Identification of SMURF1 as a possible target for 7q21.3‐22.1 amplification detected in a pancreatic cancer cell line by in‐house array‐based comparative genomic hybridization (original) (raw)
Abstract
Pancreatic cancer (PC) cell lines provide a useful starting point for the discovery and functional analysis of genes driving the genesis and progression of this lethal cancer. To increase our understanding of the gene copy number changes in pancreatic carcinomas and to identify key amplification and deletion targets, we applied genome‐wide array‐based comparative genomic hybridization using in‐house array (MCG Cancer Array‐800) to 24 PC cell lines. Overall, the analyses revealed high genomic complexity, with several copy number changes detected in each line. Homozygous deletions (log2ratio < –2) of eight genes (clones) were seen in 14 of the 24 cell lines, whereas high‐level amplifications (log2ratio > 2) of 10 genes (clones) were detected in seven lines. Among them, we focused on high‐level amplification at 7q22.1, because target genes for this alteration remain unknown. Through precise mapping of the altered region by fluorescence in situ hybridization, determination of the expression status of genes located within those regions, and functional analysis using knockdown of the gene expression or the ectopic overexpression approach in PC cell lines, as well as immunohistochemical analyses of candidates in primary tumors of PC, we successfully identified SMURF1 as having the greatest potential as a 7q21.3‐22.1 amplification target. SMURF1 may work as a growth‐promoting gene in PC through overexpression and might be a good candidate as a therapeutic target. Our results suggest that array‐based comparative genomic hybridization analysis combined with further genetic and functional examinations is a useful approach for identifying novel tumor‐associated genes involved in the pathogenesis of this lethal disease. (Cancer Sci 2008; 99: 986–994)
Pancreatic cancer (PC), mainly ductal adenocarcinoma, has the worst prognosis of all malignant solid tumors: it is the fifth leading cause of cancer death in men and the sixth in women in Japan, as well as in the USA and European countries,( 1 , 2 ) and the worldwide 5‐year patient survival rate is less than 5%.( 1 , 3 ) The low survival rate of PC patients is primarily due to the advanced stage at which most patients are diagnosed. Few patients are eligible for surgery, and the majority die within a few months of diagnosis. Therefore, new methods for early detection, better understanding of the biological mechanisms underlying cancer progression, and cancer‐targeted treatment modalities are urgently needed to reduce the mortality from this lethal disease.
Significant progress has been made in cataloguing the genetic alterations that accompany the development and progression of PC: KRAS2 (~70–90%), INK4A/ARF (~90%), TP53 (~70%), and DPC4 (~50%) are frequently mutated somatic genes;( 4 , 5 ) however, we still lack a clear understanding of the molecular genetic events that underlie tumorigenesis and progression. In addition to these alterations involving known oncogenes or tumor‐suppressor genes (TSG), cytogenetic and molecular cytogenetic studies have revealed frequent structural and numeric chromosome abnormalities in PC. Conventional comparative genomic hybridization (CGH) analyses have identified common gains affecting chromosomes 7, 8q, 17q, 19q, and 20q, and losses of 6q, 8p, 9p, and 18q in PC.( 6 , 7 , 8 ) The significantly improved resolution of the recently developed array‐based CGH (array‐CGH) technique permits highly accurate mapping of DNA copy number changes throughout the genome,( 9 , 10 ) so array‐CGH is a promising starting point for the identification of novel candidate genes affected by genomic imbalances contributing to deregulation of the expression levels of oncogenes and TSG. Although many array‐CGH‐based or single nucleotide polymorphism array‐based copy number analyses, including our own, of PC using cell lines, xenografts, or primary tumors have been reported and have successfully identified a series of copy number changes,( 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 , 19 ) few possible target genes for these alterations have been identified, suggesting that various critical genes for the pathogenesis of PC remain unidentified.
A marker of PC is the characteristic desmoplastic reaction, resulting in a low percentage (~30%) of neoplastic cells in surgically resected tumor tissue, making it difficult to carry out genetic studies,( 20 ) because molecular changes are masked by normal elements within the samples. To circumvent the issue of neoplastic purity, widespread cell lines were used. Although cell lines, particularly those that have been in passage over prolonged periods of time, show limited genetic fidelity relative to the parent tumor due to culture‐induced genetic adaptation in vitro, we have successfully shown that tumor cell lines provide valuable resources for gene discovery and functional studies by a conventional CGH‐based approach,( 21 , 22 ) probably because their molecular and cytogenetic aberrations and biological properties reflect at least a subset of primary tumors.
To increase our understanding of the complex copy number changes occurring in PC and to identify key amplification and deletion target genes, we applied array‐CGH using an in‐house genomic array comprising 800 bacterial artificial chromosome (BAC) or P1‐artificial chromosome (PAC) clones. Each was spotted in duplicate and selected specifically to contain potential tumor‐related genes,( 10 ) on a panel of 24 PC cell lines. As in recent reports of copy number analysis of this tumor using different types of array,( 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 , 19 ) our system quantitatively detected and mapped genomic copy number alterations at higher resolution than conventional CGH. In addition, our approach resulted in the identification of SMURF1 as a possible target gene for remarkable amplification, which may contribute to tumorigenesis of the pancreas, and is expected to be a diagnostic and therapeutic target for PC.
Materials and Methods
Pancreatic cancer cell lines and primary tumors. A total of 24 PC cell lines derived from adenocarcinoma, shown in Table 1, were included. All cell lines were maintained in appropriate media supplemented with 10% fetal bovine serum and 100 units/mL penicillin with 100 µg/mL streptomycin. The mutation status of SMAD4 and SMURF1 in all coding sequences with a flanking intronic sequence was determined by direct sequencing of polymerase chain reaction (PCR) products.( 5 ) The normal pancreatic duct‐derived cell line HPDE‐6 was a generous gift from Dr M.‐S. Tsao (University of Toronto), and was maintained as described previously.( 23 )
Table 1.
