Cysteinyl leukotrienes and their receptors: Bridging inflammation and colorectal cancer (original) (raw)
Abstract
Long-standing inflammation has emerged as a hallmark of neoplastic transformation of epithelial cells and may be a limiting factor of successful conventional tumor therapies. A complex milieu composed of distinct stromal and immune cells, soluble factors and inflammatory mediators plays a crucial role in supporting and promoting various types of cancers. An augmented inflammatory response can predispose a patient to colorectal cancer (CRC). Common risk factors associated with CRC development include diet and lifestyle, altered intestinal microbiota and commensals, and chronic inflammatory bowel diseases. Cysteinyl leukotrienes are potent inflammatory metabolites synthesized from arachidonic acid and have a broad range of functions involved in the etiology of various pathologies. This review discusses the important role of cysteinyl leukotriene signaling in linking inflammation and CRC.
Keywords: Eicosanoids, Cysteinyl leukotrienes, CysLT1R, CysLT2R, Inflammation, Colorectal cancer
Core tip: Despite several advances in diagnostic and therapeutic options, colorectal cancer (CRC) continues to be a major health problem and one of the leading causes of cancer-related deaths. The inflammatory milieu has been widely recognized as one of the enabling characteristics of cancer development. Cysteinyl leukotrienes are pro-inflammatory eicosanoids implicated in chronic inflammatory bowel diseases and CRC development. Hence, targeting cysteinyl leukotrienes and their receptors could provide alternative therapeutic approaches or be used in combination with existing therapies for more efficient treatment of CRC.
INTRODUCTION
Colorectal cancer (CRC) is a global health care burden, with more than 1 million new cases diagnosed every year. It is the third most common malignancy and the fourth leading cause of cancer-related deaths worldwide[1]. A diet high in fat but low in fiber, excessive alcohol consumption, obesity and lack of physical activity, disruption of normal gut microbiota and the presence of long-term inflammatory bowel diseases (IBDs) such as ulcerative colitis (UC) and Crohn’s disease (CD) predispose to CRC. Inflammation is a host-driven response to internal and external stimuli to counter non-self or self-molecules and maintain tissue homeostasis. However, chronic inflammation can be a major health problem in allergic, cardiovascular, fibrotic, local and systemic inflammatory diseases and several cancers[2-9]. In 1863, Rudolf Virchow was the first to speculate about the role of long-term inflammation in cancer based on his observations that cancerous tissues were frequently infiltrated by leukocytes[10]. Current epidemiological data indicate that more than 25% of all cancers are related to long-term infections and other types of unresolved inflammation[11-13]. Evidence from observational studies and randomized trials concerning the protective action of non-steroidal anti-inflammatory drugs (NSAIDs) such as aspirin have indicated not only a reduced long-term risk of esophageal, gastric, biliary, breast, prostate, lung, and CRC but also a lowered risk of metastasis[14-18]. Inflammation present in the tumor microenvironment is characterized by high leukocyte infiltration, ranging in size, distribution and composition, such as tumor-associated macrophages (TAMs), mast cells, dendritic cells (DCs), natural killer cells (NKs), neutrophils, eosinophils and lymphocytes[19-21]. These cells produce a variety of cytotoxic mediators such as reactive oxygen and nitrogen species, serine and cysteine proteases, membrane-perforating agents, matrix metalloproteinases, pro-inflammatory cytokines, interferons (IFNs) and increased levels of enzymes such as cyclooxygenase-2 (COX-2), 5-lipoxygenase (5-LOX) and phospholipase A2 (PLA2), hence contributing to carcinogenesis[22-25]. This review addresses the role of cysteinyl leukotrienes in inflammation-induced colorectal carcinogenesis.
EICOSANOIDS
Eicosanoids, from the Greek word “eicosa” meaning “20,” are biologically active lipophilic molecules predominantly metabolized from arachidonic acid (AA), a 20-carbon polyunsaturated essential fatty acid, that are involved in physiological processes such as inflammation[14]. AA belongs to the ω-6 family of polyunsaturated fatty acids and is usually found esterified at the second carbon position in the phospholipids of membranes. It serves as a precursor to several lipid pro-inflammatory mediators such as prostaglandins (PGs), prostacyclins, thromboxanes (TXAs), lipoxins and leukotrienes (LTs), which have individual as well as overlapping functions in acute and chronic inflammation[26]. Aberrant AA metabolism is often linked to production of pro-inflammatory eicosanoids, chronic inflammatory diseases and carcinogenesis. The first eicosanoids were discovered in the 1960s, although in 1930 scientists had found that certain substances present in biological fluids such as sputum had the potential to induce contraction and relaxation in smooth muscles; they termed them“slow-reacting substances of anaphylaxis.” Despite these early observations, it was not until 1979 that “leukotrienes” were identified and defined by Samuelsson and co-workers for their biological effects in inflammatory processes[27]. On account of their fundamental and seminal work on different eicosanoids, mainly the prostaglandins, and for their discovery of the role of anti-inflammatory compounds such as aspirin on prostaglandin metabolism, the scientists Bengt Samuelsson, John Vane and Sune Bergström were awarded the Nobel Prize for Physiology and Medicine in 1982.
AA, which is stored as diacylglycerol (DAG), is released from the phosphatidylinositol 4,5-bisphosphate (PIP2) present in the outer nuclear envelope of cells, from which it is mobilized into the cytoplasm either by activation of calcium-dependent cytosolic PLA2 or by the combined action of phospholipase C (PLC) and DAG lipase[26,28]. Once in the cytosol, AA can be enzymatically metabolized in a three-directional manner either by cytochrome P450 or by the COX pathway into prostaglandins, prostacyclins or thromboxanes, or through the 5-LOX pathway into leukotrienes A4 (LTA4), B4 (LTB4), C4 (LTC4), D4 (LTD4) and E4 (LTE4) (Figure 1). The last three alternative derivatives to LTA4 are collectively termed “cysteinyl leukotrienes” (CysLTs) owing to the presence of a cysteine residue and are structurally similar but functionally distinct. The role of these eicosanoids in maintaining intestinal epithelial cell homeostasis is well documented[29-32]. Various epidemiological, clinical, and laboratory studies have shown that dysregulation of the COX and LOX pathways results in chronic inflammation and subsequently cancer[14,29].
Figure 1.
