Structure and function of archaeal histones (original) (raw)
Abstract
The genomes of all organisms throughout the tree of life are compacted and organized in chromatin by association of chromatin proteins. Eukaryotic genomes encode histones, which are assembled on the genome into octamers, yielding nucleosomes. Post-translational modifications of the histones, which occur mostly on their N-terminal tails, define the functional state of chromatin. Like eukaryotes, most archaeal genomes encode histones, which are believed to be involved in the compaction and organization of their genomes. Instead of discrete multimers, in vivo data suggest assembly of “nucleosomes” of variable size, consisting of multiples of dimers, which are able to induce repression of transcription. Based on these data and a model derived from X-ray crystallography, it was recently proposed that archaeal histones assemble on DNA into “endless” hypernucleosomes. In this review, we discuss the amino acid determinants of hypernucleosome formation and highlight differences with the canonical eukaryotic octamer. We identify archaeal histones differing from the consensus, which are expected to be unable to assemble into hypernucleosomes. Finally, we identify atypical archaeal histones with short N- or C-terminal extensions and C-terminal tails similar to the tails of eukaryotic histones, which are subject to post-translational modification. Based on the expected characteristics of these archaeal histones, we discuss possibilities of involvement of histones in archaeal transcription regulation.
Author summary
Both Archaea and eukaryotes express histones, but whereas the tertiary structure of histones is conserved, the quaternary structure of histone–DNA complexes is very different. In a recent study, the crystal structure of the archaeal hypernucleosome was revealed to be an “endless” core of interacting histones that wraps the DNA around it in a left-handed manner. The ability to form a hypernucleosome is likely determined by dimer–dimer interactions as well as stacking interactions between individual layers of the hypernucleosome. We analyzed a wide variety of archaeal histones and found that most but not all histones possess residues able to facilitate hypernucleosome formation. Among these are histones with truncated termini or extended histone tails. Based on our analysis, we propose several possibilities of archaeal histone involvement in transcription regulation.
Introduction
Architectural chromatin proteins are found in every domain of life. Bacteria express DNA-bending and DNA-bridging proteins, such as histone-like protein from Escherichia coli strain U93 (HU) and histone-like nucleoid-structuring protein (H-NS), to structure and functionally organize the genome and to regulate genome activity [1, 2]. In eukaryotes and most archaeal lineages, histones are responsible for packaging and compaction of the DNA (Table 1). Genomic comparisons demonstrate that the Bacteria and Archaea share a common ancestor; eukaryotes are to date classified as being part of the archaeal branch [3–5]. The archaeal domain comprises single-cellular organisms found in diverse habitats. Although Archaea and Bacteria have common features, such as a circular genome and the absence of a nucleus, at the genetic level, Archaea seem to be more related to eukaryotes. Amongst others, archaeal RNA polymerase, a key component of cellular life in all domains, is more similar to RNA polymerase from eukaryotes than bacterial RNA polymerase [6, 7]. Archaeal ribosomes share their size and structural core with bacterial ribosomes but are more similar to eukaryotic ribosomes when it comes to protein and rRNA sequence and some specific domains [8–10]. Also, some cellular processes thought to be unique to eukaryotes, such as endosomal sorting and the ubiquitin system, have been identified in some archaea [11]. These observations raise the intriguing possibility that chromatin organization as we have come to understand in eukaryotes has evolved from that of the archaeal lineage. Before we describe our analysis, we briefly review current knowledge on chromatin organization in eukaryotes and Archaea and the current paradigms in the evolution of histones, the main chromatin organizing proteins.
Table 1. Phylogenetic subdivision of the archaeal domain.
| Superphylum | Phylum | Class | Histones |
|---|---|---|---|
| Asgard Archaea | Candidatus Heimdallarchaeota | Y | |
| Candidatus Lokiarchaeota | Y | ||
| Candidatus Odinarchaeota | Y | ||
| Candidatus Thorarchaeota | Y | ||
| DPANN | Candidatus Aenigmarchaeota | Y | |
| Candidatus Diapherotrites | Y | ||
| Candidatus Huberarchaea | Y | ||
| Candidatus Micrarchaeota | Y | ||
| Nanoarchaeota | Y | ||
| Candidatus Nanohaloarchaeota | Y | ||
| Candidatus Pacearchaeota | Y | ||
| Candidatus Parvarchaeota | N | ||
| Candidatus Woesearchaeota | Y | ||
| TACK | Candidatus Bathyarchaeota | Y | |
| Crenarchaeota | Y* | ||
| Candidatus Geothermarchaeota | N | ||
| Candidatus Korarchaeota | Y | ||
| Thaumarchaeota | Y | ||
| Candidatus Verstraetearchaeota | N | ||
| - | Euryarchaeota | Archaeoglobi | Y |
| Hadesarchaea | Y | ||
| Halobacteria | Y | ||
| Methanobacteria | Y | ||
| Methanococci | Y | ||
| Methanomicrobia | Y | ||
| Methanonatronarchaeia | Y | ||
| Methanopyri | Y | ||
| Theionarchaea | Y | ||
| Thermococci | Y | ||
| Thermoplasmata | Y |
The eukaryotic histone
In eukaryotes, octameric histone cores compact DNA by wrapping an approximately 150-bp unit twice around its surface, forming a nucleosome [12, 13]. Nucleosomes interact with each other, yielding an additional level of DNA organization in the form of a fibre. Besides a role in compaction, histones also play roles in genome organization, replication, repair, and expression, which highlights the nucleosome as a very important complex affecting a vast array of cellular processes. Characteristic of core histone proteins of all different origins is a common “histone fold”: two short and one long α-helix, separated by loops [14–18]. In eukaryotes, the histone core consists of two H2A-H2B dimers and a H3-H4 tetramer, around which approximately 146 bp of DNA is wrapped twice (Fig 1A). It has been suggested that smaller histone assemblies, such as tetrasomes (H3-H4 tetramers), hexasomes (H3-H4 tetramers plus one H2A/H2B dimer), and hemisomes (a H3-H4 dimer plus one H2A/H2B dimer), have functional roles as intermediate structures during, for example, transcription elongation [19–22]. The linker histone H1 (which lacks the characteristic histone fold) binds at the entry and exit points of the DNA wrapped around the octameric histone core [23, 24]. The association of histone H1 constrains an additional 20 bp of DNA and allows for the formation of the 30-nm fibre, which results in tighter compaction [25, 26]. Also, flexible N-terminal tails that protrude from eukaryotic histones contribute to tighter DNA packaging. These tails may interact with either the DNA or the histone surface on another nucleosome, which stabilizes the close association of nucleosomes [27–29]. Furthermore, post-translational modifications of amino acid residues in the N-terminal tails, such as acetylation, methylation, phosphorylation, ubiquitination, and biotinylation, are a key instrument for the cell to regulate gene expression, the DNA damage response, and many other processes [30–32]. For instance, while heterochromatin (tightly packed DNA) is typically devoid of acetylated lysines, euchromatic (lightly packed) regions typically contain histones with acetylated lysines. In general, euchromatin contains actively transcribed genes. Histone acetylation is believed to cause a locally less condensed chromatin structure in vivo, which is permissive to transcription. In particular the lysine-rich histone H4 tail seems to be crucial in the modulation of chromatin structure [27]. In vitro, H4 tails are required for higher order chromatin folding [33–35], which can be disrupted by acetylation of K16 [27]. Nucleosome function and level of genome compaction can be altered in a multitude of ways, providing flexible and versatile mechanisms for tuning the cell’s dynamic chromatin structure and transcription regulation.
Fig 1. Eukaryotic and archaeal histones.

(A) Eukaryotic nucleosome consisting of DNA wrapped around a core of a (H3-H4)2 tetramer and two H2A-H2B dimers. Yellow, H2A; red, H2B; blue, H3; green, H4. (B) Archaeal histone homodimer of HMfB. HMfB, Histone B from Methanothermus fervidus.
Architectural DNA-binding proteins in Archaea
Archaeal genomes also encode proteins that are involved in shaping DNA architecture. Genes coding for histones are found in many species throughout the domain (Table 1). In some species a homologue of the bacterial DNA bender HU was identified [36, 37]. Nucleoid-associated proteins (NAPs) from the Alba family (also known as the Sulfolobus solfataricus 10b (Sso10b) protein family) are abundant and widely conserved in Archaea. Notably, Alba family proteins have also been identified in eukaryotes [38]. Characteristic of these proteins is the formation of protein–DNA filaments and bridges between DNA duplexes [39–42]. Two Alba family proteins with different functionalities have been studied in Archaea. Alba1 cooperatively forms filaments in a sequence-independent and concentration-dependent manner in Crenarchaeota, whereas Alba2 only occurs as heterodimer with Alba1 and does not form filaments [38, 42]. Alba proteins have been shown to repress transcription in vitro [43]. In Euryarchaeota, some species express sequence-specific Alba proteins [44], which, like Alba1 homodimers at low-protein concentrations and Alba1-Alba2 heterodimers, may form loops by bridging two DNA duplexes [45]. Other proteins affecting DNA conformation are Sso10a family proteins, which are able to bend and bridge DNA as well as form filaments on DNA [46, 47] and the monomeric DNA benders Cren7 and Sul7 [48, 49]. Cren7 and Sul7 have exclusively been identified in members of the Crenarchaeota phylum, whereas Sso10a has been found in some Crenarchaeota and Euryarchaeota. Other less widespread NAPs include transcription regulator of the maltose system-like 2 (TrmBL2), methanogen chromosomal protein 1 (MC1), Methanopyrus kandleri 7 kDa protein (7kMk), Sulfolobus solfataricus protein 7c (Sso7c), and crenarchaeal chromatin protein 1 (CC1) [50–55].
