Lipid Agonism, The PIP2 Paradigm of Ligand-Gated Ion Channels (original) (raw)
. Author manuscript; available in PMC: 2016 May 1.
Published in final edited form as: Biochim Biophys Acta. 2015 Jan 26;1851(5):620–628. doi: 10.1016/j.bbalip.2015.01.011
Abstract
The past decade, membrane signaling lipids emerged as major regulators of ion channel function. However, the molecular nature of lipid binding to ion channels remained poorly described due to a lack of structural information and assays to quantify and measure lipid binding in a membrane. How does a lipid-ligand bind to a membrane protein in the plasma membrane and what does it mean for a lipid to activate or regulate an ion channel? How does lipid-binding compare to activation by soluble neurotransmitter? And how does the cell control lipid agonism? This review focuses on lipids and their interactions with membrane proteins, in particular ion channels. I discuss the intersection of membrane lipid biology and ion channel biophysics. A picture emerges of membrane lipids as bona fide agonists of ligand-gated ion channels. These freely diffusing signals reside in the plasma membrane, bind to the transmembrane domain of protein, and cause a conformational change that allosterically gates an ion channel. The system employs a catalog of diverse signaling lipids ultimately controlled by lipid enzymes and raft localization. I draw upon pharmacology, recent protein structure, and electrophysiological data to understand lipid regulation and define inward rectifying potassium channels (Kir) as a new class of PIP2 lipid-gated ion channels.
Keywords: Lipid gated, Ion channel, PIP2, Signaling lipid, G-protein, Lipid raft, Lipidomics
1. Introduction
Signaling lipids are important regulators of ion channels and exert a central role in tissue function including functional heartbeat, neuronal signaling, kidney dialysis, sight, smell, pain, and touch [1–5]. In the past, most biochemist and ion channel experts viewed lipids as unwieldy, hydrophobic molecules physically supporting ion channels in a cell membrane or liposomes but not as ligands. Recent past models of lipid signaling to ion channels suggested that the formation of anionic lipids caused a change in the plasma membrane surface charge. Little was known about how lipids engaged and disengaged the channel or how the contact of a lipid with protein might affect the conformation of ion channels in the membrane. A lack of binding constants for lipids and ion channels challenged our ability to think about lipids as ligands. Aspects of this problem remain an important hurdle.
In 1998 Hilgemann and colleagues eloquently showed that a signaling lipid could directly activate an ion channel [6]. The lipid, phosphatidylinositol 4,5-bisphosphate (PIP2), a minor constituent of the plasma membrane, was required and sufficient for the activation of a potassium channel [6]. Despite more than a decade of experimentation, the nature of PIP2 binding remained clouded by an inability to accurately measure its concentration in the membrane and directly detect binding to protein. Simple terminology such as lipid concentration and affinity are difficult to define for insoluble molecules in an aqueous environment [7]. Absent a well-characterized ligand protein interaction; initial non-specific theories of surface charge and membrane curvature dominated [8,9] but struggled to account for the specificity of signaling lipids in many systems. Recently a more accurate model emerges that includes structural and pharmacological evidence that lipids bind to and activate ion channels analogous to classic ligand-like agonist properties [10,11].
Herein a model of lipid agonism is built on PIP2 and inward rectifying potassium (Kir) channels. Aspects of many other classes of channels and signaling lipids appear to function in a similar way; select examples are included throughout this review. The intent of this review is to facilitate an understanding at the interface of ion channel activation and membrane lipid biology, although neither field is reviewed in a comprehensive way.
2. The signaling lipid PIP2 is an agonist that gates ion channels
PIP2, arguably the best-studied signaling lipid, is comprised of an inositol head group (the named feature) a phosphoglycerol backbone, and two acyl chains (Fig. 1A). PIP2 bears four negative charges and is a permanent and minor component (<1%) of the Eukaryotic plasma membrane inner leaflet [12,13].
Fig. 1.
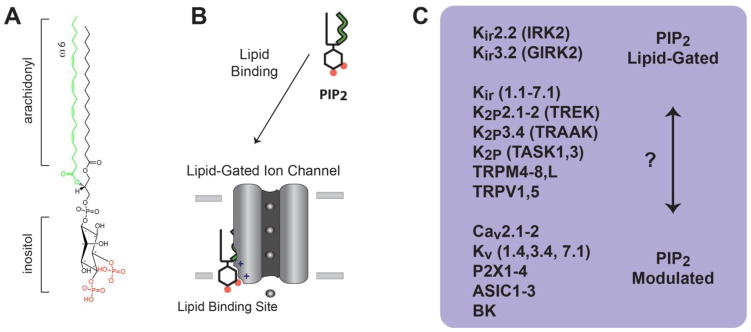
PIP2 lipid regulation of ion channels. A, The chemical structure of plasma membrane PIP2 is shown with an arachidonyl acyl chain (green) and inositol phosphates at the 4′ and 5′ position (red). B, a cartoon representation of a PIP2 lipid gated ion channel. PIP2 is shown bound to a lipid binding site in the transmembrane domain of an ion channel. C, List of ion channels with lipid gating properties. Kir2.2 and 3.2 are the most clearly “lipid-gated”. A second group appears to be dual regulated, or “PIP2 modulated”. PIP2 modulates channel gating, but gating also requires either voltage or a second ligand. A third group of channels behave similar to Kir but await definitive proof of lipid gating vs. PIP2 modulation (?). The list of channels is exemplary and not comprehensive.
2.2 PIP2 ion channel physiology
PIP2 signaling dictates the activatable state of a plethora of ion channels [2,14,15] (Fig. 1) with broad reaching cellular function. The first indication that a channel is PIP2 dependent usually arises when a channel, excised from the plasma membrane (e.g. inside out patch), steadily decreases in conductance until the channel inactivates, this is known as “rundown” [2,16]. The excised patch lacks the cytosolic factors to maintain sufficient PIP2 levels in the membrane to support ion channel function; hence the channels in the patch close. Adding ATP and Mg was shown to delay rundown [16]. Presumably, PIP2 synthesizing enzymes are excised in the patch with the channels and that these enzyme utilize the ATP to replenish PIP2 [2,16]. Adding back a soluble PIP2 analog dioctanoyl PIP2 (C8PIP2) rescues activity [2,15] of many ion channel types [17–20]. In a second method, PIP2 scavengers (e.g. polyamines or PIP2 antibodies) are used to deplete or mask PIP2 availability [21–23]. Polyamines are positively charged polymers that bind via avidity to the multiple negative charges of PIP2. More complete descriptions of PIP2 dependent ion channels and PIP2 cellular function are reviewed by Suh and Hille [2,11], Xie [5], and McLaughlin [9]. Recently, a voltage sensitive phosphatase (Ci-VSP) was shown to provide direct control over PIP2 signaling in the membrane [24–26]. When Ci-VSP is co transfected with Kir [24–26], Kv7.1 [27], Cav2 [28,29], and TRP [30,31], channels are voltage dependent consistent with Ci-VSP regulation of PIP2. This method provides better control of PIP2; however, indirect effects of PIP2 remain a possibility.
In order to directly show PIP2 modulation, an ion channel can be purified and reconstituted (reinserted) into lipid vesicles with a known lipid composition. A lack of purified ion channels limited this technique, but recent advancements in membrane protein expression and purification [32,33] has overcome this problem for select channel types [34–38]. The nAChR was among the first channels to show direct dependence on a lipid for activation, phosphatidic acid (PA) [39]. Recently PIP2 dependent channels were reconstituted into lipid vesicles and shown to respond directly to PIP2 modulation, this includes GIRK [40,41], TRPV1 [42], TRPM8 [43], and Kir2.1-2 [44] channels.
2.3 PIP2 ion channel structure
Despite robust channel modulation by indirect methods, absent a crystal structure, an understanding of the molecular action of PIP2 and the precise binding site remained speculative. In 2011 an X-ray crystal structure complex of Kir2.2 with PIP2 revealed a PIP2 binding site in the channel’s transmembrane domain [10] (Fig. 2). The glycerol backbone and 1′ phosphate of PIP2 capped the first transmembrane spanning helix (TM1) of Kir. Intimate coordination of the 5′ inositol phosphate in the distal end of the second transmembrane spanning helix (TM2) accounted for PIP2 specificity. And a conformational change appeared to initiate or open the ion conduction pathway. Basic residues on a linker between the transmembrane domain and cytoplasmic domain directly contacted PIP2, but distal basic residues proposed in the CTD [45] did not, rather they were buried and stabilized proper folding of the cytoplasmic domain structure [10]. Prior to the Kir2.2/PIP2 complex, structures of PIP2/protein complexes were limited to soluble membrane localization domains, which lack a transmembrane domain and share few if any functional similarities with ion channels. A lack of appropriate structural examples and an understanding of how lipids and proteins interact in the plasma membrane hindered a complete mechanistic interpretation of PIP2 data. Furthermore, early studies on the C-terminus of Kir included residues that turned out to be in the TMD of Kir and key to binding the 5′ inositol phosphate [6] (Fig. 2). Only with recent structural data has a model emerged where lipids bind to specific sites in the transmembrane domain of ion channels [10,46–49].
