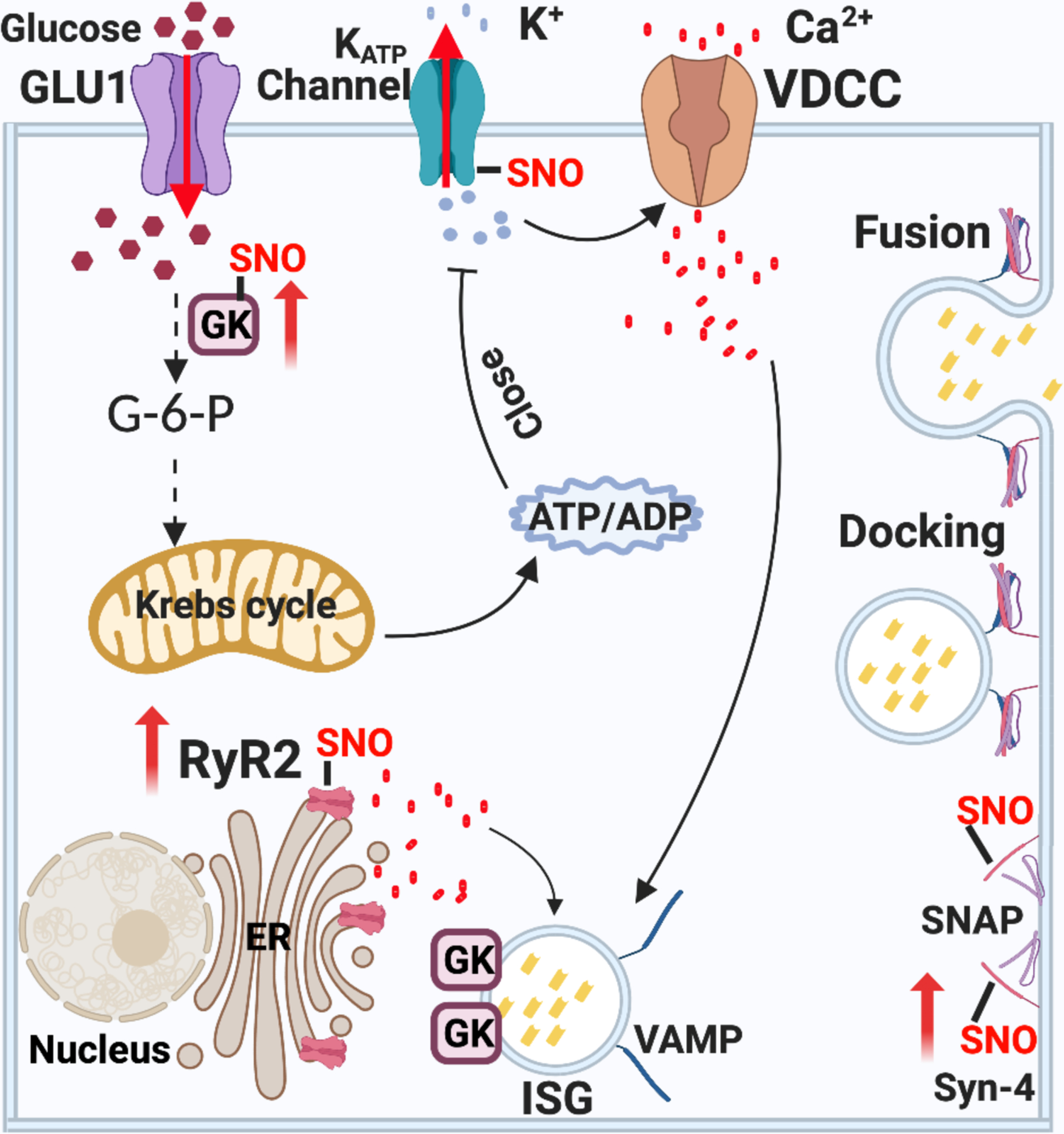
Mechanism of GSIS. Extracellular glucose enters β-cells and is phosphorylated by GCK, trapping phosphorylated glucose inside the cell and initiating its metabolism. The generation of ATP by glycolysis, the Krebs cycle and the respiratory chain in mitochondria lead to a rise in ATP, and a high ATP:ADP ratio closes ATP-sensitive KATP channels. The resulting membrane depolarization triggers the opening of voltage-dependent T-type calcium channels (VDCC) and influx of calcium ions (Ca2+). The release of intracellular ER Ca2+ is amplified by the rise of cytosolic calcium via RyR2 and IP3 receptors. Cytosolic Ca2+ induces the fusion of pre-docked insulin secretory granules (ISG) with the plasma membrane in the initial phase of insulin secretion. The rise of Ca2+ also induces new ISG to move to the plasma membrane for subsequent docking, fusion and insulin release. Interactions of VAMP on the ISG with Syn-4 and SNAP on the plasma membrane generates the active SNARE complex, mediating docking and fusion of ISG to release insulin. S-nitrosylation regulates insulin release through at least three steps: 1) S-nitrosylation of GCK increases cytosolic GCK activity by promoting the dissociation of GCK from ISG. Mutations in GCK that prevent S-nitrosylation thus cause diabetes. 2) S-nitrosylation of Syn-4 results in conformation changes of SNARE proteins and facilitates docking and fusion of ISG with the plasma membrane. 3) S-nitrosylation of RyR2 increases Ca2+ release. Hypo-nitrosylation of RyR2 causes Ca2+ leak, while hyper-nitrosylation leads to ER stress.
GSIS pathway
When the concentration of glucose in blood is increased after a meal, extracellular glucose enters β-cells via glucose transporter 2. Inside the cell, glucokinase (GCK) catalyzes the transfer of phosphate from ATP to glucose to generate glucose 6-phosphate (G6P), the rate-limiting step in glucose metabolism. G6P is rapidly metabolized through anaerobic glycolysis and mitochondrial oxidation, which each produce ATP. A high ratio of ATP/ADP closes ATP-sensitive potassium channels (KATP) in the plasma membrane of β-cells, eliciting membrane depolarization to induce the opening of L-type Ca2+ channels that allow calcium to enter the β-cells (Fig.2). This elevated cytosolic calcium stimulates exocytosis of ISG[16]. S-nitrosylation is generally stimulatory on the GSIS pathway.
S-nitrosylation of glucokinase (GCK)
GCK activity is critical to GSIS. GCK exerts nearly total control over glucose metabolism in β-cells, so small changes in GCK activity can alter glucose metabolism to change the ATP/ADP ratio and affect GSIS[13, 17, 18]. Sequestration of GCK to a biological compartment is key to its regulation. Specifically, reversible association with ISG determines the amount of active cytoplasmic GCK in β-cells. In the absence of glucose, GCK is associated with ISG and is inactive. After glucose elevation, GCK is released into the cytoplasm and undergoes a conformational change to initiate glucose metabolism[19, 20].
S-nitrosylation of GCK at Cysteine-371 promotes the disassociation of GCK from ISG and facilitates the activating conformation transition, thereby stimulating GCK enzymatic activity and increasing insulin secretion[21–23]. Mutation of Cysteine-371 to serine, which blocks S-nitrosylation of GCK, causes GCK to remain bound to ISG and inhibits insulin secretion[21]. GCK also stably interacts with neuronal NOS (nNOS), and inhibition of nNOS activity blocks both dissociation of GCK from ISG and the activating conformational change stimulated by insulin[21, 23]. Naturally-occurring, diabetes-associated GCK point mutations V367M and R369P, proximal to the S-nitrosylated Cysteine-371 site, block GCK activation by preventing S-nitrosylation of GCK at Cysteine-371. Mechanistically, V367M mutation inhibits GCK association with nNOS, thus preventing S-nitrosylation of GCK. This indicates that S-nitrosylation of GCK is a physiologically important control point for regulating glucose-stimulated insulin release[22].
S-nitrosylation of GCK is also involved in glucagon-like peptide 1 (GLP-1)-stimulated insulin secretion. GLP-1 is an incretin that is released after a meal due to sensing of glucose in the gut lumen, to promote insulin release[24, 25]. GLP-1 treatment increases GCK activity in TC3 insulinoma cells and in mouse islets, which is mediated by NOS-induced S-nitrosylation of GCK. V367M mutation in GCK, which blocks S-nitrosylation of GCK at Cysteine-371, diminished the effect of GLP-1 on exocytosis of insulin in TC3 cells by 40%[26]. It is likely that depolarization-induced entry of calcium mediated by GLP-1 serves to activate nNOS-catalyzed S-nitrosylation. An acute increase in GCK activity by S-nitrosylation-mediated disassociation of GCK from ISG provides a robust mechanism for postprandial regulation of insulin secretion (accounts for at least ~40% of the insulin secretory response)[26], whereas release of GCK to the cytoplasm could also facilitate signal termination by making GCK accessible to proteosomal degradation[23, 27, 28]. Therefore, physiological S-nitrosylation of GCK should be transient and dynamic. Investigating how physiological stimuli (such as glucose) induce acute and transient S-nitrosylation of GCK will verify this idea.
S-nitrosylation of the type 2 ryanodine receptor/Ca2+ release channel (RyR2)
A rise in cytosolic calcium in β-cells is essential for exocytosis of ISG. Although increases in calcium depend mainly on voltage-activated Ca2+ influx from the extracellular space (via L-type channels), release of calcium from the endoplasmic reticulum (ER) amplifies Ca2+-induced insulin secretion (Fig.2)[29]. RyR2 is a Ca2+-induced Ca2+ release channel on the ER (and in muscle, the sarcoplasmic reticulum) membrane and mutation of RyR2 is associated with reduced GSIS and impaired glucose clearance, involving ER stress and mitochondrial dysfunction[30]. S-nitrosylation of RyR2 promotes calcium release to stimulate insulin secretion[30–32], while hyper-nitrosylation of RyR2 in β-cells leads to ER stress, impairing GSIS and blood glucose clearance[30]. Both S-nitrosylation and oxidation of RyR2 are dramatically increased in β-cells of patients with T2DM or in ob/ob mice, leading to ER stress[30]. Similar effects in the heart result in calcium leak and arrhythmia[33].
S-nitrosylation of ATP-sensitive potassium channels (KATP)
KATP channels are inhibited by ATP and activated by ADP, providing a link between cellular energy state and electrical excitability in β-cells. ATP-sensitive KATP channels are an octameric complex of four pore-forming Kir6.2 subunits and four regulatory sulfonylurea receptor (SUR) subunits[34]. SUR1, one of these regulatory subunits, facilitates KATP channel opening via its binding of ADP[35], and is the target of antidiabetic sulfonylurea drugs that promote insulin release[36]. NO activates KATP channels in mammalian sensory neurons via S-nitrosylation of SUR1 at Cysteine-717, located at the ATP-binding site. S-nitrosylation of SUR1 inhibits ATP binding, thus preventing channel inactivation. Mutagenesis of SUR1 Cys717 to Alanine limits NO-induced KATP channel activation, confirming that S-nitrosylation of SUR1 is crucial to KATP channel regulation[37]. Dysregulation of KATP channels by SUR1 results in metabolic disease, with loss-of-function SUR1 mutations causing congenital hyperinsulinemia, but gain-of-function SUR1 mutations leading to neonatal diabetes[38]. Further work is needed to confirm such a role for S-nitrosylation of SUR1 in diabetes.
Exocytosis of ISG in islet β-cells
Secretion of ISG is mediated by exocytotic machinery consisting of the soluble N-ethylmaleimide-sensitive factor-attachment protein receptor (SNARE) complex and Munc18 (mammalian uncoordinated-18, five members in humans). The SNARE core complex includes three types of proteins: syntaxin (Syntaxin1A, Syntaxin4 or Syntaxin3), synaptosomal-associated protein (SNAP23 or SNAP25), and vesicle-associated membrane protein (VAMP2 or VAMP8) (Fig.2)[39]. Syntaxin resides in the plasma membrane through its transmembrane domain. SNAP lacks transmembrane domains, but anchors to the plasma membrane via palmitoylated cysteine residues. VAMP contains a C-terminal transmembrane domain that is embedded in the ISG membrane. Upon glucose stimulation, phosphorylated Munc18 binds to the N-terminal of syntaxin and causes a conformation change that promotes and stabilizes assembly of SNARE core complexes[40, 41].
S-nitrosylation of Syntaxins
Syntaxin 1A (Syn-1A) is required for the fusion of pre-docked ISG with the plasma membrane in the transient initial phase of insulin release. In static state, Munc18-1 interacts with and stabilizes Syn-1A in its inactive closed conformation, inhibiting Syn-1A from interacting with the other SNAREs[42]. Following glucose stimulation, phosphorylated Munc18-1 activates Syn-1A, thereby promoting its association with VAMP2 and SNAP25 to assemble the SNARE exocytotic machinery and mediate exocytosis of predocked ISG[41]. S-nitrosylation of Syn-1A at Cys145 affects the structure of the syntaxin linker region and H3 domain, thereby disrupting its interaction with Munc18-1 and promoting transition of Syn-1A from closed to open conformation, which facilitates interaction with cognate SNAREs to execute membrane fusion. Furthermore, overexpression of the Syn-1A-Cys145W mutant (mimicking nitroso-Cys145), but not Syn-1A-Cys145S mutation (resistant to S-nitrosylation), prevented its interaction with Munc18-1 in adrenal chromaffin cells, and increased quantal size of individual exocytotic fusion events[43]. Thus S-nitrosylation of Cys145 can act as molecular switch to disrupt Munc18-1 binding to Syn-1A, increasing the efficiency of the exocytotic machinery. It will be interesting to investigate if this regulatory mechanism is physiologically important in insulin secretion.