The histones found in Archaea are widespread throughout the domain but are absent in most Crenarchaeota. They have the same histone fold as eukaryotic histones, but N-terminal histone tails have not been identified (Fig 1B). Linker histones, homologous to eukaryotic H1, have not been found. Archaeal histones exist as dimers in solution, which have been shown to bend DNA [56, 57]. These histone dimers can be homodimeric or heterodimeric [58], as many archaeal species express, or at least encode, more than one histone variant. In Methanothermus fervidus (class Methanobacteria), the two histone variants are expressed at different levels and ratios at different growth phases, suggesting a distinct function for both proteins [59]. In addition to binding as dimers, archaeal histones have been reported in vivo and in vitro to bind DNA as tetramers [60–62], wrapping the DNA once. However, micrococcal nuclease (MNase) digestion patterns of Thermococcus kodakarensis (class Thermococci) chromatin suggest that histone–DNA complexes consist of discrete multiples of a dimeric histone subunit (i.e., not limited to dimers and tetramers) in vivo without obvious dependence on the DNA sequence [63]. Based on the latter observations, it was proposed that histone dimers multimerize and wrap DNA into a filament of variable length [17, 63]. The crystallography study of Luger and coworkers on histone HMfB from M. fervidus indicates that these histones assemble into an endless left-handed rod in vitro, which we propose to call a “hypernucleosome” (Fig 2). Note that these complexes were assembled on SELEX-optimized DNA previously shown to favor tetrameric nucleosome assembly [64]. The number of wraps in the hypernucleosome, which is the DNA bending 360° around the histone multimer, scales linearly with the number of histone subunits, resulting in a tight packaging of DNA. The authors also provide evidence that mutation-directed perturbation of hypernucleosome function in vivo alters response to nutrient change in T. kodakarensis, suggesting a role in transcription. Both eukaryotes and Archaea encode histone proteins, which seem to be involved in response to environmental cues by their involvement in transcription regulation.
Fig 2. Overview of the hypernucleosome structure.
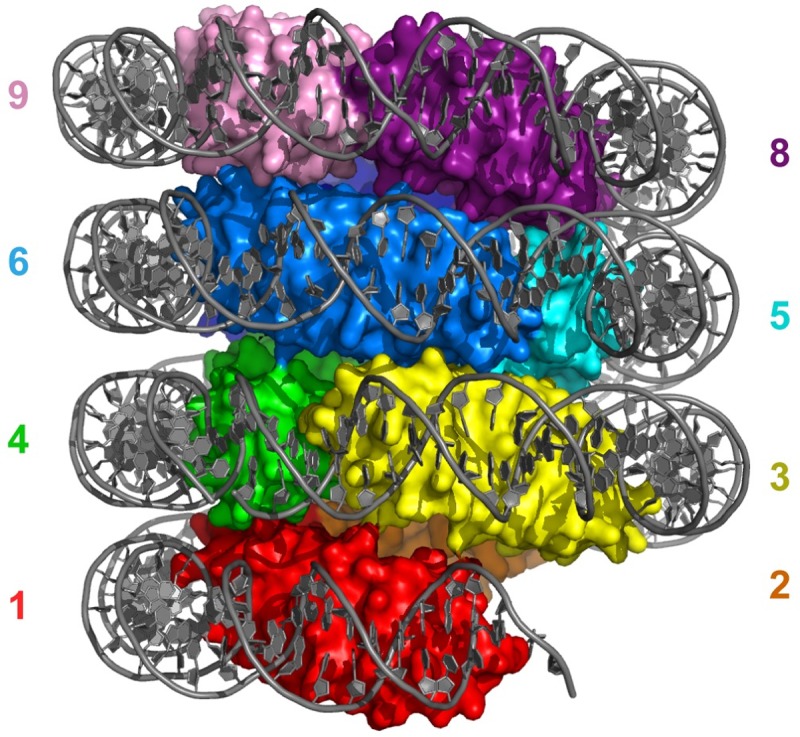
HMfB dimers stack to form a continuous, central protein core that wraps the DNA in a left-handed superhelix. Nine HMfB dimers are shown, each dimer in surface mode and in rainbow colors. Numbering indicates position of the nine histone dimers; note that dimer 5 and 6 occlude the view of dimer 7. DNA is in gray and shown as cartoon. Image generated using PDB entry 5T5K [64]. HMfB, Histone B from Methanothermus fervidus; PDB, Protein Data Bank.
Evolution of the histone protein class
It has been suggested that eukaryotic histones evolved from archaeal histones [65]. This hypothesis is supported by the high similarity at the amino acid sequence level and in secondary structure [66, 67]. Suggestive of an archaeal origin of eukaryotic histones is also the dimeric nature of archaeal histones; archaeal histone complexes are built from dimers, but members of the archaeal class Halobacteria express a “tandem histone.” In these tandem histones, the histone folds are linked end-to-end [68–70]. This implies that the histone folds always occupy the same position and role in the naturally linked dimer. This leads to the relaxation of evolutionary constraints in parts of the histone, an example of subfunctionalization [71, 72]. According to this hypothesis, the histone folds further evolved in a divergent way, leading to an asymmetric dimer. This may have been an ancestor of H3-H4, which later separated to become two individual proteins and corresponding genes [66]. The eukaryotic H3-H4 tetramer resembles the tetramer found in Archaea, and it has been suggested that H2A and H2B have arisen from H3 and H4 later on in histone evolution [66]. Indeed, H3 and H4 are more similar to archaeal histones than H2A and H2B, supporting this hypothesis. From this point, eukaryotic histones have further evolved into histone variants, highly homologous substitutes of canonical eukaryotic histones, which often play a specialist role in a wide variety of cellular processes [73]. Unlike canonical histones, which are mainly expressed during DNA replication, histone variants are expressed in a replication-independent manner [74, 75]. Histone variants of H2A and H3 are widely known and studied, whereas only a few examples have been found of diversified H2B and H4 [76]. The evolutionary pressure for the evolution of dimer-based histones to octameric histones and their subsequent variants was long believed to be DNA compaction [66]. The fact that eukaryotic cells undergo mitosis, in which chromosomes are highly compacted, together with the abundance of gene-poor regions may have favored a histone conformation that wraps DNA twice (eukaryotic octamer) instead of once (archaeal tetramer) and that via its N-terminal tails has the ability to compact DNA at a higher order. Open questions that remain are how histone evolution was driven and what the roles of archaeal histones and their variants are in genome packaging and regulation.
Here, we discuss the amino acid residues that are responsible for the formation of the hypernucleosome based on a sequence analysis of a subset of archaeal histones that includes histones from all phyla that contain genes coding for histones (Fig 3). Also, we analyze the ability of histones to form a hypernucleosome and the effects of N- or C-termini longer or shorter than the consensus on histone multimerization and transcription regulation. We emphasize the histones in species from recently discovered phyla, which are believed to be an evolutionary link to eukaryotes [11, 77]. Based on elements that archaeal histones have in common and elements that differ from that consensus, we discuss some of the open questions regarding gene regulation by archaeal histones.
Fig 3. Alignment of histones from different archaeal species and human histone H4.

Colors indicate the side chain group: R, H, K: blue; D, E: red; A, V, I, L, M: orange; F, Y, W: yellow; S, T, N, Q: green; C: turquoise; G: pink; P: purple. Symbols above the alignment indicate the dimer–dimer interface (D), the loop of the stacking interface (L), and the putative stacking interactions (S) based on HMfB). Secondary structure and numbering of HMfB is used for reference. EUKAR H4, eukaryotic (human) histone H4 NP_724344.1; HEIMDALL LC_3 HA, HB, and HC, Candidatus Heimdallarchaeota OLS22332.1, OLS24873.1, and OLS21974.1, respectively; LOKI GC14_75 HLkE and CR_4, Candidatus Lokiarchaeota KKK41979.1 and OLS16336.1, respectively; ODIN, Candidatus Odinarchaeota OLS18261.1; THOR, Candidatus Thorarchaeota KXH71038.1; WOESE, Candidatus Woesearchaeota OIO61677.1; PACE, Candidatus Pacearchaeota OIO41945.1; HUBER, Candidatus Huberarchaea CG_4_9_14_3_um_filter_31_125 HA and HB, PJB03565.1, and PJB04497.1, respectively; DIAPHERO, Candidatus Diapherotrites PJA17623.1; AENIGM, Candidatus Aenigmarchaeota OIN88081.1; MICR, Candidatus Micrarchaeota Micrarchaeum acidiphilum ARMAN-2 EET90461.1; NANOHALO, Candidatus Nanohaloarchaeota Haloredivivus sp. G17 and Nanosalina sp. J07AB43 HA and HB, EHK01841.1, EGQ42849.1, and EGQ43804.1, respectively; NANO, Nanoarchaeota Nanoarchaeum equitans Kin4-M AAR39197.1; THAUM, Thaumarchaeota Nitrososphaera gargensis Ga9.2 AFU59009.1; BATHY B23, B24, and SMTZ-80, Candidatus Bathyarchaeota KYH36356.1, KYH37304.1, and KON27866.1, respectively; CREN, Crenarchaeota Caldivirga maquilingensis IC-167, Thermofilum pendens Hrk5, and Vulcanisaeta distribute DSM14429, ABW02527.1, ABL77757.1, and ADN51226.1, respectively; EURY, Euryarchaeota Methanobrevibacter wolinii HA and HB, Methanocaldococcus jannaschii DSM2661, Methanococcoides methylutens, Thermococcus kodakarensis KOD1, and Methanothermus fervidus DSM2066, WP_42707783.1, WP_42706862.1, AAB99668.1, KGK98166.1, BAD86478.1, and ADP77985.1, respectively.