Fig. 2.
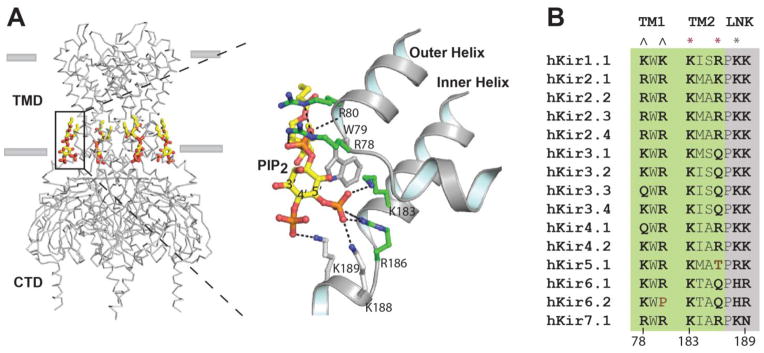
Conserved PIP2 binding site in Kir2.2. PIP2 binds the transmembrane domain (TMD) of Kir and causes a conformational change that allosterically gates the channel. A, The PIP2 binding site is specific for inositol 5′ phosphate. B, A sequence alignment of all Kir family members reveals a highly structured PIP2 binding site comprised of basic residues. Amino acid residues that directly contact PIP2 are shown in bold type. Only two residues (brown type) at the conserved site lack a positive charge. Residues originating from the TMD and a linker (LNK) are shaded green and grey respectively. ^ indicates residues that strongly coordinate the lipid backbone phosphate, * indicates the residues that strongly (red) and weakly (grey) bind the PIP2 5′ phosphate. PIP2 atoms are colored yellow, carbon, orange phosphate; red oxygen. Amino acid side chains with carbons colored green are located on transmembrane outer helix 1 (TM1) or inner helix 2 (TM2). Lysines colored grey are located on the start of a linker helix (LNK) or “tether helix” connecting the transmembrane domain (TMD) and the cytoplasmic domain (CTD). Residue numbering is according to Kir2.2.
2.4 Lipid-gating theory
Taken together these finding suggest a ligand-gating theory of PIP2 activation. In biochemistry, the term ligand refers to the reversible, specific, and dose dependent binding of a substance to a protein to form a complex. Ligands include small molecule drugs, hormones, peptides, and metabolites. Normally ligands stabilize at least two states, one bound and one unbound [50,51].
The binding of PIP2 to Kir has many features of a ligand. First, PIP2 is in low abundance [9,12]. This requires that PIP2 bind with high affinity to its targets to exert an effect. Second, PIP2 binds reversibly to ion channels in a dose dependent manner [20,23]. Third, PIP2 binds with specificity; for example, PI(4,5)P2 activates Kir2.1 and PI(3,4)P2 inhibits the same channel [52]. This specificity is striking since the two lipids are chemical isomers and only differ in the position of the 5′ phosphate. Another anionic lipid, oleoyl-CoA, competitively and reversibly inhibits all Kir’s [52] except Katp, which is specifically activated by oleoyl-CoA [53,54]. Fourth, like neurotransmitter, PIP2 is a dynamically regulated molecule [55,56]; a signaling cascade can rapidly change the concentration of PIP2 to cause the channels to open or close [57–59]. And lastly, PIP2 channel affinity determines channel function [60]. Mutations that allosterically decrease the affinity of PIP2 cause disease (e.g., Andersen-Tawil syndrome) [45,61].
The ligand-like characteristics of PIP2 binding to the entire family of inward rectifiers warrant classification of these channels as ligand-gated. The unique properties of lipids logically give rise to a lipid subclass suggested here “lipid-gated” ion channels.
3. The evolving view of PIP2
3.1 Membrane surface charge theory
PIP2 was first speculated to induce ion channel activation by non-specific avidity of negatively charged phospholipid binding to clusters of basic amino acids in the C-terminus of channels [2,5,8]. Anionic lipids were thought to accumulate on the inner leaflet and non-specifically attract positively charged residues on the surface of Kir’s cytoplasmic domain (CTD). The rational for the theory is sound and was based on data from Katp (Kir7.x) [21,62–64] and proteins like MARCKS [2,8]. However, in light of the PIP2/Kir complexes, the previous role of electrostatic theory appears inadequate for Kir. The glycerol backbone of PIP2 bound tightly to the transmembrane domain (TMD), and the inositol phosphates interacted with residues in or proximal to the TMD, not the CTD. The original influential lack of Katp’s specificity is an anomaly among Kir’s and appears to be an adaptation that allowed regulation by oleoyl-CoA [20] and not a mechanistic requirement as speculated. If non-specific anionic interactions regulate Kir, the site of anion lipid binding are likely distal to the canonical PIP2 site [65] or act synergistically with PIP2 [44,66] by binding to one of the 4 canonical sites. The notion that the cytoplasmic domain is the binding site for PIP2 and that PIP2 localizes the CTD similar to a PH domain appears to be incorrect. The Kir2.2 CTD did move toward the membrane and may reflect an evolutionary origin; but the primary mechanism appears to be an allosteric conformational change, not non-specific electrostatic attractions of the CTD to the membrane surface. The key PIP2 binding interactions were confirmed in a complex of PIP2 with GIRK2 [48] suggesting a common mechanism in related Kirs (Fig. 2B).
Voltage activated ion channels better exemplify non-specific electrostatic interaction. A well studied domain called the “voltage sensor domain” (VSD) senses and responds to changes in surface charge [33,47,67,68]. Conserved basic residues in the VSD electrostatically move towards the charge causing a conformational change that gates the channel. The charge is non-specific and can be applied by external current or by changing the charge of lipids in the plasma membrane. The latter was shown in recent bilayers studies where Kv responded symmetrically and non-specifically to anionic lipids [69]. The same study showed a distinct phosphatidic acid site in the cytoplasmic leaflet that specifically and dramatically affected Kv gating [69]. This suggests both ligand and electrostatic modes can operate in the same channel, however the structural determinants of the two are likely distinct. A similar arrangement exists in Cav2, which has a voltage sensor and a putative PIP2 specific binding site [11,70].
Few other channels currently have sufficient molecular description to definitively discriminate the mechanism of action seen in Kir and Kv. Many tetrameric channels exhibit a C-terminal charged cluster and varying degrees of specificity reminiscent of Kir, including TRP [19,42,71–75], and P2X4 [76,77] (see table 1). Typically, these charges immediately follow or are located in the last transmembrane domain. Many other channels respond to PIP2 in ways that parallel Kir responses, including Cav [70], NMDA [78], Kv [27], P2X1-3 [79] channels (see also Fig. 1C), but it is unknown if the interactions are direct with the TMD or indirect through membrane charge or other proteins. Since numerous soluble domains use polybasic clusters to target to the plasma membrane [80], some yet undefined cytoplasmic domains could utilize a membrane surface charge as previously speculated [2,8]. Future structural studies will continue to reveal the details and breadth of electrostatic theory.
Table 1.
Insositolphosphate ion channel specificity
| Channel | PIP2 effect | Selectivity over | Comments | Ref |
|---|---|---|---|---|
| TRPM8 | Activation* | PI(3,4)P2 and PIP3 | 5′ activates, 3′ inhibits | [19,43] |
| TRPV1 | Mixed | PI(4)P and PIP3 | Likely acyl chain dependence | [42,71] |
| TRPM4 | Activation | PI(4)P and PI(5)P | Modest selectivity over PIP3 | [75] |
| P2X4 | Activation | PIP3 | Modest selectivity over PIP3 | [77] |
| TRPML | Inhibition | PI(3,5)P2 (activation) | Direct competition of (3,5) with PI(4,5)P2 | [72,73] |
| Kir2.1,1.1 | Activation* | PI(3,4)P2 (inhibition) | Direct competition of (3,4) with PI(4,5)P2 | [20,52] |
| Kir3 (Girk2/4) | Activation* | PI(4)P | Gbg increases PI(4,5)P2 binding | [20] |
3.2. Cofactor theory
Lipids are sometimes viewed as co-factors. Before discussing PIP2 as a cofactor I must first define a cofactor and distinguish it from a ligand. The term cofactor stems from enzymology and generally refers to a permanent organic compound or metal that is required for the enzyme to function. A cofactor normally derives its function by remaining bound to a protein. In contrast, a ligand derives its function by binding and dissociating from its partner protein. Lipids have always existed in cells and it is reasonable to assume that some lipids may bind as cofactors. A crystal structure of Kv in a lipid like environment revealed phospholipid binding sites near the voltage sensor and some of these appear to be lipid cofactors [47]. In other words they facilitate the proper organization of the channel but at present they do not appear to initiate a change in the channel state by dynamic regulation of the lipid.