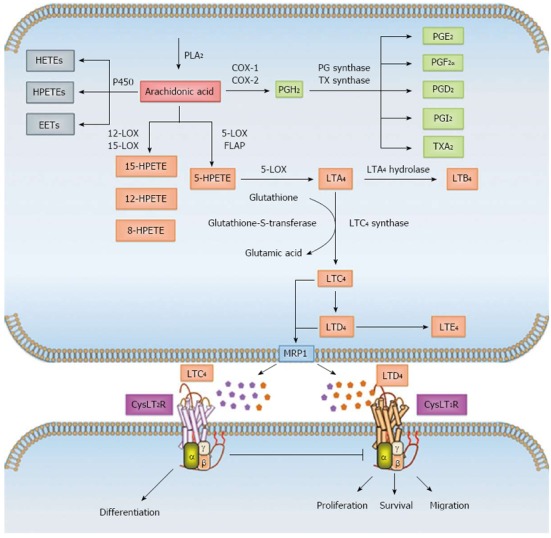
Overview of arachidonic acid metabolism. Arachidonic acid (AA) is a polyunsaturated fatty acid found in the phospholipids of cell membranes. AA is mobilized into the cytoplasm mostly by the activation of calcium-dependent cytosolic phospholipase A2 (cPLA2). Free AA in the cytosol can be enzymatically metabolized to eicosanoids through three major pathways: the cytochrome P450, cyclooxygenase (COX) and/or 5-lipoxygenase (5-LOX) pathways. In the P450 pathway, AA is metabolized to epoxyeicosatrienoic acids (EETs), hydroxyeicosatetraenoic acids (HETEs) and hydroperoxyeicosatetraenoic acids (HPETEs). In the COX pathway, AA is enzymatically converted to the intermediate prostaglandin H2 (PGH2), which is then sequentially metabolized to prostanoids, including prostaglandins (PGs), such as PGE2, PGF2, PGD2 and PGI2, and thromboxanes (TXs) such as TXA2 by specific prostaglandin and thromboxane synthases. In the LOX pathway, AA is metabolized by 12- and 15-LOX to 8-, 12- and 15-HPETE or by 5-LOX and 5-lipoxygenase activating protein (FLAP) to intermediary 5-HPETE. 5-HPETE is further processed to form leukotrienes (LTs), the first of which is the unstable leukotriene A4 (LTA4). LTA4 is subsequently converted to leukotriene B4 (LTB4) by LTA4 hydrolase or together with glutathione to leukotriene C4 (LTC4) by LTC4 synthase and glutathione-S-transferase. LTC4 is converted by ubiquitous enzymes to form leukotriene D4 (LTD4) and leukotriene E4 (LTE4). The members of the multidrug resistance-associated protein (MRP) family are efflux transporters for both PGs and LTs. The cysteinyl leukotrienes (CysLTs) LTC4, LTD4 and LTE4 act via G protein-coupled receptors CysLT1R and CysLT2R at the cell surface and induce different signaling mechanisms.
LEUKOTRIENES AND THEIR RECEPTORS
The term leukotriene is derived from the two words leuko for white blood cells and trienes, meaning three conjugated double bonds, indicating that the ability to generate LTs from AA is largely restricted to leukocytes[28].
Synthesis of LTs is initiated by 5-LOX in concert with 5-lipoxygenase-activating protein (FLAP). The latter does not exhibit any enzymatic activity but facilitates the interaction between 5-LOX and its substrate AA. The first step in this pathway is oxygenation of AA to yield unstable 5-hydroperoxyeicosatetraenoic acid (5-HPETE), which immediately undergoes dehydration to form LTA4. Further metabolism of LTA4 either generates LTB4 by the action of LTA4 hydrolase (LTA4-H) or leads to conjugation with glutathione in the presence of LTC4 synthase (LTC4-S) or glutathione S-transferase to yield LTC4. After carrier-mediated transport of LTB4 and LTC4 to the extracellular milieu, LTC4 can be further metabolized to LTD4 through the cleavage of glutamic acid from the glutathione moiety, and additional glycine cleavage yields LTE4[28,33] (Figure 1).
5-LOX is expressed predominantly by neutrophils, eosinophils, monocytes, macrophages and mast cells. Although nonleukocytes express 5-LOX and FLAP to a lesser extent and are not believed to synthesize appreciable amounts of LTs, expression of LTA4-H and/or LTC4-S, uptake of exogenous LTA4 and further metabolization is possible, via a process referred to as transcellular biosynthesis[34].
CysLT signaling is initiated upon binding of a ligand to one of the two G-protein-coupled receptors (GPCRs), CysLT1R and CysLT2R located at the plasma membrane[35,36], although the presence of other CysLT receptors such as GPR17, P2Y12, and CysLTER have also been suggested[37-39]. Both CysLT1R and CysLT2R can also be localized to the nuclear membrane, since CysLT1R has a bipartite nuclear localization sequence and CysLT2R possesses an interferon regulatory 7 (IRF7) site, which in turn carries a nuclear localization sequence domain[40-42]. While the affinity of CysLT1R for LTD4 is high, the CysLT2R has a low but an equal affinity for LTD4 and LTC4[35,36]. Functionally, CysLTs induce smooth muscle contraction, vascular leakage, eosinophil recruitment in inflammatory diseases, mucus production and chemotaxis[43-46].
LTB4 also plays a pivotal role in inflammatory processes such as leukocyte chemoattraction, particularly of granulocytes and T cells, induction of rapid invasion and recruitment of these cells to the plasma membrane of endothelial cells, production of reactive oxygen species, and induction of gene expression[47,48]. LTB4 mediates its signaling via two GPCRs: BLT1 and BLT2[49,50]. BLT1 binds to LTB4 with an affinity higher than that of the BLT2 receptor. The tissue distribution of the two receptors is quite different. Whereas BLT1 expression in both mice and humans has been reported to be predominantly restricted to peripheral leukocytes, BLT2 expression in humans appears to be fairly ubiquitous, with the highest level observed in the spleen, liver, and lymphocytes[51].
CYSTEINYL LEUKOTRIENES AND THEIR RECEPTORS IN COLORECTAL CANCER
IBD and colorectal cancer
Inflammation and CRC initiation and dissemination go hand in hand[10,52]. The most well-established connection exists between IBD-both UC and CD- and CRC[53-55]. “IBD” is a name given to a group of prolonged inflammatory disorders of the intestinal tract associated with debilitating symptoms and epithelial damage. The risk of developing CRC is 30%-50% higher in patients with IBD[56,57]. IBDs are characterized by increased leukocyte infiltration into the intestinal wall, where they can induce non-specific inflammation through activation and production of AA-derived pro-inflammatory metabolites such as LTs and PGs and subsequent tissue injury. Thus, the gastrointestinal tract is richly supplied with these eicosanoids that mediate several gastrointestinal diseases, including cancers. High levels of LTs such as LTE4 have been detected in the urine of patients with UC and CD[58,59]. Among CysLTs, the presence of LTD4 at an IBD site increases the risk of consequential cancer development, and specific LTD4 antagonists have been shown to reduce colonic inflammation[60]. Although UC is fundamentally similar to CD, a few differences exist, primarily the presentation of a cytokine profile with a T helper 2 (Th2) antibody-mediated response[61]. CD is an autoimmune disease associated with T helper 1 (Th1)-mediated cytokines such as interleukin-12 (IL-12), IFN-γ and tumor necrosis factor-alpha (TNF-α)[61,62].
Colitis-associated cancer (CAC) is known to be highly infiltrated by several cells of the innate immune system, including neutrophils, mast cells, NKs, DCs and TAMs[63]. Moreover, recent evidence supports the concept that malignant tumors also recruit a specific subpopulation of myeloid cells called myeloid-derived suppressor cells[64]. These cells share some characteristics with monocytes, macrophages, neutrophils, and DCs and help suppress any potential anti-tumor immune response and tumor angiogenesis. As in several cancers, including CRC, in which the major inflammatory cellular components are macrophages, TAMs contribute immensely to cancer growth and expansion. TAMs are macrophages that display an M2 type (alternatively activated phenotype) and secrete high levels of Th2 cytokines, growth factors and inflammatory mediators that promote tumor growth, angiogenesis, and metastasis[65,66]. We have observed a high intra-tumoral density of TAMs in colon cancer tissue compared with the adjacent normal tissue, and M2 macrophages were required for effective colon cancer cell migration via factors derived from M2 macrophages and their association with signal regulatory protein alpha (SIRP-α) through CD47[67].
