mTOR regulates the expression of DNA damage response enzymes in long‐lived Snell dwarf, GHRKO, and PAPPA‐KO mice (original) (raw)
Summary
Studies of the mTOR pathway have prompted speculation that diminished mTOR complex‐1 (mTORC1) function may be involved in controlling the aging process. Our previous studies have shown diminished mTORC1 activity in tissues of three long‐lived mutant mice: Snell dwarf mice, growth hormone receptor gene disrupted mice (GHRKO), and in this article, mice deficient in the pregnancy‐associated protein‐A (PAPPA‐KO). The ways in which lower mTOR signals slow aging and age‐related diseases are, however, not well characterized. Here, we show that Snell, GHKRO, and PAPPA‐KO mice express high levels of two proteins involved in DNA repair, O‐6‐methylguanine‐DNA methyltransferase (MGMT) and N‐myc downstream‐regulated gene 1 (NDRG1). Furthermore, we report that lowering mTOR enhances MGMT and NDRG1 protein expression via post‐transcriptional mechanisms. We show that the CCR4‐NOT complex, a post‐transcriptional regulator of gene expression, is downstream of the mTORC1 pathway and may be responsible for the upregulation of MGMT and NDRG1 in all three varieties of long‐lived mice. Our data thus suggest a novel link between DNA repair and mTOR signaling via post‐transcriptional regulation involving specific alteration in the CCR4‐NOT complex, whose modulation could control multiple aspects of the aging process.
Keywords: DNA repair, gene expression, IGF, molecular biology of aging, signal transduction
Abbreviations
DW
Snell dwarf mice
GHRKO
growth hormone receptor deletion mice
MGMT
O‐6‐methylguanine‐DNA methyltransferase
mTOR
mammalian target of rapamycin
NDRG1
N‐myc downstream‐regulated gene 1
PAPPA‐KO or KO
pregnancy‐associated protein‐A deletion mice
Introduction
Several lines of evidence have implicated the mechanistic target of rapamycin (mTOR) in the regulation of aging. In lower organisms, such as Caenorhabditis elegans and Drosophila melanogaster, inactivation or reduction of mTOR activity results in life extension (Sharp et al., 2013). In addition, reduction of mTOR activity by rapamycin (Harrison et al., 2009) or reduction in expression of mTOR kinase by hypomorphic mTOR alleles (Selman et al., 2009; Lamming et al., 2012; Wu et al., 2013) can increase mouse lifespan. mTOR is a serine/threonine kinase that forms the catalytic core of at least two complexes, mTOR complex‐1 (mTORC1) and mTOR complex‐2 (mTORC2). In vivo, the phosphorylation of ribosomal protein S6 (pS6(235)) and initiation factor 4E‐binding protein (p4E‐BP1(37)) are markers of mTORC1 activity. Phosphorylation of AKT at Ser‐473 (pAKT(473)), Ser‐450 (pAKT(450)), and the N‐myc downstream‐regulated gene (pNDRG(346)) are considered markers of mTORC2 activity. We have recently studied the phosphorylation status of these substrates in the Snell and GHRKO mice, two long‐lived genetic models, and found tissue‐specific reduction in mTORC1 activity as well as alteration in mTORC2 activity (Dominick et al., 2015). Interestingly, neither mTORC1 nor mTORC2 was altered in mice with liver‐specific deletion of GHR, mice in which lifespan is normal despite dramatically lower levels of serum IGF‐1 (Bartke, 2008; Dominick et al., 2015). The metalloproteinase PAPP‐A is thought to act by regulating local IGF‐1 action through degradation of some IGF‐1 binding proteins, particularly IGF‐BP4 (Conover & Bale, 2007). PAPP‐A deletion can extend both mean and maximum lifespan by 20–40%, with a reduced incidence of spontaneous tumors, without modulation of serum levels of GH or IGF‐1. Effects of PAPPA‐KO on mTORC1 and mTORC2 have not yet been reported. The mechanistic links between GH/IGF‐1 regulation and prolongation of lifespan in these three varieties of mice are not well understood.
O‐6‐methylguanine‐DNA methyltransferase (MGMT) is an enzyme involved in the direct reversal of DNA damage in vitro and postmitotic cells in vivo by methylating agents used clinically for chemotherapy (Iyama & Wilson, 2013; Jiang et al., 2014). It have been suggested that MGMT may play a role in regulating mouse lifespan (Meira et al., 2014), but it is not known whether long‐lived mice, such as Snell, GHRKO, and PAPPA‐KO, have altered expression of MGMT. Higher expression of NDRG1, a protein of unknown function, can promote MGMT protein stability and activity in human glioma cells by mechanisms that include protein interactions and preferential translation (Smalley et al., 2014; Weiler et al., 2014). Therefore, understanding the signaling pathways regulating MGMT and NDRG1 may provide insights into resistance to DNA damage, chemotherapy, and the aging process. We hypothesized that higher expression of MGMT and NDRG1 may be a common feature shared by Snell, GHRKO, and PAPPA‐KO mice, and that mTOR activity could be a key upstream regulator of MGMT and NDRG1 expression. Microarray analysis of the mRNA gene expression profiles of many different models of slow‐aging mice have not led to a consensus on common pathway(s) central to the aging process, including mice whose lifespan has been increased by mTOR reduction by drugs (Yu et al., 2015) or dietary interventions (Swindell, 2008). However, it is plausible that improved DNA repair may be one important common feature (Meira et al., 2014). Discrepancy between mRNA levels and protein expression in many genes complicates interpretation of mRNA patterns in tissues of slow‐aging mice, and it is likely that gene regulation by post‐transcriptional mechanisms, including changes in mRNA stability (Brewer, 2002; Lee & Gorospe, 2010), regulation by microRNAs (Bates et al., 2010; Liang et al., 2011), and sequestration to P and ER bodies may contribute to altered levels of DNA repair proteins in slow‐aging mice.
The CCR4‐NOT complex is a key regulator of mRNA stability, microRNA function, and preferential translation, and functions as a check point in the control of translation and protein degradation (Inada & Makino, 2014; Shirai et al., 2014). The CCR4‐NOT complex is comprised of at least ten subunits, but there is limited information about the function of individual components of the complex in mammals. Recent reports using in vitro methods have helped to clarify some functional roles of CNOT components (Laribee et al., 2015; Okada et al., 2015; Rodriguez‐Gil et al., 2016). Mice in which CNOT6 has been deleted are viable but, with exception of some bone abnormalities, do not have obvious changes in phenotype. However, CNOT3 heterozygous mice (Morita et al., 2011) display some phenotypes reminiscent of Snell (Bartke & Brown‐Borg, 2004; Boylston et al., 2006), Ames dwarf (Boylston et al., 2006), GHRKO (Brown‐Borg & Bartke, 2012), and PAPPA‐KO mice (Hill et al., 2015), including higher insulin sensitivity, improved glucose tolerance, and higher oxygen consumption. Here, we present evidence suggesting that Snell, GHRKO, and PAPPA‐KO mice express high levels of NDRG1 and MGMT proteins that are under the control of mTORC1 activity, and that this regulation involves post‐transcriptional mechanisms that may depend on alteration of the CCR4‐NOT complex.