In a speculative role, PIP2 was proposed to act as a ‘coincidence detector’ in order to facilitate transport of an inactive channel [2,15,81,82]. A nascent channel in the endoplasmic reticule (ER), where PIP2 is scarce, remains inactive until it arrives, at the plasma membrane where an abundance of PIP2 constitutively activates the ion channel. This fits well a definition of cofactor in the resting state. Directly demonstrating the physiological contribution remains a challenge since PIP2 is dynamically regulated [2]. For example PLC hydrolysis of PIP2 in the plasma membrane inhibits Kir [58,59], a function also consistent with ligand-like properties.
In another speculative role, PIP2 might function as a cofactor in sensing protons. The pka’s of inositol phosphates are around 6.5 and 6.9, an optimal range for sensing physiological changes in proton concentration [83]. The lipid could remain bound and simply supply the metal phosphate as a proton sensing cofactor. Ions interacting with lipids were recently shown to regulate a receptor [84]. Acid sensing ion channels (ASIC) are likely candidates for such a mechanism since they bind PIP2 and sense protons. Alternatively, PIP2 may serve as a proton sensitive ligand. An atomic structure is known for ASIC [38] but the role of PIP2 in channel activation requires further investigation.
Perhaps one reason for a slow adaptation of a “lipid-gating” model for PIP2 is the fact that the prototypical PIP2 gated channel Kir is active during the resting state of excitable cells. These channels are often considered “constitutively active” leak channels. While it is true they allow potassium out of the cell during the resting state, acetylcholine stimulation of M1 muscarinic receptor inactivates Kir [58,59]. An early study on high affinity Kir2.1 in oocytes showed resistant to ACh inactivation [60], but later studies in mammalian cells demonstrated robust and complete inhibition of Kir2.1 through activation of M1 receptor [59]. Thus neurotransmitter induced closure of Kir potassium channels is presumably synergistic with the opening of calcium, sodium and voltage-gated channels, and should result in a stronger action potential or sustained excitability.
4. Cellular regulation of PIP2 agonism
The agonist properties of lipids broaden the cell-signaling role of PIP2 regulation. Similar to neurotransmitter, the release, degradation, and localization of PIP2 must govern ion channel function.
4.1 Lipid mediated localization of PIP2 in the plasma membrane
Phosphoinositides distributes heterogeneously in the plasma membrane [85–87]. Hydrophobicity causes lipids to partition (see Fig. 5). Saturated lipid chains partition into cholesterol rich lipid rafts, often referred to as detergent resistant membranes (DRMs). Lipids with unsaturation partition into the liquid disordered phase (Ld). Mass spec of resting cells indicate that PIP2 is comprised of a polyunsaturated fatty acyl chain [88–90] and localizes in the Ld region of the membrane [87]. Quantitative studies of PIP2 suggest close to 85% of PIP2 is polyunsaturated and 70% comprised of an arachidonyl acyl chain [90]. In contrast, PIP3 is primarily comprised of saturated or monounsaturated lipid acyl chains [89]. Strikingly, arachidonyl PIP3 was not detected in quiescent cells [89]. Based on standard lipid partitioning, the saturated PIP3 is likely located in cholesterol rafts. In agreement with this arrangement, PI3 Kinase (the enzyme that generates PIP3 from PIP2) localizes to lipid rafts [91]. Taken together, these data indicate an acyl chain based localization of PIPs in the plasma membrane. Figure 4 shows a hypothetical layout of the quiescent cell based on available, but limited, mass spec, super resolution imaging, and localization studies [87–90].
Fig. 5.
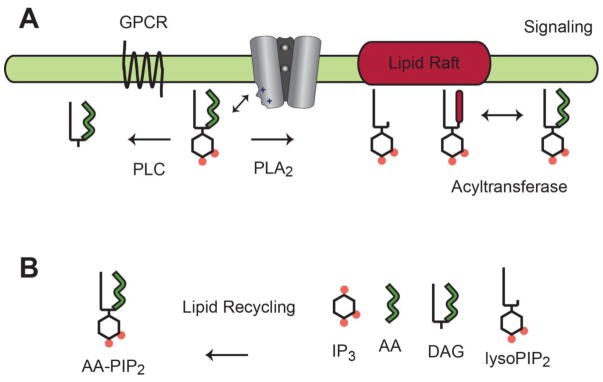
PIP2 transient signaling. A, In the proposed model, PIP2 dissociates from Kir and diffuses laterally in the plasma membrane. G proteins activate lipid-hydrolyzing enzymes that deplete PIP2 from the plasma membrane or laterally redistribute PIP2 into distinct lipid micro domains (e.g. lipid rafts). Dynamic PIP2 signaling gives rise to a transient inactivation of Kir that contributes to an action potential. B, PIP2 degradation products are taken up by endocytosis and PIP2 resynthesis returns the cell to a resting state.
Fig. 4.
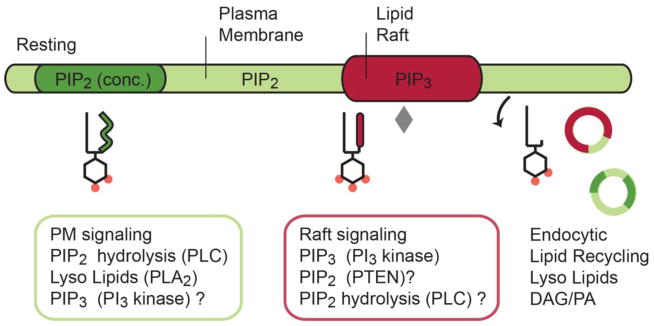
Phosphoinositide (PI) partitioning in the plasma membrane. In the absence of a stimulus, arachidonyl-PIP2 (green) localizes in the disordered region of the plasma membrane and sometimes in concentrated lipid micro domains (dark green) apart from cholesterol-rich lipid-rafts (red). Inositol lipids are distributed according to their acyl chains; hence, saturated PIP2 enters lipid rafts where PI3 kinase generates PIP3. A saturated lysoPIP2 may also associates with raft like domains. Key signaling enzymes (see colored boxes) appear localized in lipid micro domains where they are optimally positioned to remodel the PIP acyl chains and head groups during signaling. Lipid degradation products are found in endocytic vesicles, which suggest a lipid-recycling event analogous to recycling of some soluble neurotransmitters. Grey diamond represents PI3 kinase.
4.2 GPCR signaling through lipases
Famously, Gq coupled GPCRs (guanine nucleotide coupled receptors) hydrolyze PIP2 through phospholipase C (PLC) activation. G protein mediated PIP2 hydrolysis was known more than 30 years ago [92]. However, most cell biologist viewed (and many still do) PIP2 as little more than a substrate for second messenger signaling [93]. This view is inadequate for Kir channels; PIP2 must also be viewed as an ion channel activator [3,6] or agonist. Hence, hydrolysis of PIP2 by M1 muscarinic receptors should be viewed as a direct regulatory mechanism to deplete agonist. PIP2 hydrolysis inactivates both high and low affinity Kir channels [58,59]. Downstream modulation of Kir by phosphatases and kinases appear secondary to this direct PIP2 regulation [6,94], a rational also supported by the central and highly conserved role of PIP2 in channel activation as described above (2.5). PLC regulation of Cav [70], Kir [95], HCN [96], Kv7 [27], K2P [97], and TRP [98,99] channels (among others) is well-documented.
In addition to PLC, GPCR signaling activates phospholipase D [100] (PLD). PLD produces PA and free choline. PA has emerged as an important signaling lipid [101]. PA and PIP2 appear to synergistically activate Kir [44] and K2P [102] channels, in contrast the nAChR [39] and some Kv [69] respond specifically to PA and not PIP2. A third important class of lipases phospholipase A2 (PLA2) also exhibits GPCR regulation [103]. PLA2 hydrolyzes arachidonyl-lipids creating lysophospholipids and arachidonic acid. Downstream and second messenger signaling are well studied for PLA2 and PLC and include the arachidonic cascade and IP3 second messenger signaling respectively. In comparison the upstream role of the intact bioactive arachidonyl-phospholipids and PIP2 is much less understood. Nonetheless, the added role of PIP2 in directly gating ion channels solidifies a direct rout for GPCR regulation of ion channels independent of downstream kinases and calcium signaling [6,94].