Eicosanoids and colorectal cancer
Apart from its role in inflammation-associated diseases such as asthma, psoriasis, rheumatoid arthritis and IBD[68], LTB4 has pro-tumorigenic effects in breast cancer, melanoma, lymphoma, and head and neck carcinoma[69-72]. Increased expression of LTB4 and its receptor BLT1 have been demonstrated in human CRC tissue[73]. Ihara et al[73] demonstrated significant expression of BLT1 in the colon cancer cell lines Caco-2 and HT-29. Using both the 5-LOX inhibitor AA-861 and selective BLT1 antagonist U75302 in these cell lines, the authors showed induction of apoptosis and reduced proliferation[73]. LTB4-stimulated extracellular signal-regulated kinase (Erk) activation in these cancer cells was also abrogated by U75302. A subsequent study investigated the effectiveness of another LTB4 receptor antagonist (LY293111) in combination with gemcitabine, an anti-tumor adjuvant and radiosensitizer, on the proliferation rate of human colon cancer cell lines LoVo and HT-29 in an athymic heterotrophic xenograft mouse model and found a significant reduction in tumor growth due to apoptosis via the mitochondrial pathway[74]. The findings from these and several other studies emphasize the role of LTB4 signaling in colon cancer cells and warrant the use of specific LTB4 receptor antagonists to suppress CRC expansion.
Among the eicosanoids derived from the COX-pathway, PGE2 is the most abundant and extensively studied in cancer, including CRC[29]. In both the spontaneous adenomatous polyposis coli (Apc)Min/+ mouse model of intestinal cancer and the azoxymethane (AOM)-induced CRC mouse model, PGE2 has been shown to increase the tumor burden[75,76]. Selective inhibition of PGE2 synthesis, through genetic deletion of microsomal PGES (mPGES-1), significantly reduced tumor formation in an ApcMin/+ and AOM-induced mouse model of intestinal and CRC, respectively, and further established the role of PGE2 in tumorigenesis[77,78].
Increased expression of the enzymes responsible for production of PGs and LTs-COX-2 and 5-LOX, respectively has been documented in human colorectal adenocarcinomas compared with adjacent normal mucosa[79-81]. Various clinical trials over the past two decades have highlighted the use of eicosanoid-depressing and anti-inflammatory drugs in the prevention and treatment of CRC. Two groups of compounds have shown promising results: aspirin (NSAID) and celecoxib (COX-2 selective inhibitor)[82].
Cysteinyl leukotrienes, their receptors and colorectal cancer
Upregulated expression of CysLT1R has been observed in several human cancers, including transitional cell carcinoma (TCC) in the bladder, neuroblastomas, and brain, prostate, breast, and CRCs[6,80,83-86]. We have shown that high CysLT1R tumor expression is associated with a poor survival prognosis in breast and CRC patients[80,86], whereas concomitant low CysLT1R and high CysLT2R expression indicate a good prognosis in CRC patients[87]. We have also demonstrated high CysLT1R expression in established colon cancer cell lines[42,80]. The CysLT1R and CysLT2R expression ratio seems to be important in the disease etiology of CRC. Accordingly, we have shown that these two receptors are co-localized and form hetero- and homodimers in a human intestinal epithelial cell line and that LTC4 stimulation of CysLT2R negatively regulates the plasma membrane expression of CysLT1R by inducing internalization of the receptor heterodimer complex[88]. The expression of CysLT1R has also been positively correlated with the cell survival factors COX-2 and Bcl-xL in tumor specimens from patients with CRC[80].
We have previously observed that LTD4, via CysLT1R, induces upregulation of proteins associated with CRC - such as COX-2, β-catenin, and Bcl-2 - in intestinal epithelial cells[89]. Upregulation of β-catenin expression was shown to be dependent on phosphoinositide 3 kinase (PI3K)-glycogen synthase kinase 3β (GSK-3β) signaling[30]. Moreover, our previous work has shown that LTD4-induced CysLT1R signaling results in cell proliferation, survival, and migration through distinct signaling pathways. LTD4-induced CysLT1R-signaling through cAMP response element-binding protein (CREB) and p90 ribosomal s6 kinase (p90RSK) was shown to induce survival and proliferation, respectively, while inducing migration via the PI3K-Rac signaling pathway[90,91] (Figure 2).
Figure 2.
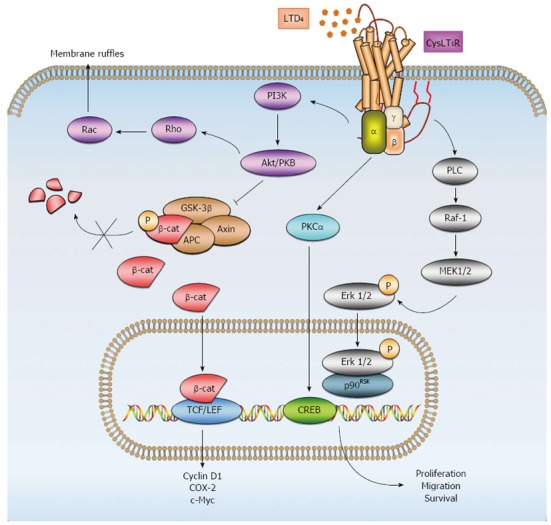
Signaling pathways of LTD4 and CysLT1R. Upon stimulation with LTD4, various downstream signaling pathways become activated. LTD4 induces cell membrane ruffles through phosphoinositide 3-kinase (PI3K) signaling, which in turn stimulates protein kinase Akt/PKB and the small GTPases Rho and Rac. Akt/PKB can inhibit glycogen synthase kinase-3 β (GSK-3β), which comprises the destruction complex for cytosolic β-catenin together with Axin and adenomatous polyposis coli (APC). Inhibition of the destruction complex leads to the accumulation of β-catenin in the cytosol and translocation to the nucleus. In the nucleus, β-catenin interacts primarily with members of the T-cell factor/lymphoid enhancer factor (TCF/LEF) family of transcription factors to activate target genes, leading to the expression of various proteins, such as cyclin D1, COX-2 and c-Myc, which contribute to diverse cellular processes, including proliferation and migration. LTD4-CysLT1R signaling also regulates cell proliferation via mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (Erk) pathways. LTD4-CysLT1R in turn activates phospholipase C (PLC), Raf-1 and mitogen-activated protein kinase kinase (MEK-1/2). This activation leads to the translocation of p-Erk from the cytosol to the nucleus and interaction with p90RSK, which can activate various transcription factors. Conversely, LTD4-CysLT1R mediates survival through protein kinase Cα (PKCα) and the transcription factor cAMP response element-binding protein (CREB). β-cat: β-catenin.
COX-2 expression has also been detected in various colon cancer cells[92], and we have shown that LTD4 via CysLT1R enhances survival by activating the mitogen-activated protein kinase kinase (Mek)/Erk signaling pathway and increasing COX-2 and subsequent Bcl-2 expression in the colon cancer cell line Caco-2[93]. Furthermore, we have demonstrated that LTD4 via CysLT1R induces increased proliferation and migration in the colon cancer cell line HCT-116, probably via the GSK-3β/β-catenin pathway with subsequent increased transcription of the target genes MYC and _CCD1_[94]. By contrast, decreased expression of CysLT2R has been observed in different colon cancer cell lines (Caco-2 and SW480) compared with an epithelial intestinal cell line, and LTC4 stimulation of CysLT2R has been shown to induce differentiation as demonstrated by increased intestinal alkaline phosphatase activity in Caco-2 cells[42]. In the same colon cancer cell line, anti-tumorigenic IFN-α was shown to induce CysLT2R promoter activity and expression, whereas mitogenic epidermal growth factor (EGF) displayed the opposite effect, suppressing CysLT2R promoter activity and expression. LTC4-mediated CysLT2R signaling suppressed EGF-induced cell migration, and IFN-α induced expression of the differentiation marker mucin-2 and alkaline phosphatase activity[95].