Summary of 24 pancreatic cancer cell lines
| No. | Name | Histology | Source of tumor | Nucleotide change (amino acid change) | |
|---|---|---|---|---|---|
| SMAD4 | SMURF1 | ||||
| 1 | AsPC‐1 | Adenocarcinoma | Ascitic fluid | G299c (r100t) | Wild type |
| 2 | BxPC‐3 | Adenocarcinoma | Primary tumor | Homozygous deletion | Wild type |
| 3 | CAPAN‐1 | Adenocarcinoma | Liver metastasis | C1028g (s343x) | Wild type |
| 4 | CAPAN‐2 | Adenocarcinoma | Primary tumor | Wild type | Wild type |
| 5 | CFPAC‐1 | Pancreatic ductal carcinoma | Liver metastasis | Homozygous deletion | Wild type |
| 6 | HPAF‐II | Adenocarcinoma | Ascitic fluid | Wild type | Wild type |
| 7 | Hs766T | Adenocarcinoma | Primary tumor | Homozygous deletion | Wild type |
| 8 | KMP2 | Adenocarcinoma | Liver metastasis | Homozygous deletion | Wild type |
| 9 | KMP3 | Adenocarcinoma | Lymph node | Wild type | Wild type |
| 10 | KMP4 | Adenocarcinoma | Primary tumor | Wild type | Wild type |
| 11 | KMP5 | Adenocarcinoma | Liver metastasis | Wild type | Wild type |
| 12 | KMP7 | Adenocarcinoma | Unknown | Wild type | Wild type |
| 13 | KMP8 | Adenocarcinoma | Unknown | Wild type | Wild type |
| 14 | KP1N | Adenocarcinoma | Liver metastasis | Wild type | Wild type |
| 15 | KP1NL | Adenocarcinoma | Liver metastasis | Wild type | Wild type |
| 16 | KP2 | Adenocarcinoma | Primary tumor | 4‐bp deletion, 1245 (frameshift) | Wild type |
| 17 | KP3 | Adenosquamous carcinoma | Liver metastasis | G1612t (e538x) | Wild type |
| 18 | KP3L | Adenosquamous carcinoma | Liver metastasis | G1612t (e538x) | Wild type |
| 19 | KP4‐4 | Pthrp‐producing human pancreatic cancer cell line | Primary tumor | Homozygous deletion | Wild type |
| 20 | MIA‐Paca‐2 | Adenocarcinoma | Primary tumor | Wild type | Wild type |
| 21 | PANC‐1 | Adenocarcinoma | Primary tumor | Wild type | Wild type |
| 22 | PSN1 | Adenocarcinoma | Primary tumor | Homozygous deletion | Wild type |
| 23 | SU.86.86 | Pancreatic ductal carcinoma | Liver metastasis | Wild type | Wild type |
| 24 | SW1990 | Adenocarcinoma | Primary tumor | Wild type | Wild type |
Primary PC samples for genomic PCR were obtained during surgery from 19 patients, who had been diagnosed and had undergone surgery at the National Cancer Center Hospital in Tokyo, Japan, with written consent obtained from each patient. Tumor cells were obtained by laser‐captured microdissection (LCM) with a PixCell II LCM system (Arcturus Engineering, Mountain View, CA, USA).( 24 ) Genomic DNA was isolated and amplified by adaptor‐ligation‐mediated PCR after end filling, as described by Tanabe et al.( 25 ) As a control for normal gene copy numbers, DNA from lymphocytes of healthy men was used. Formalin‐fixed, paraffin‐embedded tissue specimens of primary PC for immunohistochemical analysis were obtained from 106 patients, who had been diagnosed and had undergone surgery at the National Cancer Center Hospital in Tokyo, Japan, with written consent obtained from each patient. The median follow‐up period was 22 months (range 2–103 months). This study was approved by the local ethics committee.
Array‐CGH analysis. Our MCG Cancer Array‐800( 10 , 26 ) contains 800 BAC or PAC clones carrying genes or sequence‐tagged site markers of potential importance in cancer genesis or progression. Hybridization was carried out as described elsewhere.( 26 ) Hybridized slides were scanned with a GenePix 4000B (Axon Instruments, Foster City, CA, USA). Fluorescence ratios were normalized so that the mean of the middle third of log2ratios across the array was zero. Average ratios that deviated significantly (>2 SD) from zero were considered abnormal.
Fluorescence in situ hybridization. Metaphase chromosome slides were prepared from normal male lymphocytes and each PC cell line. BAC DNA was labeled with biotin‐16‐dUTP or digoxigenin‐11‐dUTP by nick‐translation (Roche Diagnostics, Tokyo, Japan). Fluorescent detection of hybridization signals was carried out as described elsewhere.( 26 ) Cells were counterstained with 4′,6‐diamidino‐2‐phenylindole.
Genomic PCR. We screened 24 cell lines and 19 primary PC for homozygous losses by genomic PCR. Genomic DNA from primary PC was prepared by LCM as described above. All primer sequences used in the present study are available on request.
Reverse transcription‐PCR. Single‐stranded cDNA was generated from total RNA, and amplified with primers specific for genes of interest. The glyceraldehyde‐3‐phosphate dehydrogenase gene was amplified at the same time to allow estimation of the efficiency of cDNA synthesis.
Knockdown of gene expression by small interfering RNA and cell‐growth assay. Small interfering RNA (siRNA) for knockdown of the expression of SMURF,( 27 ) TRRAP (sc‐36746; Santa Cruz Biotechnology, Santa Cruz, CA, USA), and the control luciferase gene,( 28 ) were introduced into cells using LipofectAMINE 2000 or RNAiMAX (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. Knockdown of the expression of each protein was confirmed 48–72 h after transfection by western blot analysis using anti‐SMURF1 (H‐60) or TRRAP (H‐300) antibody (Santa Cruz Biotechnology). To measure cell numbers, 2 × 103 cells were seeded in 96‐well plates the day before siRNA transfection, and viable cells were assessed by the colorimetric water‐soluble tetrazolium salt assay (Cell counting kit‐8; Dojindo Laboratories, Kumamoto, Japan).( 28 ) Experiments were repeated two times, and carried out in triplicate each time.
Colony‐formation assay after ectopic gene expression. A plasmid expressing MYC‐tagged SMURF1 (pCMV‐Tag3‐SMURF1) was obtained by cloning the reverse transcription (RT)‐PCR product of the full coding sequence of SMURF1 into the pCMV‐Tag3 expression vector (Stratagene, La Jolla, CA, USA) in frame along with the Myc‐epitope. pCMV‐Tag3‐SMURF1, or the empty vector (pCMV‐Tag3‐mock) control, was transfected into cells for colony‐formation assays as described elsewhere.( 28 , 29 ) Expression of Myc‐tagged SMURF1 protein in transiently transfected cells was confirmed 48 h after transfection by western blot analysis using anti‐Myc tag antibody (Cell Signaling Technology, Beverly, MA, USA).( 29 ) After 2 weeks of incubation in six‐well plates with appropriate concentrations of G418, cells were fixed with 70% ethanol and stained with crystal violet.
Immunohistochemistry. Formalin‐fixed, paraffin‐embedded surgical specimens were sliced into sections of 5 µm thickness. The sections were deparaffinized with xylene and graded ethanol, and immersed in methanol containing 0.3% hydrogen peroxide. After epitope retrieval by autoclaving, the sections were incubated with anti‐SMURF1 or anti‐TRRAP antibody at a dilution of 1 : 100. Sections were incubated with a biotinylated secondary antibody against rabbit IgG and then with Vectastain ABC reagent (Vector Laboratories, Burlingame, CA, USA). Sections were immersed in 0.05% diaminobenzidine tetrahydrochloride solution containing 0.01% hydrogen peroxide, and counterstained with hematoxylin. Tumors containing more than 50% (SMURF1) or 10% (TRRAP) positive tumor cells in the representative largest section were considered to be positive.
Statistical analysis. Differences between subgroups were tested with the Mann–Whitney _U_‐test. Correlations between SMURF1 immunoreactivity in primary PC and the clinicopathological variables pertaining to the corresponding patients were analyzed for statistical significance with χ2 or Fisher's exact test. For the analysis of overall patient survival after surgery, which was calculated from the date of surgery to the date of the latest follow‐up visit or death, Kaplan–Meier survival curves were constructed for groups based on univariate predictors, and statistical differences between the groups were tested with the log‐rank test. Differences were assessed with a two‐sided test, and considered significant at the P < 0.05 level.