Several ion channels bind G-proteins directly, this role is widely accepted for the G-protein regulated inward rectifiers (GIRK/Kir3.x) and N-type calcium channels (Cav2) [104]. A trimeric complex of GIRK with Gβγ (a G-protein) and PIP2 revealed the GIRK/Gβγ interface [40]. And biochemical studies suggest that Gβγ is important for increasing binding of PIP2 to GIRK [41]. The precise mechanism by which Gβγ enhances PIP2 activation needs further clarification.
4.3 Protein mediated localization of lipid modifying enzymes
Lipases localize with ion channels to increase the speed and specificity of PIP2 channel gating [98,105]. For example rhodopsin activated PLC hydrolyzes PIP2 opening TRPL channels. Colocalization of PLC with TRPL [98] allows for a fast 20ms response time [106]. And TRPM7 directly binds PLC to locally affect channel activation [94]. PLC functionally colocalized with NMDA receptors [78] and the IP3 receptor co-localizes with PLC to regulate calcium release [107]. PLD lipases directly localize to ion channels, including TRPM8 [108] and TREK-1 [109]. There are many subtypes of lipases; their diverse regulation and specific localization satisfies cells with the needed diversity for signaling.
4.4 Transient PIP2 signaling
The partitioning of PIPs and their modifying enzymes appears primed to deliver dynamic cell signaling. During a signaling event, G-proteins control PIP kinases, lipases, and phosphatases, to degrade PIP2 signaling. This signaling generates lipid degradation products (Fig. 5B). For example, it was shown PLC activation generates arachidonyl-diacyl-glycerol [90]. And PLA2 activation removes the arachidonyl sn2 acyl chain generating lysoPIP2 [110]. In order to return to a resting state, degradation products need to be removed from the membrane and PIP2 resynthesized.
Endocytosis recycles lipid micro domains and lipid rafts after signaling [111]. The late endosome and ER feed back into PIP2 signaling. This postsynaptic lipid reuptake would then reset the membrane for another signaling event analogous to presynaptic neurotransmitter reuptake. Further studies are needed to understand the temporal and spatial regulation of PIP2 in vivo in particular during a signaling event. However, signaling lipids are known to control the ion channel desensitization [19], voltage dependence [75], and recovery from inactivation [94], and these events correlate with ion channel rundown. Lipid regulated desensitization may prove to be a central function of many channel types. Much more data is needed to build a complete picture.
4.5 Other mechanistic considerations
Lipids localize topically by leaflets generating a lipid signal. For example phosphatidylserine (PS) is found on the inner leaflet of the plasma membrane. Enzymes known as flippases and floppases move lipids between leaflets [112]. PS signals by flipping outside the cell [113]. PS is negatively charged and movement outside the cell has the ability to change the membrane surface charge from negative inside to negative outside. Recently, asymmetric changes to the charge of lipids in a bilayer dramatically shifted the voltage midpoint potential of a Kv channel[69]. Hence lipids may “flip” as a rapid mechanism to impose a lipid induced change on the cell membrane potential, a mechanism that would have likely preceded a synapse.
In a separate mechanism, lipid acyl transferases (LAT) could signal to ion channels by changing the unsaturation of a lipid acyl chain. LAT enzymes add acyl chains to lipids or move acyl chains between existing lipids [114]. If a LAT enzyme swaps an arachidonyl acyl chain with a saturated one, the signaling lipid would most likely translocate to a lipid raft (Fig. 5). This may simply sequester the signal away from the ion channel by moving the lipid into or out of a lipid micro domain. Alternatively, the translocation could make the lipid available to other modifying enzymes that would then deplete the signal from the membrane.
Or, lipid acyl chains may directly contribute to gating of an ion channel. The acyl chains contain chemical diversity and putative specificity could determine the affinity of the lipid for the channel or cause a specific conformational change that gates the channel. Hydrophobic sites for lipid acyl chains affect PIP2 activation of Cav2.2 [11].
The four identical binding sites in Kir are positioned for PIP2 cooperatively and allosteric competition. Tetrameric channels engineered to have only one binding pocket indicated that one PIP2 molecule is sufficient to activate the channel [66]. In wild type channels with four binding sites, PIP2 in combination with PA, PG, or PS, dramatically increased channel conductance. However, absent PIP2, these lipids failed to activate Kir [44]. A structure of Kir with PA bound showed PA binding to the canonical PIP2 site [10]; a site also compatible with PG and PS. In biochemical studies, oleoyl-CoA, an endogenous inhibitor, also competes directly with PIP2 [52]. Taken together, these studies suggest that in Kir the lipid binding site is always occupied, and Kir integrates the sum total of the lipid environment in a cooperative way. At least one site must be occupied by PIP2; the remaining three canonical sites appear to be available to exert cooperative activation or inhibition through a rigid conformational change [10] in the CTD. Thus additional PIP2 binding events are poised to activate Kir with increasing affinity consistent with electrophysiology recordings [66].
Lastly, the relative abundance of diet-derived fatty acids may affect the levels of PIP2 signaling in the plasma membrane. Cells appear to incorporate the relative amounts of saturated and unsaturated fats into their cell membranes (phospholipids) [115]. It is tempting to speculate that diets with excess saturated fat would lead to saturated PIP2 signaling, which most likely favors PIP3 signaling. Diets with large amounts of polyunsaturated fats (PUFAs) would lead to more arachidonyl-PIP2 and more PIP2 signaling. This may account for the positive affect of dietary PUFAs on heart arrhythmias and insulin resistance since PIP2 channels (including Kir) are central to both these diseases. Consistent, with this model, loss of PIP2 channel activation is associated with the disease states [61]. Similar speculation could be made of chronic pain and perhaps some cancers. Understanding PIP2 acylation may shed light on these important medical problems.
5. The future of lipid Ion channel interactions
5.1 Pharmacology of lipids
Better methods are needed for assaying lipid interactions with ion channels. Most studies rely on crude pharmacological shifts of PIP2 concentrations in biological membranes; this is inadequate. Varying the concentration of lipids in a liposome is a good step in the right direction. Normally one describes a ligand in terms of an on and off rate. Certainly lipids have an affinity for ion channels, but we lack the methodology for effectively measuring lipid channel interactions. Better quantitative lipid binding assays are needed. New mass spec techniques will likely allow for quantitative measurements of lipids in vivo and in vitro. And there is no doubt lipidomics will continue to find ways to improve the quantitative, temporal, and spatial identification of lipids in a membrane.
5.2 Implications on the plasma membrane
The plasma membrane holds thousands of lipids with functions that remain largely a mystery [116]. A catalog of lipid signals appears poised to exert exquisite regulation on membrane proteins perhaps rivaled only by protein phosphorylation. Certainly the phosphodiester bonds in lipids are equally suited for rapid signaling. And lipid acyl chains may be as diverse in function as they are in chemistry. Recognizing low abundant phospholipid signaling molecules as potential ligands for membrane proteins reveals a vast pool of putative effector ligands for cellular signaling.
6. Concluding remarks
The added role of PIP2 activation presented here takes shape from the recent crystallographic Kir structures. The non-specific model of PIP2 activating Kir is gradually making room for a PIP2 site with specificity and ligand like properties. How does a lipid ligand influence its target molecule? It does so just like any other molecule; it binds in a concentration dependent manner to a binding site and elicits a conformational change in the protein. While in hindsight this seems an obvious possibility, the plasma membrane has always been a little mysterious [117] and the understanding of membrane proteins slow in coming. No doubt lipid modulation of proteins is diverse with more surprises yet to come.
Fig. 3.
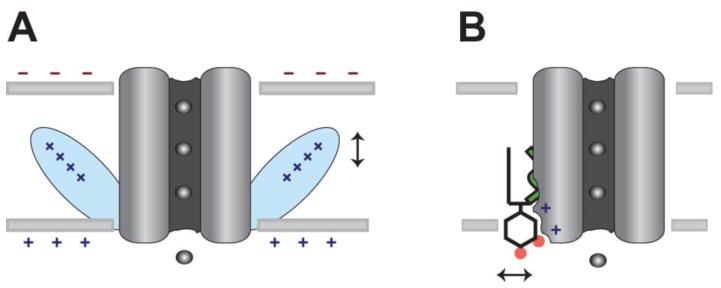
Mechanistic comparison of surface charge gating vs. direct lipid gating. A, Non-specific surface charge gates an ion channel through a charge sensor domain (blue). The vertical arrow indicates charge driven movement. B, A lipid-gated channel reversibly binds the signaling lipid PIP2 to allosterically gate the channel. A horizontal arrow indicates PIP2 dissociation from the channel.