These results indicate potential pro- and anti-tumorigenic properties conveyed by CysLT1R and CysLT2R, respectively, in CRC.
LEUKOTRIENE RECEPTOR ANTAGONISTS AND LEUKOTRIENE SYNTHESIS INHIBITORS
CysLT1R antagonists have been used in studies of inflammatory diseases such as rheumatoid arthritis and atherosclerosis[96,97]. The CysLT1R antagonists pranlukast, zafirlukast and montelukast are commercially available and are currently in clinical use to treat asthmatic patients[98]. Emerging data suggest that the pro-inflammatory CysLTs might have an important role in solid tumors.
CysLT1R antagonist treatment has been shown to inhibit tumor growth by inducing apoptosis in a variety of human urological cancer cell lines (e.g., renal cell carcinoma, bladder cancer, prostate cancer, and testicular cancer)[99]. Montelukast has been shown to induce early apoptosis in a bladder transitional cell carcinoma (TCC) cell line, as well as in three different prostate cancer cell lines[6,83]. In addition, montelukast has been shown to induce the intrinsic apoptotic pathway, resulting in cleavage of caspases 3 and 9, and cell cycle arrest in neuroblastoma cell lines[84].
Studies in CysLT1R-deficient mice have revealed its role in enhanced vascular permeability during an acute inflammatory response[100]. Pranlukast and montelukast have been shown to reduce vascular permeability by regulating vascular endothelial growth factor (VEGF) expression in allergen-induced asthmatic lungs of mice[101]. Furthermore, the two abovementioned CysLT1R antagonists have been shown to inhibit the permeability of peripheral capillaries, thereby preventing tumor metastasis in a Lewis lung carcinoma metastasis model[102]. Proliferation and migration of endothelial cells are needed to form new vessels, a process required in cancer development. Montelukast has been shown to reduce LTD4-CysLT1R-mediated migration of the endothelial cell line EA.hy926 via the Erk1/2 pathway[103]. In line with aforementioned data, we have demonstrated in a nude mouse xenograft model of colon cancer that reduction of tumor growth can be accomplished with CysLT1R antagonist treatment. The molecular mechanisms underlying the observed inhibition of tumor growth was attributed to the reduction in proliferation, induction of apoptosis and impairment of angiogenesis[104].
We have also shown that the CysLT1R antagonist ZM198,615 reduces proliferation in the colon cancer cell lines Caco-2 and SW480[105]. Cianchi et al[106] reported the additive effects of the COX-2 selective inhibitor celecoxib, when combined with either the 5-LOX inhibitor MK886 or CysLT1R antagonist LY171883, in reducing the proliferative ability of Caco-2 and HT29 cells. The combined treatment was also shown to induce apoptosis, whereas none of these compounds had any effect alone.
The COX pathway is the most extensively studied among eicosanoid pathways in CRC prevention and/or therapy. However, the cardiovascular side effects associated with long-term usage of NSAIDs and selective COX-2 inhibitors have raised some concerns. Other approaches are being explored, such as inhibition of 5-LOX activity. Simultaneous dual inhibition of COX-2 and 5-LOX activity could possibly provide a more effective and tolerable therapy than COX-inhibition alone. Accordingly, the anti-tumor effects of celecoxib in colon cancer cells were augmented when combined with inhibition of 5-LOX activity using the FLAP inhibitor MK886[106]. The combined inhibition of COX-2 and 5-LOX activity have also shown a more pronounced anti-tumor growth effect in a cigarette smoke-promoting mouse xenograft model of CRC[107]. However, targeting either the COX or LOX pathway alone resulted in a shunt toward the other pathway, except in the latter study, in which a shunt was observed when COX-2 activity was targeted with celecoxib. The abovementioned studies investigated the effects on CRC growth targeting the eicosanoid production in epithelial cells. However, the activity of 5-LOX of mast cells was also shown to be important in intestinal polyp formation in APC∆468 mice. The mast cells were found to utilize 5-LOX to promote proliferation of intestinal epithelial cells and recruit myeloid-derived suppressor cells to the polyp site[108]. Another possible effective chemopreventive option against CRC could be the modification of AA metabolism. ApcMin/+ mice with the fed diets containing highly purified ω-3 polyunsaturated fatty acids were shown to have their mucosal AA replaced, presumably with a reduction in the production of pro-inflammatory mediators. Reduced polyp formation could be observed in both the intestine and the colon of these mice. These effects were associated with significantly decreased proliferation, COX-2 expression, and nuclear β-catenin accumulation, as well as a concomitant increase in apoptosis in the intestinal epithelium[109]. A reduction in size and number of rectal polyps has also been observed in patients with hereditary CRC (familial adenomatous polyposis, FAP) who have undergone colectomy and received highly purified ω-3-polyunsaturated fatty acids[110].
CONCLUSION
Deregulated AA metabolism creates an imbalance in the tissue homeostatic events of proliferation, regeneration and repair, and host defense. Additionally, deregulated AA metabolism contributes to sustained inflammatory processes that could result in CRC development. Among the implicated inflammatory mediators are the eicosanoids, such as CysLTs. Thus, the modification of CysLT signaling could pave the way for the development of new personalized medicine for patients with CRC[104].
Footnotes
P- Reviewers: Depoortere I, He C S- Editor: Qi Y L- Editor: A E- Editor: Zhang DN
References
- 1.Tenesa A, Dunlop MG. New insights into the aetiology of colorectal cancer from genome-wide association studies. Nat Rev Genet. 2009;10:353–358. doi: 10.1038/nrg2574. [DOI] [PubMed] [Google Scholar]
- 2.Beller TC, Friend DS, Maekawa A, Lam BK, Austen KF, Kanaoka Y. Cysteinyl leukotriene 1 receptor controls the severity of chronic pulmonary inflammation and fibrosis. Proc Natl Acad Sci USA. 2004;101:3047–3052. doi: 10.1073/pnas.0400235101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Capra V, Thompson MD, Sala A, Cole DE, Folco G, Rovati GE. Cysteinyl-leukotrienes and their receptors in asthma and other inflammatory diseases: critical update and emerging trends. Med Res Rev. 2007;27:469–527. doi: 10.1002/med.20071. [DOI] [PubMed] [Google Scholar]