Results
Multiple models of slow aging, including PAPPA‐KO mice, show lower mTORC1 activity
We have recently reported that Snell and GHRKO mice show reduced mTORC1 activity in multiple tissues, accompanied by changes in both fasting and food‐induced mTORC2 activity (Dominick et al., 2015). To see whether PAPPA‐KO mice showed similar changes in mTORC1 and mTORC2 activity, we measured phosphorylation of mTORC1 substrates pS6(S235/236) [referred to as pS6 in figures and text], p4E‐BP1(T37/46) [termed p4E‐BP1 in figures and text] and mTORC2 substrates pAKT(T450) and pNDRG1(T346) [termed pNDRG1 in figures and text], either after 18 h of fasting or after 18 h of fasting followed by 6 h of access to food (Dominick et al., 2015). Figure 1A shows a representative Western blot for liver, and Fig. 1B shows mean values from three independent experiments using at least eight mice per group. We found no significant sex effects (half of the mice were males) on any of the measures reported in this article (data not shown). Table S1 shows parallel experiments for muscle, heart, and kidney of PAPPA‐KO mice for the mTORC1 substrates, with estimates of fold induction as well as the results of ANOVA testing significance of the effects of genotype (G), nutritional conditions (fasting vs. feeding, N), and GxN interactions (Int). We found that PAPPA‐KO mice show lower levels of pS6 and p4E‐BP1, independent of nutritional status, in all four tissues tested (Fig. 1 and Table S1, Supporting information). Two‐factor ANOVA confirmed the significant effect of the PAPP‐A deletion (genotype effects: WT > KO) and feeding (nutritional effects: fed > fasting), without evidence for interaction. The result is similar to our findings for Snell and GHRKO mice (Dominick et al., 2015). We found no differences in the expression of total S6 or 4E‐BP protein in any of the groups (data not shown).
Figure 1.
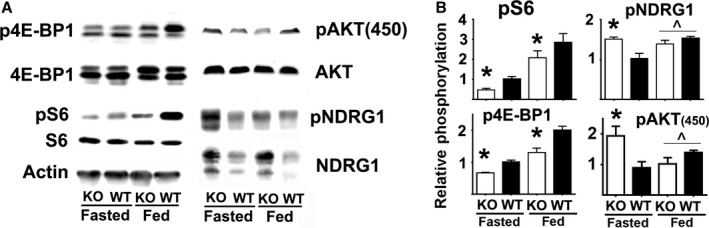
mTORC activity in liver of PAPP‐A mice. (A) Representative Western blots showing the phosphorylation of S6, 4E‐BP1, AKT, and NDRG1 in liver samples from PAPPA‐KO (KO) and control littermates (WT) under fasting (Fasted) or fed (Fed) conditions. (B) Quantification of pS6, p4E‐BP1, pAKT(450), and pNDRG1 phosphorylation as ratios to the level of the corresponding protein. All bar graphs represent the mean ± SEM from a total of eight mice (four females and four males) per group and are normalized with respect to ratio of 1 in fasted WT for each protein. The asterisk (*) indicates statistical significance relative to the fasting WT from each group.
Results for mTORC2 substrates pAKT(450) and pNDRG are shown in Fig. 1 for liver and are summarized in Table S2 (Supporting information) for all four tissues. Consistent with Snell and GHRKO mice (Dominick et al., 2015), fasted PAPPA‐KO mice have higher mTORC2 activity, and this activity decreases in response to feeding in PAPPA‐KO but not in WT control mice (Fig. 1). Statistical analysis using ANOVA confirmed that feeding has significant interaction effects for pAKT(450) and pNDRG (Fig. 1B and Table S2, Supporting information). The overall results suggest that PAPPA‐KO, Snell, and GHRKO mice share a common regulatory mechanism for mTOR activity and signaling that includes tissue‐specific declines in mTORC1 activity and differential regulation of mTORC2 activity.
Snell, GHRKO, and PAPPA‐KO mice show higher NDRG1 and MGMT protein expression, but no changes in NDRG1 or MGMT mRNA expression
Using a rabbit monoclonal from Cell Signaling (D6C2), we noted an increase in levels of total NDRG1 protein expression in liver of PAPPA‐KO mice (Fig. 1A). Higher NDRG1 expression has been associated with enhanced DNA repair through increased MGMT protein stability/activity (Weiler et al., 2014). We therefore measured NDRG1 and MGMT protein expression in liver, skeletal muscle (upper leg), and kidney from our three slow‐aging mice models fed ad lib. The right panels of Fig. 2 show representative results from liver tissue in Snell (Fig. 2A), GHRKO (Fig. 2B), and PAPPA‐KO (Fig. 2C). NDRG1 and MGMT protein expression was increased by 2× to 3× in each kind of slow‐aging mouse.
Figure 2.
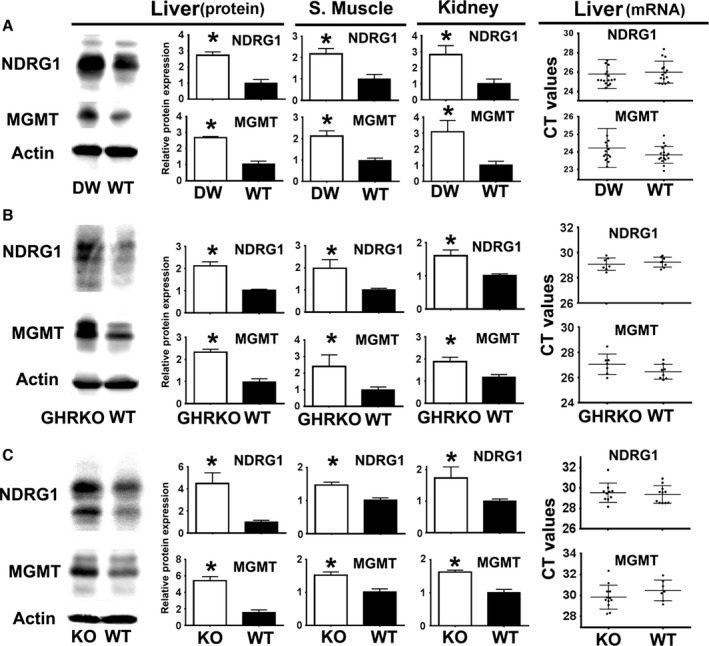
Snell mice (DW), GHRKO, and PAPP‐A (KO) mice show higher NDRG1 and MGMT protein expression but no significant change in mRNA expression. (A) Representative Western blots showing NDRG1 and MGMT expression in Snell mice (DW) from liver (left). Quantification in liver, skeletal muscle, and kidney (middle panels) is shown as mean ± SEM from three experiments, using a total of at least 12 mice (six females and six males) per group, using normalization as described in Fig. 1. The qRT–PCR CT dotplot values for NDRG1 and MGMT expression are shown in the right panels. Each symbol represents an individual mouse. Horizontal lines show mean ± SEM from a total of 16 mice (eight females and eight males) per group. The asterisk (*) indicates statistical significance relative to the WT controls of each group. (B) Results for GHRKO mice from at least 11 mice (six females and five males) per group for protein expression (middle panels) and for qRT–PCR using eight mice (four females and four males) per group (right panels). (C) Results for PAPPA‐KO (KO) mice from at least eight mice (four females and four males) per group and qRT–PCR using at least ten mice (five females and five males) per group.