Syn-4, another member of Syntaxin family, associates with Munc18-3 to form a distinct exocytotic machine. Munc18-3/Syn-4 mediates exocytosis of both pre-docked and newly-formed ISG[39]. S-nitrosylation of Syntaxin 4 plays both physiological and pathological roles in exocytosis of ISG in the β-cell line MIN6 and in human islets[44]. In response to acute glucose stimulation, Syn-4 is S-nitrosylated at Cysteine-141, which represents a transition from closed conformation to the open conformation that is more accessible to incoming VAMP2-bound insulin secretory granules (ISG), thereby facilitating ISG docking and fusion with the plasma membrane (PM) to permit insulin release from β-cells. The Syn-4 C141S mutant fails to exhibit glucose-stimulated VAMP2 binding or insulin release, confirming the role of S-nitrosylation of Syn-4 in insulin secretion. However, upon acute treatment with pro-inflammatory cytokines, aberrant S-nitrosylation of Syn-4 occurs and results in both inappropriate Syn-4 activation and insulin release in the absence of glucose stimulus[44]. Obesity, accounting for 80–85% of the risk of developing T2DM, causes insulin resistance mainly because of chronic inflammation[45]; thus investigating if aberrant S-nitrosylation of Syn-4 in obese animals affects insulin release may shed light on whether obesity-induced inflammation can promote diabetes through dysregulated exocytosis of ISG.
Another open question is what NOS is required for S-nitrosylation of Syn-1A and Syn-4. In general, NO derived from nNOS is required for normal insulin secretion in the pancreas[46]. By contrast, NO derived from inducible nitric oxide synthase (iNOS) is associated with β-cell dysfunction and death, leading to type 1 diabetes[47]. It follows, therefore, that nNOS may subserve S-nitrosylation of Syn-1A and Syn-4 in normal insulin secretion, whereas iNOS-mediated hyper-S-nitrosylation of Syn-1A and Syn-4 may cause excessive insulin secretion and ultimately damage β-cells.
S-nitrosylation of N-ethylmaleimide-sensitive factor (NSF)
NSF, a homohexameric ATPase, hydrolyzes ATP to disassemble SNARE complexes, allowing their recycling for further rounds of membrane fusion[48]. S-nitrosylation of NSF mediated by eNOS inhibits NSF disassembly activity, and has been reported to block endothelial cell exocytosis of Weibel-Palade bodies and human platelet exocytosis of dense granules, lysosomal granules and alpha-granules[49, 50]. NSF is modified at three cysteines residues (Cys 11, 91 and 264). S-nitrosylation of Cys-11 inhibits NSF interaction with the SNARE complex, while S-nitrosylation of Cys-91 and Cys-264 permits NSF interaction with the SNARE complex but blocks NSF disassembly of SNARE complex[49]. Thioredoxin (TRX1) is the protein denitrosylase acting on S-nitrosylated NSF. The interaction between TRX1 and NSF is increased three hours after exposure to NO, and is associated with reduced S-nitrosylation of NSF and in restored exocytosis[51]. Although there are no direct studies to show that S-nitrosylation of NSF play a role in exocytosis of ISG, given the common features of exocytosis, it seems very likely that S-nitrosylation of NSF will regulate ISG release.
Summary.
NO acts mainly as a mediator of insulin secretion under physiological conditions and inhibitor under pathophysiology. S-nitrosylation of multiple target proteins, mediated by different stimuli and duration of NO exposure provide both robust regulation and complexity, exemplified in feedback inhibition. Protein S-nitrosylation regulates insulin release via both the GSIS pathway and the exocytotic machinery in β-cells (Fig.2). S-nitrosylation of GCK, RyR2, Syn-1A and Syn-4 (positive group) are mechanisms by which SNO acts to elevate insulin release. However, S-nitrosylation of SUR1 and NSF (negative group) may reduce insulin secretion, as does aberrant S-nitrosylation of RyR2 and Syn-4, explaining the inhibitory role of NO in insulin release. In the future, as specific S-nitrosylases are identified, target-directed S-nitrosylation may allow pharmacological treatment of diabetes.
S-nitrosylation in transport of insulin
Before insulin can act on a target tissue, it must cross the tight endothelial barrier of the capillary wall to access target cells such as adipocytes or skeletal myocytes (unlike liver, where fenestrae in sinusoidal endothelial cells allow direct blood access to hepatocytes). This trans-endothelial transport – including endocytosis, transcytosis and exocytosis – is one of the rate-limiting steps that determine insulin availability in target tissues[52]. NO stimulates insulin trans-endothelial transport in vascular endothelial cells, through a mechanism that involves protein S-nitrosylation, not activation of sGC. Lines of evidence are as follows: First, while pretreatment of endothelial cells with NOS inhibitor L-NAME blocked insulin uptake, treatment with the NO donor SNP reversed L-NAME inhibition of endothelial cells’ insulin uptake. Second, knockdown of thioredoxin-interacting protein (TXNIP), an inhibitor of the protein denitrosylase thioredoxin, decreased overall S-nitrosylation levels in endothelial cells and completely eliminated SNP-enhanced insulin uptake, indicating that S-nitrosylation plays an important role in insulin transport[53]. Third, Wang et al. determined that S-nitrosylation of protein tyrosine phosphatase-1B (PTP1B) is a major mediator of this NO-induced insulin uptake. PTP1B is a negative regulator of insulin signaling via its action to dephosphorylate activated INSR (insulin receptor) and IRS (insulin receptor substrate) (Fig.3). Insulin activation of INSR is a required first step for insulin to be transported across endothelial cells. Therefore, inhibiting PTP1B promotes insulin uptake[54]. S-nitrosylation of PTP1B decreases its phosphatase activity, enhancing phosphorylation of tyrosine residues on INSR and IRS, enhancing insulin action and facilitating insulin uptake and transport[53].
Figure 3. Facilatory role of S-nitrosylation in the AKT branch of insulin signal transduction.
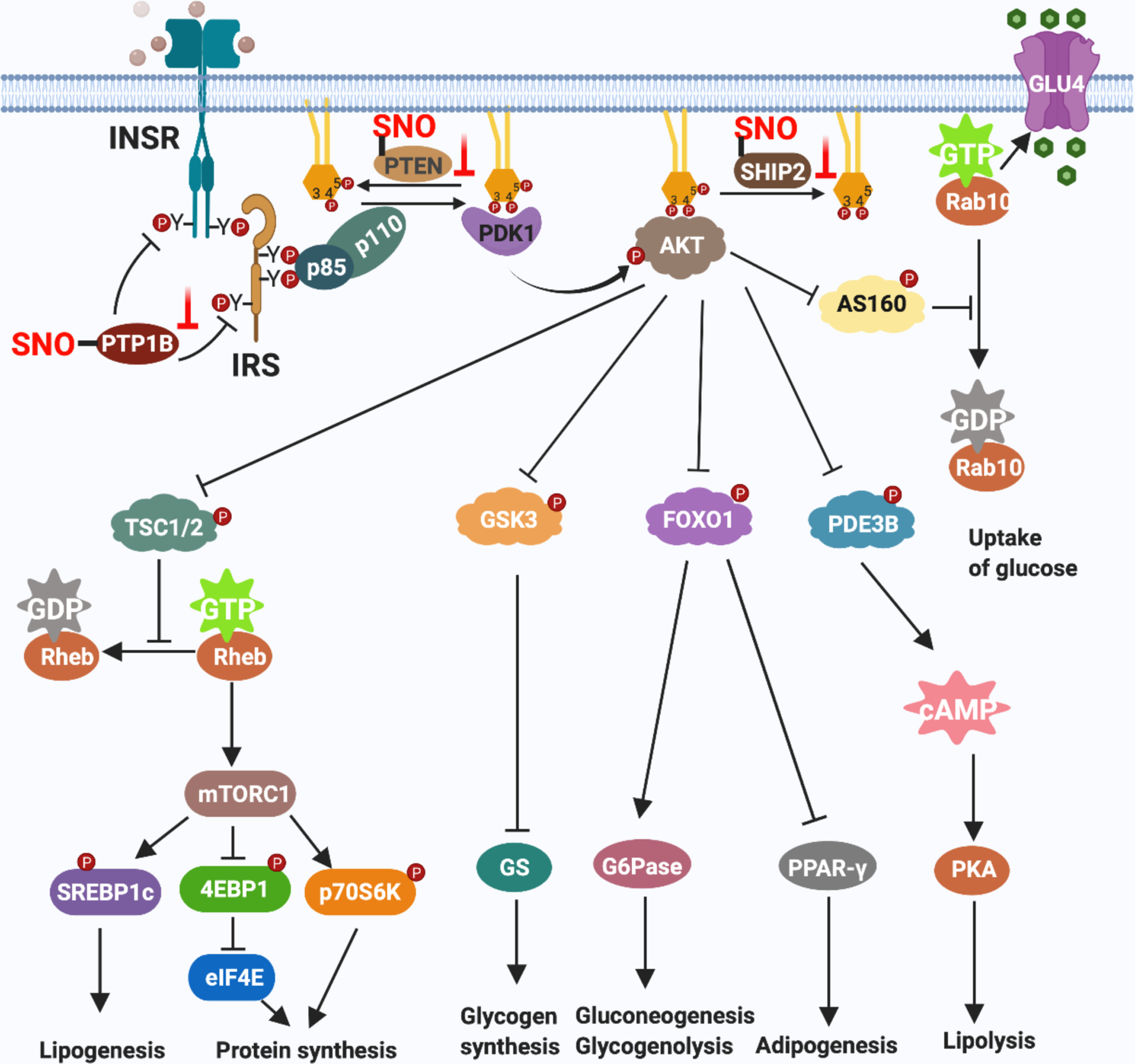
Binding of insulin to its receptor (INSR) triggers kinase activity that autophosphorylates tyrosine residues in INSR-β(pY) and phosphorylates tyrosine residues in IRS proteins. The SH2 domain of the p85 regulatory subunit of PI3K (bound to the p110 catalytic subunit of PI3K) binds to tyrosine-phosphorylated IRS to activate PI3K activity. PI3K catalyzes formation of phosphatidylinositol (3, 4, 5)-trisphosphate (PIP3) through adding a phosphate group to phosphatidylinositol (4, 5)-bisphosphate (PIP2). PIP3 binds to both PDK1 and AKT to activate these kinases. PDK1 phosphorylates AKT, thereby fully activating AKT. Activated AKT phosphorylates and inactivates TSC2, GSK3, FOXO, PDE3B and AS160 to control multiple aspects of insulin action, including the uptake of glucose, glycogen synthesis, lipogenesis, protein translation and cell proliferation. Protein S-nitrosylation elevates insulin responsiveness via modifying three phosphatases: 1) S-nitrosylation of PTP1B inhibits its phosphatase activity, prolonging activating-phosphorylation of both insulin receptor and IRS. 2) S-nitrosylation of PTEN inhibits dephosphorylation of the 3’ phosphate of the inositol ring in PIP3, leading to sustained PIP3 levels and AKT activity. 3) S-nitrosylation of SHIP2 inhibits dephosphorylation of the 5’ phosphate of the inositol ring in PIP3, also leading to sustained PIP3 levels.
S-nitrosylation of Caveolin-1 (Cav-1)
Caveolae are plasma membrane invaginations that play a key role in endocytosis and transcytosis of insulin uptake[55]. Cav-1 is the most abundant protein in caveolae, and it oligomerizes and associates with cholesterol-rich lipid rafts to form caveolae, so it is not surprising that Cav-1 is required for insulin uptake and trans-endothelial transport in vascular endothelial cells[56]. Mice lacking Cav-1 are prone to develop T2DM[57].
S-nitrosylation of Cav-1 has variable effects on caveolar activity, which may be dependent on the dose and duration of NO exposure. Transient NO production by eNOS promotes Cav-1-dependent endocytosis of both albumin and insulin in human primary umbilical vein endothelial cells[58]. S-nitrosylation of Cav-1 can account for both increases in insulin uptake and increased matrix metalloproteinase (MMP)-2 and MMP-9 secretion in endothelial cells[59], providing a major mechanism through which NO stimulates insulin (and other) trans-endothelial transport. However, prolonged stimulation of endothelial cells with TNF-α (which induces iNOS expression) or with a long-lived NO donor has the opposite effect. In this case, S-nitrosylation of Cav-1 at Cys-156 within its C-terminus, leads to disassociation of Src from Cav-1 and activation of Src; Src phosphorylates Cav-1 at tyrosine-14 in the N-terminal domain, destabilizing membrane-associated Cav-1 oligomers and promoting Cav-1 degradation[60]. Expression of wild-type Cav-1, but not C156S mutant Cav-1, restores the formation of caveolae in Cav-1-deficient endothelial cells to restore endocytosis. Whether different Cys sites in Cav-1 are involved in SNO mediated transcytosis of insulin and in opposing insulin trafficking (Cav-1 degradation) remains to be determined.
S-nitrosylation in insulin degradation
Insulin degradation is a regulated process to inactivate circulating insulin, and occurs in all insulin-sensitive cells. Degradation is as important as insulin secretion to overall insulin signaling, and abnormalities in insulin degradation are integral to T2DM[61]. After insulin triggers its downstream pathway, receptor-bound insulin is internalized into endosomes where degradation is initiated by insulin-degrading enzyme (IDE)[62–64]. IDE’s role in insulin degradation is confirmed by genetic studies. The Goto-Kakizaki (GK) rat is a polygenic non-obese Wistar substrain that rapidly develops adult T2DM. Two amino acid substitutions (H18R and A890V) in the IDE gene are found in the GK rat, which has a 31% reduction in insulin-degrading activity[65].