- 4.Riccioni G, Bäck M, Capra V. Leukotrienes and atherosclerosis. Curr Drug Targets. 2010;11:882–887. doi: 10.2174/138945010791320881. [DOI] [PubMed] [Google Scholar]
- 5.Schain F, Schain D, Mahshid Y, Liu C, Porwit A, Xu D, Claesson HE, Sundström C, Björkholm M, Sjöberg J. Differential expression of cysteinyl leukotriene receptor 1 and 15-lipoxygenase-1 in non-Hodgkin lymphomas. Clin Lymphoma Myeloma. 2008;8:340–347. doi: 10.3816/CLM.2008.n.049. [DOI] [PubMed] [Google Scholar]
- 6.Matsuyama M, Funao K, Hayama T, Tanaka T, Kawahito Y, Sano H, Takemoto Y, Nakatani T, Yoshimura R. Relationship between cysteinyl-leukotriene-1 receptor and human transitional cell carcinoma in bladder. Urology. 2009;73:916–921. doi: 10.1016/j.urology.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 7.Sperling RI. Eicosanoids in rheumatoid arthritis. Rheum Dis Clin North Am. 1995;21:741–758. [PubMed] [Google Scholar]
- 8.Nicosia S, Capra V, Rovati GE. Leukotrienes as mediators of asthma. Pulm Pharmacol Ther. 2001;14:3–19. doi: 10.1006/pupt.2000.0262. [DOI] [PubMed] [Google Scholar]
- 9.Hendel J, Nielsen OH. Expression of cyclooxygenase-2 mRNA in active inflammatory bowel disease. Am J Gastroenterol. 1997;92:1170–1173. [PubMed] [Google Scholar]
- 10.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rakoff-Nahoum S. Why cancer and inflammation? Yale J Biol Med. 2006;79:123–130. [PMC free article] [PubMed] [Google Scholar]
- 12.Karin M, Lawrence T, Nizet V. Innate immunity gone awry: linking microbial infections to chronic inflammation and cancer. Cell. 2006;124:823–835. doi: 10.1016/j.cell.2006.02.016. [DOI] [PubMed] [Google Scholar]
- 13.Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539–545. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- 14.Wang D, Dubois RN. Eicosanoids and cancer. Nat Rev Cancer. 2010;10:181–193. doi: 10.1038/nrc2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Algra AM, Rothwell PM. Effects of regular aspirin on long-term cancer incidence and metastasis: a systematic comparison of evidence from observational studies versus randomised trials. Lancet Oncol. 2012;13:518–527. doi: 10.1016/S1470-2045(12)70112-2. [DOI] [PubMed] [Google Scholar]
- 16.Burn J, Gerdes AM, Macrae F, Mecklin JP, Moeslein G, Olschwang S, Eccles D, Evans DG, Maher ER, Bertario L, et al. Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: an analysis from the CAPP2 randomised controlled trial. Lancet. 2011;378:2081–2087. doi: 10.1016/S0140-6736(11)61049-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moysich KB, Menezes RJ, Ronsani A, Swede H, Reid ME, Cummings KM, Falkner KL, Loewen GM, Bepler G. Regular aspirin use and lung cancer risk. BMC Cancer. 2002;2:31. doi: 10.1186/1471-2407-2-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cossack M, Ghaffary C, Watson P, Snyder C, Lynch H. Aspirin Use is Associated with Lower Prostate Cancer Risk in Male Carriers of BRCA Mutations. J Genet Couns. 2013:Epub ahead of print. doi: 10.1007/s10897-013-9629-8. [DOI] [PubMed] [Google Scholar]
- 19.Negus RP, Stamp GW, Hadley J, Balkwill FR. Quantitative assessment of the leukocyte infiltrate in ovarian cancer and its relationship to the expression of C-C chemokines. Am J Pathol. 1997;150:1723–1734. [PMC free article] [PubMed] [Google Scholar]
- 20.Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124:263–266. doi: 10.1016/j.cell.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 21.Egeblad M, Littlepage LE, Werb Z. The fibroblastic coconspirator in cancer progression. Cold Spring Harb Symp Quant Biol. 2005;70:383–388. doi: 10.1101/sqb.2005.70.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:161–174. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 23.Wang D, Dubois RN. Prostaglandins and cancer. Gut. 2006;55:115–122. doi: 10.1136/gut.2004.047100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zlotnik A. Chemokines and cancer. Int J Cancer. 2006;119:2026–2029. doi: 10.1002/ijc.22024. [DOI] [PubMed] [Google Scholar]
- 25.Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5:749–759. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- 26.Singh RK, Gupta S, Dastidar S, Ray A. Cysteinyl leukotrienes and their receptors: molecular and functional characteristics. Pharmacology. 2010;85:336–349. doi: 10.1159/000312669. [DOI] [PubMed] [Google Scholar]
- 27.Samuelsson B. The discovery of the leukotrienes. Am J Respir Crit Care Med. 2000;161:S2–S6. doi: 10.1164/ajrccm.161.supplement_1.ltta-1. [DOI] [PubMed] [Google Scholar]
- 28.Peters-Golden M, Henderson WR. Leukotrienes. N Engl J Med. 2007;357:1841–1854. doi: 10.1056/NEJMra071371. [DOI] [PubMed] [Google Scholar]
- 29.Cathcart MC, Lysaght J, Pidgeon GP. Eicosanoid signalling pathways in the development and progression of colorectal cancer: novel approaches for prevention/intervention. Cancer Metastasis Rev. 2011;30:363–385. doi: 10.1007/s10555-011-9324-x. [DOI] [PubMed] [Google Scholar]
- 30.Mezhybovska M, Wikström K, Ohd JF, Sjölander A. The inflammatory mediator leukotriene D4 induces beta-catenin signaling and its association with antiapoptotic Bcl-2 in intestinal epithelial cells. J Biol Chem. 2006;281:6776–6784. doi: 10.1074/jbc.M509999200. [DOI] [PubMed] [Google Scholar]
- 31.Yudina Y, Parhamifar L, Bengtsson AM, Juhas M, Sjölander A. Regulation of the eicosanoid pathway by tumour necrosis factor alpha and leukotriene D4 in intestinal epithelial cells. Prostaglandins Leukot Essent Fatty Acids. 2008;79:223–231. doi: 10.1016/j.plefa.2008.09.024. [DOI] [PubMed] [Google Scholar]
- 32.Mezhybovska M, Yudina Y, Abhyankar A, Sjölander A. Beta-catenin is involved in alterations in mitochondrial activity in non-transformed intestinal epithelial and colon cancer cells. Br J Cancer. 2009;101:1596–1605. doi: 10.1038/sj.bjc.6605342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lam BK, Austen KF. Leukotriene C4 synthase: a pivotal enzyme in cellular biosynthesis of the cysteinyl leukotrienes. Prostaglandins Other Lipid Mediat. 2002;68-69:511–520. doi: 10.1016/s0090-6980(02)00052-7. [DOI] [PubMed] [Google Scholar]
- 34.Fabre JE, Goulet JL, Riche E, Nguyen M, Coggins K, Offenbacher S, Koller BH. Transcellular biosynthesis contributes to the production of leukotrienes during inflammatory responses in vivo. J Clin Invest. 2002;109:1373–1380. doi: 10.1172/JCI14869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lynch KR, O’Neill GP, Liu Q, Im DS, Sawyer N, Metters KM, Coulombe N, Abramovitz M, Figueroa DJ, Zeng Z, et al. Characterization of the human cysteinyl leukotriene CysLT1 receptor. Nature. 1999;399:789–793. doi: 10.1038/21658. [DOI] [PubMed] [Google Scholar]