Results
Array‐CGH analysis of PC cell lines. We assessed copy number alterations among the 24 PC cell lines using the same batch of in‐house MCG Cancer Array‐800. Figure 1 shows the frequencies of copy number gains and losses across the entire genomes of all 24 lines. 2, 3 list the clones that had the most frequent gains or losses in this series as well as those with high‐level amplifications or homozygous deletions, respectively. Some degree of gain or loss was seen in every cell line. Our array‐CGH predicted frequent copy number gains for 1p, 3q, 6p, 7p, 8q, 11q, 12p, 20p, and 20q, and frequent losses for 3p, 4q, 6p, 6q, 8p, 9p, 10p, 13q, 17p, and 18q. High‐level amplifications (log2ratio > 2) were detected in 7 of the 24 PC cell lines, and 10 genes (clones) were represented. Among them, two genes, SUPT5H and AKT2 (19q13.2), were detected as high‐level amplifications in more than two cell lines each. Homozygous deletions (log2ratio < –2) were seen in 14 cell lines. Of them, MTAP and CDKN2A/p16 at 9p21.3 were observed in 10 and 12 cell lines, respectively, N33 (8p22) in two cell lines, and TEK (9p21.2), MLLT3 (9p21.3), DEC1 (9q33.1), CDH23 (10q22.1), and SMAD4 (18q21.1) in one cell line. These homozygous losses were confirmed by genomic PCR in cell lines (data not shown). In addition, homozygous deletion of those genes was frequently (15.8–47.4%) observed in our panel of primary PC tumors, although only a small number of cases were available for genomic PCR (Fig. 1b), suggesting that homozygous losses detected in cell lines are not likely to be changes simply occurring during their establishment and passage of cultures.
Figure 1.
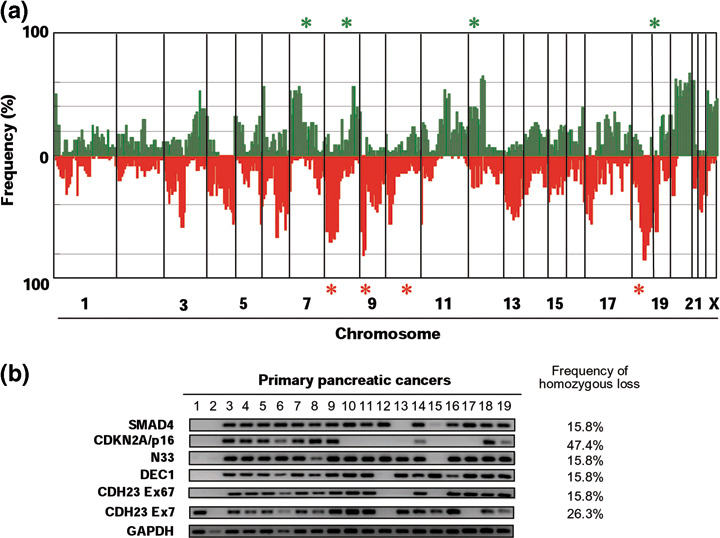
(a) Copy number analysis of pancreatic cancer (PC) cell lines using array‐based comparative genomic hybridization (array‐CGH) and (b) confirmation of homozygously deleted regions in primary tumors of PC. (a) Genome‐wide frequencies of copy number gains (above 0, green) and losses (below 0, red) in 24 PC cell lines. Clones are ordered from chromosomes 1–22, X, and Y, and within each chromosome on the basis of the UCSC mapping position (http://genome.ucsc.edu/, version May, 2004). Green asterisks, clones with at least one high‐level amplification; red asterisks, clones with at least one homozygous deletion. (b) Representative images from genomic polymerase chain reaction experiments showing glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH), the functional control, and SMAD4, p16, N33, DEC1, and CDH23 exons 7 and 67 in 19 laser‐captured microdissection (LCM)‐treated primary PC. The frequency of homozygous deletion in each suppressor gene is shown.
Table 2.
Most frequently gained and lost genes (clones) detected by in‐house bacterial artificial chromosome (BAC) array among 24 pancreatic cancer cell lines †
| Alteration | Gene/Marker | Locus | Frequency (%) |
|---|---|---|---|
| Gain | CDC2L1 | 1p36.33 | 52 |
| EIF4G | 3q27.1 | 54 | |
| DEK | 6p22.3 | 58 | |
| PMS2 | 7p22.1 | 54 | |
| CDC10 | 7p14.2 | 52 | |
| TCRG | 7p14.1 | 52 | |
| GLI3 | 7p14.2 | 52 | |
| IGFBP1 | 7p13 | 58 | |
| EGFR | 7p11.2 | 56 | |
| MYC | 8q24.21 | 58 | |
| PVT1 | 8q24.21 | 56 | |
| PPP1CA | 11q13.1 | 52 | |
| KRAS | 12p12.1 | 62 | |
| SSPN (KRAG) | 12p12.1 | 64 | |
| PTHLH | 12p11.22 | 62 | |
| BCLX | 20q11.21 | 60 | |
| HCK | 20q11.21 | 54 | |
| TGIF2 | 20q11.23 | 56 | |
| SRC | 20q11.23 | 50 | |
| MYBL2 | 20q13.12 | 54 | |
| CD40 (TNFRSF5) | 20q13.12 | 56 | |
| ELMO2 | 20q13.12 | 54 | |
| PREX1 | 20q13.13 | 56 | |
| PTPN1 | 20q13.13 | 64 | |
| ZNF217 | 20q13.2 | 52 | |
| BCAS1 | 20q13.2 | 56 | |
| TFAP2C | 20q13.31 | 56 | |
| BIRC7 | 20q13.33 | 58 | |
| TNFRSF6B | 20q13.33 | 56 | |
| PCTK1 | Xp11.3 | 54 | |
| Loss | PTPRG | 3p14.2 | 52 |
| MAGI1 (BAIAP1) | 3p14.1 | 58 | |
| CASP3 | 4q35.1 | 56 | |
| EEF1E1 | 6p24.3 | 50 | |
| MAP3K7 | 6q15 | 60 | |
| ESR1 | 6q25.1 | 50 | |
| D8S504 | 8ptel | 64 | |
| BLK | 8p23.1 | 58 | |
| DLC1 | 8p22 | 60 | |
| NAT1 (AAC) | 8p22 | 72 | |
| NAT2 | 8p22 | 64 | |
| TUSC3 (N33) | 8p22 | 72 | |
| LZTS1 | 8p21.3 | 64 | |
| TNFRSF10B | 8p21.3 | 64 | |
| LPL | 8p21.3 | 58 | |
| NKX3‐1 (NKXA) | 8p21.2 | 56 | |
| NRG1 | 8p12 | 58 | |
| D9S913 | 9ptel | 64 | |
| JMJD2C (GASC1) | 9p24.1 | 64 | |
| JAK2 | 9p24.1 | 54 | |
| MTAP | 9p21.3 | 82 | |
| MLLT3 | 9p21.3 | 58 | |
| CDKN2A/p16 | 9p21.3 | 78 | |
| TEK | 9p21.2 | 68 | |
| VIM | 10p12.33 | 52 | |
| stSG27915 | 10qtel | 52 | |
| KLF12 | 13q22.1 | 50 | |
| RH68621 | 17p11.2 | 52 | |
| SS18 | 18q11.2 | 56 | |
| SMAD2 | 18q21.1 | 58 | |
| SMAD7 | 18q21.1 | 56 | |
| DCC | 18q21.2 | 82 | |
| SMAD4 | 18q21.2 | 70 | |
| GRP | 18q21.32 | 70 | |
| MALT1 | 18q21.32 | 60 | |
| BCL2 | 18q21.33 | 56 | |
| FVT1 | 18q21.33 | 64 | |
| SCCA1, SCCA2 | 18q21.33 | 58 | |
| WI‐7336 (PI5) | 18q21.33 | 56 | |
| CTDP1, SHGC‐145820 | 18qtel | 52 | |
| stSG42796 | 19p13.2 | 60 | |
| Loss | BAIAP1 | 3p14.1 | 58 |
| PTPRG | 3p14.2 | 52 | |
| CASP3 | 4q35.1 | 56 | |
| EEF1E1 | 6p24.3 | 50 | |
| MAP3K7 | 6q15 | 60 | |
| ESR1 | 6q25 | 50 | |
| NKX3A | 8p21 | 56 | |
| N33 | 8p22 | 72 | |
| LZTS1 | 8p22 | 64 | |
| LPL | 8p22 | 58 | |
| NRG1 | 8p22‐p11 | 58 | |
| TNFRSF10B | 8p22‐p21 | 64 | |
| DLC1 | 8p22‐p21.3 | 60 | |
| BLK | 8p23.1 | 58 | |
| AAC1 | 8p23.1‐p21.3 | 72 | |
| NAT2 | 8p23.1‐p21.3 | 64 | |
| D8S504 | 8ptel | 64 | |
| CDKN2A/p16 | 9p21 | 78 | |
| TEK | 9p21 | 68 | |
| MTAP | 9p21.3 | 82 | |
| MLLT3 | 9p22 | 58 | |
| GASC1 | 9p23 | 64 | |
| JAK2 | 9p24 | 54 | |
| D9S913 | 9ptel | 64 | |
| VIM | 10p13 | 52 | |
| stSG27915 | 10qtel | 52 | |
| KLF12 | 13q22.1 | 50 | |
| RH68621 | 17p11.2 | 52 | |
| SSXT | 18q11.2 | 56 | |
| DCC | 18q21 | 82 | |
| SMAD4 | 18q21 | 70 | |
| GRP | 18q21 | 70 | |
| MALT1 | 18q21 | 60 | |
| MADH2 | 18q21 | 58 | |
| SMAD7 | 18q21 | 56 | |
| MLL1 | 18q21 | 56 | |
| FVT1 | 18q21.3 | 64 | |
| SCCA1, SCCA2 | 18q21.3 | 58 | |
| PI5 | 18q21.3 | 56 | |
| BCL2 | 18q22 | 56 | |
| CTDP1, SHGC‐145820 | 18qtel | 52 | |
| stSG42796 | 19p12‐p13 | 60 |
Table 3.