IDE is S-nitrosylated after oxidative burst or S-nitrosoglutathione (GSNO) treatment in BV-2 microglial cells, leading to reduced activity. Mechanistically, S-nitrosylation increases IDE oligomerization and decreases IDE thermostability[66], thereby inhibiting both insulin- and β-amyloid peptide degrading activities of IDE in vitro, (of note, insulin-degrading activity appeared more sensitive to NO inhibition than β-amyloid peptide-degrading activity)[67]. Surprisingly, IDE contains five SNO sites with different functions. S-nitrosylation of Cys-110, Cys-178 and Cys-819 regulates catalytic activity, while S-nitrosylation of Cys-789 and Cys-966 increases oligomerization. Cys-110 is near the zinc-binding catalytic center and is normally buried. S-nitrosylation of Cys-819 allows Cys-110 to be nitrosylated, leading to complete inactivation of IDE. Cys-789 is spatially adjacent to Cys-966, and their S-nitrosylation together triggers oligomerization and inhibition of IDE[68]. Aberrant S-nitrosylation of IDE in the brains of patients with T2DM plus Alzheimer’s disease, inhibits both insulin and Aβ degradation, leading to additional increases in glucose and β-amyloid peptide levels, which are thought responsible for dysfunctional synaptic plasticity and synapse loss in cortical and hippocampal neurons[66]. These results provide mechanistic insight into aberrant S-nitrosylation of IDE causing hippocampal-dependent learning and memory abnormalities observed in such models of T2DM with Alzheimer’s disease, and suggest that this regulation of IDE will also have widespread effects on insulin homeostasis.
S-nitrosylation in insulin responsiveness
Insulin signal transduction
The diverse cellular actions of insulin are initiated by its binding to INSR on the cell surface. INSR is a heterotetramer composed of two extracellular insulin-binding α subunits, covalently linked by disulfide bonds to two transmembrane β subunits. Binding of insulin to the α subunit outside the cell induces a conformational change that activates tyrosine kinase activity of the β subunits inside the cell, leading to autophosphorylation of several tyrosine residues in the β subunits and to phosphorylation of insulin receptor substrate (IRS) proteins. There are two major branches of the insulin signaling pathway mediated by distinct phosphorylation sites: the ERK and AKT branches[69, 70]. The ERK branch mainly mediates the cell proliferation effects of insulin, through activating the rat sarcoma-mitogen-activated protein kinase/ERK (Ras-MAPK/ERK) signaling pathway. The AKT branch (shown in detail in Fig.3) mediates both metabolic and proliferation effects of insulin[66].
S-nitrosylation in insulin signaling
NO derived from eNOS plays a pivotal role in the physiological effects of insulin. Genetic deletion of eNOS leads to insulin resistance and peripheral organ-specific abnormalities in mice, suggesting that insulin responsiveness requires eNOS-derived NO, that this NO acts within endothelial cells, and that eNOS deficiency is a critical determinant of hyperglycemia-induced organ-specific complications in diabetes[71–73]. Treatment of mice with pharmacological inhibitors of eNOS confirms NO’s constitutive role in insulin signaling[74]. One main function of eNOS-derived NO is to promote insulin responsiveness via S-nitrosylation of negative regulators of insulin signaling in endothelial cells (Fig.3). Several of these regulators are phosphatases and active-site S-nitrosylation in uniformly inhibitory.
S-nitrosylation of protein tyrosine phosphatase 1B (PTP1B)
PTP1B is a negative regulator of insulin signal transduction. PTPB1 interacts with and dephosphorylates activated INSR and IRS. Mice lacking PTP1B are more sensitive to insulin than wild-type mice, and have improved glycemic control and resistance to diet-induced obesity (Fig.3)[75]. S-nitrosylation of PTP1B is a major regulatory point in NO-mediated enhancement of insulin responsiveness. Endogenous PTP1B is S-nitrosylated at cysteine 215 in both MS-1 cells and eNOS-transfected COS-7 cells after stimulation by insulin. S-nitrosylated PTP1B co-localizes with activated INSR at the cell periphery in an eNOS-dependent manner. Ectopic expression of mutant PTP1B C215S (mimicking S-nitrosylation) restores insulin responsiveness in eNOS-deficient cells, which are otherwise insensitive to insulin stimulation[76]. This study highlights a positive effect of NO to promote insulin responsiveness through inhibitory S-nitrosylation of PTP1B. S-nitrosylation of PTP1B also protects PTP1B from permanent inactivation caused by H2O2-induced oxidative stress, since the Cys-215 residue in PTP1B can be modified by either reversible S-nitrosylation or irreversible oxidation. Increasing the level of cellular NO by pretreating cells with NO donor (SNAP) or by ectopic expression of eNOS inhibits H2O2-induced irreversible oxidation of PTP1B, protecting PTP1B from damage caused by oxidative stress[77, 78].
S-nitrosylation of lipid phosphatase
Phosphatase and tensin homolog protein (PTEN) acts as a lipid phosphatase to specifically remove 3’ phosphate from the inositol ring in PIP3, thereby regenerating PIP2 and turning off the AKT branch of insulin signaling (Fig.3). PTEN provides a negative feedback loop to fine-tune insulin signal transduction[79]. Deletion of PTEN in mice lead to enhanced insulin sensitivity, as well as resistance to HFD-induced T2DM[80].
S-nitrosylation of PTEN is an important mechanism to control PI3K–AKT signaling and is known to play a role in ischemic brain injury, neurodegeneration, and autophagy[81–84]. S-nitrosylation of PTEN at Cys-83 by either nNOS or eNOS inhibits PTEN activity, allowing accumulation of PIP3 and prolonging downstream AKT cascades[84]. Moreover, S-nitrosylation of PTEN can lead to its degradation via the ubiquitin-proteasome system through NEDD4-1-mediated ubiquitination[83]. S-nitrosylation of PTEN is catalyzed by the S-nitrosylase DJ-1 (protein/nucleic acid deglycase), which transfers the NO group from Cys106 of DJ-1 to PTEN. Mutation of Cys106 in DJ-1 blocks this transnitrosylation. DJ-1 has been identified as one of several recessively inherited genes whose mutation can cause familial Parkinson’s disease, and inactivation of DJ-1 renders neurons more susceptible to oxidative stress and cell death. S-nitrosylation of PTEN mediated by DJ-1 decreases its phosphatase activity, thus promoting cell survival[85]. This newly discovered enzymatic role for DJ-1 in S-nitrosylation of PTEN suggests it should augment insulin signaling.
SHIP2 (src homology 2-domain-containing inositol 5-phosphatase), another lipid phosphatase, removes the 5’ phosphate of the inositol ring in PIP3 to generate phosphatidyl inositol (3,4)-bisphosphate, which antagonizes PI3K-stimulated insulin signaling (Fig.3)[86]. S-nitrosylation of SHIP2 induced by mild oxidative stress such as ionizing radiation or low concentrations of H2O2 is dependent on NOS activity and inhibits its activity in CHO cells[78]. However, the role of S-nitrosylation of SHIP2 in insulin signaling remains unexplored.
Summary:
S-nitrosylation of major phosphatases (PTP1B, PTEN and SHIP2) inhibits their phosphatase activity and thereby potentially increases insulin responsiveness (Fig.3). Crosstalk between S-nitrosylation and phosphorylation manifested by S-nitrosylation-mediated suppression of phosphatases provides a quick way to amplify insulin signaling, as mediated by eNOS-derived NO. In turn, activated insulin signaling increases eNOS activity through phosphorylation of eNOS[87]. S-nitrosylation of phosphatases thus constitutes a positive feedback loop, which amplifies insulin responsiveness.
S-nitrosylation in insulin resistance
Insulin resistance (IR) is a pathological condition in which the insulin signaling pathway fails to respond sufficiently to insulin, leading to elevated blood glucose and to tissue dysfunction. IR is a principal feature of T2DM. At early stages of IR, β cells in the endocrine pancreas increase their production of insulin to compensate for elevated blood glucose. However, at late stages of IR, when insensitivity to insulin is high, β cells become damaged and fail to produce sufficient insulin. This dysfunction of β cells to produce sufficient insulin to control hyperglycemia is what characterizes the transition from pre-diabetic IR to T2DM[88].
Mechanism of obesity-linked IR
Obesity induces chronic inflammation through ER stress, adipokine dysregulation and elevated circulating pro-inflammatory cytokines and free fatty acids (FFAs), which causes and/or exacerbates insulin resistance[45]. Obesity results in high levels of FFAs in the circulation, secretion of adipokines, and infiltration of immune cells into adipose tissue. The FFAs and pro-inflammatory cytokines (from both hypertrophic adipocytes and activated immunocytes in adipose tissue) induce skeletal myocyte and hepatic inflammation, which leads to local secretion of pro-inflammatory cytokines by immunocytes in muscle and liver as well. These FFAs and cytokines (including TNFα, IL-1β, IL-6, IFN-γ and LTB4) from either local or systematic immunocytes ultimately result in insulin resistance via paracrine effects[89]. Mechanistically, there are four ways by which inflammatory signaling mediates IR, as summarized in Fig.4[89–94].
Figure 4. Mechanisms of inflammation-mediated insulin resistance.
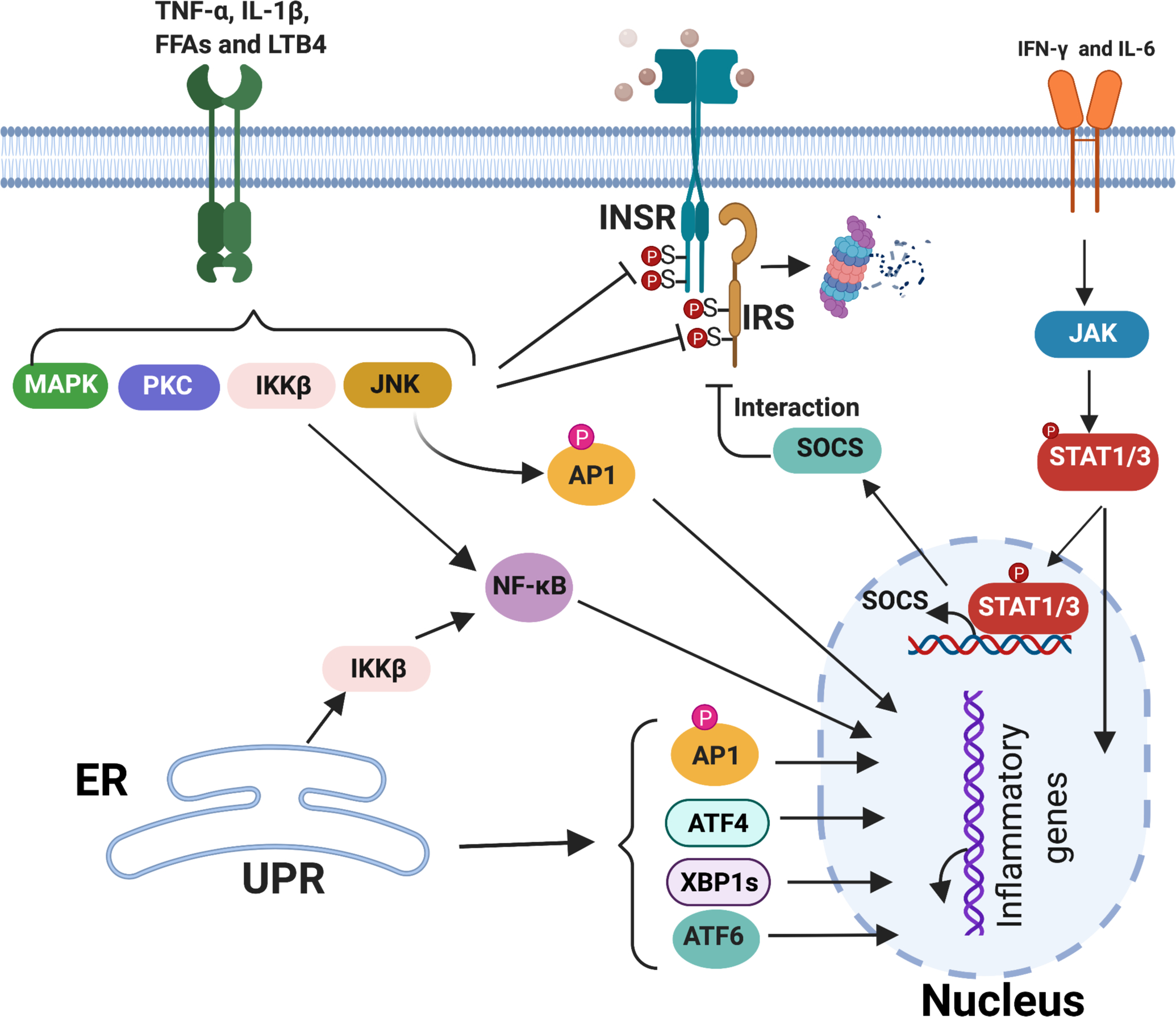
Four mechanisms contribute to inflammatory signaling leading to insulin resistance. 1) TNF-α and IL-1β (from M1-like macrophages), FFAs (derived from adipocyte lipolysis), and LTB4 (from leukocytes) bind to receptors to activate downstream kinases such as c-Jun N-terminal kinase (JNK), IκB kinase β (IKK), protein kinase C (PKC), and ERK (MAPK). These activated kinases impair insulin signal transduction via insulin-independent serine/threonine phosphorylation of INSR and IRS, which reduces insulin-induced tyrosine phosphorylation of IRS. 2) IFN-γ from Th1 cells and IL-6 from M1-like macrophages activate JAK/STAT1/3 pathways that impair insulin signaling by increasing the expression of suppressor of cytokine signaling protein (SOCS), which interrupts the interaction of INSR with IRS and promotes their degradation. 3) JNK, IKKβ and JAK kinases, activated by either inflammatory mediators or sustained ER stress, trigger translocation of the transcriptional factors AP-1, NF-κB and STAT1/3 to the nucleus, where they initiate inflammatory gene expression to maintain the chronic inflammatory state that contributes to insulin resistance. 4) The transcription factors XBP1, ATF4 and ATF6 activated by the unfolded protein response (UPR) during FFA-induced ER stress initiate inflammatory gene expression, increase the inflammatory response and lead to insulin resistance.