Highlights.
- Membrane resident lipids bind to and activate ion channels with ligand like properties.
- Inward rectifier potassium channel Kir2.2 is a PIP2 lipid-gated ion channel
- Lipid microdomains compartmentalize lipid signals.
- Lipases and endocytosis terminate lipid signaling to ion channels.
Acknowledgments
I thank Andrew S. Hansen for helpful discussion and comments on the manuscript. This work was supported by a Director’s New Innovator Award to SBH (1DP2NS087943-01) from the NIH Common Fund and The National Institute of Neurological Disorders and Stroke (NINDS).
Abbreviations
AA
arachidonic acid
ASIC
acid sensing ion channel
ATP
adenosine triphosphate
BK
big conductance potassium channel
Cav
voltage-dependent calcium channel or VDCC
Ci-VSP
Ciona intestinalis voltage sensitive phosphatase
CoA
coenzyme A
CTD
cytoplasmic domain
C8PIP2
dioctanoyl PIP2
DAG
diacylglycerol
DRM
detergent resistant membrane
ER
endoplasmic reticulum
GIRK
G-protein inward rectifying potassium channel or Kir3
Gβγ
G-protein beta gamma subunit
GPCR
G-protein coupled receptor
HCN
hyperpolarization-activated cyclic nucleotide-gated
IP3
inositol triphosphate
Katp
ATP- sensitive potassium channel or Kir6
Kir
inward rectifying potassium channel
Kv
voltage-gated potassium channel
K2P
two pore domain potassium channel
LAT
lipid acyl transferase
Ld
liquid disordered phase
MARCKS
myristoylated alanine-rich C-kinase substrate
Mg
magnesium
NMDA
N-methyl-D-aspartate receptor
nAChR
nicotinic acetylcholine receptor
PA
phosphatidic acid
PH
pleckstrin homology
PI
phosphoinositide
PIP2
phosphatidylinositol 4,5-bisphosphate
PIP3
phosphatidylinositol 3,4,5-triphosphate
PI3 kinase
phosphatidylinositol-4,5-bisphosphate 3-kinase
PLA2
phospholipase A2
PLC
phospholipase C
PLD
phospholipase D
PS
phosphatidylserine
PTEN
phosphatase and tensin homolog
PUFA
polyunsaturated fatty acid
P2X
purinergic receptors
Sn2
stereospecific numbering position 2 or the second hydroxyl group of glycerol
TMD
transmembrane domain
TM1
transmembrane helix 1
TREK
TWIK related potassium channel or K2P2.1
TRP
transient receptor potential channel
VSD
voltage sensor domain
Footnotes
Conflict of interest.
The author declares no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibliography
- 1.Hilgemann DW. Local PIP(2) signals: when, where, and how? Pflugers Arch. 2007;455:55–67. doi: 10.1007/s00424-007-0280-9. [DOI] [PubMed] [Google Scholar]
- 2.Suh B-C, Hille B. PIP2 is a necessary cofactor for ion channel function: how and why? Annu Rev Biophys. 2008;37:175–195. doi: 10.1146/annurev.biophys.37.032807.125859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huang C-L. Complex roles of PIP2 in the regulation of ion channels and transporters. Am J Physiol Renal Physiol. 2007;293:F1761–5. doi: 10.1152/ajprenal.00400.2007. [DOI] [PubMed] [Google Scholar]
- 4.Gamper N, Shapiro MS. Regulation of ion transport proteins by membrane phosphoinositides. Nat Rev Neurosci. 2007;8:921–934. doi: 10.1038/nrn2257. [DOI] [PubMed] [Google Scholar]
- 5.Xie L-H, John Sa, Ribalet B, Weiss JN. Activation of inwardly rectifying potassium (Kir) channels by phosphatidylinosital-4,5-bisphosphate (PIP2): interaction with other regulatory ligands. Prog Biophys Mol Biol. 2007;94:320–35. doi: 10.1016/j.pbiomolbio.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 6.Huang CL, Feng S, Hilgemann DW. Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gbetagamma. Nature. 1998;391:803–6. doi: 10.1038/35882. [DOI] [PubMed] [Google Scholar]
- 7.Gamper N, Shapiro MS. Target-specific PIP(2) signalling: how might it work? J Physiol. 2007;582:967–75. doi: 10.1113/jphysiol.2007.132787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McLaughlin S, Wang J, Gambhir A, Murray D. PIP(2) and proteins: interactions, organization, and information flow. Annu Rev Biophys Biomol Struct. 2002;31:151–75. doi: 10.1146/annurev.biophys.31.082901.134259. [DOI] [PubMed] [Google Scholar]
- 9.McLaughlin S, Murray D. Plasma membrane phosphoinositide organization by protein electrostatics. Nature. 2005;438:605–11. doi: 10.1038/nature04398. [DOI] [PubMed] [Google Scholar]
- 10.Hansen SB, Tao X, MacKinnon R. Structural basis of PIP2 activation of the classical inward rectifier K+ channel Kir22. Nature. 2011;477:495–8. doi: 10.1038/nature10370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hille B, Dickson EJ, Kruse M, Vivas O, Suh B. Phosphoinositides regulate ion channels. Biochim Biophys Acta. 2014 doi: 10.1016/j.bbalip.2014.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Allen Ja, Halverson-Tamboli Ra, Rasenick MM. Lipid raft microdomains and neurotransmitter signalling. Nat Rev Neurosci. 2007;8:128–40. doi: 10.1038/nrn2059. [DOI] [PubMed] [Google Scholar]
- 13.McLaughlin S, Murray D. Plasma membrane phosphoinositide organization by protein electrostatics. Nature. 2005;438:605–11. doi: 10.1038/nature04398. [DOI] [PubMed] [Google Scholar]
- 14.Suh B-C, Hille B. Regulation of ion channels by phosphatidylinositol 4,5-bisphosphate. Curr Opin Neurobiol. 2005;15:370–8. doi: 10.1016/j.conb.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 15.Hilgemann DW, Feng S, Nasuhoglu C. The complex and intriguing lives of PIP2 with ion channels and transporters. Sci STKE. 2001;2001:re19. doi: 10.1126/stke.2001.111.re19. [DOI] [PubMed] [Google Scholar]
- 16.Hilgemann DW, Ball R. Regulation of cardiac Na+,Ca2+ exchange and KATP potassium channels by PIP2. Science. 1996;273:956–9. doi: 10.1126/science.273.5277.956. [DOI] [PubMed] [Google Scholar]
- 17.Li Y, Gamper N, Hilgemann DW, Shapiro MS. Regulation of Kv7 (KCNQ) K+ channel open probability by phosphatidylinositol 4,5-bisphosphate. J Neurosci. 2005;25:9825–35. doi: 10.1523/JNEUROSCI.2597-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haider S, Tarasov AI, Craig TJ, Sansom MSP, Ashcroft FM. Identification of the PIP2-binding site on Kir62 by molecular modelling and functional analysis. EMBO J. 2007;26:3749–59. doi: 10.1038/sj.emboj.7601809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rohács T, Lopes CMB, Michailidis I, Logothetis DE. PI(4,5)P2 regulates the activation and desensitization of TRPM8 channels through the TRP domain. Nat Neurosci. 2005;8:626–34. doi: 10.1038/nn1451. [DOI] [PubMed] [Google Scholar]
- 20.Rohács T, Lopes CMB, Jin T, Ramdya PP, Molnár Z, Logothetis DE. Specificity of activation by phosphoinositides determines lipid regulation of Kir channels. Proc Natl Acad Sci U S A. 2003;100:745–750. doi: 10.1073/pnas.0236364100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fan Z, Makielski JC. Anionic phospholipids activate ATP-sensitive potassium channels. J Biol Chem. 1997;272:5388–95. doi: 10.1074/jbc.272.9.5388. [DOI] [PubMed] [Google Scholar]
- 22.Rodriguez N, Amarouch MY, Montnach J, Piron J, Labro AJ, Charpentier F, Mérot J, Baró I, Loussouarn G. Phosphatidylinositol-4,5-bisphosphate (PIP(2)) stabilizes the open pore conformation of the Kv111 (hERG) channel. Biophys J. 2010;99:1110–8. doi: 10.1016/j.bpj.2010.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Enkvetchakul D, Jeliazkova I, Nichols CG. Direct modulation of Kir channel gating by membrane phosphatidylinositol 4,5-bisphosphate. J Biol Chem. 2005;280:35785–8. doi: 10.1074/jbc.C500355200. [DOI] [PubMed] [Google Scholar]
- 24.Murata Y, Iwasaki H, Sasaki M, Inaba K, Okamura Y. Phosphoinositide phosphatase activity coupled to an intrinsic voltage sensor. Nature. 2005;435:1239–43. doi: 10.1038/nature03650. [DOI] [PubMed] [Google Scholar]