Histones are found in some newly discovered Archaea
With the widespread use of metagenomic sequencing, entire new branches within the archaeal domain have been discovered. Next to Euryarchaeota, the phylum that has been known since the establishment of Archaea as one of the domains of life [78], the superphyla Thaumarchaeota, Aigarchaeota, Crenarchaeota, Korarchaeota (TACK), Diapherotrites, Pacearchaeota, Aenigmarchaeota, Nanoarchaeota, Nanohaloarchaeota (DPANN), and Asgard Archaea are part of the most recent representation of the tree of life [79]. Genomes of the recently discovered archaeal superphylum Asgard Archaea and candidate phyla Bathyarchaeota, Woesearchaeota, Pacearchaeota, Aenigmarchaeota, Diapherotrites, Huberarchaea, and Micrarchaeota encode histones [11, 77, 80–82](Table 1). With the publication of the genome sequences of these organisms, we were able to scrutinize the sequence divergence of histones by comparing sequences of histones from Archaea throughout the domain (Fig 3). The selection of histones shown here is based on the presence of histone-coding genes in different phyla. Since many of those phyla were only discovered in the last three years, our selection includes a relatively large number of histones that have not yet been studied in vivo or in vitro.
We found that in genome LC_3 of the candidate phylum Heimdallarchaeota, 10 different histones are encoded, which is the highest number of histones found in one archaeal genome [83]. We have not found any histones in the genomes of candidate phyla Parvarchaeota, Geothermarchaeota, and Verstraetearchaeota, although it should be noted that abundance of available genomes and completeness of the genomes differs. The majority of available genomes from the phyla that do not seem to encode histones have an estimated completeness of between 70% and 99% [84–87]. This means that we cannot rule out the possibility that any of those genomes does contain one or more genes coding for histones. The absence of histones suggests that other NAPs may be involved in genome compaction. In that light, it is notable that in genomes from Candidatus Parvarchaeota, as well as in genomes from the candidate phyla from Asgard Archaea, Woesearchaeota, Bathyarchaeota, Pacearchaeota, Aenigmarchaeota, and Micrarchaeota, genes coding for the DNA-bridging protein Alba1 (and in some cases, Alba2) are present. Like histones, Alba (or Sso10b) proteins are likely involved in transcription repression. They are highly abundant in the nonhistone-coding Crenarchaeota [88], possibly taking the functional role of histones as found in other Archaea. Some Parvarchaeota genomes encode Alba but not histones, and their genomes may therefore be shaped or regulated in a similar way as in Crenarchaeota. For Geothermarchaeota and Verstraetearchaeota, we were not able to identify any protein that clearly resembles known chromatin proteins. Furthermore, we found that only Candidatus Thorarchaeota contains an HU gene (a DNA-bending protein generally found in Bacteria and Archaea without histones only [67]). The genome of Candidatus Huberarchaea encodes an MC1 homologue, which is a monomeric DNA-bending protein often found in organisms from the euryarchaeal class Halobacteria. Genes coding for other known archaeal NAPs [50, 89, 90] were not found.
Some archaeal histones have eukaryote-like N-terminal tails
A striking finding based on the amino acid sequence comparison reveals that two histones from Candidatus Heimdallarchaeota archaeon LC_3 (only Histone A [HA] shown in Fig 3), one from Candidatus Huberarchaea archaeon CG_4_9_14_3_um_filter_31_125 and one from Candidatus Bathyarchaeota archaeon B23, contain an N-terminal tail, which was previously thought to exist only in eukaryotic histones and only recently reported for Heimdallarchaeota [64]. In eukaryotes, these tails stabilize a higher order of compaction by interacting with either the DNA or another nucleosome. The tails of the two histones from Heimdallarchaeota and Huberarchaea are of roughly the same length and sequence composition as eukaryotic H4 tails (see Fig 3). Prompted by the importance of the eukaryotic histone tails in modulating chromatin structure and function [27, 32], we constructed a molecular model of a hypernucleosome formed by Histone A (HA) from Heimdallarchaeota LC_3 to investigate its potential function (see Methods section).
The model illustrates how three subsequent arginines (R17–R19) could facilitate passing of the tails through the DNA gyres (Fig 4). The tails exit the hypernucleosome through DNA minor grooves, similar to eukaryotic histone tails, and might position their lysine side chains to bind to the hypernucleosomal DNA or to other DNA close by, facilitating (long-range) genomic interactions in trans. Like the H4 tail that is subject to acetylation of lysines K5, K8, K12, and K16 [91], lysines in the Heimdallarchaeal histone tail may well be subject to acetylation. Archaeal genomes are known to have several candidate lysine acetyltransferase and deacetylase enzymes, including proteins belonging to the ELP3 superfamily, to which transcription elongation factor and histone acetyltransferase ELP3 belongs [92–94]. Searches using the ProSite database (http://prosite.expasy.org, [95]) and Protein Information Resource (http://pir.georgetown.edu, [96]) further reveal that the Heimdallarchaeota LC_3 genome contains multiple gene products containing the Gcn5-related N-acetyltransferase domain, which is present in many histone acetyltransferases [97]. Interestingly, a potential “reader” protein that binds modified lysines can also be identified. This protein, HeimC3_47440, contains a YEATS-domain, which has recently been shown to bind histone tails that carry acetylated or crotonylated lysines [98–101]. Comparison with the closest homolog of known 3D structure, YEATS2 (35% identity, PDB-id 5IQL, [102]), shows that the binding site for the modified lysine side chain is strictly conserved in the archaeal protein. Notably, only Candidatus Bathyarchaeota, which also features tailed histones, contains a detectable homolog of HeimC3_47440. The presence of lysine-containing N-terminal tails in combination with histone modification writers and readers suggests that Archaea use post-translational modifications in a similar way to Eukaryotes as modulators of genome compaction and gene activity. The tail of the Huberarchaea histone also contains lysine residues that are found at the same position as some of the lysines of the H4 tail. However, no proteins involved in post-translational modification of histone tails have been identified in this phylum.
Fig 4. Model of a Heimdall LC_3 hypernucleosome with N-terminal tails.

(A) View showing histone tails protruding through the DNA minor grooves. The R17 Cα-atom is shown as a blue sphere to mark the exit point of the tail. (B) Close up of the histone tails with lysine and arginine residues shown as sticks, and N-terminal lysines are labeled. Homodimers of Heimdall LC_3 histone HA are shown in teal; one dimer is highlighted in darker colors. Models are based on the structure of HMfB (PDB entry 5T5K); the tail in the top (bottom) of panel B is modeled in the H3 (H4) tail conformation (PDB entry 1KX5). HA, Histone A; HMfB, Histone B from Methanothermus fervidus; PDB, Protein Data Bank.
Other histones, for example from Candidatus Lokiarchaeota CR_4, Candidatus Odinarchaeota LBC_4, Nanoarchaeum equitans, and Thermofilum pendens, contain a short N-terminal tail of 5–10 residues. Also, histones with a C-terminal tail have been found. The histone from the euryarchaeal species Methanocaldococcus jannaschii (class Methanococci) has a 28-residue tail, which seems to be unique among archaeal histones. Other C-terminal tails are up to 11 residues long (as compared to Methanothermus fervidus HMfB) and appear in Caldiarchaeum subterraneum, Candidatus Bathyarchaeota SMTZ-80, Candidatus Heimdallarchaeota LC_3, Candidatus Lokiarchaeota CR_4, and all histones found in Crenarchaeota. These short C-terminal tails are similar in length to the H4 C-terminal tail, that is reported to play a role in the promotion of histone octamer formation in eukaryotes [103]. The genomes of some archaeal species contain genes for histone truncates. The histone from Haloredivivus sp. G17, member of the candidate phylum Nanohaloarchaeota, and the histone from Candidatus Bathyarchaeota archaeon B24 both lack part of the N-terminal α-helix (α1), and one histone from Candidatus Lokiarchaeota GC14-75 is reduced in length at the C-terminus. The remainder of the C-terminal amino acids likely does not form a C-terminal helix (α3) in this histone from Candidatus Lokiarchaeota. Although histones of reduced length or containing tails lack part of the histone fold, they likely still possess DNA-binding properties. Therefore, they possibly have functional roles in the regulation of genes.
Multimerization of histones
Both eukaryotic histones and HMfB form dimers, a process that is driven by a hydrophobic core (involving residues A24, L28, L32, I39, and A43 in HMfB) as well as a crucial salt bridge for a stable histone fold (R52-D59 in HMfB) [14]. These hydrophobic residues and the salt bridge are conserved among Archaea. This indicates that archaeal histones have very similar tertiary structures [14, 104]. Also, residues that play an important role in DNA binding are present in all examined histones, including the arginines that anchor archaeal histone dimers to the DNA minor grooves (R10 and R19 in HMfB) [14]. Both eukaryotic H3-H4-dimers and HMfB dimers can form tetramers by hydrogen bonding of H49 and D59 (HMfB) and additional hydrophobic interactions in the interface (L46 and L62 in HMfB) [105], pairs of residues that, too, are generally conserved among archaeal histones (Fig 3).