Aberrant S-nitrosylation causes IR
It is well known that iNOS plays an important role in IR. Overproduction of NO by iNOS, a major mediator of inflammation, induces nitrosative stress and leads to tissue damage. Expression of iNOS is increased in obesity, associated with marked impairment in insulin-stimulated glucose transport in skeletal muscle, liver and adipose tissue. Genetic disruption of iNOS or iNOS inhibitor treatment prevents impairments in insulin-stimulated activation of PI3K and AKT in both muscle and liver, thus protecting against obesity-linked IR[95, 96]. Conversely, liver-specific overexpression of iNOS markedly attenuates insulin-stimulated phosphorylation of IRS1 and AKT to cause hepatic IR and mild hyperglycemia in mice[97]. Mechanistically, iNOS causes aberrant and excessive protein S-nitrosylation, leading to IR by disrupting insulin signaling, impairing function of adipocytes, and maintaining an inflammatory state, as summarized in Fig.5.
Figure 5. Mechanisms of insulin resistance mediated by aberrant S-nitrosylation of proteins in insulin signaling cascades.
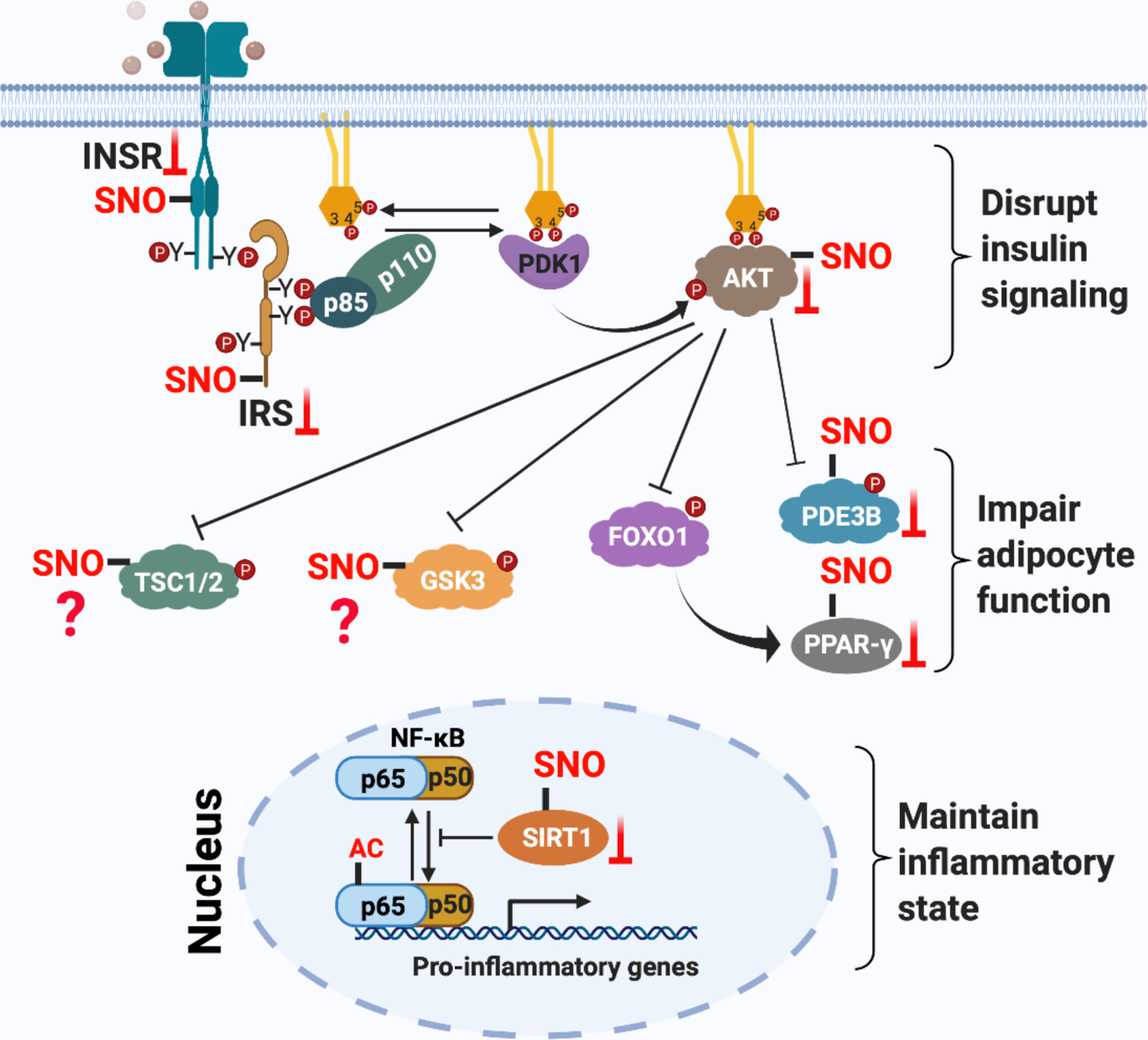
Three mechanisms are known for how insulin signal transduction is regulated by protein S-nitrosylation to promote insulin resistance. 1) S-nitrosylation of INSR-β, IRS1 and AKT1 inhibit their activity or promote their degradation, disrupting insulin signaling. 2) S-nitrosylation of PPARγ and PDE3B inhibit their activity and impair adipocyte function, thereby leading to insulin resistance of adipocytes. 3) S-nitrosylation of SIRT1 blocks its deacetylase activity and augments acetylation of NF-kB p65, thereby activating NF-κB transcriptional activity. Activated NF-κB initiates the expression of pro-inflammatory genes to maintain the inflammatory state that reinforces insulin resistance. NF-κB and IKK can also be S-nitrosylated, but whether this plays a role in IR is not known.
Protein S-nitrosylation disrupts insulin signaling
S-nitrosylation of multiple proteins in the insulin signaling cascade, including INSR-β, IRS1 and AKT, have been demonstrated in skeletal muscle of obesity-linked diabetic mice. S-nitrosylation of INSR-β and AKT reduce their kinase activity, while S-nitrosylation of IRS-1 reduces its expression[98–100]. Treatment with GSNO (a SNO donor) directly induces IR in both isolated soleus muscle and in intact Wistar rats, and enhances S-nitrosylation of INSR-β, IRS1 and AKT. In contrast, targeted disruption of iNOS in high fat-fed mice or pharmacological inhibition of iNOS in Wistar rats increased insulin sensitivity in muscle, correlating with reduced S-nitrosylation of INSR-β, IRS-1 and AKT in soleus muscle. These studies indicate that S-nitrosylation of INSR-β, IRS-1 and AKT are molecular mediators of iNOS-induced insulin resistance[100, 101]. This mechanism is further confirmed by another study demonstrating that S-nitrosylation of INSR-β, IRS-1 and AKT mediated by iNOS participates in aging-induced insulin resistance[102]. Genetic deletion of iNOS, reduction of iNOS expression via exercise, or inhibition of iNOS activity by pharmacological treatment reduced S-nitrosylation of INSR-β, IRS-1 and AKT in skeletal muscle of aged mice, thereby leading to improved insulin signaling and insulin sensitivity[102]. It should be noted that more work has been done on S-nitrosylation of AKT than either IRS1 or INSR-β. AKT1 is S-nitrosylated at cysteine-224, and mutation of Cys-224 to serine eliminated NO donor-induced S-nitrosylation and led to AKT inactivation[99]. S-nitrosylation of AKT is also involved in streptozotocin-induced insulin resistance in neurons, functional disorders in aged skeletal muscle, and hypoxia/HIF-1-induced insulin resistance in obesity[103–105].
As S-nitrosylation of proteins in the insulin signaling cascade is critical in the pathogenesis of iNOS-induced IR, several studies have examined whether reducing S-nitrosylation of INSR, IRS1 and AKT can protect against diabetes. First, aspirin treatment attenuated IR in muscle of diet-induced obese rats by inhibiting iNOS expression and S-nitrosylation of INSR-β, IRS-1 and AKT[106]. This may explain how high-dose aspirin improves fasting- and postprandial-hyperglycemia in patients with T2DM[107]. Second, exercise can efficiently restore insulin sensitivity via reduced S-nitrosylation. Voluntary exercise improves IR by reducing iNOS-mediated S-nitrosylation of AKT in the liver of obese rats[108]. Acute physical exercise reverses S-nitrosylation of INSR-β, IRS-1 and AKT in diet-induced obese Wistar rats, and increases insulin sensitivity, in parallel with a decrease in iNOS expression (Fig.5)[109].
IR is frequently associated with endothelial dysfunction and has been proposed to play a major role in cardiovascular diseases[110]. The vascular response to insulin is determined in part by the balance between NO-dependent vasodilator action and endothelin-dependent vasoconstrictor action, which are regulated by IRS/PI3K/AKT-associated and RAF/MEK/MAPK-associated insulin-signaling pathways, respectively[111]. Under healthy conditions, NO production by insulin-stimulated eNOS in vascular endothelium increases blood flow and endothelial insulin transcytosis to enhance glucose uptake into skeletal muscle. Under IR, IRS/PI3K/AKT- (but not RAF/MEK/MAPK) signaling is impaired in the endothelium and NO bioavailability is reduced. Moreover, production of endothelin and cell adhesion molecules via RAF/MEK/MAPK is preserved, further promoting endothelial dysfunction [112, 113]. The mechanisms by which IR causes pathway-specific inhibition in endothelium may relate to S-nitrosylation status. As discussed above, multiple components of the IRS/PI3K/AKT pathway (e.g. IRS1, AKT) can be S-nitrosylated in primary insulin target tissues (adipose, liver and skeletal muscle), and S-nitrosylation of IRS1 and AKT is induced by IR to inhibit IRS/PI3K/AKT signaling. So this mechanism may explain pathway-specific insulin resistance in the vasculature.
GSK3β and TSC2 are two important downstream mediators of insulin signaling (Fig.3)[114, 115]. Both GSK3β and TSC2 are modified by S-nitrosylation. S-nitrosylation of GSK-3β has been characterized in H9C2 myoblasts, primary neonatal rat ventricular myocytes, and heart tissue from an animal model of heart failure and sudden cardiac death. S-nitrosylation of GSK-3β significantly inhibits its kinase activity and promotes its nuclear translocation, where it can interact with splicing machinery to coordinate DNA damage response and cell cycle regulation[116]. S-nitrosylation of TSC2 plays an important role in iNOS-driven survival and proliferation of human melanoma cells. S-nitrosylation of TSC2 inhibits its GAP activity by blocking the dimerization of TSC2 with its partner TSC1, thereby enhancing the GTP-dependent activity of its target Rheb to activate mTOR signaling[117]. The implied role of S-nitrosylation of GSK3β or TSC2 in insulin-mediated physiology remains to be determined (Fig.5).
S-nitrosylation impairs adipocyte function
FOXO1 and PDE3B are two additional important downstream mediators of insulin signaling, particularly in insulin-regulated metabolism in adipocytes(Fig.3)[118, 119]. Adipose tissue is an endocrine organ that influences both glucose and lipid metabolism. Dysfunction and/or dysregulation of adipose tissue contributes to both local and systematic IR by impairing the release of adipokines and releasing excessive inflammatory cytokines and FFAs[120]. PPARγ is a ligand-activated nuclear receptor and a master regulator of adipocyte differentiation. PPARγ increases insulin sensitivity by regulating genes controlling fatty acid storage and glucose metabolism. iNOS-derived NO secreted by the macrophage cell line RAW264.7 mediates S-nitrosylation of PPARγ in co-cultured 3T3-L1 preadipocytes, suppressing its transcriptional activity and providing a potential mechanism for how macrophages contribute to IR[121]. Mechanistically, S-nitrosylation of PPARγ at cysteine-168 disrupts the conformational flexibility of the DNA-binding domain to impair its interaction with promoters of target genes, thereby reducing production of such targets as adiponectin and adipocyte protein 2 to block adipogenic differentiation.
Anti-lipolysis is one major consequence of insulin stimulation of adipocytes. Dysregulated lipolysis is involved in insulin resistance and T2DM through elevating blood levels of FFAs[122]. The anti-lipolytic action of insulin was markedly impaired in both cultured adipocytes and mice treated with GSNO prior to insulin, indicating that protein S-nitrosylation induced by GSNO is involved in dysregulated lipolysis-induced IR. Phosphodiesterase 3B (PDE3B) plays a key role in the anti-lipolytic action of insulin by reducing cellular cAMP levels (Fig.3). Therefore, altered expression and/or regulation of PDE3B is a potential mediator of systemic IR[123]. Rudich et al found that PDE3B is modified by S-nitrosylation in GSNO-treated cultured adipocytes or in adipose tissue from high-fat fed mice, and that this interferes with the anti-lipolytic action of insulin in adipocytes and contributes to obesity-associated IR. Two S-nitrosylation sites were identified in PDE3B (Cys-768 and -1040), which are located adjacent to the cAMP substrate-binding pocket in the catalytic domain of PDE3B, suggesting that S-nitrosylation of PDE3B may directly inhibit its activity, to promote lipolysis (Fig.5)[124].