- 25.Murata Y, Okamura Y. Depolarization activates the phosphoinositide phosphatase Ci-VSP, as detected in Xenopus oocytes coexpressing sensors of PIP 2. 2007;3:875–889. doi: 10.1113/jphysiol.2007.134775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iwasaki H, Murata Y, Kim Y, Hossain MI, Worby Ca, Dixon JE, McCormack T, Sasaki T, Okamura Y. A voltage-sensing phosphatase, Ci-VSP, which shares sequence identity with PTEN, dephosphorylates phosphatidylinositol 4,5-bisphosphate. Proc Natl Acad Sci U S A. 2008;105:7970–7975. doi: 10.1073/pnas.0803936105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zaydman MA, Silva JR, Delaloye K, Li Y, Liang H, Larsson HP, Shi J, Cui J. Kv71 ion channels require a lipid to couple voltage sensing to pore opening. Proc Natl Acad Sci U S A. 2013;110:13180–5. doi: 10.1073/pnas.1305167110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Suh B-C, Leal K, Hille B. Modulation of high-voltage activated Ca(2+) channels by membrane phosphatidylinositol 4,5-bisphosphate. Neuron. 2010;67:224–38. doi: 10.1016/j.neuron.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Suh B-C, Kim D-I, Falkenburger BH, Hille B. Membrane-localized β-subunits alter the PIP2 regulation of high-voltage activated Ca2+ channels. Proc Natl Acad Sci U S A. 2012;109:3161–6. doi: 10.1073/pnas.1121434109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li X, Wang X, Zhang X, Zhao M, Tsang WL, Zhang Y, Yau RGW, Weisman LS, Xu H. Genetically encoded fluorescent probe to visualize intracellular phosphatidylinositol 3,5-bisphosphate localization and dynamics. Proc Natl Acad Sci U S A. 2013;110:21165–70. doi: 10.1073/pnas.1311864110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xie J, Sun B, Du J, Yang W, Chen H-C, Overton JD, Runnels LW, Yue L. Phosphatidylinositol 4,5-bisphosphate (PIP(2)) controls magnesium gatekeeper TRPM6 activity. Sci Rep. 2011;1:146. doi: 10.1038/srep00146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kawate T, Gouaux E. Fluorescence-detection size-exclusion chromatography for precrystallization screening of integral membrane proteins. Structure. 2006;14:673–81. doi: 10.1016/j.str.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 33.Long SB, Campbell EB, Mackinnon R. Voltage sensor of Kv12: structural basis of electromechanical coupling. Science. 2005;309:903–8. doi: 10.1126/science.1116270. [DOI] [PubMed] [Google Scholar]
- 34.Hibbs RE, Gouaux E. Principles of activation and permeation in an anion-selective Cys-loop receptor. Nature. 2011 doi: 10.1038/nature10139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tao X, Avalos JL, Chen J, MacKinnon R. Crystal structure of the eukaryotic strong inward-rectifier K+ channel Kir22 at 31 A resolution. Science. 2009;326:1668–74. doi: 10.1126/science.1180310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Furukawa H, Singh SK, Mancusso R, Gouaux E. Subunit arrangement and function in NMDA receptors. Nature. 2005;438:185–92. doi: 10.1038/nature04089. [DOI] [PubMed] [Google Scholar]
- 37.Liao M, Cao E, Julius D, Cheng Y. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature. 2013;504:107–12. doi: 10.1038/nature12822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jasti J, Furukawa H, Gonzales EB, Gouaux E. Structure of acid-sensing ion channel 1 at 19 A resolution and low pH. Nature. 2007;449:316–23. doi: 10.1038/nature06163. [DOI] [PubMed] [Google Scholar]
- 39.Fong TM, McNamee MG. Correlation between acetylcholine receptor function and structural properties of membranes. Biochemistry. 1986;25:830–40. doi: 10.1021/bi00352a015. [DOI] [PubMed] [Google Scholar]
- 40.Whorton MR, MacKinnon R. X-ray structure of the mammalian GIRK2–βγ G-protein complex. Nature. 2013;498:190–197. doi: 10.1038/nature12241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang W, Whorton MR, MacKinnon R. Quantitative analysis of mammalian GIRK2 channel regulation by G proteins, the signaling lipid PIP2 and Na+ in a reconstituted system. Elife. 2014;3:e03671. doi: 10.7554/eLife.03671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cao E, Cordero-Morales JF, Liu B, Qin F, Julius D. TRPV1 Channels Are Intrinsically Heat Sensitive and Negatively Regulated by Phosphoinositide Lipids. Neuron. 2013;77:667–79. doi: 10.1016/j.neuron.2012.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zakharian E, Cao C, Rohacs T. Gating of transient receptor potential melastatin 8 (TRPM8) channels activated by cold and chemical agonists in planar lipid bilayers. J Neurosci. 2010;30:12526–34. doi: 10.1523/JNEUROSCI.3189-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cheng WWL, D’Avanzo N, Doyle Da, Nichols CG. Dual-mode phospholipid regulation of human inward rectifying potassium channels. Biophys J. 2011;100:620–8. doi: 10.1016/j.bpj.2010.12.3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lopes CMB, Zhang H, Rohacs T, Jin T, Yang J, Logothetis DE. Alterations in conserved Kir channel-PIP2 interactions underlie channelopathies. Neuron. 2002;34:933–944. doi: 10.1016/s0896-6273(02)00725-0. [DOI] [PubMed] [Google Scholar]
- 46.Laganowsky A, Reading E, Allison TM, Ulmschneider MB, Degiacomi MT, Baldwin AJ, Robinson CV. Membrane proteins bind lipids selectively to modulate their structure and function. Nature. 2014;510:172–175. doi: 10.1038/nature13419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Long SB, Tao X, Campbell EB, MacKinnon R. Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature. 2007;450:376–382. doi: 10.1038/nature06265. [DOI] [PubMed] [Google Scholar]
- 48.Whorton M, MacKinnon R. Crystal Structure of the Mammalian GIRK2 K+ Channel and Gating Regulation by G Proteins, PIP 2, and Sodium. Cell. 2011;147:199–208. doi: 10.1016/j.cell.2011.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gonen T, Cheng Y, Sliz P, Hiroaki Y, Fujiyoshi Y, Harrison SC, Walz T. Lipid-protein interactions in double-layered two-dimensional AQP0 crystals. Nature. 2005;438:633–8. doi: 10.1038/nature04321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.MONOD J, WYMAN J, CHANGEUX JP. ON THE NATURE OF ALLOSTERIC TRANSITIONS: A PLAUSIBLE MODEL. J Mol Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 51.Changeux J-P, Edelstein SJ. Allosteric mechanisms of signal transduction. Science. 2005;308:1424–8. doi: 10.1126/science.1108595. [DOI] [PubMed] [Google Scholar]
- 52.Rapedius M, Soom M, Shumilina E, Schulze D, Schönherr R, Kirsch C, Lang F, Tucker SJ, Baukrowitz T. Long chain CoA esters as competitive antagonists of phosphatidylinositol 4,5-bisphosphate activation in Kir channels. J Biol Chem. 2005;280:30760–30767. doi: 10.1074/jbc.M503503200. [DOI] [PubMed] [Google Scholar]
- 53.Gribble FM, Proks P, Corkey BE, Ashcroft FM. Mechanism of cloned ATP-sensitive potassium channel activation by oleoyl-CoA. J Biol Chem. 1998;273:26383–7. doi: 10.1074/jbc.273.41.26383. [DOI] [PubMed] [Google Scholar]
- 54.Schulze D, Rapedius M, Krauter T, Baukrowitz T. Long-chain acyl-CoA esters and phosphatidylinositol phosphates modulate ATP inhibition of KATP channels by the same mechanism. J Physiol. 2003;552:357–67. doi: 10.1113/jphysiol.2003.047035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Czech MP. PIP2 and PIP3: complex roles at the cell surface. Cell. 2000;100:603–6. doi: 10.1016/s0092-8674(00)80696-0. [DOI] [PubMed] [Google Scholar]