- 36.Heise CE, O’Dowd BF, Figueroa DJ, Sawyer N, Nguyen T, Im DS, Stocco R, Bellefeuille JN, Abramovitz M, Cheng R, et al. Characterization of the human cysteinyl leukotriene 2 receptor. J Biol Chem. 2000;275:30531–30536. doi: 10.1074/jbc.M003490200. [DOI] [PubMed] [Google Scholar]
- 37.Ciana P, Fumagalli M, Trincavelli ML, Verderio C, Rosa P, Lecca D, Ferrario S, Parravicini C, Capra V, Gelosa P, et al. The orphan receptor GPR17 identified as a new dual uracil nucleotides/cysteinyl-leukotrienes receptor. EMBO J. 2006;25:4615–4627. doi: 10.1038/sj.emboj.7601341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Paruchuri S, Tashimo H, Feng C, Maekawa A, Xing W, Jiang Y, Kanaoka Y, Conley P, Boyce JA. Leukotriene E4-induced pulmonary inflammation is mediated by the P2Y12 receptor. J Exp Med. 2009;206:2543–2555. doi: 10.1084/jem.20091240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maekawa A, Kanaoka Y, Xing W, Austen KF. Functional recognition of a distinct receptor preferential for leukotriene E4 in mice lacking the cysteinyl leukotriene 1 and 2 receptors. Proc Natl Acad Sci USA. 2008;105:16695–16700. doi: 10.1073/pnas.0808993105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jans DA, Xiao CY, Lam MH. Nuclear targeting signal recognition: a key control point in nuclear transport? Bioessays. 2000;22:532–544. doi: 10.1002/(SICI)1521-1878(200006)22:6<532::AID-BIES6>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 41.Servant MJ, Tenoever B, Lin R. Overlapping and distinct mechanisms regulating IRF-3 and IRF-7 function. J Interferon Cytokine Res. 2002;22:49–58. doi: 10.1089/107999002753452656. [DOI] [PubMed] [Google Scholar]
- 42.Magnusson C, Ehrnström R, Olsen J, Sjölander A. An increased expression of cysteinyl leukotriene 2 receptor in colorectal adenocarcinomas correlates with high differentiation. Cancer Res. 2007;67:9190–9198. doi: 10.1158/0008-5472.CAN-07-0771. [DOI] [PubMed] [Google Scholar]
- 43.Chan CC, McKee K, Tagari P, Chee P, Ford-Hutchinson A. Eosinophil-eicosanoid interactions: inhibition of eosinophil chemotaxis in vivo by a LTD4-receptor antagonist. Eur J Pharmacol. 1990;191:273–280. doi: 10.1016/0014-2999(90)94159-u. [DOI] [PubMed] [Google Scholar]
- 44.Barnes NC, Piper PJ, Costello JF. Comparative effects of inhaled leukotriene C4, leukotriene D4, and histamine in normal human subjects. Thorax. 1984;39:500–504. doi: 10.1136/thx.39.7.500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Drazen JM, Austen KF, Lewis RA, Clark DA, Goto G, Marfat A, Corey EJ. Comparative airway and vascular activities of leukotrienes C-1 and D in vivo and in vitro. Proc Natl Acad Sci USA. 1980;77:4354–4358. doi: 10.1073/pnas.77.7.4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marom Z, Shelhamer JH, Bach MK, Morton DR, Kaliner M. Slow-reacting substances, leukotrienes C4 and D4, increase the release of mucus from human airways in vitro. Am Rev Respir Dis. 1982;126:449–451. doi: 10.1164/arrd.1982.126.3.449. [DOI] [PubMed] [Google Scholar]
- 47.Ford-Hutchinson AW. Leukotriene B4 in inflammation. Crit Rev Immunol. 1990;10:1–12. [PubMed] [Google Scholar]
- 48.Funk CD. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science. 2001;294:1871–1875. doi: 10.1126/science.294.5548.1871. [DOI] [PubMed] [Google Scholar]
- 49.Yokomizo T, Izumi T, Chang K, Takuwa Y, Shimizu T. A G-protein-coupled receptor for leukotriene B4 that mediates chemotaxis. Nature. 1997;387:620–624. doi: 10.1038/42506. [DOI] [PubMed] [Google Scholar]
- 50.Yokomizo T, Kato K, Terawaki K, Izumi T, Shimizu T. A second leukotriene B(4) receptor, BLT2. A new therapeutic target in inflammation and immunological disorders. J Exp Med. 2000;192:421–432. doi: 10.1084/jem.192.3.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yokomizo T, Izumi T, Shimizu T. Leukotriene B4: metabolism and signal transduction. Arch Biochem Biophys. 2001;385:231–241. doi: 10.1006/abbi.2000.2168. [DOI] [PubMed] [Google Scholar]
- 52.Hussain SP, Hofseth LJ, Harris CC. Radical causes of cancer. Nat Rev Cancer. 2003;3:276–285. doi: 10.1038/nrc1046. [DOI] [PubMed] [Google Scholar]
- 53.Bernstein CN, Blanchard JF, Kliewer E, Wajda A. Cancer risk in patients with inflammatory bowel disease: a population-based study. Cancer. 2001;91:854–862. doi: 10.1002/1097-0142(20010215)91:4<854::aid-cncr1073>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 54.Hammerbeck DM, Brown DR. Presence of immunocytes and sulfidopeptide leukotrienes in the inflamed guinea pig distal colon. Inflammation. 1996;20:413–425. doi: 10.1007/BF01486743. [DOI] [PubMed] [Google Scholar]
- 55.Rubin DC, Shaker A, Levin MS. Chronic intestinal inflammation: inflammatory bowel disease and colitis-associated colon cancer. Front Immunol. 2012;3:107. doi: 10.3389/fimmu.2012.00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Eaden J. Review article: colorectal carcinoma and inflammatory bowel disease. Aliment Pharmacol Ther. 2004;20 Suppl 4:24–30. doi: 10.1111/j.1365-2036.2004.02046.x. [DOI] [PubMed] [Google Scholar]
- 57.Ekbom A, Helmick C, Zack M, Adami HO. Ulcerative colitis and colorectal cancer. A population-based study. N Engl J Med. 1990;323:1228–1233. doi: 10.1056/NEJM199011013231802. [DOI] [PubMed] [Google Scholar]
- 58.Sharon P, Stenson WF. Enhanced synthesis of leukotriene B4 by colonic mucosa in inflammatory bowel disease. Gastroenterology. 1984;86:453–460. [PubMed] [Google Scholar]
- 59.Kim JH, Tagari P, Griffiths AM, Ford-Hutchinson A, Smith C, Sherman PM. Levels of peptidoleukotriene E4 are elevated in active Crohn’s disease. J Pediatr Gastroenterol Nutr. 1995;20:403–407. doi: 10.1097/00005176-199505000-00005. [DOI] [PubMed] [Google Scholar]
- 60.Nishikawa M, Hikasa Y, Hori K, Tanida N, Shimoyama T. Effect of leukotriene C4D4 antagonist on colonic damage induced by intracolonic administration of trinitrobenzene sulfonic acid in rats. J Gastroenterol. 1995;30:34–40. doi: 10.1007/BF01211372. [DOI] [PubMed] [Google Scholar]
- 61.Shanahan F. Inflammatory bowel disease: immunodiagnostics, immunotherapeutics, and ecotherapeutics. Gastroenterology. 2001;120:622–635. doi: 10.1053/gast.2001.22122. [DOI] [PubMed] [Google Scholar]