Genes (clones) showing high‐level amplification and homozygous deletions detected by in‐house bacterial artificial chromosome (BAC) array among 24 pancreatic cancer cell lines
| Alteration | Gene | Locus | No. † | Cell line | % |
|---|---|---|---|---|---|
| High‐level amplification (log2 > 2.0) | TRRAP | 7q22.1 | 1 | AsPC1 | 4 |
| SMURF1 | 7q22.1 | 1 | AsPC1 | 4 | |
| PDAP1 | 7q22.1 | 1 | AsPC1 | 4 | |
| MYC | 8q24.21 | 1 | PSN1 | 4 | |
| PVT1 | 8q24.21 | 1 | PSN1 | 4 | |
| KRAS | 12p12.1 | 1 | PSN1 | 4 | |
| SSPN (KRAG) | 12p12.1 | 1 | HPAF2 | 4 | |
| SUPT5H | 19q13.2 | 4 | KP1N, KP1NL, PANC1, SU.86.86 | 17 | |
| AKT2 | 19q13.2 | 4 | KP1N, KP1NL, PANC1, SU.86.86 | 17 | |
| MIA | 19q13.2 | 1 | SU.86.86 | 4 | |
| Homozygous deletion (log2 < –2.0) | TUSC3 (N33) | 8p22 | 2 | KMP‐3, MIA PaCa‐2 | 8 |
| MLLT3 | 9p21.3 | 1 | CAPAN2 | 4 | |
| MTAP | 9p21.3 | 10 | BxPC‐3, CAPAN2, KMP‐3, KMP‐4, KMP‐5, KMP‐7, KMP‐8, KP4‐4, MIA PaCa‐2, SU.86.86 | 42 | |
| CDKN2A/p16 | 9p21.3 | 12 | BxPC‐3, CAPAN2, KMP‐3, KMP‐4, KMP‐5, KMP‐7, KMP‐8, KP1N, KP1NL, KP4‐4, MIA PaCa‐2, SU.86.86 | 50 | |
| TEK | 9p21.2 | 1 | KMP‐5 | 4 | |
| DEC1 | 9q33.1 | 1 | BxPC3 | 4 | |
| CDH23 | 10q22.1 | 1 | BxPC3 | 4 | |
| SMAD4 | 18q21.2 | 1 | KMP‐5 | 4 |
Copy number aberrations revealed through array‐CGH were mostly consistent with those of other reports using conventional CGH;( 6 , 7 , 8 ) however, our array‐CGH analysis also disclosed additional regions that had never been pointed out by conventional CGH, such as small gains, losses, and homozygous deletions, as reported in recent studies using various microarray‐based methods, including BAC array‐CGH.( 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 , 19 ) To identify novel candidates for PC‐associated genes, we paid attention to high‐level amplifications, which are likely to be markers of oncogenes.
Possible target genes identified in 7q21.3‐22.1 amplified regions. Among the amplified genes or regions identified in 24 PC cell lines, several high‐level amplifications corresponded to known oncogenes. Amplifications of MYC at 8q24,( 30 ) KRAS at 12p12.1,( 30 ) and AKT2 at 19q13.2( 31 ) have been reported in PC. In addition, KRAG, PVT, and SUPT5H are likely to be coamplified with KRAS, MYC, and AKT2, respectively, because clones containing KRAS, MYC, or AKT2 genes were always affected in cell lines with KRAG, PVT, or SUPT5H amplification, respectively (Table 3). However, no target gene within the 7q22.1 amplicon detected in AsPC‐1 cells has ever been noted, although this amplification in the same cell line has been reported previously.( 12 , 15 ) Therefore, we focused on the 7q22.1 amplification around three BAC clones containing SMURF1, TRRAP, and PDAP1, which was spotted on our MCG Cancer Array‐800 (Fig. 2a).
Figure 2.