To assess whether higher protein levels were the result of upregulation of NDRG1 and MGMT mRNA transcripts, we performed qRT–PCR for both genes in liver. In contrast to the protein data, there were no differences in the levels of NDRG1 and MGMT mRNA in any of the mouse models (Fig. 2 right panels). We confirmed these results using three other differently designed probes for MGMT and NDRG1 (data not shown). We also found no differences in the GAPDH mRNA expression (Fig. S1, Supporting information). This suggested that discrepancies between protein expression and mRNA levels might reflect differential post‐transcriptional regulation of MGMT and NDRG1 in slow‐aging mice.
mTOR signaling regulates NDRG1 and MGMT protein expression but not their mRNA expression
In some human cell lines, rapamycin (an mTOR inhibitor) can enhance MGMT protein expression, and this increase is the result of preferential translation of MGMT transcripts (Smalley et al., 2014). mTOR can also regulate stress resistance by preferential translation of certain mRNAs (Reiter et al., 2004), including ATF‐4 (Ait et al., 2012). Hence, we hypothesized that changes in mTOR function in Snell, GHRKO, and PAPPA‐KO mice could increase NDRG1 and MGMT via a post‐transcriptional pathway. To test this idea, we treated mouse fibroblasts from a genetically heterogeneous stock of mice (Harrison et al., 2009) with rapamycin for 48 h and analyzed the expression of NDRG1 and MGMT using Western blots and qRT–PCR. The data (Fig. 3) show that rapamycin enhances NDRG1 and MGMT protein expression, at a concentration that impairs phosphorylation of the mTORC1 substrates 4E‐BP1 and S6. Statistical analysis confirmed significant increases in NDRG1 and MGMT protein in fibroblasts treated for 48 h with rapamycin (Fig. 3A middle panel). Parallel mRNA analysis found no differences in NDRG1 and MGMT mRNA expression (or in GAPDH mRNA) in fibroblasts exposed to rapamycin for 24 or 72 h (not shown). These results suggest that mTOR regulates fibroblast NDRG1 and MGMT protein expression through a post‐transcriptional mechanism. However, extended rapamycin treatments can diminish expression and phosphorylation of some of the mTORC2 substrates [pAKT/AKT in Fig. 3A and (Sarbassov et al., 2006; Lamming et al., 2012)], suggesting that lower mTORC2 function may, in part, be responsible for some of the effects we see in MGMT and NDRG1 expression. The overall results suggest that mTOR plays a key role, but more work will be needed to determine the specific role of each complex (mTORC1 and mTORC2) in these animals.
Figure 3.
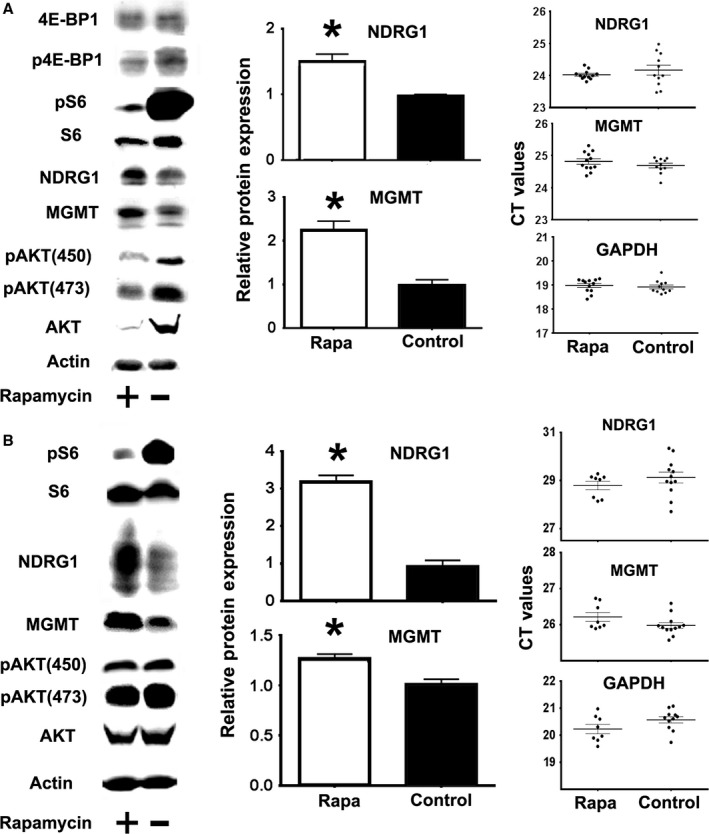
Rapamycin enhances MGMT and NDRG1 protein expression but does not significantly alter their mRNA expression. (A) Representative Western blots showing pS6, p4E‐BP1, and pAKT phosphorylation (in parallel to total S6, 4E‐BP1 and AKT protein), as well as NDRG1 and MGMT expression in mouse fibroblasts, either untreated (−, Control) or treated with rapamycin (+, Rapa). Bar graphs represent the mean ± SEM from six independent experiments normalized with respect to WT controls. The CT value dotplots show mRNA levels from six independent experiments. The (*) indicates statistical significance. (B) Analysis of rapamycin effects in liver of UM‐HET mice from at least eight mice per group.
We also tested the in vivo effects of rapamycin on NDRG1 and MGMT in liver. A single rapamycin injection increases NDRG1 and MGMT protein expression in liver (Fig. 3B). Rapamycin leads to the expected reduction in mTORC1 activity (indicated by pS6), and no apparent changes in mTORC2 activity (as measured by phosphorylation of AKT450 and AKT473). Statistical analysis showed significant increases in NDRG1 and MGMT protein expression by rapamycin. Consistent with the fibroblast experiments, there are no significant changes in NDRG1, MGMT mRNA levels. The in vivo results suggest that mTOR can regulate NDRG1 and MGMT expression through post‐transcriptional mechanisms, and that lower mTORC1 activity in Snell, GHRKO, and PAPPA‐KO mice may upregulate MGMT and NDRG1. However, Schreiber et al. (2015) have suggested that rapamycin effects on mTORC2 signaling may be difficult to detect, and we cannot rule out the idea that mTORC2 could also play an important role in regulating MGMT and NDRG1 expression in the slow‐aging mice.
Snell, GHRKO, and PAPPA‐KO mice show similar alterations in the expression of components of the CCR4‐NOT complex
To see whether changes in components of the CCR4‐NOT complex could be involved in differential post‐transcriptional regulation, we tested expression of CNOT1, CNOT3, CNOT6, and CNOT6L in liver of Snell, GHRKO, and PAPPA‐KO mice. CNOT3 was selected because its decline leads to some features shared with slow‐aging mice (Morita et al., 2011). CNOT6 and CNOT6L were tested because they may regulate DNA repair (Sanchez‐Perez et al., 2009). As shown in Fig. 4, CNOT1 and CNOT3 levels are lower, and CNOT6 and CNOT6L higher, in liver from each variety of slow‐aging mice.