The HMfB–DNA cocrystal structure reveals left-handed wrapping of DNA around a histone-multimer core [64] (Fig 2). This structure supports the model in which HMfB dimers multimerize along DNA into an “infinite” hypernucleosome, thereby linearly compacting the DNA approximately ten-fold. It is likely that hypernucleosomes grow or shrink by association or dissociation of dimers at both ends. The resolution of the crystal structure allowed us to identify several interacting residues between layers of dimers that may be important for stabilizing the complex (Fig 5). Based on this structural information, the propensity of different archaeal histones to multimerize can be predicted.
Fig 5. Stacking interface between HMfB dimers in the hypernucleosome.
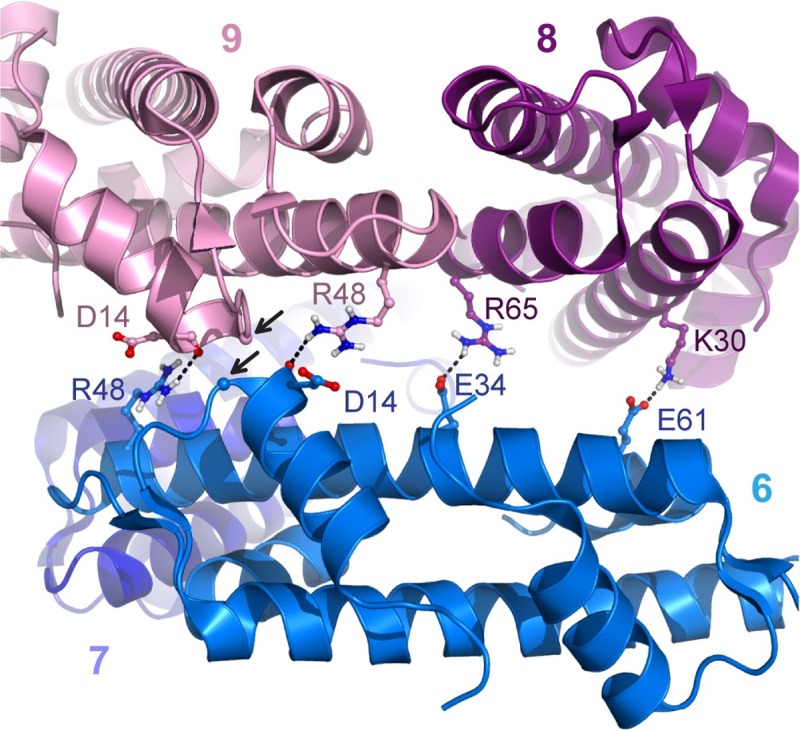
Each dimer i forms stacking interactions with dimer i+2 and i+3, shown here for dimer 6. Residues deemed important are shown in ball-and-sticks and labeled; close stacking of G16 in dimer 6 and 9 is indicated by arrows (Cα shown as spheres); hydrogen-bonds are indicated with dashes. Image generated using PDB entry 5T5K [64]. HMfB; Histone B from Methanothermus fervidus; PDB, Protein Data Bank.
In Table 2, we set out three criteria for hypernucleosome formation by archaeal histones. Firstly, conservation of residues in the dimer–dimer interface (L46, H49, D59, and L62 in HMfB) is required, as forming a tetramer is the first step in multimerization. Secondly, residue G16, which is positioned at the stacking interface of the hypernucleosome (Fig 5), is crucial in permitting formation of the hypernucleosome [64]. Bulkier residues at this position interfere with multimerization [64]. Lastly, favorable interactions between histone dimers i and i+2 and i+3, here termed stacking interactions, will contribute to stability of the compacted hypernucleosome. The HMfB hypernucleosome crystal structure shows three stacking interactions, hydrogen bonds from K30 to E61, E34 to R65, and R48 to D14 (Figs 3 and 5).
Table 2. Assessment of possible hypernucleosome formation by archaeal histones.
| Dimer–dimer interfacea | Stacking interfaceb | Potentialstacking interactionsc | Hypernucleosome formation | Histonefeatures | ||
|---|---|---|---|---|---|---|
| Heimdall | LC_3 HA | ± | + | 3 (E14-R48, K26-E57, R41-E45) | ± | N-terminal tail |
| Heimdall | LC_3 HB | + | + | 5 (E30-K61, Q14-R48, R13-Q18, K27-E57, K37-E45) | + | |
| Heimdall | LC_3 HC | ± | + | 3 (N34-R65, T15-K41, Y14-Q53) | ± | |
| Loki | GC14_75 (HLkE) | − | + | 2 (D14-R48, K34-E45) | − | Truncated C-term |
| Loki | CR_4 | + | + | 2 (Q14-D48, Q41-Q41) | + | |
| Odin | LCB_4 | + | − | 3 (K30-Q61, K14-E18, E38-R41) | ± | |
| Thor | SMTZ1-45 | + | + | 5 (Q30-D61, E34-K65, K14-E48, E37-R41, E26-K58) | + | |
| Woese | CG1_02_33_12 | + | + | 4 (R14-T48, R34-E61, E26-K57, E37-R41) | + | |
| Pace | CG1_02_31_27 | + | + | 4 (S30-K61, E34-K65, K14-T48, E37-K45) | + | |
| Huber | CG_4_9_14_3_um_filter_31_125 (HA) | ± | + | 2 (E14-K48, E14-K18) | ± | |
| Huber | CG_4_9_14_3_um_filter_31_125 (HB) | + | + | 2 (N30-R61, K34-E65) | + | N-terminal tail |
| Diaphero | CG_4_10_14_0_2_um_filter_31_5 | ± | ± | 4 (E33-R48, E37-R41, E37-R48, E27-K61) | ± | |
| Aenigm | CG1_02_38_14 | + | + | 5 (E30-K61, D34-R65, E14-H48, A15-K41, E37-K41) | + | |
| Micr | M. acidiphilum ARMAN-2 | + | + | 3 (E30-K61, K34-Q65, Y2-K48) | + | |
| Nanohalo | Haloredivivus sp. G17 | ± | − | 2 (E27-R61, Q37-E45) | − | Truncated N-term |
| Nanohalo | Nanosalina sp. J07AB43 (HA) | + | + | 5 (Q30-K61, D34-R65, K14-E48, K14-E18, Q37-Q45) | + | |
| Nanohalo | Nanosalina sp. J07AB43 (HB) | − | ± | 2 (Q30-R61, D14-K18) | − | |
| Nano | N. equitans Kin4-M | + | + | 4 (E30-R61, Q14-K48, Q14(bb)-R41, K37-E45) | + | |
| Kor | ARK-16 (HA) | ± | ± | 7 (R30-E61, Q14-K48, K15-E41, E26-R58, E27-R57, E33-K48, R38-E41) | ± | |
| Kor | ARK-16 (HB) | + | + | 3 (Y30-E61, D14-K48, R27-E61) | + | |
| Thaum | N. gargensis Ga9.2 | + | + | 4 (E34-K65, K14-E18, E27-R61, E37-K41) | + | |
| Bathy | B23 | ± | ± | 4 (R14-V44, E34-K61, E37-R41, E26-R58) | ± | N-terminal tail |
| Bathy | B24 | + | + | 3 (E34-R65, K14-E18, E27-R61) | + | Truncated N-term |
| Bathy | SMTZ-80 | + | + | 3 (E34-K65, K41-E45, E27-R61) | + | |
| Cren | C. maquilingensis IC-167 | + | + | 4 (D30-K61, N34-R65, K14-E18, Y37-K48) | + | |
| Cren | T. pendens Hrk5 | + | + | 4 (E30-K61, S14-R48, R37-E45, R13-E18) | + | |
| Cren | V. distributa DSM14429 | + | + | 4 (D30-K61, Y34-R65, K14(bb)-R48, K14-E18) | + | |
| Eury | M. wolinii (HA) | ± | + | 4 (N30-E61, E34-K65, E14-K48, K41-E45) | ± | |
| Eury | M. wolinii (HB) | + | + | 5 (E30-K61, E34-K65, N14-R48, N14-Q18, Q41-Q41) | + | |
| Eury | M. jannaschii DSM2661 | + | − | 4 (N30-K61, Q14-R48, K37-Q45, D26-R58) | ± | C-terminal tail |
| Eury | M. methylutens | ± | − | 2 (D30-K61, S14-E18) | − | |
| Eury | T. kodakarensis KOD1 (HTkB) | + | + | 4 (E30-K61, E34-K65, K14-Q48, K26-E58) | + | |
| Eury | M. fervidus DSM2088 (HMfB) | + | + | 3 (K30-E61, E34-R65, D14-R48) | + |
Scrutiny of histone sequences reveals that most archaeal histones meet these criteria and are thus likely to form hypernucleosomes (Table 2, marked +). We identified two to seven potential stacking interactions for this group of histones, which may affect hypernucleosome stability and compactness. Fewer interactions may allow for more “breathing” of the hypernucleosome structure, yielding hypernucleosomes that are more flexible or “floppy.” We predict such structures to be formed also by a number of archaeal histones that do not fully meet our criteria (Table 2, marked ±). For example, Candidatus Heimdallarchaeota LC_3 HA and Candidatus Lokiarchaeota GC14_75 HLkE have H49N and D59S substitutions, respectively, which likely weakens the crucial hydrogen-bonding interaction at the dimer–dimer interface [105]. Similarly, substitution of the hydrophobic residues 46 and 62 for more hydrophilic or bulkier ones would lead to a less stable dimer–dimer interface, as for Candidatus Heimdallarchaeota LC_3 HC and Candidatus Bathyarchaeota B23. In the presence of the canonical dimer–dimer interface, bulky substitutions at position 16 likely also result in a more open hypernucleosome structure, as for Candidatus Odinarchaeota LCB_4.