- 62.Fuss IJ, Neurath M, Boirivant M, Klein JS, de la Motte C, Strong SA, Fiocchi C, Strober W. Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn’s disease LP cells manifest increased secretion of IFN-gamma, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. J Immunol. 1996;157:1261–1270. [PubMed] [Google Scholar]
- 63.Atreya I, Neurath MF. Immune cells in colorectal cancer: prognostic relevance and therapeutic strategies. Expert Rev Anticancer Ther. 2008;8:561–572. doi: 10.1586/14737140.8.4.561. [DOI] [PubMed] [Google Scholar]
- 64.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Solinas G, Germano G, Mantovani A, Allavena P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J Leukoc Biol. 2009;86:1065–1073. doi: 10.1189/jlb.0609385. [DOI] [PubMed] [Google Scholar]
- 66.Sica A, Schioppa T, Mantovani A, Allavena P. Tumour-associated macrophages are a distinct M2 polarised population promoting tumour progression: potential targets of anti-cancer therapy. Eur J Cancer. 2006;42:717–727. doi: 10.1016/j.ejca.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 67.Zhang Y, Sime W, Juhas M, Sjölander A. Crosstalk between colon cancer cells and macrophages via inflammatory mediators and CD47 promotes tumour cell migration. Eur J Cancer. 2013;49:3320–3334. doi: 10.1016/j.ejca.2013.06.005. [DOI] [PubMed] [Google Scholar]
- 68.Sampson AP. The role of eosinophils and neutrophils in inflammation. Clin Exp Allergy. 2000;30 Suppl 1:22–27. doi: 10.1046/j.1365-2222.2000.00092.x. [DOI] [PubMed] [Google Scholar]
- 69.Earashi M, Noguchi M, Tanaka M. In vitro effects of eicosanoid synthesis inhibitors in the presence of linoleic acid on MDA-MB-231 human breast cancer cells. Breast Cancer Res Treat. 1996;37:29–37. doi: 10.1007/BF01806629. [DOI] [PubMed] [Google Scholar]
- 70.Okano-Mitani H, Ikai K, Imamura S. Human melanoma cells generate leukotrienes B4 and C4 from leukotriene A4. Arch Dermatol Res. 1997;289:347–351. doi: 10.1007/s004030050203. [DOI] [PubMed] [Google Scholar]
- 71.Bittner S, Wielckens K. Glucocorticoid-induced lymphoma cell growth inhibition: the role of leukotriene B4. Endocrinology. 1988;123:991–1000. doi: 10.1210/endo-123-2-991. [DOI] [PubMed] [Google Scholar]
- 72.el-Hakim IE, Langdon JD, Zakrzewski JT, Costello JF. Leukotriene B4 and oral cancer. Br J Oral Maxillofac Surg. 1990;28:155–159. doi: 10.1016/0266-4356(90)90078-y. [DOI] [PubMed] [Google Scholar]
- 73.Ihara A, Wada K, Yoneda M, Fujisawa N, Takahashi H, Nakajima A. Blockade of leukotriene B4 signaling pathway induces apoptosis and suppresses cell proliferation in colon cancer. J Pharmacol Sci. 2007;103:24–32. doi: 10.1254/jphs.fp0060651. [DOI] [PubMed] [Google Scholar]
- 74.Hennig R, Ding XZ, Tong WG, Witt RC, Jovanovic BD, Adrian TE. Effect of LY293111 in combination with gemcitabine in colonic cancer. Cancer Lett. 2004;210:41–46. doi: 10.1016/j.canlet.2004.02.023. [DOI] [PubMed] [Google Scholar]
- 75.Wang D, Wang H, Shi Q, Katkuri S, Walhi W, Desvergne B, Das SK, Dey SK, DuBois RN. Prostaglandin E(2) promotes colorectal adenoma growth via transactivation of the nuclear peroxisome proliferator-activated receptor delta. Cancer Cell. 2004;6:285–295. doi: 10.1016/j.ccr.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 76.Kawamori T, Uchiya N, Sugimura T, Wakabayashi K. Enhancement of colon carcinogenesis by prostaglandin E2 administration. Carcinogenesis. 2003;24:985–990. doi: 10.1093/carcin/bgg033. [DOI] [PubMed] [Google Scholar]
- 77.Nakanishi M, Montrose DC, Clark P, Nambiar PR, Belinsky GS, Claffey KP, Xu D, Rosenberg DW. Genetic deletion of mPGES-1 suppresses intestinal tumorigenesis. Cancer Res. 2008;68:3251–3259. doi: 10.1158/0008-5472.CAN-07-6100. [DOI] [PubMed] [Google Scholar]
- 78.Nakanishi M, Menoret A, Tanaka T, Miyamoto S, Montrose DC, Vella AT, Rosenberg DW. Selective PGE(2) suppression inhibits colon carcinogenesis and modifies local mucosal immunity. Cancer Prev Res (Phila) 2011;4:1198–1208. doi: 10.1158/1940-6207.CAPR-11-0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dreyling KW, Hoppe U, Peskar BA, Morgenroth K, Kozuschek W, Peskar BM. Leukotriene synthesis by human gastrointestinal tissues. Biochim Biophys Acta. 1986;878:184–193. doi: 10.1016/0005-2760(86)90145-1. [DOI] [PubMed] [Google Scholar]
- 80.Ohd JF, Nielsen CK, Campbell J, Landberg G, Löfberg H, Sjölander A. Expression of the leukotriene D4 receptor CysLT1, COX-2, and other cell survival factors in colorectal adenocarcinomas. Gastroenterology. 2003;124:57–70. doi: 10.1053/gast.2003.50011. [DOI] [PubMed] [Google Scholar]
- 81.Soumaoro LT, Iida S, Uetake H, Ishiguro M, Takagi Y, Higuchi T, Yasuno M, Enomoto M, Sugihara K. Expression of 5-lipoxygenase in human colorectal cancer. World J Gastroenterol. 2006;12:6355–6360. doi: 10.3748/wjg.v12.i39.6355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang D, DuBois RN. The role of anti-inflammatory drugs in colorectal cancer. Annu Rev Med. 2013;64:131–144. doi: 10.1146/annurev-med-112211-154330. [DOI] [PubMed] [Google Scholar]
- 83.Matsuyama M, Hayama T, Funao K, Kawahito Y, Sano H, Takemoto Y, Nakatani T, Yoshimura R. Overexpression of cysteinyl LT1 receptor in prostate cancer and CysLT1R antagonist inhibits prostate cancer cell growth through apoptosis. Oncol Rep. 2007;18:99–104. [PubMed] [Google Scholar]
- 84.Sveinbjörnsson B, Rasmuson A, Baryawno N, Wan M, Pettersen I, Ponthan F, Orrego A, Haeggström JZ, Johnsen JI, Kogner P. Expression of enzymes and receptors of the leukotriene pathway in human neuroblastoma promotes tumor survival and provides a target for therapy. FASEB J. 2008;22:3525–3536. doi: 10.1096/fj.07-103457. [DOI] [PubMed] [Google Scholar]
- 85.Zhang WP, Hu H, Zhang L, Ding W, Yao HT, Chen KD, Sheng WW, Chen Z, Wei EQ. Expression of cysteinyl leukotriene receptor 1 in human traumatic brain injury and brain tumors. Neurosci Lett. 2004;363:247–251. doi: 10.1016/j.neulet.2004.03.088. [DOI] [PubMed] [Google Scholar]
- 86.Magnusson C, Liu J, Ehrnström R, Manjer J, Jirström K, Andersson T, Sjölander A. Cysteinyl leukotriene receptor expression pattern affects migration of breast cancer cells and survival of breast cancer patients. Int J Cancer. 2011;129:9–22. doi: 10.1002/ijc.25648. [DOI] [PubMed] [Google Scholar]
- 87.Magnusson C, Mezhybovska M, Lörinc E, Fernebro E, Nilbert M, Sjölander A. Low expression of CysLT1R and high expression of CysLT2R mediate good prognosis in colorectal cancer. Eur J Cancer. 2010;46:826–835. doi: 10.1016/j.ejca.2009.12.022. [DOI] [PubMed] [Google Scholar]