S-nitrosylation maintains the inflammatory state
SIRT1 is a protein deacetylase that removes acetyl groups from lysine residues of target proteins, such as the p65 subunit of the NF-kB transcription factor. Acetylation of p65 activates NF-kB-dependent transcription of many pro-inflammatory genes. In contrast, deacetylation of p65 by SIRT1 inhibits NF-kB activity and suppresses inflammation[125]. Burn injury can lead to IR and hyperglycemia because it induces robust pro-inflammatory gene expression in skeletal muscle[126]. Knockout of iNOS in mice inhibits burn-induced inflammatory gene expression and apoptosis, suggesting a role in burn-mediated IR[127]. Furthermore, Shinozaki et al confirmed that S-nitrosylation of SIRT1, mediated by iNOS, controlled burn-induced inflammatory gene expression and was involved in burn-induced IR. Mechanistically, S-nitrosylation of SIRT1 after burn injury occurs within zinc finger CXXC motifs, inhibiting SIRT1 deacetylase activity by disrupting its ability to bind zinc, thereby promoting increased acetylation of p65 and transcriptional activity of NF-κB in skeletal myocytes. Activated NF-κB induces transcription of pro-inflammatory genes and enhances the inflammatory response, promoting burn injury-induced IR[127]. The role of S-nitrosylated SIRT1 in diabetic IR remains to be tested (Fig.5).
In addition to the role of IR-associated hyperlipidemia and inflammation within insulin target tissues, insulin-producing β-cells are extremely susceptible to hyperlipidemia and inflammation and have minimal regenerative capacity during adulthood [128]. Chronic hyperlipidemia injures β-cells via generation of cytotoxic intracellular metabolites (saturated FFAs, ceramides, reactive oxidative species) that activate inflammatory pathways leading to β-cell dysfunction and death[129]. Pancreatic islets also show abundant expression of proteins involved in insulin production that may serve as substrates for S-nitrosylation on the one hand [130–132] and act as potential mediators in hyperlipidemia and inflammation-mediated pancreatic IR on the other, including PPARγ, PDE3B and SIRT1 (Fig 5).
S-nitrosylation in ER stress-induced IR
The endoplasmic reticulum (ER) is a large membrane-enclosed cellular organelle in all eukaryotes. Proper folding of transmembrane and secreted proteins is one major role of ER. A high demand for folding of newly synthesized proteins compared to the capacity of the ER to assist protein folding leads to accumulation of unfolded/misfolded proteins in the ER lumen, thereby causing ER stress[133]. Signal transduction pathways known as the unfolded protein response (UPR) will sense and attempt to resolve this ER stress by decreasing unfolded protein load, amplifying folding chaperone capacity, and accelerating degradation of misfolded proteins[134].
UPR pathway
The UPR is mediated by three transmembrane proteins: inositol-requiring enzyme 1 (IRE1), PKR-like ER kinase (PERK), and activating transcription factor 6 (ATF6). In unstressed conditions, ATF6, IRE1, and PERK are associated with the chaperone BiP (binding immunoglobulin protein), and are kept in their inactive state. Upon ER stress, BiP interacts instead with unfolded/misfolded proteins, releasing IRE1, PERK and ATF6 to activate the UPR pathway, as summarized in Fig.6[135, 136].
Figure 6. The role of aberrant protein S-nitrosylation in ER stress-mediated insulin resistance.
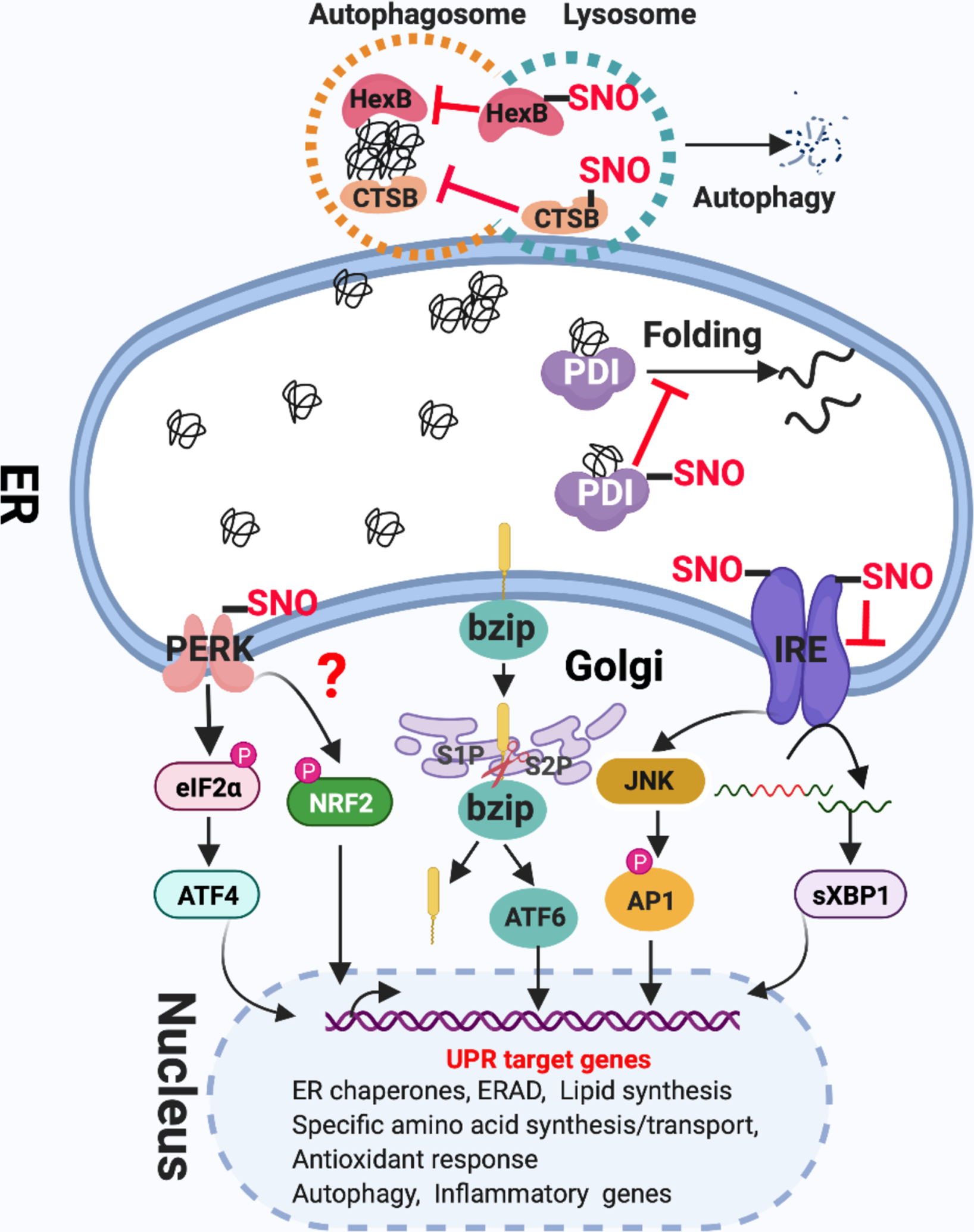
The UPR pathway is mediated by three transmembrane proteins: IRE1, PERK, and ATF6. Upon ER stress, 1) PERK dimerizes to activate its kinase, and phosphorylates eIF2α to decrease global protein translation, but selectively activates the translation of ATF4. PERK also phosphorylates NRF2. ATF4 and NRF2 initiate the transcription of genes involved in the antioxidant response and amino acid transport, thereby alleviating ER stress; 2) ATF-6α traffics from the ER to the Golgi, where it is proteolytically activated to release the cytosolic B-ZIP transcription factor domain of ATF-6α that translocates to the nucleus, where it acts as a transcriptional factor to upregulate UPR target gene expression to protect the cell from ER stress; 3) IRE1 dimerization and autophosphorylation activate its endoribonuclease activity, which is responsible for removing 26 nucleotides from the X box-binding protein 1 (XBP1) mRNA via an unconventional splicing event. Spliced Xbp-1 encodes a potent transcription factor of the basic-leucine zipper (B-ZIP) family, which induces ERAD proteins and chaperones. The ER protein quality control system includes protein re-folding apparatus (ER-resident folding chaperones), ERAD and autophagy. There are at least three mechanisms for how protein S-nitrosylation affects the UPR pathway and protein quality control systems in ER stress, eventually leading to IR. 1) S-nitrosylation of IRE1 prevents the generation of sXBP1 by inhibiting IRE1 endoribonuclease activity. 2) S-nitrosylation of the chaperone PDI inhibits its enzymatic activity, leading to accumulation of misfolded proteins. 3) S-nitrosylation of the lysosomal proteins HexB and CTSB inhibit these autophagy-related lysosomal enzymes, blocking autophagy. Although PEAK, BiP and UBE2D1 also can be S-nitrosylated in certain conditions or cell types, the link to IR remains unclear.
S-nitrosylation of proteins of the UPR pathway in IR
The UPR is considered an acute mechanism that re-establishes cellular homeostasis, and specifically reduces ER stress. When dysfunctional, the UPR loses the ability to respond to ER stress; sustained ER stress will cause chronic low-grade inflammation and result in IR[137]. As shown in Fig.6, all of three branches of the UPR are linked to inflammation by activating the transcription factors NF-kB or AP-1, which are required for the expression of many pro-inflammatory genes. After ER stress, PERK–eIF2a promotes nuclear NF-kB translocation by inhibiting translation of IkB (inhibitor of NF-kB). The IRE1a pathway activates JNK and IKKβ by recruiting TNF-receptor-associated factor 2 (TRAF2) and its signaling complex, and by this means then activates the transcription factors AP-1 and NF-kB. Finally, the ATF6 branch also induces inflammation through NF-kB activation[92].
Yang et al recently demonstrated that obesity-related chronic inflammation induced expression of iNOS, and iNOS-derived NO impaired the IRE1A pathway through S-nitrosylation of IRE1A, resulting in IR[138]. IRE1A is S-nitrosylated at cysteine-931 and cysteine-951 in its RNAse domain, inhibiting its ribonuclease activity (but not kinase activity), which thereby prevents generation of sXBP1. This dysfunctional IRE1A-sXBP1 pathway in obese mice loses the ability to respond to inflammation-induced ER stress, prolonging ER stress in the presence of chronic metabolic and inflammatory stress, and leading to IR. Exogenous expression of either S-nitrosylation-resistant mutant IRE1a (C931A/C951A) or pre-spliced XBP1 in the liver of ob/ob mice rescues insulin sensitivity and glucose tolerance, confirming that iNOS-mediated S-nitrosylation of IRE1A is a critical component contributing to impairment of the UPR and IR. It is well known that dysfunctional UPR is also involved in neurodegenerative and cardiac disorders. Thus, it is not surprising that two additional studies have demonstrated that S-nitrosylation of IRE1A leads to dysfunctional ER stress signaling in settings of neuronal cell death and heart failure with preserved ejection fraction (HFpEF)[139, 140]. To ensure the proper secretion of insulin by the pancreas, UPR also functions in β-cells to alleviate ER stress associated with IR, as caused by heavy insulin secretory load, amyloid aggregates and FFA exposure[141, 142]. It will therefore be informative to investigate whether S-nitrosylation of IRE1A is involved in pancreatic causes of IR.
Another UPR pathway protein, PERK, also has been reported to be S-nitrosylated in cell-based models of Parkinson’s disease. S-nitrosylation of PERK activated its kinase activity and increased phosphorylation of eIF2α, thereby inhibiting the protein translation activity of eIF2α. It will be interesting to investigate what role S-nitrosylation of PERK may play in IR[139].
ER Protein Quality Control systems
After the UPR is activated in response to accumulation of unfolded/misfolded proteins, cells immediately deal with these proteins using a three-step quality control pathway to avert tissue damage[143]. As summarized in Fig.6, firstly, molecular chaperones will speed up folding of unfolded proteins and will attempt to refold misfolded proteins. Secondly, if unfolded/misfolded proteins are unable to be restored to their normal conformation and function, they will be targeted for proteasomal degradation via the ER-associated protein degradation (ERAD) pathway. Lastly, if unfolded/misfolded proteins cannot be re-solubilized by molecular chaperones or degraded by the ERAD, they will form protein aggregates, which will be trafficked to autolysosomes and cleared by autophagy[144]. S-nitrosylation of multiple proteins involved in this three-step protein quality control pathway have been implicated in IR (Fig.6).
S-nitrosylation of chaperones and ERAD-associated protein
Chronic hyperglycemia causes ER stress in β-cells, and can suppress insulin production and secretion. BiP and PDI, two important UPR-induced chaperones, are required for optimal insulin production and secretion under both normal glucose levels and chronic hyperglycemia[145, 146]. BiP can actively fold its substrate or restrict that substrate from aggregating via direct binding. Overexpression of BiP in the INS-1832/13 β-cell line partially relieved chronic hyperglycemia-mediated suppression of steady-state proinsulin levels and insulin secretion. In contrast, knockdown of BiP under basal glucose conditions reduced cellular insulin levels and glucose-stimulated insulin secretion[145]. PDIs are thiol oxidoreductases that catalyze thiol–disulfide exchange, thus facilitating disulfide bond formation and rearrangement/pairing reactions. They also have a chaperone function[147]. In pancreatic β cells, PDIA1 contributes to oxidative maturation of proinsulin disulfide bonds in the ER to support insulin production. In the absence of PDIA1, the ratio of intracellular proinsulin/insulin is increased, and aggregated proinsulin complexes accumulate. PDIA1 deletion specifically in β-cells in high fat diet-fed young mice or in aged mice also aggravated glucose intolerance due to inadequate insulin release[146].
S-nitrosylation of BiP was identified in HCT-116RC cells. High-glucose treatment resulted in a significant decrease in S-nitrosylated BiP, suggesting that S-nitrosylation of BiP may be involved in GSIS[148]. S-nitrosylation of PDI, first identified in brains of sporadic Parkinson’s or Alzheimer’s disease patients, inhibits its enzymatic activity and leads to the accumulation of misfolded proteins and to activation of the unfolded protein response[149]. S-nitrosylation of PDI occurs at both N-terminal (Cys-36, Cys-39) and C-terminal (Cys-383, Cys-386) sites. To further clarify the relationship between S-nitrosylation of PDI and ER stress, Uehara et al showed that overexpression of wild-type PDI largely abrogated ER stress by triggering the UPR pathway to protect against neuronal cell death, whereas SNO-site SNO mimetic mutant PDI was unable to reduce ER stress or trigger the UPR. This protective effect of wild-type PDI was reversed by exposure to NO donor Cys-NO[149]. Given that PDIs are required for optimal insulin production and secretion under chronic hyperglycemia in β-cells, these findings suggest similar mechanisms that may alter insulin production in IR and diabetes.