- 56.Willars GB. Differential Regulation of Muscarinic Acetylcholine Receptor-sensitive Polyphosphoinositide Pools and Consequences for Signaling in Human Neuroblastoma Cells. J Biol Chem. 1998;273:5037–5046. doi: 10.1074/jbc.273.9.5037. [DOI] [PubMed] [Google Scholar]
- 57.Murata Y, Okamura Y. Depolarization activates the phosphoinositide phosphatase Ci-VSP, as detected in Xenopus oocytes coexpressing sensors of PIP2. J Physiol. 2007;583:875–89. doi: 10.1113/jphysiol.2007.134775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Carr DB, Surmeier DJ. M1 muscarinic receptor modulation of Kir2 channels enhances temporal summation of excitatory synaptic potentials in prefrontal cortex pyramidal neurons. J Neurophysiol. 2007;97:3432–8. doi: 10.1152/jn.00828.2006. [DOI] [PubMed] [Google Scholar]
- 59.Rossignol TM, Jones SVP. Regulation of a family of inwardly rectifying potassium channels (Kir2) by the m1 muscarinic receptor and the small GTPase Rho. Pflugers Arch. 2006;452:164–174. doi: 10.1007/s00424-005-0014-9. [DOI] [PubMed] [Google Scholar]
- 60.Du X, Zhang H, Lopes C, Mirshahi T, Rohacs T, Logothetis DE. Characteristic interactions with phosphatidylinositol 4,5-bisphosphate determine regulation of kir channels by diverse modulators. J Biol Chem. 2004;279:37271–81. doi: 10.1074/jbc.M403413200. [DOI] [PubMed] [Google Scholar]
- 61.Plaster NM, Tawil R, Tristani-Firouzi M, Canún S, Bendahhou S, Tsunoda A, Donaldson MR, Iannaccone ST, Brunt E, Barohn R. Mutations in Kir21 Cause the Developmental and Episodic Electrical Phenotypes of Andersen’s Syndrome. Cell. 2001;105:511–519. doi: 10.1016/s0092-8674(01)00342-7. [DOI] [PubMed] [Google Scholar]
- 62.Baukrowitz T. PIP2 and PIP as Determinants for ATP Inhibition of KATP Channels. Science (80-) 1998;282:1141–1144. doi: 10.1126/science.282.5391.1141. [DOI] [PubMed] [Google Scholar]
- 63.Shyng S. Membrane Phospholipid Control of Nucleotide Sensitivity of KATP Channels. Science (80-) 1998;282:1138–1141. doi: 10.1126/science.282.5391.1138. [DOI] [PubMed] [Google Scholar]
- 64.Krauter T, Ruppersberg JP, Baukrowitz T. Phospholipids as modulators of K(ATP) channels: distinct mechanisms for control of sensitivity to sulphonylureas, K(+) channel openers, and ATP. Mol Pharmacol. 2001;59:1086–93. [PubMed] [Google Scholar]
- 65.Lee S-J, Wang S, Borschel W, Heyman S, Gyore J, Nichols CG. Secondary anionic phospholipid binding site and gating mechanism in Kir21 inward rectifier channels. Nat Commun. 2013;4:2786. doi: 10.1038/ncomms3786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xie L-H, John Sa, Ribalet B, Weiss JN. Phosphatidylinositol-4,5-bisphosphate (PIP2) regulation of strong inward rectifier Kir21 channels: multilevel positive cooperativity. J Physiol. 2008;586:1833–48. doi: 10.1113/jphysiol.2007.147868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jiang Y, Lee A, Chen J, Ruta V, Cadene M, Chait BT, MacKinnon R. X-ray structure of a voltage-dependent K+ channel. Nature. 2003;423:33–41. doi: 10.1038/nature01580. [DOI] [PubMed] [Google Scholar]
- 68.Long SB, Campbell EB, Mackinnon R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 2005;309:897–903. doi: 10.1126/science.1116269. [DOI] [PubMed] [Google Scholar]
- 69.Hite RK, Butterwick JA, MacKinnon R. Phosphatidic acid modulation of Kv channel voltage sensor function. Elife. 2014;3 doi: 10.7554/eLife.04366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gamper N, Reznikov V, Yamada Y, Yang J, Shapiro MS. Phosphatidylinositol 4,5-bisphosphate signals underlie receptor-specific Gq/11-mediated modulation of N-type Ca2+ channels. J Neurosci. 2004;24:10980–92. doi: 10.1523/JNEUROSCI.3869-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ufret-Vincenty Ca, Klein RM, Hua L, Angueyra J, Gordon SE. Localization of the PIP2 sensor of TRPV1 ion channels. J Biol Chem. 2011;286:9688–98. doi: 10.1074/jbc.M110.192526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Feng X, Huang Y, Lu Y, Xiong J, Wong C-O, Yang P, Xia J, Chen D, Du G, Venkatachalam K, Xia X, Zhu MX. Drosophila TRPML forms PI(3,5)P2-activated cation channels in both endolysosomes and plasma membrane. J Biol Chem. 2014;289:4262–72. doi: 10.1074/jbc.M113.506501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang X, Li X, Xu H. Phosphoinositide isoforms determine compartment-specific ion channel activity. Proc Natl Acad Sci U S A. 2012;109:11384–9. doi: 10.1073/pnas.1202194109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rohacs T. Phosphoinositide regulation of non-canonical transient receptor potential channels. Cell Calcium. 2009;45:554–65. doi: 10.1016/j.ceca.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nilius B, Mahieu F, Prenen J, Janssens A, Owsianik G, Vennekens R, Voets T. The Ca2+-activated cation channel TRPM4 is regulated by phosphatidylinositol 4,5-biphosphate. EMBO J. 2006;25:467–78. doi: 10.1038/sj.emboj.7600963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ase AR, Bernier L-P, Blais D, Pankratov Y, Séguéla P. Modulation of heteromeric P2X1/5 receptors by phosphoinositides in astrocytes depends on the P2X1 subunit. J Neurochem. 2010;113:1676–84. doi: 10.1111/j.1471-4159.2010.06734.x. [DOI] [PubMed] [Google Scholar]
- 77.Bernier L-P, Ase AR, Chevallier S, Blais D, Zhao Q, Boué-Grabot E, Logothetis D, Séguéla P. Phosphoinositides regulate P2X4 ATP-gated channels through direct interactions. J Neurosci. 2008;28:12938–45. doi: 10.1523/JNEUROSCI.3038-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Michailidis IE, Helton TD, Petrou VI, Mirshahi T, Ehlers MD, Logothetis DE. Phosphatidylinositol-4,5-bisphosphate regulates NMDA receptor activity through alpha-actinin. J Neurosci. 2007;27:5523–32. doi: 10.1523/JNEUROSCI.4378-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mo G, Bernier L-P, Zhao Q, Chabot-Doré A-J, Ase AR, Logothetis D, Cao C-Q, Séguéla P. Subtype-specific regulation of P2X3 and P2X2/3 receptors by phosphoinositides in peripheral nociceptors. Mol Pain. 2009;5:47. doi: 10.1186/1744-8069-5-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Do Heo W, Inoue T, Park WS, Kim ML, Park BO, Wandless TJ, Meyer T. PI(3,4,5)P3 and PI(4,5)P2 lipids target proteins with polybasic clusters to the plasma membrane. Science. 2006;314:1458–61. doi: 10.1126/science.1134389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Di Paolo G, De Camilli P. Phosphoinositides in cell regulation and membrane dynamics. Nature. 2006;443:651–7. doi: 10.1038/nature05185. [DOI] [PubMed] [Google Scholar]
- 82.Zhang X, Li X, Xu H. Phosphoinositide isoforms determine compartment-specific ion channel activity. Proc Natl Acad Sci U S A. 2012;109:11384–9. doi: 10.1073/pnas.1202194109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kooijman EE, Burger KNJ. Biophysics and function of phosphatidic acid: a molecular perspective. Biochim Biophys Acta. 2009;1791:881–8. doi: 10.1016/j.bbalip.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 84.Shi X, Bi Y, Yang W, Guo X, Jiang Y, Wan C, Li L, Bai Y, Guo J, Wang Y, Chen X, Wu B, Sun H, Liu W, Wang J, Xu C. Ca2+ regulates T-cell receptor activation by modulating the charge property of lipids. Nature. 2013;493:111–5. doi: 10.1038/nature11699. [DOI] [PubMed] [Google Scholar]
- 85.Simons K, Ikonen E. Functional rafts in cell membranes. Nature. 1997;387:569–72. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- 86.Lingwood D, Simons K. Lipid rafts as a membrane-organizing principle. Science. 2010;327:46–50. doi: 10.1126/science.1174621. [DOI] [PubMed] [Google Scholar]