Figure 4.
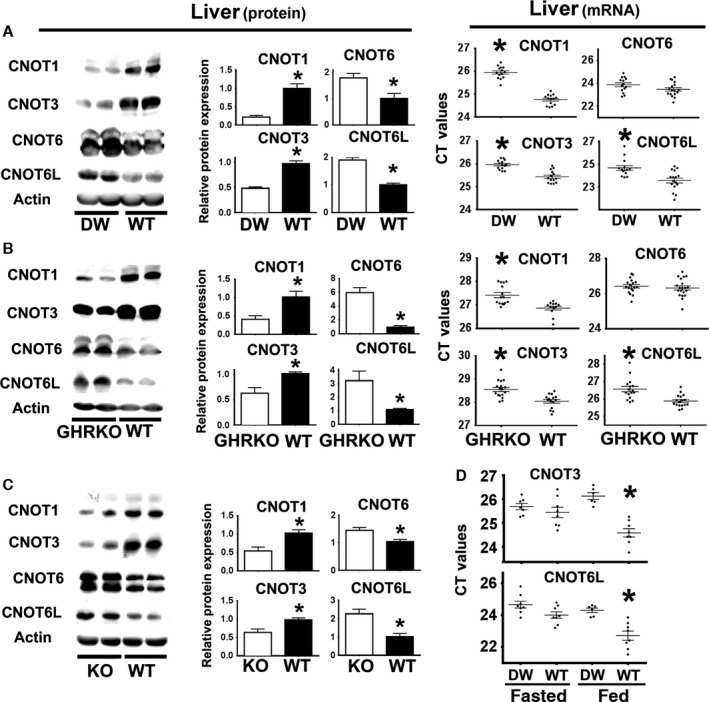
Livers of Snell mice (DW), GHRKO, and PAPPA‐KO (KO) mice show declines in CNOT1 and CNOT3 accompanied by increases in CNOT6L and CNOT6 protein. (A) Left panels represent Western blots of CCR4‐CNOT complex components in Snell mice (DW) and control mice (WT) from liver. Quantification shown as mean ± SEM of three experiments in middle panels from a total of 12 mice (six females and six males) per group using normalization as described in Fig. 1. Right panel shows the qRT–PCR CT values for CCR4‐CNOT mRNA levels in livers from at least 14 mice (seven females and seven males) per group. The (*) indicates statistical significance relative to the WT controls of each group. (B) Data for protein levels in GHRKO mice (left and middle panels), using six mice (three females and three males) per group. qRT–PCR CT data (right panels) from at least 12 mice (six females and six males) per group. (C) Data for protein in PAPPA‐KO (KO) mice, using nine mice (five females and four males) per group. (D) Effect of fasting and feeding on mRNA expression of CNOT3 and CNOT6L in livers of Snell (DW) and WT control mice from at least six mice (1/2 females and 1/2 males) per group. The (*) indicates statistical significance relative to Snell (DW).
CNOT1 and CNOT3 mRNA CT values are higher in Snell and GHRKO liver (Fig. 4) in good agreement with the protein data. In contrast, we found no changes in the CNOT6 mRNA levels with respect to WT controls and, unexpectedly, found a significant reduction in CNOT6L mRNA (Fig. 4A,B). The results of CNOT6 and CNOT6L do not correspond with the level of their protein expression. We do not know the reason for this discrepancy but it suggests that CNOT components may also themselves be subject to post‐transcriptional regulation. CNOT4, which does not associate with CCR4‐NOT in mammals and does not regulate mRNA stability or translation (Shirai et al., 2014), did not differ between long‐lived and littermate control mice (Fig. S2, Supporting information).
Feeding mice increases CNOT3 expression (Morita et al., 2011). Interesting, provision of food after a period of fasting increased CNOT3 and CNOT6L mRNA expression in control mice, but not in Snell littermates (Fig. 4D). These results could be explained by a higher level of mTORC1 activity in control mice (WT) that could contribute to the differential upregulation of CNOT3 and CNOT6L mRNA in response to food.
Rapamycin inhibition of mTORC1 activity mimics some of the changes in the CCR4‐CNOT complex expression found in Snell, GHRKO, and PAPPA‐KO mice
To test the hypothesis that mTOR can regulate the expression of components of the CCR4‐NOT complex, we treated primary mouse fibroblasts with rapamycin and analyzed the expression of CNOT1, CNOT3, CNOT6, and CNOT6L using Western blots and qRT–PCR. Rapamycin significantly decreased CNOT1 and CNOT3 protein expression and enhanced expression of CNOT6L; however, CNOT6 protein was not affected (Fig. 5A). Rapamycin significantly diminished CNOT1, CNOT3, and CNOT6L mRNA but not CNOT6 mRNA (Fig. S3, Supporting information). This pattern of consistent changes in protein and mRNA data for CNOT1, CNOT3, and CNOT6L is similar to that noted for Snell, GHRKO, and PAPPA‐KO mice (Fig. 4). For in vivo confirmation, we treated mice with rapamycin for 48 h and analyzed the expression of CCR4‐NOT components in liver. Rapamycin reduced CNOT1 and CNOT3 and enhanced CNOT6L, but did not change CNOT6 (Fig. 5B), consistent with the fibroblast results. Thus, CNOT1, CNOT3, and CNOT6L are under the control of mTOR signaling at the level of transcription and post‐transcriptional mechanisms. In contrast, CNOT6 is not affected by rapamycin at the dose and time used.
Figure 5.
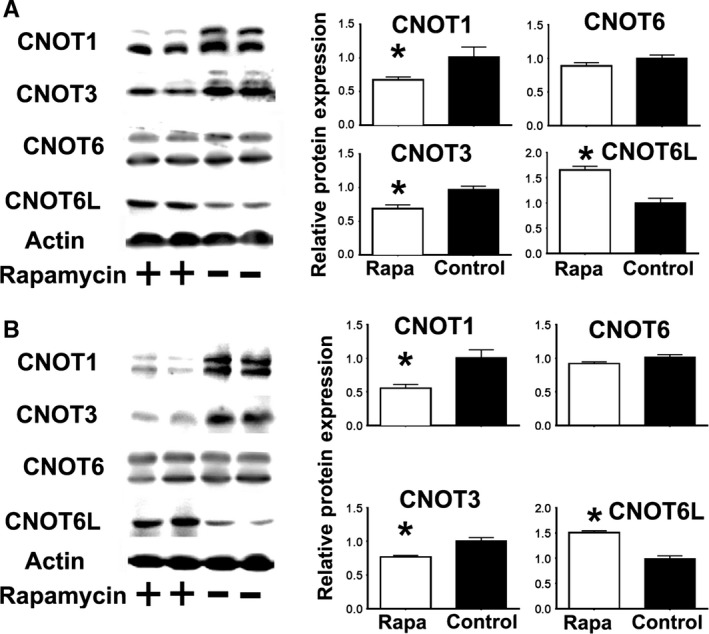
Rapamycin treatment decreases CNOT1 and CNOT3 expression and increases CNOT6L protein expression, but does not affect CNOT6 expression. (A) Representative Western blots showing expression of components of the CCR4‐CNOT complex in mouse fibroblasts untreated (−, Control) or treated with rapamycin (+, Rapa). Bar graphs represent the mean ± SEM from a total of six independent experiments normalized with respect to WT controls. The (*) indicates statistical significance. (B) Analysis of rapamycin effects on CNOT components in mouse livers, either untreated (−, Control) or treated with rapamycin (+, Rapa) from at least eight mice per group (50% males).