ERAD, an evolutionarily conserved protein quality control mechanism required to prevent the accumulation of unfolded or misfolded proteins in the ER, has a critical role in supporting glucose-stimulated insulin secretion in β-cells[150]. Ubiquitin-conjugating enzyme E2 D1 (UBE2D1), an ERAD-associated protein, is involved in ERAD-mediated protein degradation[151]. Intriguingly, single nucleotide polymorphisms (SNPs) near the UBE2D1 gene were genetically associated with diabetic retinopathy, suggesting that abnormal protein degradation mediated by UBE2D1 plays a role [152]. UBE2D1 is S-nitrosylated at its active site Cys-85 in HEK293T cells, which inhibits ubiquitin-conjugating activity and attenuates ubiquitination of ERAD substrates, thereby potentially leading to accumulation of ERAD substrates and sustained ER stress[153]. The role of this regulation in β-cells in insulin resistance remains untested, but would be expected to increase ER stress and accumulation of unfolded protein aggregates (including insulin itself).
S-nitrosylation impairs autophagy
Autophagy plays an important role in protein quality control and the maintenance of cellular metabolic homeostasis. Disruption of autophagy is associated with hepatic IR and affects insulin homeostasis in β-cells[154, 155]. Conversely, restoration of autophagy via either overexpression of autophagy regulators or treatment with autophagy inducers ameliorates obesity-induced IR[156, 157]. In the liver, autophagy participates in the basal turnover of lipids by engulfing and degrading lipid droplets; thus reduced autophagic activity leads to hepatic steatosis and insulin resistance[156, 158]. Recently, Yang et al revealed that S-nitrosylation can impact autophagy by inhibiting autophagy-related lysosomal enzyme activities[159]. They found that diet-induced obesity promoted S-nitrosylation of two lysosomal proteins, HexB (at Cys-530) and CTSB (at cCys-211) in mouse liver, impairing lysosomal function. This critical regulation is dependent on both obesity-induced inflammation and the enzyme S-nitrosoglutathione reductase (GSNOR). GSNOR is a glutathione-dependent denitrosylase, and the absence of GSNOR leads to increased protein S-nitrosylation[160]. In GSNOR knockout mice, obesity impairs autophagy in the liver, thus leading to hepatic insulin resistance by aberrant S-nitrosylation of lysosomal enzymes. Conversely, AAV-mediated overexpression of GSNOR in the liver of obese mice markedly enhances autophagy and improves insulin action by decreasing S-nitrosylation of lysosomal enzymes. Furthermore, AAV-mediated overexpression of S-nitrosylation-resistant variants of HexB (C530A) or CTSB (C211A) enhances autophagy, indicating that S-nitrosylation of lysosomal enzymes regulated by GSNOR causes defective autophagy[159]. Thus, in short, obesity-induced protein S-nitrosylation plays a key role in compromising hepatic autophagy, thereby contributing to hepatic insulin resistance.
Functional autophagy is critical for insulin production and survival of β-cells. Appropriate induction of autophagy is required to protect β-cells from stress conditions, including inflammation, oxidative stress, ER stress, nutrient depletion, mitochondrial damage, and hypoxia, thereby blocking β-cell apoptosis [161]. Accumulation of autophagic vacuoles and autophagosomes has been observed in human and rodent type 2 diabetic β-cells[162, 163]. This enhanced autophagy induced by diabetes provides a protective mechanism against the dysfunction and death of β-cells[155]. Notably, S-nitrosylation of multiple proteins (IKKβ, JNK, BCL-2 and TSC2) was shown to inhibit autophagosome formation by impairing two different pro-autophagy pathways: JNK-BCL-2-BECLIN1 and IKKβ/AMPK/mTOR[117, 164, 165]. Future investigation of protein S-nitrosylation (e.g., IKKβ, JNK, BCL-2, TSC2, HexB, and CTSB) in regulation of β-cell autophagy is warranted.
Summary
Aberrant protein S-nitrosylation is a major mechanism underlying insulin resistance caused by iNOS-derived NO. S-nitrosylation of INSR-β, IRS1, AKT, PPARγ, PDE3B and SIRT contribute to insulin resistance by disrupting insulin signaling, impairing adipocyte function, and maintaining the inflammatory state (Fig.5). S-nitrosylation of IRE1, PDI, HexB and CTSB play important roles in ER-stress-induced insulin resistance (Fig.6). It should be noted that in addition to hyper-S-nitrosylation of the above proteins under stress condition, many are also S-nitrosylated under normal conditions[100, 121, 124, 138, 159], suggesting physiological roles in insulin signaling. One idea is that S-nitrosylation serves as a negative feedback loop to rapidly turn off insulin in order to keep blood glucose within a normal range. In obesity, hyper- or aberrant S-nitrosylation of these targets in response to chronic inflammation improperly blocks insulin signaling, leading to IR and T2DM.
Remarks and future perspectives
Protein S-nitrosylation regulates the entire span of insulin actions, including processing and secretion from β-cells, transport across endothelia, signal transduction cascades and insulin degradation at target organs. Thus, S-nitrosylation plays important roles in both the physiological effects of insulin in primary target tissues and the pathophysiology of IR and diabetes. What is surprising, perhaps, is just how many essential proteins involved in insulin action are known to be regulated by S-nitrosylation (Table1) and how strong the evidence is (including human genetic mutations in GCK) for aberrant protein S-nitrosylation in IR and T2DM. Aside from its characteristic effects on blood glucose uptake by insulin target tissues, IR has been also identified as a risk factor for Alzheimer’s disease (AD) and Parkinson’s disease (PD)[166, 167]. Many pathologies of AD and PD may be explained by dysregulated insulin signaling with impairments in signal transduction and gene expression, supporting the concept that AD and PD should be regarded as metabolic diseases of brain, where insulin resistance or deficiency play causal roles[168, 169]. More generally, S-nitrosylation may link IR (or insulin deficiency) with neurodegeneration through a common set of proteins (IDE, DJ-1, PEAK, IREa1 and PDI). Tantalizing new evidence indicates that S-nitrosylation is enzymatic, involving multiple classes of enzymes, drawing analogy to, ubiquitinylation[10](Fig.1). Identifying these enzymes, including both S-nitrosylases and denitrosylases, holds exciting promise to shed new light on the life of insulin and may offer new therapeutic approaches for T2DM.
Table 1:
S-nitrosylated proteins in the insulin pathway
| SNO-Protein | SNO site | Role of S-nitrosylation in the insulin pathway | Established or potential effect of S-nitrosylation on insulin action | Is aberrant SNO involved in diabetes? | Reference |
|---|---|---|---|---|---|
| GK | C371 | Insulin secretion | Activation | Yes | 21–23, 26 |
| RyR2 | C3602 | Insulin secretion | Activation | Yes | 31–33 |
| SUR1 | C717 | Insulin secretion | Inhibition | Unknown | 37 |
| Syn-1A | C145 | Insulin secretion | Activation | Unknown | 43 |
| Syn-4 | C141 | Insulin secretion | Activation | Yes | 44 |
| NSF | C11, 91,264 | Insulin secretion | Inhibition | Unknown | 49–51 |
| Cav-1 | C156 | Insulin transport | Activation/Inhibition | Unknown | 58–60 |
| IDE | C110,178,819 | Insulin degradation | Activation | Yes | 66–68 |
| PTP1B | C215 | Insulin signaling/Transport | Activation | Unknown | 53, 76–78 |
| PTEN | C83 | Insulin signalling | Activation | Unknown | 81–85 |
| SHIP2 | Unknown | Insulin signalling | Activation | Unknown | 78 |
| INSR-β | Unknown | Insulin resistance | Inhibition | Yes | 98, 100–102, 106–109 |
| IRS1 | Unknown | Insulin resistance | Inhibition | Yes | 98, 100–102, 106–109 |
| AKT | C224 | Insulin resistance | Inhibition | Yes | 98–109 |
| GSK3 | C76,199,317 | Insulin resistance | Activation | Unknown | 112 |
| PPARγ | C168 | Insulin resistance | Inhibition | Unknown | 117 |
| PDE3B | C768, 1040 | Insulin resistance | Inhibition | Yes | 120 |
| SIRT1 | C371,374,395,398 | Insulin resistance | Inhibition | Yes | 123 |
| IRE1 | C931/951 | Insulin resistance | Inhibition | Yes | 129–131 |
| PDI | C36,39,383,386 | Insulin resistance | Inhibition | Unknown | 138 |
| UBE2D1 | C85 | Insulin resistance | Inhibition | Unknown | 142 |
| HexB | C530 | Insulin resistance | Inhibition | Yes | 148 |
| CTSB | C211 | Insulin resistance | Inhibition | Yes | 148 |
Footnotes
Competing interests
We declare that the authors have no competing financial or non-financial interests as defined by Nature journals’ policy.
Reference
- 1.Sonksen P and Sonksen J (2000) Insulin: understanding its action in health and disease. Br J Anaesth 85 (1), 69–79. [DOI] [PubMed] [Google Scholar]
- 2.Weiss M et al. (2000) Insulin Biosynthesis, Secretion, Structure, and Structure-Activity Relationships. In Endotext (Feingold KR et al. eds). [PubMed] [Google Scholar]
- 3.Fu Z et al. (2013) Regulation of insulin synthesis and secretion and pancreatic Beta-cell dysfunction in diabetes. Curr Diabetes Rev 9 (1), 25–53. [PMC free article] [PubMed] [Google Scholar]
- 4.Saklayen MG (2018) The Global Epidemic of the Metabolic Syndrome. Curr Hypertens Rep 20 (2), 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kurohane Kaneko Y and Ishikawa T (2013) Dual role of nitric oxide in pancreatic beta-cells. J Pharmacol Sci 123 (4), 295–300. [DOI] [PubMed] [Google Scholar]
- 6.Kapur S et al. (2000) Nitric oxide: a new player in the modulation of energy metabolism. Int J Obes Relat Metab Disord 24 Suppl 4, S36–40. [DOI] [PubMed] [Google Scholar]
- 7.Bahadoran Z et al. (2020) Role of Nitric Oxide in Insulin Secretion and Glucose Metabolism. Trends Endocrinol Metab 31 (2), 118–130. [DOI] [PubMed] [Google Scholar]
- 8.Sansbury BE and Hill BG (2014) Regulation of obesity and insulin resistance by nitric oxide. Free Radic Biol Med 73, 383–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hess DT et al. (2005) Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol 6 (2), 150–66. [DOI] [PubMed] [Google Scholar]
- 10.Seth D et al. (2018) A Multiplex Enzymatic Machinery for Cellular Protein S-nitrosylation. Mol Cell 69 (3), 451–464 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stomberski CT et al. (2019) Protein S-Nitrosylation: Determinants of Specificity and Enzymatic Regulation of S-Nitrosothiol-Based Signaling. Antioxid Redox Signal 30 (10), 1331–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gaisano HY (2017) Recent new insights into the role of SNARE and associated proteins in insulin granule exocytosis. Diabetes Obes Metab 19 Suppl 1, 115–123. [DOI] [PubMed] [Google Scholar]
- 13.Byrne MM et al. (1994) Insulin secretory abnormalities in subjects with hyperglycemia due to glucokinase mutations. J Clin Invest 93 (3), 1120–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cherrington AD et al. (2002) Physiological consequences of phasic insulin release in the normal animal. Diabetes 51 Suppl 1, S103–8. [DOI] [PubMed] [Google Scholar]
- 15.Smukler SR et al. (2002) Exogenous nitric oxide and endogenous glucose-stimulated beta-cell nitric oxide augment insulin release. Diabetes 51 (12), 3450–60. [DOI] [PubMed] [Google Scholar]
- 16.Roder PV et al. (2016) Pancreatic regulation of glucose homeostasis. Exp Mol Med 48, e219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Postic C et al. (1999) Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J Biol Chem 274 (1), 305–15. [DOI] [PubMed] [Google Scholar]
- 18.Matschinsky FM (2002) Regulation of pancreatic beta-cell glucokinase: from basics to therapeutics. Diabetes 51 Suppl 3, S394–404. [DOI] [PubMed] [Google Scholar]
- 19.Rizzo MA et al. (2002) A functional link between glucokinase binding to insulin granules and conformational alterations in response to glucose and insulin. J Biol Chem 277 (37), 34168–75. [DOI] [PubMed] [Google Scholar]
- 20.Stubbs M et al. (2000) Subcellular localization, mobility, and kinetic activity of glucokinase in glucose-responsive insulin-secreting cells. Diabetes 49 (12), 2048–55. [DOI] [PubMed] [Google Scholar]
- 21.Rizzo MA and Piston DW (2003) Regulation of beta cell glucokinase by S-nitrosylation and association with nitric oxide synthase. J Cell Biol 161 (2), 243–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ding SY et al. (2010) Naturally occurring glucokinase mutations are associated with defects in posttranslational S-nitrosylation. Mol Endocrinol 24 (1), 171–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Markwardt ML et al. (2012) Association with nitric oxide synthase on insulin secretory granules regulates glucokinase protein levels. Mol Endocrinol 26 (9), 1617–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Holst JJ and Gromada J (2004) Role of incretin hormones in the regulation of insulin secretion in diabetic and nondiabetic humans. Am J Physiol Endocrinol Metab 287 (2), E199–206. [DOI] [PubMed] [Google Scholar]
- 25.Muller TD et al. (2019) Glucagon-like peptide 1 (GLP-1). Mol Metab 30, 72–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ding SY et al. (2011) Glucagon-like peptide 1 stimulates post-translational activation of glucokinase in pancreatic beta cells. J Biol Chem 286 (19), 16768–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bjorkhaug L et al. (2007) Allosteric activation of human glucokinase by free polyubiquitin chains and its ubiquitin-dependent cotranslational proteasomal degradation. J Biol Chem 282 (31), 22757–64. [DOI] [PubMed] [Google Scholar]