- 88.Parhamifar L, Sime W, Yudina Y, Vilhardt F, Mörgelin M, Sjölander A. Ligand-induced tyrosine phosphorylation of cysteinyl leukotriene receptor 1 triggers internalization and signaling in intestinal epithelial cells. PLoS One. 2010;5:e14439. doi: 10.1371/journal.pone.0014439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ohd JF, Wikström K, Sjölander A. Leukotrienes induce cell-survival signaling in intestinal epithelial cells. Gastroenterology. 2000;119:1007–1018. doi: 10.1053/gast.2000.18141. [DOI] [PubMed] [Google Scholar]
- 90.Paruchuri S, Sjölander A. Leukotriene D4 mediates survival and proliferation via separate but parallel pathways in the human intestinal epithelial cell line Int 407. J Biol Chem. 2003;278:45577–45585. doi: 10.1074/jbc.M302881200. [DOI] [PubMed] [Google Scholar]
- 91.Paruchuri S, Broom O, Dib K, Sjölander A. The pro-inflammatory mediator leukotriene D4 induces phosphatidylinositol 3-kinase and Rac-dependent migration of intestinal epithelial cells. J Biol Chem. 2005;280:13538–13544. doi: 10.1074/jbc.M409811200. [DOI] [PubMed] [Google Scholar]
- 92.Parker J, Kaplon MK, Alvarez CJ, Krishnaswamy G. Prostaglandin H synthase expression is variable in human colorectal adenocarcinoma cell lines. Exp Cell Res. 1997;236:321–329. doi: 10.1006/excr.1997.3741. [DOI] [PubMed] [Google Scholar]
- 93.Wikström K, Ohd JF, Sjölander A. Regulation of leukotriene-dependent induction of cyclooxygenase-2 and Bcl-2. Biochem Biophys Res Commun. 2003;302:330–335. doi: 10.1016/s0006-291x(03)00187-6. [DOI] [PubMed] [Google Scholar]
- 94.Salim T, Sand-Dejmek J, Sjölander A. The inflammatory mediator leukotriene D4 induces subcellular β-catenin translocation and migration of colon cancer cells. Exp Cell Res. 2013:Epub ahead of print. doi: 10.1016/j.yexcr.2013.10.021. [DOI] [PubMed] [Google Scholar]
- 95.Magnusson C, Bengtsson AM, Liu M, Liu J, Ceder Y, Ehrnström R, Sjölander A. Regulation of cysteinyl leukotriene receptor 2 expression--a potential anti-tumor mechanism. PLoS One. 2011;6:e29060. doi: 10.1371/journal.pone.0029060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shiota N, Shimoura K, Okunishi H. Pathophysiological role of mast cells in collagen-induced arthritis: study with a cysteinyl leukotriene receptor antagonist, montelukast. Eur J Pharmacol. 2006;548:158–166. doi: 10.1016/j.ejphar.2006.07.046. [DOI] [PubMed] [Google Scholar]
- 97.Mueller CF, Wassmann K, Widder JD, Wassmann S, Chen CH, Keuler B, Kudin A, Kunz WS, Nickenig G. Multidrug resistance protein-1 affects oxidative stress, endothelial dysfunction, and atherogenesis via leukotriene C4 export. Circulation. 2008;117:2912–2918. doi: 10.1161/CIRCULATIONAHA.107.747667. [DOI] [PubMed] [Google Scholar]
- 98.Bateman ED, Hurd SS, Barnes PJ, Bousquet J, Drazen JM, FitzGerald M, Gibson P, Ohta K, O’Byrne P, Pedersen SE, et al. Global strategy for asthma management and prevention: GINA executive summary. Eur Respir J. 2008;31:143–178. doi: 10.1183/09031936.00138707. [DOI] [PubMed] [Google Scholar]
- 99.Matsuyama M, Yoshimura R. Cysteinyl-leukotriene1 receptor is a potent target for the prevention and treatment of human urological cancer. Mol Med Rep. 2010;3:245–251. doi: 10.3892/mmr_00000247. [DOI] [PubMed] [Google Scholar]
- 100.Maekawa A, Austen KF, Kanaoka Y. Targeted gene disruption reveals the role of cysteinyl leukotriene 1 receptor in the enhanced vascular permeability of mice undergoing acute inflammatory responses. J Biol Chem. 2002;277:20820–20824. doi: 10.1074/jbc.M203163200. [DOI] [PubMed] [Google Scholar]
- 101.Lee KS, Kim SR, Park HS, Jin GY, Lee YC. Cysteinyl leukotriene receptor antagonist regulates vascular permeability by reducing vascular endothelial growth factor expression. J Allergy Clin Immunol. 2004;114:1093–1099. doi: 10.1016/j.jaci.2004.07.039. [DOI] [PubMed] [Google Scholar]
- 102.Nozaki M, Yoshikawa M, Ishitani K, Kobayashi H, Houkin K, Imai K, Ito Y, Muraki T. Cysteinyl leukotriene receptor antagonists inhibit tumor metastasis by inhibiting capillary permeability. Keio J Med. 2010;59:10–18. doi: 10.2302/kjm.59.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yuan YM, Fang SH, Qian XD, Liu LY, Xu LH, Shi WZ, Zhang LH, Lu YB, Zhang WP, Wei EQ. Leukotriene D4 stimulates the migration but not proliferation of endothelial cells mediated by the cysteinyl leukotriene cyslt(1) receptor via the extracellular signal-regulated kinase pathway. J Pharmacol Sci. 2009;109:285–292. doi: 10.1254/jphs.08321fp. [DOI] [PubMed] [Google Scholar]
- 104.Savari S, Liu M, Zhang Y, Sime W, Sjölander A. CysLT(1)R antagonists inhibit tumor growth in a xenograft model of colon cancer. PLoS One. 2013;8:e73466. doi: 10.1371/journal.pone.0073466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Paruchuri S, Mezhybovska M, Juhas M, Sjölander A. Endogenous production of leukotriene D4 mediates autocrine survival and proliferation via CysLT1 receptor signalling in intestinal epithelial cells. Oncogene. 2006;25:6660–6665. doi: 10.1038/sj.onc.1209666. [DOI] [PubMed] [Google Scholar]
- 106.Cianchi F, Cortesini C, Magnelli L, Fanti E, Papucci L, Schiavone N, Messerini L, Vannacci A, Capaccioli S, Perna F, et al. Inhibition of 5-lipoxygenase by MK886 augments the antitumor activity of celecoxib in human colon cancer cells. Mol Cancer Ther. 2006;5:2716–2726. doi: 10.1158/1535-7163.MCT-06-0318. [DOI] [PubMed] [Google Scholar]
- 107.Ye YN, Wu WK, Shin VY, Bruce IC, Wong BC, Cho CH. Dual inhibition of 5-LOX and COX-2 suppresses colon cancer formation promoted by cigarette smoke. Carcinogenesis. 2005;26:827–834. doi: 10.1093/carcin/bgi012. [DOI] [PubMed] [Google Scholar]
- 108.Cheon EC, Khazaie K, Khan MW, Strouch MJ, Krantz SB, Phillips J, Blatner NR, Hix LM, Zhang M, Dennis KL, et al. Mast cell 5-lipoxygenase activity promotes intestinal polyposis in APCDelta468 mice. Cancer Res. 2011;71:1627–1636. doi: 10.1158/0008-5472.CAN-10-1923. [DOI] [PubMed] [Google Scholar]
- 109.Fini L, Piazzi G, Ceccarelli C, Daoud Y, Belluzzi A, Munarini A, Graziani G, Fogliano V, Selgrad M, Garcia M, et al. Highly purified eicosapentaenoic acid as free fatty acids strongly suppresses polyps in Apc(Min/+) mice. Clin Cancer Res. 2010;16:5703–5711. doi: 10.1158/1078-0432.CCR-10-1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.West NJ, Clark SK, Phillips RK, Hutchinson JM, Leicester RJ, Belluzzi A, Hull MA. Eicosapentaenoic acid reduces rectal polyp number and size in familial adenomatous polyposis. Gut. 2010;59:918–925. doi: 10.1136/gut.2009.200642. [DOI] [PubMed] [Google Scholar]