CNOT3 regulates NDRG1 and MGMT protein expression
To see whether CNOT3 or CNOT6L might be involved in mTORC1 regulation of NDRG1 and MGMT protein expression, we transfected mouse fibroblasts with constructs overexpressing human CNOT3 and CNOT6L (GFP‐CNOT3, GFP‐CNOT6L). Human CNOT3 and CNOT6L share 95–99% of their amino acid sequence with their mouse homologs and thus are likely to function in mouse cells. In addition, we tested the effects of reduction of CNOT3 and CNOT6L expression using silencing constructs (shRNAs) tailored to their respective mouse genes. Mouse fibroblasts were transfected, and efficiency was measured using green fluorescent protein (Fig. S4A, Supporting information). Quantification showed a range from 10 to 30% efficiency, depending on the construct (data not shown). Immunoblotting shows the expected increases or declines in CNOT3 and CNOT6L protein at 72 h after transfection (Fig. S4B, Supporting information). Overexpression of CNOT3 led to reduction in NDRG1 and MGMT protein expression, while CNOT3 downregulation increased NDRG1 and MGMT (Fig. 6A, top panel). Statistical analysis confirmed significant effects in NDRG1 and MGMT expression by CNOT3 (Fig. 6B). Neither overexpression nor reduction of CNOT6L protein affected NDRG1 or MGMT protein expression. Furthermore, changes in CNOT3 and CNOT6L did not affect mTOR activity (no changes in phosphorylation of S6 or AKT(473)), suggesting that CCR4‐NOT is downstream of mTOR in regulating NDRG1 and MGMT expression (Fig. 6A). These results suggest that CNOT3 is a post‐transcriptional regulator of NDRG1 and MGMT protein expression and that declines in CNOT3 in Snell, GHRKO, and PAPPA‐KO mice may be involved in the enhanced expression of these DNA repair enzymes.
Figure 6.
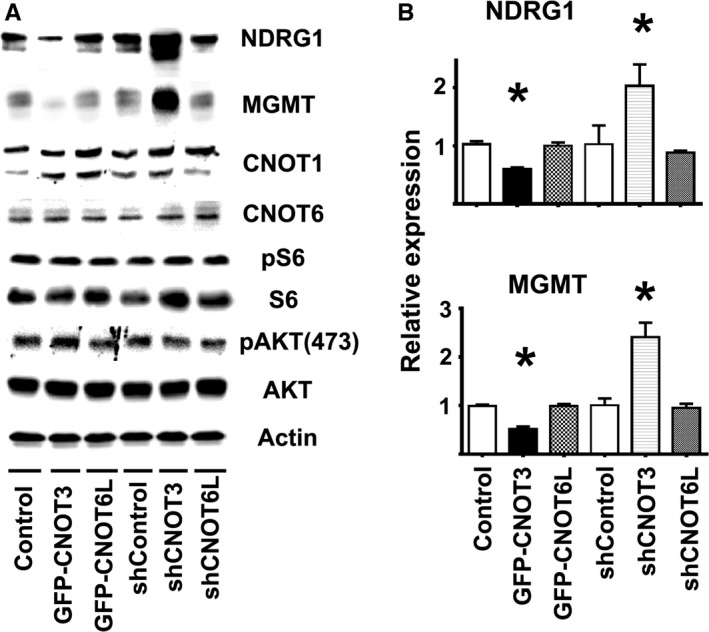
Effect of overexpressing or downregulating CNOT3 and CNOT6L protein on NDRG1 and MGMT protein expression in mouse fibroblasts. (A) Representative Western blots for NDRG1 and MGMT expression after 72 h of transfection, together with pS6, pAKT, CNOT1, CNOT6 in the same samples. (B) Quantification of the NDRG1 and MGMT protein expression after each transfection shown as mean ± SEM normalized to controls for six independent experiments; the (*) indicates statistical significance.
Discussion
Our data showing reduction of mTORC1 in multiple tissues of Snell, GHRKO (Dominick et al., 2015), and now PAPPA‐KO mice support the hypothesis that mTORC1 controls some of the intracellular events leading to lifespan extension in mammals. It is still unclear, however, how lower mTORC1 action might lead to improved health and better survival in aging mice. Many lines of evidence (Lombard & Miller, 2012) suggest that increases in stress resistance (Murakami, 2006), including better DNA repair (De Luca et al., 2013), may be involved in longevity and disease resistance. This idea is now supported by our observation of increases in MGMT and NDRG1, which are involved in direct reversal of DNA damage, in multiple tissues of three stocks of slow‐aging mice. Our evidence that reducing mTORC1 activity leads to enhancement in MGMT and NDRG1 protein expression, both in cultured cells and in mice, strongly suggests a link between mTOR signaling and the DNA repair pathway (Strozyk & Kulms, 2013) that may be central in delaying the aging process. Our data also suggest a possible involvement of mTORC1‐mediated alteration in post‐transcriptional regulation of MGMT and NDRG1 translation via changes in elements of the CCR4‐CNOT complex.
Comparative microarray analysis among several genetic models (Svensson et al., 2011; Hofmann et al., 2015), pharmacological interventions (Miller et al., 2014; Yu et al., 2015), and dietary interventions (Bartke et al., 2007) known to extend lifespan has not revealed consistent shared features at the level of transcriptional changes, including mRNA levels for enzymes from the DNA repair response such as MGMT and NDRG1. These observations are consistent with our qRT–PCR analysis, which showed no significant changes in NDRG1 and MGMT mRNA expression in the Snell, GHRKO, and PAPPA‐KO mice with respect to controls (Fig. 2). The discrepancy between the mRNA and protein levels suggested that a post‐transcriptional mechanism may be involved in the physiology of these mouse models and in the control of longevity and age‐dependent diseases.
Our data suggest that the CCR4‐NOT complex, a key regulator of post‐transcriptional gene expression, may link mTOR changes to DNA repair in these slow‐aging mice. Snell, GHRKO, and PAPPA‐KO mice show a consistent pattern of changes in CNOT1, CNOT3, CNOT6, and CNOT6L, both in cultured cells and in intact mice, a pattern recapitulated in control cells and in control mice by short‐term exposure to rapamycin. Thus, regulation of key CCR4‐NOT expression components seems to be under the control of mTORC1 (such as CNOT1, CNOT3 and CNOT6L shown in Fig. 5), and down regulation of CNOT3 expression seems to upregulate NDRG1 and MGMT protein expression (Fig. 6). The CCR4‐NOT complex has been studied in yeast (Shirai et al., 2014), but little is known about the function of individual components in higher organisms such as mice (Shirai et al., 2014).