Three archaeal histone species fail multiple criteria in our analysis, indicating that these cannot form hypernucleosomes. These histone species are Haloredivivus sp G17, Nanosalina J07AB43 HB, and Euryarchaeal Methanococcoides methylutens (class Methanomicrobia) that all combine defects in the dimer interface with a bulky substitution at position 16 and few potential stacking interactions (Table 2, marked–). In particular, Nanosalina J07AB43 Histone B (HB) shows a H49D substitution and a glutamic acid at position 62, making the dimer surface highly negatively charged and thus very unlikely to interact with another dimer.
It is remarkable that most of the histones having N- or C-terminal tails or N- or C-terminal truncations additionally have substitutions in the dimer–dimer and/or stacking interface that will affect hypernucleosome formation. Histones with reduced ability to form compact hypernucleosomes are expected to exhibit different roles in shaping the genome, like simple DNA bending or site-specific interference with histone multimerization. Interestingly, the genomes of several organisms encode histones that we predict are able to multimerize as well as histones that probably do not multimerize. This suggests that they may, in addition to directly binding to promoters, also be able to affect gene regulation by multimerization.
Histones in genome regulation
MNase-seq experiments have shown that histones position upstream and downstream of a promoter region [106]. This, in combination with knock-out studies showing both up- and down-regulation of transcription levels, leads to the hypothesis that histones are important for transcription regulation in the relatively well-studied phylum Euryarchaeota [45, 69, 107, 108] and may play a similar role in other histone-coding phyla. The exact mechanisms by which histones act in regulation are at this moment largely unknown. What is the mechanistic role of histones in the regulation of gene expression? Is the hypernucleosome, with a mechanism analogous to that in bacterial gene repression, able to block promoter regions and other regulatory elements, thereby making them inaccessible to the transcription machinery [109–112]? In Bacteria, such a mechanism exists for H-NS and partition protein B (ParB) proteins, in which filaments laterally spread from a nucleation site, often a high-affinity DNA sequence [113–116]. Specific high-affinity sites have been identified both in vivo and in vitro in Archaea [61, 106, 117, 118]. The role of such high-affinity sites may be to position the hypernucleosome on the genome and could be a key feature in archaeal genome regulation. In Archaea, cooperative lateral spreading of filaments has been reported for Alba proteins [40, 42, 119, 120]. Also, promoter occlusion mechanisms and competitive binding of archaeal NAPs and transcription factors have been reported [45, 121, 122].
In addition, how dynamic are hypernucleosomes, and how does the cell control the size of the hypernucleosome in order for it to be functional? Is up- and down-regulation of histone expression important in fine tuning this process? Another option for control of hypernucleosome size is heteromerization of histone variants with different stacking propensity. Heteromerization of such histone variants, for instance HA and HB from Nanosalina J07AB43 (Table 2), could restrict hypernucleosome size to fewer subunits. Distinct expression patterns of histone variants at different growth phases or as a result of environmental cues such as osmolarity [59, 107], may alter the composition and size of the hypernucleosome. However, so far, histone variants have been poorly studied in Archaea. The results of our predictions on hypernucleosome formation clearly point out the need for in vitro and in vivo studies explicitly addressing all of these questions.
Conclusion
Histones from Archaea and eukaryotes are similar in tertiary but not in quaternary structure when bound to DNA. While eukaryotic histones form octamers on the DNA, archaeal histones form filaments of variable size: hypernucleosomes. Important residues responsible for DNA binding, dimer–dimer interactions, and stacking interactions are mostly conserved among Archaea, including Asgard Archaea, Bathyarchaeota, and other newly discovered Archaea. In these recently discovered Archaeal phyla, histone tails and truncated histone variants were also found. In terms of evolution, it appears that, based on fragmentary data derived from extant lineages, the hypernucleosome has progressively become more flexible as histones with N-terminal and C-terminal tails and additional terminal helices (like in H2A and H2B in the nucleosome) developed. Furthermore, the appearance of additional DNA-binding residues and positively charged N-terminal tails may have increased the affinity of histones for DNA [123]. These changes in dimer structure and DNA affinity may have stabilized octameric nucleosomes and disfavored multimerization. Specifically, the emergence of the eukaryotic H2A-H2B heterodimer blocked hypernucleosome formation since H2A lacks the dimer–dimer interface, and H2B contains an additional helix at its C-terminus that blocks the stacking interface.
The histone tails from Candidatus Heimdallarchaeota are likely to function in similar ways as those of eukaryotic histones. They are lysine rich and potentially subject to post-translational modification, thereby possibly affecting the histone’s interactions with other actors. Alternatively, they may provide stabilization of the hypernucleosome via interactions with DNA in cis or in trans. Since it is believed that eukaryotes share their latest common ancestor with Candidatus Heimdallarchaeota, eukaryotic histones may have evolved from the predecessors of the tail-containing Heimdallarchaeal histones. As some histone proteins that have an N-terminal tail (Candidatus Heimdallarchaeota LC_3 HA and Bathyarchaeota archaeon B23) seem to form less stable hypernucleosomes, these histones may represent an evolutionary transition towards a different mechanism of gene regulation, switching from regulation by multimerization and compaction toward regulation by histone tail modifications.
Although the hypernucleosome structure is suggestive of stacking interactions between dimers in adjacent turns, experimental evidence for such interactions is lacking. Also, the functional role of tails, as well as truncates, has yet to be proven experimentally. In vitro hypernucleosome reconstitution experiments and in vivo foot-printing assays of species expressing nonstandard histones combined with mutation of the residues proposed to be involved in stacking interactions could answer these questions. Lastly, the existence of post-translational modifications of residues in archaeal histone tails, as well as their effect on transcription regulation, remains to be discovered and would give an important insight into the evolution of transcription regulation and genome folding from Archaea to eukaryotes.
Methods
Selection and alignment of archaeal histone sequences
We have included histones from every histone-encoding (candidate) phylum within the archaeal domain in our analysis. We show different histones from the same organism if the predicted stacking properties are very dissimilar. Sequences were aligned with Clustal Omega [124] using default parameters, removing gaps.
Analysis of potential hypernucleosome formation
Structural analysis of the selected archaeal histones and assessment of potential hypernucleosome formation was done by inspecting the conservation of residues that are important for multimerization in the published HMfB hypernucleosome structure [64]. Comparative multichain modeling was performed in MODELLER [125] using default parameters to construct dimer models of the archaeal histones. These models were superimposed onto HMfB dimers in the hypernucleosome crystal structure to assess whether alternative or additional interactions were possible in the different archaeal histone complexes.
Model of Heimdall HA tails in hypernucleosome
The molecular model of the histone HA dimer from the Heimdallarchaeota LC_3 genome was constructed by multitemplate modeling in MODELLER [125] using otherwise default parameters. The HMfB dimer in the hypernucleosome [64] was used as a structural template for the histone fold and eukaryotic histone H3 and H4 as structural templates for the N-terminal tails. An initial model for the Heimdall HA hypernucleosome was obtained by superimposing the HA dimer model onto HMfB in the hypernucleosome crystal structure, with either an H3-like or an H4-like tail conformation. To optimize the path of the tails through the DNA gyres and remove major steric clashes, the HA dimer model and surrounding DNA was excised from the initial model and water refined separately using High-Ambiguity Driven Docking (HADDOCK) [126], imposing ambiguous interaction restraints between HA residues 14–19 and the surrounding 3-bp section of DNA, using otherwise default parameters.
Funding Statement
This work was supported by the Netherlands Organisation for Scientific Research (NWO) (VICI 016.160.613 to RTD and VIDI 723.013.010 to HvI) and the Human Frontier Science Program (HFSP) (RGP0014/2014 to RTD). The funders had no role in the preparation of the article.