- 28.Tiedge M et al. (2000) Importance of cysteine residues for the stability and catalytic activity of human pancreatic beta cell glucokinase. Arch Biochem Biophys 375 (2), 251–60. [DOI] [PubMed] [Google Scholar]
- 29.Graves TK and Hinkle PM (2003) Ca(2+)-induced Ca(2+) release in the pancreatic beta-cell: direct evidence of endoplasmic reticulum Ca(2+) release. Endocrinology 144 (8), 3565–74. [DOI] [PubMed] [Google Scholar]
- 30.Santulli G et al. (2015) Calcium release channel RyR2 regulates insulin release and glucose homeostasis. J Clin Invest 125 (11), 4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Llanos P et al. (2015) Glucose-Dependent Insulin Secretion in Pancreatic beta-Cell Islets from Male Rats Requires Ca2+ Release via ROS-Stimulated Ryanodine Receptors. PLoS One 10 (6), e0129238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun J et al. (2008) Regulation of the cardiac muscle ryanodine receptor by O(2) tension and S-nitrosoglutathione. Biochemistry 47 (52), 13985–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gonzalez DR et al. (2007) Deficient ryanodine receptor S-nitrosylation increases sarcoplasmic reticulum calcium leak and arrhythmogenesis in cardiomyocytes. Proc Natl Acad Sci U S A 104 (51), 20612–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li N et al. (2017) Structure of a Pancreatic ATP-Sensitive Potassium Channel. Cell 168 (1–2), 101–110 e10. [DOI] [PubMed] [Google Scholar]
- 35.Aittoniemi J et al. (2009) Review. SUR1: a unique ATP-binding cassette protein that functions as an ion channel regulator. Philos Trans R Soc Lond B Biol Sci 364 (1514), 257–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aquilante CL (2010) Sulfonylurea pharmacogenomics in Type 2 diabetes: the influence of drug target and diabetes risk polymorphisms. Expert Rev Cardiovasc Ther 8 (3), 359–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kawano T et al. (2009) Nitric oxide activates ATP-sensitive potassium channels in mammalian sensory neurons: action by direct S-nitrosylation. Mol Pain 5, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nichols CG (2006) KATP channels as molecular sensors of cellular metabolism. Nature 440 (7083), 470–6. [DOI] [PubMed] [Google Scholar]
- 39.Jewell JL et al. (2010) Exocytosis mechanisms underlying insulin release and glucose uptake: conserved roles for Munc18c and syntaxin 4. Am J Physiol Regul Integr Comp Physiol 298 (3), R517–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hou JC et al. (2009) Insulin granule biogenesis, trafficking and exocytosis. Vitam Horm 80, 473–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kasai H et al. (2012) Distinct initial SNARE configurations underlying the diversity of exocytosis. Physiol Rev 92 (4), 1915–64. [DOI] [PubMed] [Google Scholar]
- 42.Yang B et al. (2000) nSec1 binds a closed conformation of syntaxin1A. J Cell Biol 148 (2), 247–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Palmer ZJ et al. (2008) S-nitrosylation of syntaxin 1 at Cys(145) is a regulatory switch controlling Munc18-1 binding. Biochem J 413 (3), 479–91. [DOI] [PubMed] [Google Scholar]
- 44.Wiseman DA et al. (2011) Stimulus-induced S-nitrosylation of Syntaxin 4 impacts insulin granule exocytosis. J Biol Chem 286 (18), 16344–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hardy OT et al. (2012) What causes the insulin resistance underlying obesity? Curr Opin Endocrinol Diabetes Obes 19 (2), 81–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Henningsson R et al. (2002) Role of nitric oxide synthase isoforms in glucose-stimulated insulin release. Am J Physiol Cell Physiol 283 (1), C296–304. [DOI] [PubMed] [Google Scholar]
- 47.Muhammed SJ et al. (2012) Pancreatic beta-cell dysfunction, expression of iNOS and the effect of phosphodiesterase inhibitors in human pancreatic islets of type 2 diabetes. Diabetes Obes Metab 14 (11), 1010–9. [DOI] [PubMed] [Google Scholar]
- 48.Zhao C et al. (2012) Requirements for the catalytic cycle of the N-ethylmaleimide-Sensitive Factor (NSF). Biochim Biophys Acta 1823 (1), 159–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Matsushita K et al. (2003) Nitric oxide regulates exocytosis by S-nitrosylation of N-ethylmaleimide-sensitive factor. Cell 115 (2), 139–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morrell CN et al. (2005) Regulation of platelet granule exocytosis by S-nitrosylation. Proc Natl Acad Sci U S A 102 (10), 3782–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ito T et al. (2011) Thioredoxin increases exocytosis by denitrosylating N-ethylmaleimide-sensitive factor. J Biol Chem 286 (13), 11179–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Barrett EJ et al. (2011) Insulin regulates its own delivery to skeletal muscle by feed-forward actions on the vasculature. Am J Physiol Endocrinol Metab 301 (2), E252–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang H et al. (2013) Nitric oxide directly promotes vascular endothelial insulin transport. Diabetes 62 (12), 4030–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang H et al. (2008) Insulin signaling stimulates insulin transport by bovine aortic endothelial cells. Diabetes 57 (3), 540–7. [DOI] [PubMed] [Google Scholar]
- 55.Wang H et al. (2011) Caveolin-1 is required for vascular endothelial insulin uptake. Am J Physiol Endocrinol Metab 300 (1), E134–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Haddad D et al. (2020) Role of Caveolin-1 in Diabetes and Its Complications. Oxid Med Cell Longev 2020, 9761539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cohen AW et al. (2003) Caveolin-1-deficient mice show insulin resistance and defective insulin receptor protein expression in adipose tissue. Am J Physiol Cell Physiol 285 (1), C222–35. [DOI] [PubMed] [Google Scholar]
- 58.Chen Z et al. (2018) Reciprocal regulation of eNOS and caveolin-1 functions in endothelial cells. Mol Biol Cell 29 (10), 1190–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Song H et al. (2016) Release of Matrix Metalloproteinases-2 and 9 by S-Nitrosylated Caveolin-1 Contributes to Degradation of Extracellular Matrix in tPA-Treated Hypoxic Endothelial Cells. PLoS One 11 (2), e0149269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bakhshi FR et al. (2013) Nitrosation-dependent caveolin 1 phosphorylation, ubiquitination, and degradation and its association with idiopathic pulmonary arterial hypertension. Pulm Circ 3 (4), 816–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tang WJ (2016) Targeting Insulin-Degrading Enzyme to Treat Type 2 Diabetes Mellitus. Trends Endocrinol Metab 27 (1), 24–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pivovarova O et al. (2016) Insulin-degrading enzyme: new therapeutic target for diabetes and Alzheimer’s disease? Ann Med 48 (8), 614–624. [DOI] [PubMed] [Google Scholar]
- 63.Farris W et al. (2003) Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci U S A 100 (7), 4162–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wroblewski VJ et al. (1992) Mechanisms involved in degradation of human insulin by cytosolic fractions of human, monkey, and rat liver. Diabetes 41 (4), 539–47. [DOI] [PubMed] [Google Scholar]
- 65.Fakhrai-Rad H et al. (2000) Insulin-degrading enzyme identified as a candidate diabetes susceptibility gene in GK rats. Hum Mol Genet 9 (14), 2149–58. [DOI] [PubMed] [Google Scholar]
- 66.Akhtar MW et al. (2016) Elevated glucose and oligomeric beta-amyloid disrupt synapses via a common pathway of aberrant protein S-nitrosylation. Nat Commun 7, 10242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cordes CM et al. (2009) Nitric oxide inhibits insulin-degrading enzyme activity and function through S-nitrosylation. Biochem Pharmacol 77 (6), 1064–73. [DOI] [PubMed] [Google Scholar]
- 68.Ralat LA et al. (2009) Protective role of Cys-178 against the inactivation and oligomerization of human insulin-degrading enzyme by oxidation and nitrosylation. J Biol Chem 284 (49), 34005–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kadowaki T et al. (2012) SnapShot: Insulin signaling pathways. Cell 148 (3), 624, 624 e1. [DOI] [PubMed] [Google Scholar]
- 70.Kadowaki T et al. (2012) SnapShot: physiology of insulin signaling. Cell 148 (4), 834–834 e1. [DOI] [PubMed] [Google Scholar]
- 71.Duplain H et al. (2001) Insulin resistance, hyperlipidemia, and hypertension in mice lacking endothelial nitric oxide synthase. Circulation 104 (3), 342–5. [DOI] [PubMed] [Google Scholar]
- 72.Vecoli C et al. (2014) Partial deletion of eNOS gene causes hyperinsulinemic state, unbalance of cardiac insulin signaling pathways and coronary dysfunction independently of high fat diet. PLoS One 9 (8), e104156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nakagawa T et al. (2007) Diabetic endothelial nitric oxide synthase knockout mice develop advanced diabetic nephropathy. J Am Soc Nephrol 18 (2), 539–50. [DOI] [PubMed] [Google Scholar]
- 74.Kashyap SR et al. (2005) Insulin resistance is associated with impaired nitric oxide synthase activity in skeletal muscle of type 2 diabetic subjects. J Clin Endocrinol Metab 90 (2), 1100–5. [DOI] [PubMed] [Google Scholar]
- 75.Elchebly M et al. (1999) Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science 283 (5407), 1544–8. [DOI] [PubMed] [Google Scholar]
- 76.Hsu MF and Meng TC (2010) Enhancement of insulin responsiveness by nitric oxide-mediated inactivation of protein-tyrosine phosphatases. J Biol Chem 285 (11), 7919–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chen YY et al. (2008) Cysteine S-nitrosylation protects protein-tyrosine phosphatase 1B against oxidation-induced permanent inactivation. J Biol Chem 283 (50), 35265–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Barrett DM et al. (2005) Inhibition of protein-tyrosine phosphatases by mild oxidative stresses is dependent on S-nitrosylation. J Biol Chem 280 (15), 14453–61. [DOI] [PubMed] [Google Scholar]
- 79.Chen CY et al. (2018) PTEN: Tumor Suppressor and Metabolic Regulator. Front Endocrinol (Lausanne) 9, 338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kurlawalla-Martinez C et al. (2005) Insulin hypersensitivity and resistance to streptozotocin-induced diabetes in mice lacking PTEN in adipose tissue. Mol Cell Biol 25 (6), 2498–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pei DS et al. (2009) S-nitrosylation of PTEN Invovled in ischemic brain injury in rat hippocampal CA1 region. Neurochem Res 34 (8), 1507–12. [DOI] [PubMed] [Google Scholar]
- 82.Zhu L et al. (2019) PTEN S-nitrosylation by NOS1 inhibits autophagy in NPC cells. Cell Death Dis 10 (4), 306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kwak YD et al. (2010) NO signaling and S-nitrosylation regulate PTEN inhibition in neurodegeneration. Mol Neurodegener 5, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Numajiri N et al. (2011) On-off system for PI3-kinase-Akt signaling through S-nitrosylation of phosphatase with sequence homology to tensin (PTEN). Proc Natl Acad Sci U S A 108 (25), 10349–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Choi MS et al. (2014) Transnitrosylation from DJ-1 to PTEN attenuates neuronal cell death in parkinson’s disease models. J Neurosci 34 (45), 15123–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Clement S et al. (2001) The lipid phosphatase SHIP2 controls insulin sensitivity. Nature 409 (6816), 92–7. [DOI] [PubMed] [Google Scholar]
- 87.Montagnani M et al. (2001) Insulin-stimulated activation of eNOS is independent of Ca2+ but requires phosphorylation by Akt at Ser(1179). J Biol Chem 276 (32), 30392–8. [DOI] [PubMed] [Google Scholar]
- 88.Cerf ME (2013) Beta cell dysfunction and insulin resistance. Front Endocrinol (Lausanne) 4, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kahn SE et al. (2006) Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 444 (7121), 840–6. [DOI] [PubMed] [Google Scholar]
- 90.Lackey DE and Olefsky JM (2016) Regulation of metabolism by the innate immune system. Nat Rev Endocrinol 12 (1), 15–28. [DOI] [PubMed] [Google Scholar]
- 91.Kim JH et al. (2009) Interleukin-6 and insulin resistance. Vitam Horm 80, 613–33. [DOI] [PubMed] [Google Scholar]
- 92.Salvado L et al. (2015) Targeting endoplasmic reticulum stress in insulin resistance. Trends Endocrinol Metab 26 (8), 438–48. [DOI] [PubMed] [Google Scholar]
- 93.Boucher J et al. (2014) Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb Perspect Biol 6 (1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Copps KD and White MF (2012) Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia 55 (10), 2565–2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Perreault M and Marette A (2001) Targeted disruption of inducible nitric oxide synthase protects against obesity-linked insulin resistance in muscle. Nat Med 7 (10), 1138–43. [DOI] [PubMed] [Google Scholar]
- 96.Katashima CK et al. (2017) iNOS promotes hypothalamic insulin resistance associated with deregulation of energy balance and obesity in rodents. Sci Rep 7 (1), 9265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Shinozaki S et al. (2011) Liver-specific inducible nitric-oxide synthase expression is sufficient to cause hepatic insulin resistance and mild hyperglycemia in mice. J Biol Chem 286 (40), 34959–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kaneki M et al. (2007) Nitrosative stress and pathogenesis of insulin resistance. Antioxid Redox Signal 9 (3), 319–29. [DOI] [PubMed] [Google Scholar]
- 99.Yasukawa T et al. (2005) S-nitrosylation-dependent inactivation of Akt/protein kinase B in insulin resistance. J Biol Chem 280 (9), 7511–8. [DOI] [PubMed] [Google Scholar]