CCR4‐NOT components, including CNOT3, CNOT6, and CNOT6L, can regulate mRNA stability, microRNA function, localization to the P/Stress bodies, and preferential translation (Shirai et al., 2014). However, this regulation involves the interaction of a wide range of tissue‐specific mRNA‐binding proteins (Reese, 2013; Temme et al., 2014), and thus more multifaceted work will be needed to determine how the changes in CNOT protein levels shown in Fig. 4 contribute to protection against age‐related changes in specific cell and tissue types. Recent studies in Snell, GHRKO, and PAPPA‐KO mice have shown a common physiology that may control aging rate and multiple age‐related diseases (Murakami et al., 2003; Boylston et al., 2006; Brown‐Borg, 2006). These mice all exhibit high insulin sensitivity and an increased oxygen consumption with high levels of peroxisome proliferator‐associated receptor γ coactivator 1 (Pgc1α) from oxidative phosphorylation (Corton & Brown‐Borg, 2005; Stauber et al., 2005). Similarly, CNOT3 heterozygous mice (Morita et al., 2011) have been shown to have increased insulin sensitivity, oxidative phosphorylation (including higher Pgc1α expression), and oxygen consumption. Our data suggest that CCR4‐NOT complex, including CNOT3, could be one of the signaling pathways downstream of mTORC1 regulating some of these changes (Fig. 2). Therefore, it would be interesting to test if declines in CNOT3 [such as in the CNOT3 heterozygous mice (Morita _et al_., 2011)], along with changes in other components of the CCR4‐NOT, could themselves extend lifespan. It would also be interesting to define what specific mechanism the CCR4‐NOT components are using to enhance expression of the MGMT and NDRG1 DNA repair genes, such as regulation by microRNA or preferential translation (or both). Because many of the CCR4‐NOT components are essential for development, future studies of CCR4‐NOT and its effects on DNA repair and the aging process would require the use of conditional knockouts triggered in adulthood. Of particular interest in the aging processes would be CCR4‐NOT components that affect post‐transcriptional mechanisms, such as CNOT6 and CNOT6L. Alternatively, when available, the analysis of CNOT6 and CNOT6L +/− heterozygous mice would also shed some light on possible functions of CNOT in aging. On the other hand, it is also possible that changes in proteasomal function in slow‐aging mice could contribute to the elevation of MGMT, NDRG1, and CNOT6L (Wang & Miller, 2012; Zhang et al., 2014; Zhang & Manning, 2015).
Increases in CNOT6L have been suggested to increase the ATM‐dependent DNA repair response (Sanchez‐Perez et al., 2009). In this context, it will be interesting to determine whether tissues from slow‐aging mice have increases in other DNA repair mechanisms, or, alternatively, whether being heterozygous for CNOT components would lead to changes in DNA repair that may affect the aging process. Overall, the data presented here suggest a signaling pathway in which alterations in mTOR, including declines in mTORC1, lead to alterations in post‐transcriptional regulation of gene expression, including genes involved in DNA repair, via changes in composition and function of the CCR4‐NOT complex.
Material and methods
Reagents, mice, and fasting and feeding conditions
Snell (DW) were produced as previously described (Dominick et al., 2015). Breeding stock for GHRKO mice was provided by Andrzej Bartke (Southern Illinois University, IL) and John Kopchick (Edison Biotechnology Institute of Ohio University, OH) and mice were produced as previously described (Dominick et al., 2015). Breeding stock for PAPPA‐KO mice was provided by Cheryl Conover (Mayo Clinic, Rochester, MN) and mice were produced as previously described (Conover et al., 2004). DW, GHRKO, and PAPPA‐KO mice were used at ages from 5 to 6 months. Genetically heterogeneous mice (UM‐HET3) ranging from 10 to 12 months of age were produced by a cross between two different F1 parents: (BALB/cByJ × C57BL/6J)F1 mothers and (C3H/HeJ × DBA/2J)F1 fathers as previously described (Harrison et al., 2009). All experiments were approved by the University of Michigan University Committee on Use and Care of Animals. Sentinel animals were examined quarterly for evidence of viral infection; all tests were negative. Mice used were fed ad lib or, as indicated in the text, fasted for 18 h (fasted mice) followed by ad lib feeding with a regular chow diet for 6 h (fed mice). All mice had free access to water during the course of treatments. Chemical reagents were purchased from Sigma (www.sigmaldrich.com). Rapamycin was purchased from LC laboratories (www.lclabs.com). The source of antibodies and phospho‐specific antibodies are described in Table S3. Primary mouse fibroblasts from UM‐HET3 mice were cultured as described (Murakami et al., 2003). Clones corresponding to human CNOT3 (pT7‐EGFP‐HsNOT3, Addgene plasmid 37369) and CNOT6L (pT7‐EGFP‐HsNOT6L, Addgene plasmid 37372) were a gift from Elisa Izaurralde (Max Planck Institute for Developmental Biology, Germany). The shRNA for mouse CNOT3 (NM_146176.2) and mouse CNOT6L (NM_144910.1) were purchased from Genecopoeia (www.genecopoeia.com). Plasmid transfections were carried out with lipofectamine 3000 as described by the manufacturer (www.thermofisher.com). qRT–PCR was performed as described (Li et al., 2013). Probes were manufactured by IDT (www.idtdna.com) corresponding to the sequences shown in Table S4.
Rapamycin treatments, tissue preparation, Western blot analysis
Acute rapamycin treatments in UM‐HET3 mouse fibroblasts were performed at 0.1 μm. In vivo rapamycin treatments were performed with a single intraperitoneal injection of rapamycin at 5 mg kg−1 to each UM‐HET mouse (ranging from 10 to 12 months of age) as previously described (Houde et al., 2010). Tissues from each mouse were collected, frozen with liquid nitrogen, and stored at −70 °C. The tissue samples were then processed to obtain cell lyses for Western blots (Dominick et al., 2015) or qRT–PCR analysis (Li et al., 2013). Mouse fibroblasts were collected and lysed, and equal amounts of protein were loaded for Western blot analysis (Murakami et al., 2003).
Statistical analysis
Unless indicated otherwise, results are presented as mean ± standard error of the mean (SEM). Statistical tests for genotype, fasting and fed conditions (nutritional), or interaction effects were assessed by two‐way analysis of variance (ANOVA) using P = 0.05 as the threshold for significant effects. To compare specific effects between groups, an unpaired t‐test was used with P = 0.05 as the criterion for significance.
Funding
This work was supported by grants from the National Institutes of Health AG019899 and AG024824 and the University of Michigan Paul Glenn Center for the Biology of Aging. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of interest
The authors have declared that no conflict of interest exist.
Supporting information
Fig. S1 The CT dotplot graphs for GAPDH mRNA used for normalization controls from liver samples from Snell (DW), GHRKO (B) or PAPPA (C) show no statistically significant differences.