References
- 1.Dorman CJ. Genome architecture and global gene regulation in bacteria: making progress towards a unified model? Nat Rev Microbiol. 2013;11(5):349–55. 10.1038/nrmicro3007 . [DOI] [PubMed] [Google Scholar]
- 2.Dame RT, Tark-Dame M. Bacterial chromatin: converging views at different scales. Curr Opin Cell Biol. 2016;40:60–5. 10.1016/j.ceb.2016.02.015 . [DOI] [PubMed] [Google Scholar]
- 3.Eme L, Spang A, Lombard J, Stairs CW, Ettema TJG. Archaea and the origin of eukaryotes. Nat Rev Microbiol. 2017;15(12):711–23. 10.1038/nrmicro.2017.133 . [DOI] [PubMed] [Google Scholar]
- 4.Williams TA, Foster PG, Cox CJ, Embley TM. An archaeal origin of eukaryotes supports only two primary domains of life. Nature. 2013;504(7479):231–6. 10.1038/nature12779 . [DOI] [PubMed] [Google Scholar]
- 5.Cox CJ, Foster PG, Hirt RP, Harris SR, Embley TM. The archaebacterial origin of eukaryotes. Proc Natl Acad Sci U S A. 2008;105(51):20356–61. 10.1073/pnas.0810647105 ; PubMed Central PMCID: PMCPMC2629343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huet J, Schnabel R, Sentenac A, Zillig W. Archaebacteria and eukaryotes possess DNA-dependent RNA polymerases of a common type. EMBO J. 1983;2(8):1291–4. ; PubMed Central PMCID: PMCPMC555274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lane WJ, Darst SA. Molecular evolution of multisubunit RNA polymerases: structural analysis. J Mol Biol. 2010;395(4):686–704. 10.1016/j.jmb.2009.10.063 ; PubMed Central PMCID: PMCPMC2813324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Armache JP, Anger AM, Marquez V, Franckenberg S, Frohlich T, Villa E, et al. Promiscuous behaviour of archaeal ribosomal proteins: implications for eukaryotic ribosome evolution. Nucleic Acids Res. 2013;41(2):1284–93. 10.1093/nar/gks1259 ; PubMed Central PMCID: PMCPMC3553981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Petrov AS, Bernier CR, Hsiao C, Norris AM, Kovacs NA, Waterbury CC, et al. Evolution of the ribosome at atomic resolution. Proc Natl Acad Sci U S A. 2014;111(28):10251–6. 10.1073/pnas.1407205111 ; PubMed Central PMCID: PMCPMC4104869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yutin N, Puigbo P, Koonin EV, Wolf YI. Phylogenomics of prokaryotic ribosomal proteins. PLoS ONE. 2012;7(5):e36972 10.1371/journal.pone.0036972 ; PubMed Central PMCID: PMCPMC3353972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zaremba-Niedzwiedzka K, Caceres EF, Saw JH, Backstrom D, Juzokaite L, Vancaester E, et al. Asgard archaea illuminate the origin of eukaryotic cellular complexity. Nature. 2017;541(7637):353–8. 10.1038/nature21031 . [DOI] [PubMed] [Google Scholar]
- 12.Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389(6648):251–60. 10.1038/38444 . [DOI] [PubMed] [Google Scholar]
- 13.Kornberg RD. Chromatin structure: a repeating unit of histones and DNA. Science. 1974;184(4139):868–71. . [DOI] [PubMed] [Google Scholar]
- 14.Decanniere K, Babu AM, Sandman K, Reeve JN, Heinemann U. Crystal structures of recombinant histones HMfA and HMfB from the hyperthermophilic archaeon Methanothermus fervidus. J Mol Biol. 2000;303(1):35–47. 10.1006/jmbi.2000.4104 . [DOI] [PubMed] [Google Scholar]
- 15.Sandman K, Reeve JN. Archaeal histones and the origin of the histone fold. Curr Opin Microbiol. 2006;9(5):520–5. 10.1016/j.mib.2006.08.003 . [DOI] [PubMed] [Google Scholar]
- 16.Arents G, Burlingame RW, Wang BC, Love WE, Moudrianakis EN. The nucleosomal core histone octamer at 3.1 A resolution: a tripartite protein assembly and a left-handed superhelix. Proc Natl Acad Sci U S A. 1991;88(22):10148–52. ; PubMed Central PMCID: PMCPMC52885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Henneman B, Dame RT. Archaeal histones: dynamic and versatile genome architects. AIMS Microbiol. 2015;1(1):72–81. 10.3934/microbiol.2015.1.72 PubMed PMID: WOS:000215290400005. [DOI] [Google Scholar]
- 18.Malik HS, Henikoff S. Phylogenomics of the nucleosome. Nat Struct Biol. 2003;10(11):882–91. 10.1038/nsb996 PubMed PMID: WOS:000186229100006. [DOI] [PubMed] [Google Scholar]
- 19.Katan AJ, Vlijm R, Lusser A, Dekker C. Dynamics of nucleosomal structures measured by high-speed atomic force microscopy. Small. 2015;11(8):976–84. 10.1002/smll.201401318 . [DOI] [PubMed] [Google Scholar]
- 20.Levchenko V, Jackson B, Jackson V. Histone release during transcription: displacement of the two H2A-H2B dimers in the nucleosome is dependent on different levels of transcription-induced positive stress. Biochemistry. 2005;44(14):5357–72. 10.1021/bi047786o . [DOI] [PubMed] [Google Scholar]
- 21.Hamiche A, Carot V, Alilat M, De Lucia F, O'Donohue MF, Revet B, et al. Interaction of the histone (H3-H4)2 tetramer of the nucleosome with positively supercoiled DNA minicircles: Potential flipping of the protein from a left- to a right-handed superhelical form. Proc Natl Acad Sci U S A. 1996;93(15):7588–93. ; PubMed Central PMCID: PMCPMC38790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arimura Y, Tachiwana H, Oda T, Sato M, Kurumizaka H. Structural analysis of the hexasome, lacking one histone H2A/H2B dimer from the conventional nucleosome. Biochemistry. 2012;51(15):3302–9. 10.1021/bi300129b [DOI] [PubMed] [Google Scholar]
- 23.Bednar J, Garcia-Saez I, Boopathi R, Cutter AR, Papai G, Reymer A, et al. Structure and Dynamics of a 197 bp Nucleosome in Complex with Linker Histone H1. Mol Cell. 2017;66(3):384–97 e8. 10.1016/j.molcel.2017.04.012 ; PubMed Central PMCID: PMCPMC5508712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou BR, Feng HQ, Kato H, Dai L, Yang YD, Zhou YQ, et al. Structural insights into the histone H1-nucleosome complex. P Natl Acad Sci USA. 2013;110(48):19390–5. 10.1073/pnas.1314905110 PubMed PMID: WOS:000327390400064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cutter AR, Hayes JJ. Linker histones: novel insights into structure-specific recognition of the nucleosome. Biochem Cell Biol. 2017;95(2):171–8. 10.1139/bcb-2016-0097 ; PubMed Central PMCID: PMCPMC5654525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robinson PJ, Rhodes D. Structure of the '30 nm' chromatin fibre: a key role for the linker histone. Curr Opin Struct Biol. 2006;16(3):336–43. 10.1016/j.sbi.2006.05.007 . [DOI] [PubMed] [Google Scholar]
- 27.Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR, Peterson CL. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science. 2006;311(5762):844–7. 10.1126/science.1124000 . [DOI] [PubMed] [Google Scholar]
- 28.Pepenella S, Murphy KJ, Hayes JJ. A distinct switch in interactions of the histone H4 tail domain upon salt-dependent folding of nucleosome arrays. J Biol Chem. 2014;289(39):27342–51. 10.1074/jbc.M114.595140 ; PubMed Central PMCID: PMCPMC4175364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang R, Erler J, Langowski J. Histone Acetylation Regulates Chromatin Accessibility: Role of H4K16 in Inter-nucleosome Interaction. Biophys J. 2017;112(3):450–9. 10.1016/j.bpj.2016.11.015 ; PubMed Central PMCID: PMCPMC5300776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao Y, Garcia BA. Comprehensive Catalog of Currently Documented Histone Modifications. Cold Spring Harb Perspect Biol. 2015;7(9):a025064 10.1101/cshperspect.a025064 ; PubMed Central PMCID: PMCPMC4563710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293(5532):1074–80. 10.1126/science.1063127 . [DOI] [PubMed] [Google Scholar]
- 32.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705. 10.1016/j.cell.2007.02.005 . [DOI] [PubMed] [Google Scholar]
- 33.Kalashnikova AA, Porter-Goff ME, Muthurajan UM, Luger K, Hansen JC. The role of the nucleosome acidic patch in modulating higher order chromatin structure. J R Soc Interface. 2013;10(82):20121022 10.1098/rsif.2012.1022 ; PubMed Central PMCID: PMCPMC3627075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou J, Fan JY, Rangasamy D, Tremethick DJ. The nucleosome surface regulates chromatin compaction and couples it with transcriptional repression. Nat Struct Mol Biol. 2007;14(11):1070–6. 10.1038/nsmb1323 . [DOI] [PubMed] [Google Scholar]
- 35.Dorigo B, Schalch T, Bystricky K, Richmond TJ. Chromatin fiber folding: requirement for the histone H4 N-terminal tail. J Mol Biol. 2003;327(1):85–96. . [DOI] [PubMed] [Google Scholar]
- 36.Stein DB, Searcy DG. Physiologically important stabilization of DNA by a prokaryotic histone-like protein. Science. 1978;202(4364):219–21. . [DOI] [PubMed] [Google Scholar]
- 37.Searcy DG, Delange RJ. Thermoplasma acidophilum histone-like protein. Partial amino acid sequence suggestive of homology to eukaryotic histones. Biochim Biophys Acta. 1980;609(1):197–200. . [DOI] [PubMed] [Google Scholar]