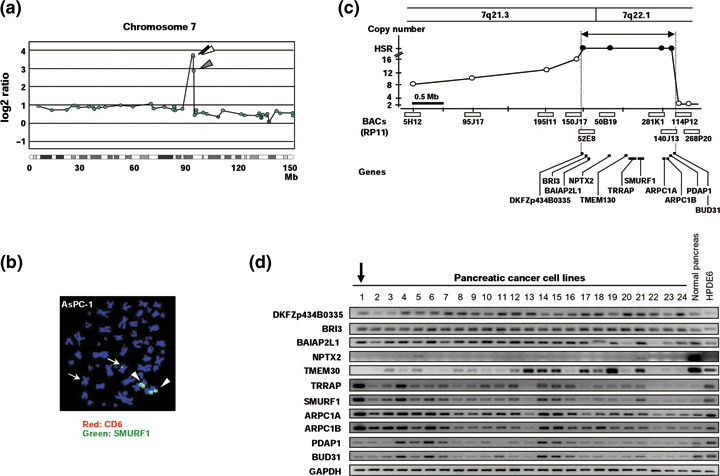
Amplification at 7q21.3‐22.1 in pancreatic cancer (PC) cell line. (a) Representative copy number profiles of chromosome 7 of AsPC‐1 cells in which array‐based comparative genomic hybridization (array‐CGH) analysis identified high‐level amplifications of SMURF1, TRRAP, and PDAP1 at 7q22.1 (arrowheads). Blue closed circles indicate genes (clones) showing a normal copy number ratio (–0.4 ≤ log2ratio ≤ 0.4), whereas green closed circles indicate those showing an increased copy number ratio (log2ratio > 0.4). (b) Representative fluorescence in situ hybridization (FISH) images with a bacterial artificial chromosome (BAC) clone containing the SMUFF1 gene (green signal, arrows and arrowheads) and a control BAC clone containing the CD6 gene (red signals, arrows) hybridized to metaphase chromosomes from AsPC‐1 cells, which showed a remarkably increased copy number of SMURF1 (arrow heads) with a homogeneously staining region (HSR) pattern. (c) Amplicon map of 7q21.3‐22.1 in the AsPC‐1 cell line. BAC used for FISH and their copy number are indicated as open bars and circles, respectively: those with >20 copies within the peak of HSR and outside of HSR in AsPC‐1 cells are shown in closed and open circles, respectively. Eleven genes located within the peak of amplification (closed arrow) showing an HSR pattern in AsPC‐1 cells are indicated as bars. (d) Expression of 11 genes located within the 7q21.3‐22.1 amplicon in 24 PC cell lines determined by reverse transcription–polymerase chain reaction. 1, AsPC‐1; 2, BxPC‐3; 3, Capan‐1; 4, Capan‐2; 5, CFPAC‐1; 6, HPAF‐II; 7, Hs766T; 8, KMP2; 9, KMP3; 10, KMP4; 11, KMP5; 12, KMP7; 13, KMP8; 14, KP1N; 15, KP1NL; 16, KP2; 17, KP3; 18, KP3L; 19, KP4‐4; 20, MIA‐Paca‐2; 21, PANC‐1; 22, PSN1; 23, SU.86.86; and 24, SW1990. Note that four genes, TRRAP, SMURF1, ARPC1A, and ARPC1B, showed remarkably increased expression in AsPC‐1 cells (arrow), and increased expression in several other cell lines compared to normal pancreas and normal pancreatic ductal cell‐derived line HPDE6.
In order to construct a detailed amplicon map, we carried out a series of fluorescence in situ hybridization (FISH) analyses using the AsPC‐1 cell line (Fig. 2b,c). Four BAC (RP11–52E8, 50B19, 81K1, and 140J13) produced the highest number of signals as homogeneously staining regions (HSR) on marker chromosomes (Fig. 2b). Fewer signals were detected with the remaining six BAC (RP11–5H12, 95J17, 95I14, 150J17, 114P12, and 268P20), suggesting that they are located outside the amplicon (Fig. 2c). Therefore, the peak of the amplified region could be defined between BAC RP11‐52E8 and 140J13 at approximately 1.5 around 7q21.3‐22.1, and contained 11 transcripts, obtained from the public databases (http://www.ncbi.nlm.nih.gov/ and http://genome.ucsc.edu/, Fig. 2c). To identify the most likely target genes, we determined the correlation between gene amplification and the expression status of each gene. Among 11 transcripts, only four genes, SMURF1, TRRAP, ARPC1A, and ARPC1B, were remarkably overexpressed in the AsPC‐1 cell line compared with other cell lines, normal pancreas, and the HPDE6 cell line by RT‐PCR (Fig. 2d). In addition, the expression levels of TRRAP and SMURF1 mRNA revealed a statistically significant difference between cell lines with amplification or gain and those without gain according to array‐CGH (P = 0.005 and P = 0.032, respectively, Mann–Whitney _U_‐test); the other nine genes showed none (data not shown), as targets for gene amplification are likely to show a positive correlation between the copy number and expression,( 14 , 21 , 22 , 26 ) suggesting that TRRAP and SMURF1 might be the most likely target genes activated through the gene amplification mechanism.
To further disclose the potential role of SMURF1 and TRRAP in pancreas tumorigenesis, we first investigated their functional influences on PC cell growth in vitro by means of: (a) a cell‐growth assay after knockdown of gene expression using specific siRNA; and (b) a colony‐formation assay after transient transfection of an expression construct.( 29 ) Knockdown of SMURF1 by siRNA against this gene (_SMURF1_‐siRNA) at 50 nM showed a suppressive effect on SMURF1 protein expression and the growth of AsPC‐1 cells compared with mock transfection (transfection reagent alone) as well as control siRNA against luciferase (_Luc_‐siRNA) (Fig. 3a). A significant growth‐suppressing effect by SMURF1 knockdown was observed on the KMP4 cell line expressing a lower level of SMURF1 mRNA and protein compared with the AsPC‐1 cell line, but there was little effect on the KP3 cell line showing almost no expression of SMURF1 mRNA and protein (2, 3). Knockdown of TRRAP expression by siRNA against this gene (_TRRAP_‐siRNA) at 50 nM also showed a suppressive effect on TRRAP protein expression and the growth of AsPC‐1 cells compared with mock transfection and Luc‐_siRNA, whereas there was little effect on the KP3 cell line showing almost no expression of TRRAP mRNA and protein (2, 3). Because the expression construct was available only for the SMURF1 gene due to the size of the coding region of genes, we carried out the colony‐formation assay only for SMURF1. Transiently transfected pCMV‐Tag3‐_SMURF1 ectopically expressed Myc‐tagged SMURF1 protein and produced markedly more colonies than control plasmids (pCMV‐Tag3‐empty vector) 2 weeks after transfection and following selection by an appropriate concentration of G418 in KP‐3 and PSN1 cell lines, which showed a relatively lower expression level of SMURF1 mRNA (Fig. 4e). These results suggest that, in these two genes, at least SMURF1 exerts a growth‐promoting effect on PC cells, and its overexpression through gene amplification or other mechanisms may contribute to pancreatic carcinogenesis. No somatic mutation of SMURF1 was identified in PC cell lines (Table 1).
Figure 3.