- 87.van den Bogaart G, Meyenberg K, Risselada HJ, Amin H, Willig KI, Hubrich BE, Dier M, Hell SW, Grubmüller H, Diederichsen U, Jahn R. Membrane protein sequestering by ionic protein-lipid interactions. Nature. 2011;479:552–5. doi: 10.1038/nature10545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wenk MR, Lucast L, Di Paolo G, Romanelli AJ, Suchy SF, Nussbaum RL, Cline GW, Shulman GI, McMurray W, De Camilli P. Phosphoinositide profiling in complex lipid mixtures using electrospray ionization mass spectrometry. Nat Biotechnol. 2003;21:813–7. doi: 10.1038/nbt837. [DOI] [PubMed] [Google Scholar]
- 89.Milne SB, Ivanova PT, DeCamp D, Hsueh RC, Brown HA. A targeted mass spectrometric analysis of phosphatidylinositol phosphate species. J Lipid Res. 2005;46:1796–802. doi: 10.1194/jlr.D500010-JLR200. [DOI] [PubMed] [Google Scholar]
- 90.Haag M, Schmidt A, Sachsenheimer T, Brügger B. Quantification of Signaling Lipids by Nano-Electrospray Ionization Tandem Mass Spectrometry (Nano-ESI MS/MS) Metabolites. 2012;2:57–76. doi: 10.3390/metabo2010057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Peres C, Yart A, Perret B, Salles J-P, Raynal P. Modulation of phosphoinositide 3-kinase activation by cholesterol level suggests a novel positive role for lipid rafts in lysophosphatidic acid signalling. FEBS Lett. 2003;534:164–168. doi: 10.1016/s0014-5793(02)03832-2. [DOI] [PubMed] [Google Scholar]
- 92.Birnbaumer L, Abramowitz J, Brown AM. Receptor-effector coupling by G proteins. Biochim Biophys Acta. 1990;1031:163–224. doi: 10.1016/0304-4157(90)90007-y. [DOI] [PubMed] [Google Scholar]
- 93.Berridge MJ, Irvine RF. Inositol trisphosphate, a novel second messenger in cellular signal transduction. Nature. 1984;312:315–21. doi: 10.1038/312315a0. [DOI] [PubMed] [Google Scholar]
- 94.Runnels LW, Yue L, Clapham DE. The TRPM7 channel is inactivated by PIP(2) hydrolysis. Nat Cell Biol. 2002;4:329–36. doi: 10.1038/ncb781. [DOI] [PubMed] [Google Scholar]
- 95.Carr DB, Surmeier DJ, Dembrow NC, Chitwood RA, Johnston D, Shirey JK, Brady AE, Jones PJ, Davis AA, Bridges TM, Kennedy P, Jadhav SB, Menon UN, Xiang Z, Watson ML, Christian EP, Doherty JJ, Quirk MC, Snyder DH, Lah JJ, et al. M 1 Muscarinic Receptor Modulation of Kir2 Channels Enhances Temporal Summation of Excitatory Synaptic Potentials in Prefrontal Cortex Pyramidal Neurons M 1 Muscarinic Receptor Modulation of Kir2 Channels Enhances Temporal Summation of Excitatory Synaptic. J Neurophysiol. 2011 doi: 10.1152/jn.00828.2006. [DOI] [PubMed] [Google Scholar]
- 96.Pian P, Bucchi A, Decostanzo A, Robinson RB, Siegelbaum Sa. Modulation of cyclic nucleotide-regulated HCN channels by PIP(2) and receptors coupled to phospholipase C. Pflugers Arch. 2007;455:125–45. doi: 10.1007/s00424-007-0295-2. [DOI] [PubMed] [Google Scholar]
- 97.Enyedi P, Czirják G. Molecular background of leak K+ currents: two-pore domain potassium channels. Physiol Rev. 2010;90:559–605. doi: 10.1152/physrev.00029.2009. [DOI] [PubMed] [Google Scholar]
- 98.Delmas P, Crest M, Brown Da. Functional organization of PLC signaling microdomains in neurons. Trends Neurosci. 2004;27:41–7. doi: 10.1016/j.tins.2003.10.013. [DOI] [PubMed] [Google Scholar]
- 99.Rohacs T. Regulation of transient receptor potential channels by the phospholipase C pathway. Adv Biol Regul. 2013;53:341–55. doi: 10.1016/j.jbior.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Exton JH. Regulation of phospholipase D. Biochim Biophys Acta. 1999;1439:121–33. doi: 10.1016/s1388-1981(99)00089-x. [DOI] [PubMed] [Google Scholar]
- 101.Wang X, Devaiah SP, Zhang W, Welti R. Signaling functions of phosphatidic acid. Prog Lipid Res. 2006;45:250–78. doi: 10.1016/j.plipres.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 102.Chemin J, Patel AJ, Duprat F, Lauritzen I, Lazdunski M, Honoré E. A phospholipid sensor controls mechanogating of the K+ channel TREK-1. EMBO J. 2005;24:44–53. doi: 10.1038/sj.emboj.7600494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kurrasch-Orbaugh DM, Parrish JC, Watts VJ, Nichols DE. A complex signaling cascade links the serotonin2A receptor to phospholipase A2 activation: the involvement of MAP kinases. J Neurochem. 2003;86:980–91. doi: 10.1046/j.1471-4159.2003.01921.x. [DOI] [PubMed] [Google Scholar]
- 104.Tedford HW, Zamponi GW. Direct G protein modulation of Cav2 calcium channels. Pharmacol Rev. 2006;58:837–62. doi: 10.1124/pr.58.4.11. [DOI] [PubMed] [Google Scholar]
- 105.Delmas P, Coste B, Gamper N, Shapiro MS. Phosphoinositide lipid second messengers: new paradigms for calcium channel modulation. Neuron. 2005;47:179–82. doi: 10.1016/j.neuron.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 106.Ranganathan R, Harris GL, Stevens CF, Zuker CS. A Drosophila mutant defective in extracellular calcium-dependent photoreceptor deactivation and rapid desensitization. Nature. 1991;354:230–2. doi: 10.1038/354230a0. [DOI] [PubMed] [Google Scholar]
- 107.Yuan Z, Cai T, Tian J, Ivanov AV, Giovannucci DR, Xie Z. Na/K-ATPase tethers phospholipase C and IP3 receptor into a calcium-regulatory complex. Mol Biol Cell. 2005;16:4034–45. doi: 10.1091/mbc.E05-04-0295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Vinuela-Fernandez I, Sun L, Jerina H, Curtis J, Allchorne A, Gooding H, Rosie R, Holland P, Tas B, Mitchell R, Fleetwood-Walker S. The TRPM8 channel forms a complex with the 5-HT1B receptor and phospholipase D that amplifies its reversal of pain hypersensitivity. Neuropharmacology. 2014;79:136–51. doi: 10.1016/j.neuropharm.2013.11.006. [DOI] [PubMed] [Google Scholar]
- 109.Comoglio Y, Levitz J, Kienzler Ma, Lesage F, Isacoff EY, Sandoz G. Phospholipase D2 specifically regulates TREK potassium channels via direct interaction and local production of phosphatidic acid. Proc Natl Acad Sci U S A. 2014 doi: 10.1073/pnas.1407160111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Capestrano M, Mariggio S, Perinetti G, Egorova AV, Iacobacci S, Santoro M, Di Pentima A, Iurisci C, Egorov MV, Di Tullio G, Buccione R, Luini A, Polishchuk RS. Cytosolic phospholipase A2ε drives recycling through the clathrin-independent endocytic route. J Cell Sci. 2014;127:977–93. doi: 10.1242/jcs.136598. [DOI] [PubMed] [Google Scholar]
- 111.Maxfield FR, McGraw TE. Endocytic recycling. Nat Rev Mol Cell Biol. 2004;5:121–32. doi: 10.1038/nrm1315. [DOI] [PubMed] [Google Scholar]
- 112.Clark MR. Flippin’ lipids. Nat Immunol. 2011;12:373–5. doi: 10.1038/ni.2024. [DOI] [PubMed] [Google Scholar]
- 113.Leventis Pa, Grinstein S. The distribution and function of phosphatidylserine in cellular membranes. Annu Rev Biophys. 2010;39:407–27. doi: 10.1146/annurev.biophys.093008.131234. [DOI] [PubMed] [Google Scholar]
- 114.Schmidt A, Wolde M, Thiele C, Fest W, Kratzin H, Podtelejnikov AV, Witke W, Huttner WB, Söling HD. Endophilin I mediates synaptic vesicle formation by transfer of arachidonate to lysophosphatidic acid. Nature. 1999;401:133–41. doi: 10.1038/43613. [DOI] [PubMed] [Google Scholar]
- 115.Mclennan L. Relative effects polyunsaturated of dietary saturated, fatty acids on cardiac arrhythmias and in rats1. 1993. [DOI] [PubMed] [Google Scholar]
- 116.van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol. 2008;9:112–24. doi: 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Singer SJ, Nicolson GL. The fluid mosaic model of the structure of cell membranes. Science. 1972;175:720–31. doi: 10.1126/science.175.4023.720. [DOI] [PubMed] [Google Scholar]