- 38.Goyal M, Banerjee C, Nag S, Bandyopadhyay U. The Alba protein family: Structure and function. Biochim Biophys Acta. 2016;1864(5):570–83. 10.1016/j.bbapap.2016.02.015 . [DOI] [PubMed] [Google Scholar]
- 39.Forterre P, Confalonieri F, Knapp S. Identification of the gene encoding archeal-specific DNA-binding proteins of the Sac10b family. Mol Microbiol. 1999;32(3):669–70. . [DOI] [PubMed] [Google Scholar]
- 40.Jelinska C, Petrovic-Stojanovska B, Ingledew WJ, White MF. Dimer-dimer stacking interactions are important for nucleic acid binding by the archaeal chromatin protein Alba. Biochem J. 2010;427(1):49–55. 10.1042/BJ20091841 ; PubMed Central PMCID: PMCPMC2841500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lurz R, Grote M, Dijk J, Reinhardt R, Dobrinski B. Electron microscopic study of DNA complexes with proteins from the Archaebacterium Sulfolobus acidocaldarius. EMBO J. 1986;5(13):3715–21. ; PubMed Central PMCID: PMCPMC1167416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Laurens N, Driessen RP, Heller I, Vorselen D, Noom MC, Hol FJ, et al. Alba shapes the archaeal genome using a delicate balance of bridging and stiffening the DNA. Nat Commun. 2012;3:1328 10.1038/ncomms2330 ; PubMed Central PMCID: PMCPMC3535426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bell SD, Botting CH, Wardleworth BN, Jackson SP, White MF. The interaction of Alba, a conserved archaeal chromatin protein, with Sir2 and its regulation by acetylation. Science. 2002;296(5565):148–51. 10.1126/science.1070506 . [DOI] [PubMed] [Google Scholar]
- 44.Liu Y, Guo L, Guo R, Wong RL, Hernandez H, Hu J, et al. The Sac10b homolog in Methanococcus maripaludis binds DNA at specific sites. J Bacteriol. 2009;191(7):2315–29. 10.1128/JB.01534-08 ; PubMed Central PMCID: PMCPMC2655493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peeters E, Driessen RP, Werner F, Dame RT. The interplay between nucleoid organization and transcription in archaeal genomes. Nat Rev Microbiol. 2015;13(6):333–41. 10.1038/nrmicro3467 . [DOI] [PubMed] [Google Scholar]
- 46.Driessen RP, Lin SN, Waterreus WJ, van der Meulen AL, van der Valk RA, Laurens N, et al. Diverse architectural properties of Sso10a proteins: Evidence for a role in chromatin compaction and organization. Sci Rep. 2016;6:29422 10.1038/srep29422 ; PubMed Central PMCID: PMCPMC4941522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kahsai MA, Vogler B, Clark AT, Edmondson SP, Shriver JW. Solution structure, stability, and flexibility of Sso10a: a hyperthermophile coiled-coil DNA-binding protein. Biochemistry. 2005;44(8):2822–32. 10.1021/bi047669t [DOI] [PubMed] [Google Scholar]
- 48.Edmondson SP, Shriver JW. DNA binding proteins Sac7d and Sso7d from Sulfolobus. Methods Enzymol. 2001;334:129–45. . [DOI] [PubMed] [Google Scholar]
- 49.Guo L, Feng Y, Zhang Z, Yao H, Luo Y, Wang J, et al. Biochemical and structural characterization of Cren7, a novel chromatin protein conserved among Crenarchaea. Nucleic Acids Res. 2008;36(4):1129–37. 10.1093/nar/gkm1128 ; PubMed Central PMCID: PMCPMC2275093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Driessen RP, Dame RT. Nucleoid-associated proteins in Crenarchaea. Biochem Soc Trans. 2011;39(1):116–21. 10.1042/BST0390116 . [DOI] [PubMed] [Google Scholar]
- 51.Maruyama H, Shin M, Oda T, Matsumi R, Ohniwa RL, Itoh T, et al. Histone and TK0471/TrmBL2 form a novel heterogeneous genome architecture in the hyperthermophilic archaeon Thermococcus kodakarensis. Mol Biol Cell. 2011;22(3):386–98. 10.1091/mbc.E10-08-0668 ; PubMed Central PMCID: PMCPMC3031468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Culard F, Laine B, Sautiere P, Maurizot JC. Stoichiometry of the binding of chromosomal protein MC1 from the archaebacterium, Methanosarcina spp. CHTI55, to DNA. FEBS Lett. 1993;315(3):335–9. . [DOI] [PubMed] [Google Scholar]
- 53.Pavlov NA, Cherny DI, Nazimov IV, Slesarev AI, Subramaniam V. Identification, cloning and characterization of a new DNA-binding protein from the hyperthermophilic methanogen Methanopyrus kandleri. Nucleic Acids Res. 2002;30(3):685–94. ; PubMed Central PMCID: PMCPMC100301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Oppermann UC, Knapp S, Bonetto V, Ladenstein R, Jornvall H. Isolation and structure of repressor-like proteins from the archaeon Sulfolobus solfataricus. Co-purification of RNase A with Sso7c. FEBS Lett. 1998;432(3):141–4. . [DOI] [PubMed] [Google Scholar]
- 55.Luo X, Schwarz-Linek U, Botting CH, Hensel R, Siebers B, White MF. CC1, a novel crenarchaeal DNA binding protein. J Bacteriol. 2007;189(2):403–9. 10.1128/JB.01246-06 ; PubMed Central PMCID: PMCPMC1797387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bailey KA, Marc F, Sandman K, Reeve JN. Both DNA and histone fold sequences contribute to archaeal nucleosome stability. J Biol Chem. 2002;277(11):9293–301. 10.1074/jbc.M110029200 . [DOI] [PubMed] [Google Scholar]
- 57.Sandman K, Krzycki JA, Dobrinski B, Lurz R, Reeve JN. HMf, a DNA-binding protein isolated from the hyperthermophilic archaeon Methanothermus fervidus, is most closely related to histones. Proc Natl Acad Sci U S A. 1990;87(15):5788–91. ; PubMed Central PMCID: PMCPMC54413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nishida H, Oshima T. Archaeal histone distribution is associated with archaeal genome base composition. J Gen Appl Microbiol. 2017;63(1):28–35. 10.2323/jgam.2016.07.003 . [DOI] [PubMed] [Google Scholar]
- 59.Sandman K, Grayling RA, Dobrinski B, Lurz R, Reeve JN. Growth-phase-dependent synthesis of histones in the archaeon Methanothermus fervidus. Proc Natl Acad Sci U S A. 1994;91(26):12624–8. ; PubMed Central PMCID: PMCPMC45491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pereira SL, Grayling RA, Lurz R, Reeve JN. Archaeal nucleosomes. Proc Natl Acad Sci U S A. 1997;94(23):12633–7. ; PubMed Central PMCID: PMCPMC25063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bailey KA, Pereira SL, Widom J, Reeve JN. Archaeal histone selection of nucleosome positioning sequences and the procaryotic origin of histone-dependent genome evolution. J Mol Biol. 2000;303(1):25–34. 10.1006/jmbi.2000.4128 . [DOI] [PubMed] [Google Scholar]
- 62.van der Valk RA, Laurens N, Dame RT. Tethered Particle Motion Analysis of the DNA Binding Properties of Architectural Proteins. Methods Mol Biol. 2017;1624:127–43. 10.1007/978-1-4939-7098-8_11 . [DOI] [PubMed] [Google Scholar]
- 63.Maruyama H, Harwood JC, Moore KM, Paszkiewicz K, Durley SC, Fukushima H, et al. An alternative beads-on-a-string chromatin architecture in Thermococcus kodakarensis. EMBO Rep. 2013;14(8):711–7. 10.1038/embor.2013.94 ; PubMed Central PMCID: PMCPMC3736136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mattiroli F, Bhattacharyya S, Dyer PN, White AE, Sandman K, Burkhart BW, et al. Structure of histone-based chromatin in Archaea. Science. 2017;357(6351):609–12. 10.1126/science.aaj1849 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Arents G, Moudrianakis EN. The histone fold: a ubiquitous architectural motif utilized in DNA compaction and protein dimerization. Proc Natl Acad Sci U S A. 1995;92(24):11170–4. ; PubMed Central PMCID: PMCPMC40593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Malik HS, Henikoff S. Phylogenomics of the nucleosome. Nat Struct Biol. 2003;10(11):882–91. 10.1038/nsb996 . [DOI] [PubMed] [Google Scholar]
- 67.Sandman K, Pereira SL, Reeve JN. Diversity of prokaryotic chromosomal proteins and the origin of the nucleosome. Cell Mol Life Sci. 1998;54(12):1350–64. 10.1007/s000180050259 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ng WV, Kennedy SP, Mahairas GG, Berquist B, Pan M, Shukla HD, et al. Genome sequence of Halobacterium species NRC-1. Proc Natl Acad Sci U S A. 2000;97(22):12176–81. 10.1073/pnas.190337797 ; PubMed Central PMCID: PMCPMC17314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dulmage KA, Todor H, Schmid AK. Growth-Phase-Specific Modulation of Cell Morphology and Gene Expression by an Archaeal Histone Protein. MBio. 2015;6(5):e00649–15. 10.1128/mBio.00649-15 ; PubMed Central PMCID: PMCPMC4600100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Becker EA, Seitzer PM, Tritt A, Larsen D, Krusor M, Yao AI, et al. Phylogenetically driven sequencing of extremely halophilic archaea reveals strategies for static and dynamic osmo-response. PLoS Genet. 2014;10(11):e1004784 10.1371/journal.pgen.1004784 ; PubMed Central PMCID: PMCPMC4230888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stoltzfus A. On the possibility of constructive neutral evolution. J Mol Evol. 1999;49(2):169–81. . [DOI] [PubMed] [Google Scholar]