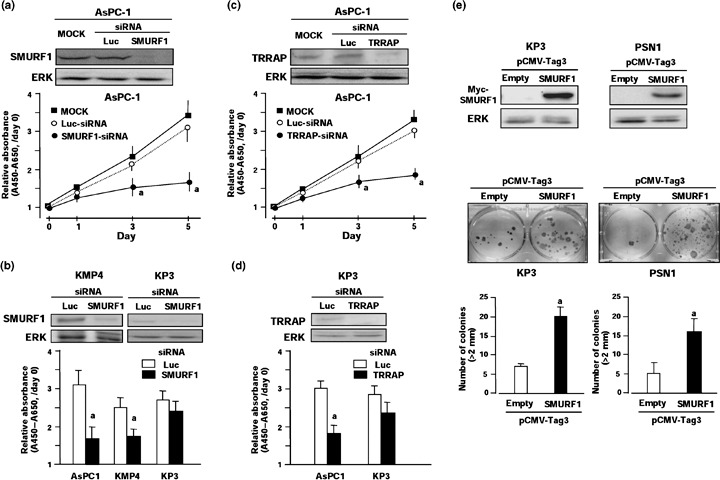
Growth‐promoting effects of SMURF1 and TRRAP1 on pancreatic cancer (PC) cell lines. (a) Effect of _SMURF1_‐small interfering RNA (siRNA) on growth of PC cell lines. AsPC‐1 cells were treated with 50 nM siRNA for SMURF1 (_SMURF1_‐siRNA) or control luciferase (_Luc_‐siRNA), or transfection reagent alone (mock). Upper, expression levels of SMURF1 protein 3 days after transfection were determined by western blotting using a specific antibody. Lower, relative cell number was determined by water‐soluble tetrazolium salt (WST) assay at the indicated times after transfection. Relative absorbance levels were calculated against the absorbance of cells before transfection (day 0). Data are the means ± SD of three separate experiments, each carried out in triplicate. Statistical analysis used the Mann–Whitney _U_‐test: (a) _SMURF_‐siRNA versus _Luc_‐siRNA, P < 0.05. (b) KMP4 and KP3 cell lines were treated with 50 nM _SMURF1_‐siRNA or control _Luc‐_siRNA, and analyses were carried out as described in (a). Relative absorbance levels of these cell lines 5 days after transfection are shown with the result of AsPC‐1 cell line. (c) Effect of _TRRAP_‐siRNA on the number of AsPC‐1 cells. AsPC‐1 cells were treated with 50 nM siRNA for _TRRAP_ (_TRRAP_‐siRNA) or control _luciferase_ (_Luc_‐siRNA), or transfection reagent alone (mock). Analyses were carried out as described in (a). (d) KP3 cells were treated with 50 nM _TRRAP_‐siRNA or control _Luc‐_siRNA, and analyses were carried out as described in (a). Relative absorbance levels in KP3 cell line 5 days after transfection are shown with the result of AsPC‐1 cell line. (e) Colony‐formation assay after transient ectopic expression of SMURF1 protein in KP3 and PSN1 cell lines. Myc‐tagged constructs containing the full coding sequence of _SMURF1_ (pCMV‐Tag3‐_SMURF1_) or empty vector (pCMV‐Tag3‐mock) as a control were transfected into cells, which showed a relatively low expression level of _SMURF1_. Upper, western blot analysis was carried out using 10 µg of protein extract and anti‐Myc tag antibody 48 h after transfection. Lower, 2 weeks after transfection of expression construct and subsequent selection of drug‐resistant colonies with appropriate concentrations of G418 in six‐well plates, the colonies formed were stained. Colonies >2 mm were counted, and the results are presented as the mean ± SD of three separate experiments, each carried out in triplicate. Statistical analysis used the Mann–Whitney U_‐test: (a) pCMV‐Tag3‐_SMURF1 versus pCMV‐Tag3‐mock, P < 0.05.
Figure 4.
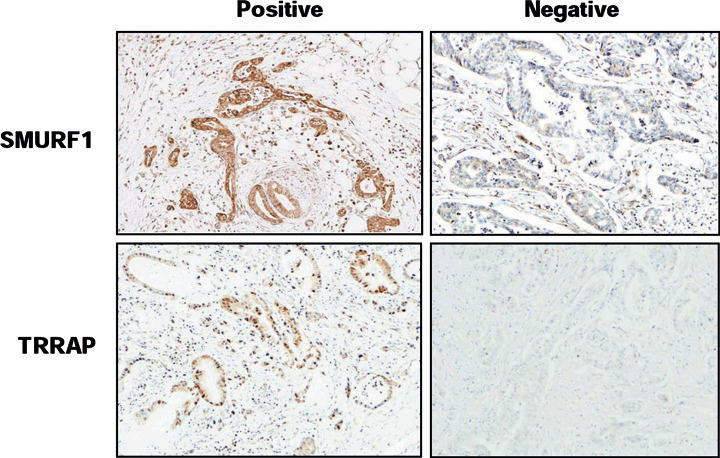
Expression of SMURF1 and TRRAP1 in primary pancreatic cancer (PC). Representative images of positive (left) and negative (right) immunohistochemical staining of SMURF1 (upper) and TRRAP (lower) proteins in primary tumors of PC (×100).
As antibodies specific to SMURF1 and TRRAP proteins useful for immunohistochemistry were available, we next examined their protein expression in primary PC by immunohistochemistry (Fig. 4a). Of 106 primary PC tumors, cytoplasmic SMURF1 immunoreactivity was clearly observed in 49 tumors (46%), although weak SMURF1 immunoreactivity was also detected in the cytoplasm of normal pancreatic ductal cells (data not shown). A diffuse staining pattern of SMURF1 was observed in most SMURF1‐positive PC tumors. No significant relationship was found between the level of SMURF1 expression and the age (<65 or 65 years, P = 0.797) or sex (P = 0.237) of patients, degree of differentiation (well, moderate, or poor, P = 0.671), or venous, lymph vessel, or perineural invasion status (negative or positive; P = 0.601, 910, and 0.488, respectively). Kaplan–Meier survival curves for 106 cases of PC showed no association between increased SMURF1 expression in PC and overall survival (P = 0.366, data not shown). In TRRAP immunoreactivity, however, preliminary immunostaining using a small number of samples detected only 2 of 33 PC tumors (6%); therefore, we did not carry out further analysis of all available cases using TRRAP.
Discussion
Pancreatic cancer remains a highly insidious and deadly malignancy, despite attempts to elucidate the genetic determinants responsible for tumorigenesis in the ductal epithelium. Understanding the molecular basis of the initiation and progression of PC is an important step toward developing successful strategies for early detection of this lethal neoplasm, and for the identification of novel cellular targets for therapy. To define the location of previously unrecognized candidate oncogenes and TSG, we carried out array‐based CGH using our in‐house BAC or PAC‐array MCG Cancer Array‐800( 10 ) to map genome‐wide DNA copy number alterations in a panel of 24 PC cell lines, half of which were established from Japanese patients. Because PC produces a strong desmoplastic reaction, resulting in an underestimation of copy number alterations, the present analysis was carried out on PC cell lines. Indeed, several recent reports of microarray‐based copy number analyses in PC focused only on cell lines or xenografts because of the difficulty of avoiding contamination of normal cells.( 11 , 12 , 13 , 14 , 15 , 16 , 17 ) In addition, because remarkable chromosome imbalances, such as local high‐level amplifications and homozygous deletions, could be markers for oncogenes and TSG, respectively, we focused on those alterations.