Fig. S2 The CT dotplot graphs for CNOT4 and GAPDH mRNA used as normalizing controls from liver samples of Snell (A) and GHRKO (B) show no statistically significant differences.
Fig. S3 The CT value graphs representing the CNOT mRNA levels in mouse fibroblasts untreated (−, control) or treated with Rapamycin (+, Rapa) from 6 independent experiments. The (*) indicates statistical significance.
Fig. S4 (A) Representative phase contrast and green fluorescence channels of mouse fibroblasts overexpressing the human GFP‐CNOT3 or GFP‐CNOT6L, as well as the silencing shCNOT3, shCNOT6L and respective control vectors.
Table S1 Effects of genotype and feeding on the ratio of pS6 and p4E‐BP1 in different tissues from PAPP‐A (KO) mice compared to control mice (WT).
Table S2 Effects of genotype and feeding on the ratio of pNDRG1 and pAKT(450) in different tissues from PAPP‐A (KO) mice compared to control mice (WT).
Table S3 Source of antibodies.
Table S4 qRT‐PCR probes.
Acknowledgments
We thank Dr. Andrzej Bartke and John Kopchick for GHRKO breeders and Dr. Cheryl Conover for PAPPA‐KO breeders. We thank Dr. David Lombard and Dr. Diane Fingar for comments on the manuscript and Sabrina Van Roekel, Amanda Keedle, and Natalie Perry for expert animal husbandry.
References
- Ait Ghezala H, Jolles B, Salhi S, Castrillo K, Carpentier W, Cagnard N, et al (2012) Translation termination efficiency modulates ATF4 response by regulating ATF4 mRNA translation at 5′ short ORFs. Nucleic Acids Res. 40, 9557–9570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartke A (2008) Impact of reduced insulin‐like growth factor‐1/insulin signaling on aging in mammals: novel findings. Aging Cell 7, 285–290. [DOI] [PubMed] [Google Scholar]
- Bartke A, Brown‐Borg H (2004) Life extension in the dwarf mouse. Curr. Top. Dev. Biol. 63, 189–225. [DOI] [PubMed] [Google Scholar]
- Bartke A, Masternak MM, Al‐Regaiey KA, Bonkowski MS (2007) Effects of dietary restriction on the expression of insulin‐signaling‐related genes in long‐lived mutant mice. Interdiscip. Top. Gerontol. 35, 69–82. [DOI] [PubMed] [Google Scholar]
- Bates DJ, Li N, Liang R, Sarojini H, An J, Masternak MM, et al (2010) MicroRNA regulation in Ames dwarf mouse liver may contribute to delayed aging. Aging Cell 9, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boylston WH, DeFord JH, Papaconstantinou J (2006) Identification of longevity‐associated genes in long‐lived Snell and Ames dwarf mice. Age 28, 125–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer G (2002) Messenger RNA decay during aging and development. Ageing Res. Rev. 1, 607–625. [DOI] [PubMed] [Google Scholar]
- Brown‐Borg HM (2006) Longevity in mice: is stress resistance a common factor? Age 28, 145–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown‐Borg HM, Bartke A (2012) GH and IGF1: roles in energy metabolism of long‐living GH mutant mice. J. Gerontol. A Biol. Sci. Med. Sci. 67, 652–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conover CA, Bale LK (2007) Loss of pregnancy‐associated plasma protein A extends lifespan in mice. Aging Cell 6, 727–729. [DOI] [PubMed] [Google Scholar]
- Conover CA, Bale LK, Overgaard MT, Johnstone EW, Laursen UH, Fuchtbauer EM, et al (2004) Metalloproteinase pregnancy‐associated plasma protein A is a critical growth regulatory factor during fetal development. Development 131, 1187–1194. [DOI] [PubMed] [Google Scholar]
- Corton JC, Brown‐Borg HM (2005) Peroxisome proliferator‐activated receptor gamma coactivator 1 in caloric restriction and other models of longevity. J. Gerontol. A Biol. Sci. Med. Sci. 60, 1494–1509. [DOI] [PubMed] [Google Scholar]
- De Luca G, Ventura I, Sanghez V, Russo MT, Ajmone‐Cat MA, Cacci E, et al (2013) Prolonged lifespan with enhanced exploratory behavior in mice overexpressing the oxidized nucleoside triphosphatase hMTH1. Aging Cell 12, 695–705. [DOI] [PubMed] [Google Scholar]
- Dominick G, Berryman DE, List EO, Kopchick JJ, Li X, Miller RA, et al (2015) Regulation of mTOR activity in Snell dwarf and GH receptor gene‐disrupted mice. Endocrinology 156, 565–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA (2009) Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 460, 392–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill CM, Arum O, Boparai RK, Wang F, Fang Y, Sun LY, et al (2015) Female PAPP‐A knockout mice are resistant to metabolic dysfunction induced by high‐fat/high‐sucrose feeding at middle age. Age 37, 9765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann JW, Zhao X, De Cecco M, Peterson AL, Pagliaroli L, Manivannan J, et al (2015) Reduced expression of MYC increases longevity and enhances healthspan. Cell 160, 477–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houde VP, Brule S, Festuccia WT, Blanchard PG, Bellmann K, Deshaies Y, et al (2010) Chronic rapamycin treatment causes glucose intolerance and hyperlipidemia by upregulating hepatic gluconeogenesis and impairing lipid deposition in adipose tissue. Diabetes 59, 1338–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inada T, Makino S (2014) Novel roles of the multi‐functional CCR4‐NOT complex in post‐transcriptional regulation. Front Genet. 5, 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyama T, Wilson DM III (2013) DNA repair mechanisms in dividing and non‐dividing cells. DNA Repair 12, 620–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang G, Jiang AJ, Xin Y, Li LT, Cheng Q, Zheng JN (2014) Progression of O(6)‐methylguanine‐DNA methyltransferase and temozolomide resistance in cancer research. Mol. Biol. Rep. 41, 6659–6665. [DOI] [PubMed] [Google Scholar]
- Lamming DW, Ye L, Katajisto P, Goncalves MD, Saitoh M, Stevens DM, et al (2012) Rapamycin‐induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science 335, 1638–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laribee RN, Hosni‐Ahmed A, Workman JJ, Chen H (2015) Ccr4‐not regulates RNA polymerase I transcription and couples nutrient signaling to the control of ribosomal RNA biogenesis. PLoS Genet. 11, e1005113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EK, Gorospe M (2010) Minireview: posttranscriptional regulation of the insulin and insulin‐like growth factor systems. Endocrinology 151, 1403–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Bartke A, Berryman DE, Funk K, Kopchick JJ, List EO, et al (2013) Direct and indirect effects of growth hormone receptor ablation on liver expression of xenobiotic metabolizing genes. Am. J. Physiol. Endocrinol. Metab. 305, E942–E950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang R, Khanna A, Muthusamy S, Li N, Sarojini H, Kopchick JJ, et al (2011) Post‐transcriptional regulation of IGF1R by key microRNAs in long‐lived mutant mice. Aging Cell 10, 1080–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombard DB, Miller RA (2012) Ageing: sorting out the sirtuins. Nature 483, 166–167. [DOI] [PubMed] [Google Scholar]