- 100.Carvalho-Filho MA et al. (2005) S-nitrosation of the insulin receptor, insulin receptor substrate 1, and protein kinase B/Akt: a novel mechanism of insulin resistance. Diabetes 54 (4), 959–67. [DOI] [PubMed] [Google Scholar]
- 101.Carvalho-Filho MA et al. (2006) Targeted disruption of iNOS prevents LPS-induced S-nitrosation of IRbeta/IRS-1 and Akt and insulin resistance in muscle of mice. Am J Physiol Endocrinol Metab 291 (3), E476–82. [DOI] [PubMed] [Google Scholar]
- 102.Ropelle ER et al. (2013) Targeted disruption of inducible nitric oxide synthase protects against aging, S-nitrosation, and insulin resistance in muscle of male mice. Diabetes 62 (2), 466–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Crunfli F et al. (2018) NO-Dependent Akt Inactivation by S-Nitrosylation as a Possible Mechanism of STZ-Induced Neuronal Insulin Resistance. J Alzheimers Dis 65 (4), 1427–1443. [DOI] [PubMed] [Google Scholar]
- 104.Wu M et al. (2009) Aging-associated dysfunction of Akt/protein kinase B: S-nitrosylation and acetaminophen intervention. PLoS One 4 (7), e6430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lee YS et al. (2014) Increased adipocyte O2 consumption triggers HIF-1alpha, causing inflammation and insulin resistance in obesity. Cell 157 (6), 1339–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Carvalho-Filho MA et al. (2009) Aspirin attenuates insulin resistance in muscle of diet-induced obese rats by inhibiting inducible nitric oxide synthase production and S-nitrosylation of IRbeta/IRS-1 and Akt. Diabetologia 52 (11), 2425–34. [DOI] [PubMed] [Google Scholar]
- 107.Hundal RS et al. (2002) Mechanism by which high-dose aspirin improves glucose metabolism in type 2 diabetes. J Clin Invest 109 (10), 1321–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tsuzuki T et al. (2015) Voluntary Exercise Can Ameliorate Insulin Resistance by Reducing iNOS-Mediated S-Nitrosylation of Akt in the Liver in Obese Rats. PLoS One 10 (7), e0132029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Pauli JR et al. (2008) Acute physical exercise reverses S-nitrosation of the insulin receptor, insulin receptor substrate 1 and protein kinase B/Akt in diet-induced obese Wistar rats. J Physiol 586 (2), 659–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Muniyappa R and Sowers JR (2013) Role of insulin resistance in endothelial dysfunction. Rev Endocr Metab Disord 14 (1), 5–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Potenza MA et al. (2005) Insulin resistance in spontaneously hypertensive rats is associated with endothelial dysfunction characterized by imbalance between NO and ET-1 production. Am J Physiol Heart Circ Physiol 289 (2), H813–22. [DOI] [PubMed] [Google Scholar]
- 112.Montagnani M et al. (2002) Inhibition of phosphatidylinositol 3-kinase enhances mitogenic actions of insulin in endothelial cells. J Biol Chem 277 (3), 1794–9. [DOI] [PubMed] [Google Scholar]
- 113.Mukai Y et al. (2007) Phosphatidylinositol 3-kinase/protein kinase Akt negatively regulates plasminogen activator inhibitor type 1 expression in vascular endothelial cells. Am J Physiol Heart Circ Physiol 292 (4), H1937–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Patel P and Woodgett JR (2017) Glycogen Synthase Kinase 3: A Kinase for All Pathways? Curr Top Dev Biol 123, 277–302. [DOI] [PubMed] [Google Scholar]
- 115.Dibble CC and Cantley LC (2015) Regulation of mTORC1 by PI3K signaling. Trends Cell Biol 25 (9), 545–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wang SB et al. (2018) Protein S-Nitrosylation Controls Glycogen Synthase Kinase 3beta Function Independent of Its Phosphorylation State. Circ Res 122 (11), 1517–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lopez-Rivera E et al. (2014) Inducible nitric oxide synthase drives mTOR pathway activation and proliferation of human melanoma by reversible nitrosylation of TSC2. Cancer Res 74 (4), 1067–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lee S and Dong HH (2017) FoxO integration of insulin signaling with glucose and lipid metabolism. J Endocrinol 233 (2), R67–R79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.DiPilato LM et al. (2015) The Role of PDE3B Phosphorylation in the Inhibition of Lipolysis by Insulin. Mol Cell Biol 35 (16), 2752–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Scherer PE (2016) The Multifaceted Roles of Adipose Tissue-Therapeutic Targets for Diabetes and Beyond: The 2015 Banting Lecture. Diabetes 65 (6), 1452–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yin R et al. (2015) Pro-inflammatory Macrophages suppress PPARgamma activity in Adipocytes via S-nitrosylation. Free Radic Biol Med 89, 895–905. [DOI] [PubMed] [Google Scholar]
- 122.Guilherme A et al. (2008) Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat Rev Mol Cell Biol 9 (5), 367–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Choi YH et al. (2006) Alterations in regulation of energy homeostasis in cyclic nucleotide phosphodiesterase 3B-null mice. J Clin Invest 116 (12), 3240–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ovadia H et al. (2011) Increased adipocyte S-nitrosylation targets anti-lipolytic action of insulin: relevance to adipose tissue dysfunction in obesity. J Biol Chem 286 (35), 30433–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yeung F et al. (2004) Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J 23 (12), 2369–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gauglitz GG et al. (2010) Post-burn hepatic insulin resistance is associated with endoplasmic reticulum (ER) stress. Shock 33 (3), 299–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Shinozaki S et al. (2014) Inflammatory stimuli induce inhibitory S-nitrosylation of the deacetylase SIRT1 to increase acetylation and activation of p53 and p65. Sci Signal 7 (351), ra106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Poitout V et al. (2010) Glucolipotoxicity of the pancreatic beta cell. Biochim Biophys Acta 1801 (3), 289–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ye R et al. (2019) Lipotoxicity and beta Cell Maintenance in Obesity and Type 2 Diabetes. J Endocr Soc 3 (3), 617–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Heimann E et al. (2010) Expression and regulation of cyclic nucleotide phosphodiesterases in human and rat pancreatic islets. PLoS One 5 (12), e14191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Dubois M et al. (2000) Expression of peroxisome proliferator-activated receptor gamma (PPARgamma) in normal human pancreatic islet cells. Diabetologia 43 (9), 1165–9. [DOI] [PubMed] [Google Scholar]
- 132.Bordone L et al. (2006) Sirt1 regulates insulin secretion by repressing UCP2 in pancreatic beta cells. PLoS Biol 4 (2), e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lin JH et al. (2008) Endoplasmic reticulum stress in disease pathogenesis. Annu Rev Pathol 3, 399–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Walter P and Ron D (2011) The unfolded protein response: from stress pathway to homeostatic regulation. Science 334 (6059), 1081–6. [DOI] [PubMed] [Google Scholar]
- 135.Hetz C (2012) The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol 13 (2), 89–102. [DOI] [PubMed] [Google Scholar]
- 136.Anholt RR and Carbone MA (2013) A molecular mechanism for glaucoma: endoplasmic reticulum stress and the unfolded protein response. Trends Mol Med 19 (10), 586–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ghosh R et al. (2019) Endoplasmic reticulum stress, degeneration of pancreatic islet beta-cells, and therapeutic modulation of the unfolded protein response in diabetes. Mol Metab 27S, S60–S68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Yang L et al. (2015) METABOLISM. S-Nitrosylation links obesity-associated inflammation to endoplasmic reticulum dysfunction. Science 349 (6247), 500–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Nakato R et al. (2015) Regulation of the unfolded protein response via S-nitrosylation of sensors of endoplasmic reticulum stress. Sci Rep 5, 14812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Schiattarella GG et al. (2019) Nitrosative stress drives heart failure with preserved ejection fraction. Nature 568 (7752), 351–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Fonseca SG et al. (2009) Endoplasmic reticulum stress in beta-cells and development of diabetes. Curr Opin Pharmacol 9 (6), 763–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Huang CJ et al. (2007) High expression rates of human islet amyloid polypeptide induce endoplasmic reticulum stress mediated beta-cell apoptosis, a characteristic of humans with type 2 but not type 1 diabetes. Diabetes 56 (8), 2016–27. [DOI] [PubMed] [Google Scholar]
- 143.Higuchi-Sanabria R et al. (2018) A Futile Battle? Protein Quality Control and the Stress of Aging. Dev Cell 44 (2), 139–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Volpi VG et al. (2016) Endoplasmic Reticulum Protein Quality Control Failure in Myelin Disorders. Front Mol Neurosci 9, 162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Zhang L et al. (2009) GRP78, but Not Protein-disulfide Isomerase, Partially Reverses Hyperglycemia-induced Inhibition of Insulin Synthesis and Secretion in Pancreatic {beta}-Cells. J Biol Chem 284 (8), 5289–98. [DOI] [PubMed] [Google Scholar]
- 146.Jang I et al. (2019) PDIA1/P4HB is required for efficient proinsulin maturation and ss cell health in response to diet induced obesity. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wilkinson B and Gilbert HF (2004) Protein disulfide isomerase. Biochim Biophys Acta 1699 (1–2), 35–44. [DOI] [PubMed] [Google Scholar]
- 148.Wadham C et al. (2007) High glucose attenuates protein S-nitrosylation in endothelial cells: role of oxidative stress. Diabetes 56 (11), 2715–21. [DOI] [PubMed] [Google Scholar]
- 149.Uehara T et al. (2006) S-nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature 441 (7092), 513–7. [DOI] [PubMed] [Google Scholar]
- 150.Hu Y et al. (2019) Endoplasmic Reticulum-Associated Degradation (ERAD) Has a Critical Role in Supporting Glucose-Stimulated Insulin Secretion in Pancreatic beta-Cells. Diabetes 68 (4), 733–746. [DOI] [PubMed] [Google Scholar]
- 151.Lopata A et al. (2020) Ubiquitination in the ERAD Process. Int J Mol Sci 21 (15). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Azzam SK et al. (2019) Genetic Associations With Diabetic Retinopathy and Coronary Artery Disease in Emirati Patients With Type-2 Diabetes Mellitus. Front Endocrinol (Lausanne) 10, 283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Fujikawa K et al. (2020) S-Nitrosylation at the active site decreases the ubiquitin conjugating activity of ubiquitin-conjugating enzyme E2 D1 (UBE2D1), an ERAD-associated protein. Biochem Biophys Res Commun 524 (4), 910–915. [DOI] [PubMed] [Google Scholar]
- 154.Choi AM et al. (2013) Autophagy in human health and disease. N Engl J Med 368 (19), 1845–6. [DOI] [PubMed] [Google Scholar]
- 155.Watada H and Fujitani Y (2015) Minireview: Autophagy in pancreatic beta-cells and its implication in diabetes. Mol Endocrinol 29 (3), 338–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Yang L et al. (2010) Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab 11 (6), 467–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Arai C et al. (2013) Trehalose prevents adipocyte hypertrophy and mitigates insulin resistance in mice with established obesity. J Nutr Sci Vitaminol (Tokyo) 59 (5), 393–401. [DOI] [PubMed] [Google Scholar]
- 158.Singh R et al. (2009) Autophagy regulates lipid metabolism. Nature 458 (7242), 1131–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Qian Q et al. (2018) S-Nitrosoglutathione Reductase Dysfunction Contributes to Obesity-Associated Hepatic Insulin Resistance via Regulating Autophagy. Diabetes 67 (2), 193–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Liu L et al. (2004) Essential roles of S-nitrosothiols in vascular homeostasis and endotoxic shock. Cell 116 (4), 617–28. [DOI] [PubMed] [Google Scholar]
- 161.Kroemer G et al. (2010) Autophagy and the integrated stress response. Mol Cell 40 (2), 280–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Ebato C et al. (2008) Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab 8 (4), 325–32. [DOI] [PubMed] [Google Scholar]
- 163.Masini M et al. (2009) Autophagy in human type 2 diabetes pancreatic beta cells. Diabetologia 52 (6), 1083–6. [DOI] [PubMed] [Google Scholar]
- 164.Wright C et al. (2016) S-Nitrosylation of Bcl-2 Negatively Affects Autophagy in Lung Epithelial Cells. J Cell Biochem 117 (2), 521–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Montagna C et al. (2016) To eat, or NOt to eat: S-nitrosylation signaling in autophagy. FEBS J 283 (21), 3857–3869. [DOI] [PubMed] [Google Scholar]
- 166.Luchsinger JA et al. (2004) Hyperinsulinemia and risk of Alzheimer disease. Neurology 63 (7), 1187–92. [DOI] [PubMed] [Google Scholar]
- 167.Sergi D et al. (2019) Diabetes, a Contemporary Risk for Parkinson’s Disease: Epidemiological and Cellular Evidences. Front Aging Neurosci 11, 302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Lee S et al. (2013) CSF and Brain Indices of Insulin Resistance, Oxidative Stress and Neuro-Inflammation in Early versus Late Alzheimer’s Disease. J Alzheimers Dis Parkinsonism 3, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.de la Monte SM and Wands JR (2008) Alzheimer’s disease is type 3 diabetes-evidence reviewed. J Diabetes Sci Technol 2 (6), 1101–13. [DOI] [PMC free article] [PubMed] [Google Scholar]