As noted above, several microarray‐based copy number analyses of PC, especially PC cell lines, have been reported recently.( 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 , 19 ) For example, Holzmann et al. surveyed 13 cell lines (four in common with our study) using BAC arrays with low‐resolution coverage (498 clones).( 13 ) Aguirre et al.( 11 ) and Bashyam et al.( 15 ) profiled 24 and 22 cell lines, respectively, using cDNA arrays (10 and 12 lines, respectively, in common with our study). Heidenblad et al. studied 31 cell lines (nine in common with our study) using both high‐density BAC array (3565 clones) and cDNA microarray.( 12 ) Recently, high‐density single nucleotide polymorphism array was also applied to genome‐wide copy number analysis in PC.( 19 ) The most striking difference between these reported studies and our study is that, although our findings are in general agreement for the common set of cell lines investigated, we focused on aberrations of interest. We tried to explore candidate targets for those aberrations as novel PC‐associated genes, which might be useful molecular markers for diagnosis as well as therapeutic targets of this lethal disease, by not only combing with expression analysis but also further functional analyses. Using this approach for various tumors, we have successfully identified a series of target genes for activation and inactivation from amplified regions and homozygously deleted regions, respectively, in various tumors.( 21 , 22 , 26 , 28 , 29 , 32 , 33 )
Among genes within the 7q21.3‐22.1 amplicon detected in the AsPC‐1 cell line, SMURF1 and TRRAP were pointed out as the most probable targets, which are overexpressed in a genomic copy number‐dependent manner. In our previous array‐CGH study analyzing 44 primary PC tumors using the same in‐house array platform (MCG Cancer Array‐800),( 18 ) SMURF1 and TRRAP were also identified as frequently amplified genes or loci (6/44, 14% and 5/44, 11%, respectively), and both genes were overexpressed through amplification or gain mechanisms in xenografts. Indeed, 18 of 19 primary PC cases used for homozygous deletion analysis in the present study (Fig. 1b) were analyzed in this array‐CGH analysis, and three cases (cases 12, 14, and 17; 17%) showed amplification of SMURF1. Recently, the same 7q21.3‐22.1 amplicon in the AsPC‐1 cell line was identified using array‐CGH, and SMURF1 was noted as a possible target gene,( 12 , 15 ) although neither detailed expression analyses in cell lines and primary tumors of PC nor any functional analyses of this gene were carried out in those studies. In addition, the 7q22.1 amplicon contains many genes other than SMURF1 and TRRAP, suggesting that additional experimental evidence needs to be provided to determine possible targets for 7q22.1 amplification. In our study, we constructed a precise amplicon map using FISH, listed all known genes within this region, and carried out expression analysis, identifying SMURF1 and TRRAP as the most probable targets by comparing the expression status with the copy number status. Knockdown of both SMURF1 and TRRAP using siRNA inhibited the growth of PC cells overexpressing those genes, and ectopic expression of SMURF1 promoted the growth of PC cells with low expression, suggesting that these genes, especially SMURF1, may exert a growth‐promoting effect on PC cells. Notably, growth‐inhibiting effects in SMURF1 and TRRAP knockdown experiments were observed in an expression level‐dependent manner, suggesting that PC cells overexpressing those genes might show ‘oncogene addiction’ to those genes.( 34 ) Of these two genes, combined with an overexpression experiment and frequent overexpression in primary PC, SMURF1 is likely to be the most promising PC‐associated gene located within 7q21.3‐22.1 and may be a good therapeutic target for this disease, although SMURF1 expression status in primary PC did not correlate with survival.
As SMURF1 is known as an E3 ubiquitin ligase and negative regulator of the transforming growth factor (TGF)‐β signaling pathway through the degradation of TGF‐β receptor type I with SMAD7,( 35 ) and that of SMAD4 with SMAD2 and SMAD7,( 36 ) SMURF1 overexpression may contribute to suppression of the growth‐inhibitory effects of TGFβ in epithelial cells, including pancreatic ductal cells; however, the effects of both knockdown and overexpression of SMURF1 on the growth of PC cell lines were observed in a SMAD4 mutation status‐independent manner (Table 1; Fig. 3a,c), suggesting that SMURF1 may regulate cell growth through a TGF‐β receptor–SMAD4 signaling pathway‐independent mechanism in PC cells. However, it was also reported that TGF‐β can regulate p21 expression and cell growth through a SMAD4‐independent mechanism.( 37 ) Therefore, it is still possible for SMURF1 to show growth‐promoting activity in PC cells in the TGF‐β‐related pathway synergistically to the alteration of SMAD4 or other molecules. In addition, it was recently reported that SMURF1 regulates tumor cell plasticity and motility through the degradation of RhoA.( 38 ) Taken together, SMURF1 may contribute to both the development and malignant progression of PC. Further examination will be needed to clarify the functional role and clinical significance of SMURF1 in this disease.
TRRAP was reported to be an essential histone acetyltransferase cofactor for both the c‐myc and E1A/E2F oncogenic transcription factor pathways.( 39 ) As previously shown in normal cells,( 40 ) we demonstrated that knockdown of TRRAP expression inhibited cell growth in PC cells, suggesting that TRRAP is essential for both normal and neoplastic cell proliferation. However, our immunohistochemical analysis demonstrated infrequent overexpression of TRRAP protein in primary tumors of PC and no expression in normal pancreatic ductal cells, suggesting that TRRAP is likely to have less potential as a target gene within this amplicon.
Acknowledgments
We are grateful to Professor Yusuke Nakamura (Human Genome Center, Institute of Medical Science, The University of Tokyo) for continuous encouragement throughout this work, and Dr Ming‐Sound Tsao (University of Toronto) for providing the HPDE6 cell line. We also thank Ayako Takahashi and Rumi Mori for technical assistance. This work was supported by Grants‐in‐Aid for Scientific Research, Scientific Research on Priority Areas, and a 21st Century Center of Excellence Program for Molecular Destruction and Reconstitution of Tooth and Bone from the Ministry of Education, Culture, Sports, Science, and Technology, Japan; a grant from the Pancreas Research Foundation of Japan; a grant from Core Research for Evolutional Science and Technology of Japan Science and Technology Corporation; and a grant from the New Energy and Industrial Technology Development Organization.
References
- 1.Bardeesy N, DePinho RA. Pancreatic cancer biology and genetics. Nat Rev Cancer 2002; 2: 897–909. [DOI] [PubMed] [Google Scholar]
- 2.Matsuno S, Egawa S, Fukuyama S_et al_. Pancreatic cancer registry in Japan: 20 years of experience. Pancreas 2004; 28: 219–30. [DOI] [PubMed] [Google Scholar]
- 3.Murr MM, Sarr MG, Oishi AJ, Van Heereden JA. Pancreatic cancer. CA Cancer J Clin 1994; 44: 304–18. [DOI] [PubMed] [Google Scholar]
- 4.Grunewald K, Lyons J, Frohlich A_et al_. High frequency of Ki‐ras codon 12 mutations in pancreatic adenocarcinomas. Int J Cancer 1989; 43: 1037–41. [DOI] [PubMed] [Google Scholar]
- 5.Moore PS, Sipos B, Orlandini S_et al_. Genetic profile of 22 pancreatic carcinoma cell lines. Analysis of K‐ras, p53, p16 and DPC4/Smad4. Virchows Arch 2001; 439: 798–802. [DOI] [PubMed] [Google Scholar]
- 6.Solinas‐Toldo S, Wallrapp C, Muller‐Pillasch F, Bentz M, Gress T, Lichter P. Mapping of chromosomal imbalances in pancreatic carcinoma by comparative genomic hybridization. Cancer Res 1996; 56: 3803–7. [PubMed] [Google Scholar]