- Meira LB, Calvo JA, Shah D, Klapacz J, Moroski‐Erkul CA, Bronson RT, et al (2014) Repair of endogenous DNA base lesions modulate lifespan in mice. DNA Repair 21, 78–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RA, Harrison DE, Astle CM, Fernandez E, Flurkey K, Han M, et al (2014) Rapamycin‐mediated lifespan increase in mice is dose and sex dependent and metabolically distinct from dietary restriction. Aging Cell 13, 468–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita M, Oike Y, Nagashima T, Kadomatsu T, Tabata M, Suzuki T, et al (2011) Obesity resistance and increased hepatic expression of catabolism‐related mRNAs in Cnot3+/− mice. EMBO J. 30, 4678–4691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami S (2006) Stress resistance in long‐lived mouse models. Exp. Gerontol. 41, 1014–1019. [DOI] [PubMed] [Google Scholar]
- Murakami S, Salmon A, Miller RA (2003) Multiplex stress resistance in cells from long‐lived dwarf mice. FASEB J. 17, 1565–1566. [DOI] [PubMed] [Google Scholar]
- Okada H, Schittenhelm RB, Straessle A, Hafen E (2015) Multi‐functional regulation of 4E‐BP gene expression by the Ccr4‐Not complex. PLoS ONE 10, e0113902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese JC (2013) The control of elongation by the yeast Ccr4‐not complex. Biochim. Biophys. Acta 1829, 127–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter AK, Anthony TG, Anthony JC, Jefferson LS, Kimball SR (2004) The mTOR signaling pathway mediates control of ribosomal protein mRNA translation in rat liver. Int. J. Biochem. Cell Biol. 36, 2169–2179. [DOI] [PubMed] [Google Scholar]
- Rodriguez‐Gil A, Ritter O, Hornung J, Stekman H, Kruger M, Braun T, et al (2016) HIPK family kinases bind and regulate the function of the CCR4‐NOT complex. Mol. Biol. Cell 27, 1969–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez‐Perez I, Manguan‐Garcia C, Menacho‐Marquez M, Murguia JR, Perona R (2009) hCCR4/cNOT6 targets DNA‐damage response proteins. Cancer Lett. 273, 281–291. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, et al (2006) Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 22, 159–168. [DOI] [PubMed] [Google Scholar]
- Schreiber KH, Ortiz D, Academia EC, Anies AC, Liao CY, Kennedy BK (2015) Rapamycin‐mediated mTORC2 inhibition is determined by the relative expression of FK506‐binding proteins. Aging Cell 14, 265–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selman C, Tullet JMA, Wieser D, Irvine E, Lingard SJ, Choudhury AI, et al (2009) Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science 326, 140–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp ZD, Curiel TJ, Livi CB (2013) Chronic mechanistic target of rapamycin inhibition: preventing cancer to delay aging, or vice versa? Interdiscip. Top. Gerontol. 38, 1–16. [DOI] [PubMed] [Google Scholar]
- Shirai YT, Suzuki T, Morita M, Takahashi A, Yamamoto T (2014) Multifunctional roles of the mammalian CCR4‐NOT complex in physiological phenomena. Front Genet. 5, 286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smalley S, Chalmers AJ, Morley SJ (2014) mTOR inhibition and levels of the DNA repair protein MGMT in T98G glioblastoma cells. Mol. Cancer. 13, 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stauber AJ, Brown‐Borg H, Liu J, Waalkes MP, Laughter A, Staben RA, et al (2005) Constitutive expression of peroxisome proliferator‐activated receptor alpha‐regulated genes in dwarf mice. Mol. Pharmacol. 67, 681–694. [DOI] [PubMed] [Google Scholar]
- Strozyk E, Kulms D (2013) The role of AKT/mTOR pathway in stress response to UV‐irradiation: implication in skin carcinogenesis by regulation of apoptosis, autophagy and senescence. Int. J. Mol. Sci. 14, 15260–15285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson J, Sjogren K, Faldt J, Andersson N, Isaksson O, Jansson JO, et al (2011) Liver‐derived IGF‐I regulates mean life span in mice. PLoS ONE 6, e22640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swindell WR (2008) Comparative analysis of microarray data identifies common responses to caloric restriction among mouse tissues. Mech. Ageing Dev. 129, 138–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temme C, Simonelig M, Wahle E (2014) Deadenylation of mRNA by the CCR4‐NOT complex in Drosophila: molecular and developmental aspects. Front Genet. 5, 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Miller RA (2012) Augmented autophagy pathways and MTOR modulation in fibroblasts from long‐lived mutant mice. Autophagy 8, 1273–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiler M, Blaes J, Pusch S, Sahm F, Czabanka M, Luger S, et al (2014) mTOR target NDRG1 confers MGMT‐dependent resistance to alkylating chemotherapy. Proc. Natl Acad. Sci. USA 111, 409–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JJ, Liu J, Chen EB, Wang JJ, Cao L, Narayan N, et al (2013) Increased mammalian lifespan and a segmental and tissue‐specific slowing of aging after genetic reduction of mTOR expression. Cell Rep. 4, 913–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z, Sunchu B, Fok WC, Alshaikh N, Perez VI (2015) Gene expression in the liver of female, but not male mice treated with rapamycin resembles changes observed under dietary restriction. SpringerPlus 4, 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Manning BD (2015) mTORC1 signaling activates NRF1 to increase cellular proteasome levels. Cell Cycle 14, 2011–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Nicholatos J, Dreier JR, Ricoult SJ, Widenmaier SB, Hotamisligil GS, et al (2014) Coordinated regulation of protein synthesis and degradation by mTORC1. Nature 513, 440–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 The CT dotplot graphs for GAPDH mRNA used for normalization controls from liver samples from Snell (DW), GHRKO (B) or PAPPA (C) show no statistically significant differences.
Fig. S2 The CT dotplot graphs for CNOT4 and GAPDH mRNA used as normalizing controls from liver samples of Snell (A) and GHRKO (B) show no statistically significant differences.
Fig. S3 The CT value graphs representing the CNOT mRNA levels in mouse fibroblasts untreated (−, control) or treated with Rapamycin (+, Rapa) from 6 independent experiments. The (*) indicates statistical significance.
Fig. S4 (A) Representative phase contrast and green fluorescence channels of mouse fibroblasts overexpressing the human GFP‐CNOT3 or GFP‐CNOT6L, as well as the silencing shCNOT3, shCNOT6L and respective control vectors.
Table S1 Effects of genotype and feeding on the ratio of pS6 and p4E‐BP1 in different tissues from PAPP‐A (KO) mice compared to control mice (WT).
Table S2 Effects of genotype and feeding on the ratio of pNDRG1 and pAKT(450) in different tissues from PAPP‐A (KO) mice compared to control mice (WT).
Table S3 Source of antibodies.
Table S4 qRT‐PCR probes.