- 7.Mahlamaki EH, Hoglund M, Gorunova L_et al_. Comparative genomic hybridization reveals frequent gains of 20q, 8q, 11q, 12p, and 17q, and losses of 18q, 9p, and 15q in pancreatic cancer. Genes Chromosomes Cancer 1997; 20: 383–91. [DOI] [PubMed] [Google Scholar]
- 8.Schleger C, Arens N, Zentgraf H, Bleyl U, Verbeke C. Identification of frequent chromosomal aberrations in ductal adenocarcinomas of the pancreas by comparative genomic hybridization (CGH). J Pathol 2000; 191: 27–32. [DOI] [PubMed] [Google Scholar]
- 9.Pinkel D, Segraves R, Sudar D_et al_. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat Genet 1998; 20: 207–11. [DOI] [PubMed] [Google Scholar]
- 10.Inazawa J, Inoue J, Imoto I. Comparative genomic hybridization (CGH)‐arrays pave the way for identification of novel cancer‐related genes. Cancer Sci 2004; 95: 559–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aguirre AJ, Brennan C, Bailey G_et al_. High‐resolution characterization of the pancreatic adenocarcinoma genome. Proc Natl Acad Sci USA 2004; 101: 9067–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heidenblad M, Schoenmakers EF, Jonson T_et al_. Genome‐wide array‐based comparative genomic hybridization reveals multiple amplification targets and novel homozygous deletions in pancreatic carcinoma cell lines. Cancer Res 2004; 64: 3052–9. [DOI] [PubMed] [Google Scholar]
- 13.Holzmann K, Kohlhammer H, Schwaenen C_et al_. Genomic DNA‐chip hybridization reveals a higher incidence of genomic amplifications in pancreatic cancer than conventional comparative genomic hybridization and leads to the identification of novel candidate genes. Cancer Res 2004; 64: 4428–33. [DOI] [PubMed] [Google Scholar]
- 14.Mahlamaki EH, Kauraniemi P, Monni O, Wolf M, Hautaniemi S, Kallioniemi A. High‐resolution genomic and expression profiling reveals 105 putative amplification target genes in pancreatic cancer. Neoplasia 2004; 6: 432–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bashyam MD, Bair R, Kim YH_et al_. Array‐based comparative genomic hybridization identifies localized DNA amplifications and homozygous deletions in pancreatic cancer. Neoplasia 2005; 6: 556–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nowak NJ, Gaile D, Conroy JM_et al_. Genome‐wide aberrations in pancreatic adenocarcinoma. Cancer Genet Cytogenet 2005; 161: 36–50. [DOI] [PubMed] [Google Scholar]
- 17.Gysin S, Rickert P, Kastury K, McMahon M. Analysis of genomic DNA alterations and mRNA expression patterns in a panel of human pancreatic cancer cell lines. Genes Chromosomes Cancer 2005; 44: 37–51. [DOI] [PubMed] [Google Scholar]
- 18.Loukopoulos P, Shibata T, Katoh H_et al_. Genome‐wide array‐based comparative genomic hybridization analysis of pancreatic adenocarcinoma: identification of genetic indicators that predict patient outcome. Cancer Sci 2007; 98: 392–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harada T, Chelala C, Bhakta V_et al_. Genome‐wide DNA copy number analysis in pancreatic cancer using high‐density single nucleotide polymorphism arrays. Oncogene 2007: [Epub ahead of print]. [DOI] [PMC free article] [PubMed]
- 20.Maitra A, Wistuba II, Virmani AK_et al_. Enrichment of epithelial cells for molecular studies. Nat Med 1999; 5: 459–63. [DOI] [PubMed] [Google Scholar]
- 21.Imoto I, Yang ZQ, Pimkhaokham A_et al_. Identification of cIAP1 as a candidate target gene within an amplicon at 11q22 in esophageal squamous cell carcinomas. Cancer Res 2001; 61: 6629–34. [PubMed] [Google Scholar]
- 22.Saito‐Ohara F, Imoto I, Inoue J_et al_. PPM1D is a potential target for 17q gain in neuroblastoma. Cancer Res 2003; 63: 1876–83. [PubMed] [Google Scholar]
- 23.Furukawa T, Duguid WP, Rosenberg L, Viallet J, Galloway DA, Tsao MS. Long‐term culture and immortalization of epithelial cells from normal adult human pancreatic ducts transfected by the E6E7 gene of human papilloma virus 16. Am J Pathol 1996; 148: 1763–70. [PMC free article] [PubMed] [Google Scholar]
- 24.Noguchi M, Furuya S, Takeuchi T, Hirohashi S. Modified formalin and methanol fixation methods for molecular biological and morphological analyses. Pathol Int 1997; 47: 685–91. [DOI] [PubMed] [Google Scholar]
- 25.Tanabe C, Aoyagi K, Sakiyama T_et al_. Evaluation of a whole‐genome amplification method based on adaptor‐ligation PCR of randomly sheared genomic DNA. Genes Chromosomes Cancer 2003; 38: 168–76. [DOI] [PubMed] [Google Scholar]
- 26.Takada H, Imoto I, Tsuda H_et al_. Screening of DNA copy‐number aberrations in gastric cancer cell lines by array‐based comparative genomic hybridization. Cancer Sci 2005; 96: 100–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang HR, Zhang Y, Ozdamar B_et al_. Regulation of cell polarity and protrusion formation by targeting RhoA for degradation. Science 2003; 302: 1775–9. [DOI] [PubMed] [Google Scholar]
- 28.Yu W, Imoto I, Inoue J, Onda M, Emi M, Inazawa J. A novel amplification target, DUSP26, promotes anaplastic thyroid cancer cell growth by inhibiting p38 MAPK activity. Oncogene 2007; 26: 1178–87. [DOI] [PubMed] [Google Scholar]
- 29.Sonoda I, Imoto I, Inoue J_et al_. Frequent silencing of low density lipoprotein receptor‐related protein 1B (LRP1B) expression by genetic and epigenetic mechanisms in esophageal squamous‐cell carcinoma. Cancer Res 2004; 64: 3741–7. [DOI] [PubMed] [Google Scholar]
- 30.Yamada H, Sakamoto H, Taira M_et al_. Amplifications of both c‐Ki‐ras with a point mutation and c‐myc in a primary pancreatic cancer and its metastatic tumors in lymph nodes. Jpn J Cancer Res 1986; 77: 370–5. [PubMed] [Google Scholar]
- 31.Cheng JQ, Ruggeri B, Klein WM_et al_. Amplification of AKT2 in human pancreatic cells and inhibition of AKT2 expression and tumorigenicity by antisense RNA. Proc Natl Acad Sci USA 1996; 93: 3636–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Izumi H, Inoue J, Yokoi S_et al_. Frequent silencing of DBC1 is by genetic or epigenetic mechanisms in non‐small cell lung cancers. Hum Mol Genet 2005; 14: 1–11. [DOI] [PubMed] [Google Scholar]
- 33.Kikuchi R, Tsuda H, Kanai Y_et al_. Promoter hypermethylation contributes to frequent inactivation of a putative conditional tumor‐suppressor gene connective tissue growth factor in ovarian cancer. Cancer Res 2007; 67: 7095–105. [DOI] [PubMed] [Google Scholar]
- 34.Garber K. New insights into oncogene addiction found. J Natl Cancer Inst 2007; 99: 264–5. [DOI] [PubMed] [Google Scholar]
- 35.Ebisawa T, Fukuchi M, Murakami G_et al_. Smurf1 interacts with transforming growth factor‐β type I receptor through Smad7 and induces receptor degradation. J Biol Chem 2001; 276: 12 477–80. [DOI] [PubMed] [Google Scholar]
- 36.Moren A, Imamura T, Miyazono K, Heldin CH, Moustakas A. Degradation of the tumor suppressor Smad4 by WW and HECT domain ubiquitin ligases. J Biol Chem 2005; 280: 22 115–23. [DOI] [PubMed] [Google Scholar]
- 37.Ijichi H, Otsuka M, Tateishi K_et al_. Smad4‐independent regulation of p21/WAF1 by transforming growth factor‐beta. Oncogene 2004; 23: 1043–51. [DOI] [PubMed] [Google Scholar]
- 38.Sahai E, Garcia‐Medina R, Pouyssegur J, Vial E. Smurf1 regulates tumor cell plasticity and motility through degradation of RhoA leading to localized inhibition of contractility. J Cell Biol 2007; 176: 35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McMahon SB, Van Buskirk HA, Dugan KA, Copeland TD, Cole MD. The novel ATM‐related protein TRRAP is an essential cofactor for the c‐Myc and E2F oncoproteins. Cell 1998; 94: 363–74. [DOI] [PubMed] [Google Scholar]
- 40.Herceg Z, Hulla W, Gell D_et al_. Disruption of Trrap causes early embryonic lethality and defects in cell cycle progression. Nat Genet 2001; 29: 206–11. [DOI] [PubMed] [Google Scholar]