- 72.Lynch M, Force A. The probability of duplicate gene preservation by subfunctionalization. Genetics. 2000;154(1):459–73. ; PubMed Central PMCID: PMCPMC1460895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ausio J. Histone variants—the structure behind the function. Brief Funct Genomic Proteomic. 2006;5(3):228–43. 10.1093/bfgp/ell020 . [DOI] [PubMed] [Google Scholar]
- 74.Marzluff WF, Duronio RJ. Histone mRNA expression: multiple levels of cell cycle regulation and important developmental consequences. Curr Opin Cell Biol. 2002;14(6):692–9. . [DOI] [PubMed] [Google Scholar]
- 75.Weber CM, Henikoff S. Histone variants: dynamic punctuation in transcription. Genes Dev. 2014;28(7):672–82. 10.1101/gad.238873.114 ; PubMed Central PMCID: PMCPMC4015494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Henikoff S, Smith MM. Histone variants and epigenetics. Cold Spring Harb Perspect Biol. 2015;7(1):a019364 10.1101/cshperspect.a019364 ; PubMed Central PMCID: PMCPMC4292162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Spang A, Saw JH, Jorgensen SL, Zaremba-Niedzwiedzka K, Martijn J, Lind AE, et al. Complex archaea that bridge the gap between prokaryotes and eukaryotes. Nature. 2015;521(7551):173–9. 10.1038/nature14447 ; PubMed Central PMCID: PMCPMC4444528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Woese CR, Fox GE. Phylogenetic structure of the prokaryotic domain: the primary kingdoms. Proc Natl Acad Sci U S A. 1977;74(11):5088–90. ; PubMed Central PMCID: PMCPMC432104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Spang A, Caceres EF, Ettema TJG. Genomic exploration of the diversity, ecology, and evolution of the archaeal domain of life. Science. 2017;357(6351). 10.1126/science.aaf3883 . [DOI] [PubMed] [Google Scholar]
- 80.He Y, Li M, Perumal V, Feng X, Fang J, Xie J, et al. Genomic and enzymatic evidence for acetogenesis among multiple lineages of the archaeal phylum Bathyarchaeota widespread in marine sediments. Nat Microbiol. 2016;1(6):16035 10.1038/nmicrobiol.2016.35 . [DOI] [PubMed] [Google Scholar]
- 81.Probst AJ, Castelle CJ, Singh A, Brown CT, Anantharaman K, Sharon I, et al. Genomic resolution of a cold subsurface aquifer community provides metabolic insights for novel microbes adapted to high CO2 concentrations. Environ Microbiol. 2017;19(2):459–74. 10.1111/1462-2920.13362 . [DOI] [PubMed] [Google Scholar]
- 82.Probst AJ, Ladd B, Jarett JK, Geller-McGrath DE, Sieber CMK, Emerson JB, et al. Differential depth distribution of microbial function and putative symbionts through sediment-hosted aquifers in the deep terrestrial subsurface. Nat Microbiol. 2018;3(3):328–36. 10.1038/s41564-017-0098-y . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fricke WF, Seedorf H, Henne A, Kruer M, Liesegang H, Hedderich R, et al. The genome sequence of Methanosphaera stadtmanae reveals why this human intestinal archaeon is restricted to methanol and H2 for methane formation and ATP synthesis. J Bacteriol. 2006;188(2):642–58. 10.1128/JB.188.2.642-658.2006 ; PubMed Central PMCID: PMCPMC1347301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rinke C, Schwientek P, Sczyrba A, Ivanova NN, Anderson IJ, Cheng JF, et al. Insights into the phylogeny and coding potential of microbial dark matter. Nature. 2013;499(7459):431–7. 10.1038/nature12352 . [DOI] [PubMed] [Google Scholar]
- 85.Parks DH, Rinke C, Chuvochina M, Chaumeil PA, Woodcroft BJ, Evans PN, et al. Recovery of nearly 8,000 metagenome-assembled genomes substantially expands the tree of life. Nat Microbiol. 2017;2(11):1533–42. 10.1038/s41564-017-0012-7 . [DOI] [PubMed] [Google Scholar]
- 86.Jungbluth SP, Amend JP, Rappe MS. Metagenome sequencing and 98 microbial genomes from Juan de Fuca Ridge flank subsurface fluids. Sci Data. 2017;4:170037 10.1038/sdata.2017.37 ; PubMed Central PMCID: PMCPMC5369317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Vanwonterghem I, Evans PN, Parks DH, Jensen PD, Woodcroft BJ, Hugenholtz P, et al. Methylotrophic methanogenesis discovered in the archaeal phylum Verstraetearchaeota. Nat Microbiol. 2016;1:16170 10.1038/nmicrobiol.2016.170 . [DOI] [PubMed] [Google Scholar]
- 88.Xue H, Guo R, Wen YF, Liu DX, Huang L. An abundant DNA binding protein from the hyperthermophilic archaeon Sulfolobus shibatae affects DNA supercoiling in a temperature-dependent fashion. Journal of Bacteriology. 2000;182(14):3929–33. PubMed PMID: WOS:000087938500007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Driessen RP, Dame RT. Structure and dynamics of the crenarchaeal nucleoid. Biochem Soc Trans. 2013;41(1):321–5. 10.1042/BST20120336 . [DOI] [PubMed] [Google Scholar]
- 90.White MF, Bell SD. Holding it together: chromatin in the Archaea. Trends Genet. 2002;18(12):621–6. . [DOI] [PubMed] [Google Scholar]
- 91.O'Neill LP, Turner BM. Histone H4 acetylation distinguishes coding regions of the human genome from heterochromatin in a differentiation-dependent but transcription-independent manner. EMBO J. 1995;14(16):3946–57. ; PubMed Central PMCID: PMCPMC394473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Reeve JN. Archaeal chromatin and transcription. Mol Microbiol. 2003;48(3):587–98. . [DOI] [PubMed] [Google Scholar]
- 93.Eichler J, Adams MWW. Posttranslational protein modification in Archaea. Microbiol Mol Biol R. 2005;69(3):393–425. PubMed PMID: WOS:000231838800002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Beltrao P, Bork P, Krogan NJ, van Noort V. Evolution and functional cross-talk of protein post-translational modifications. Mol Syst Biol. 2013;9. doi: ARTN 714 10.1002/msb.201304521 PubMed PMID: WOS:000342502000002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sigrist CJA, Cerutti L, de Castro E, Langendijk-Genevaux PS, Bulliard V, Bairoch A, et al. PROSITE, a protein domain database for functional characterization and annotation. Nucleic Acids Research. 2010;38:D161–D6. 10.1093/nar/gkp885 PubMed PMID: WOS:000276399100026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wu CH, Yeh LSL, Huang HZ, Arminski L, Castro-Alvear J, Chen YX, et al. The Protein Information Resource. Nucleic Acids Research. 2003;31(1):345–7. 10.1093/nar/gkg040 PubMed PMID: WOS:000181079700083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Berger SL. Gene activation by histone and factor acetyltransferases. Curr Opin Cell Biol. 1999;11(3):336–41. 10.1016/S0955-0674(99)80046-5 PubMed PMID: WOS:000080799100007. [DOI] [PubMed] [Google Scholar]
- 98.Li YY, Wen H, Xi YX, Tanaka K, Wang HB, Peng DN, et al. AF9 YEATS Domain Links Histone Acetylation to DOT1L-Mediated H3K79 Methylation. Cell. 2014;159(3):558–71. 10.1016/j.cell.2014.09.049 PubMed PMID: WOS:000344521700012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Shanle EK, Andrews FH, Meriesh H, McDaniel SL, Dronamraju R, DiFiore JV, et al. Association of Taf14 with acetylated histone H3 directs gene transcription and the DNA damage response. Gene Dev. 2015;29(17):1795–800. 10.1101/gad.269977.115 PubMed PMID: WOS:000361415700003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Andrews FH, Shinsky SA, Shanle EK, Bridgers JB, Gest A, Tsun IK, et al. The Taf14 YEATS domain is a reader of histone crotonylation. Nat Chem Biol. 2016;12(6):396–U33. 10.1038/nchembio.2065 PubMed PMID: WOS:000376160600007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Li YY, Sabari BR, Panchenko T, Wen H, Zhao D, Guan HP, et al. Molecular Coupling of Histone Crotonylation and Active Transcription by AF9 YEATS Domain. Mol Cell. 2016;62(2):181–93. 10.1016/j.molcel.2016.03.028 PubMed PMID: WOS:000374643900005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhao D, Guan HP, Zhao S, Mi WY, Wen H, Li YY, et al. YEATS2 is a selective histone crotonylation reader. Cell Res. 2016;26(5):629–32. PubMed PMID: WOS:000377449200011. 10.1038/cr.2016.49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chavez MS, Scorgie JK, Dennehey BK, Noone S, Tyler JK, Churchill ME. The conformational flexibility of the C-terminus of histone H4 promotes histone octamer and nucleosome stability and yeast viability. Epigenetics Chromatin. 2012;5(1):5 10.1186/1756-8935-5-5 ; PubMed Central PMCID: PMCPMC3439350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Fahrner RL, Cascio D, Lake JA, Slesarev A. An ancestral nuclear protein assembly: crystal structure of the Methanopyrus kandleri histone. Protein Sci. 2001;10(10):2002–7. 10.1110/ps.10901 ; PubMed Central PMCID: PMCPMC2374223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Marc F, Sandman K, Lurz R, Reeve JN. Archaeal histone tetramerization determines DNA affinity and the direction of DNA supercoiling. J Biol Chem. 2002;277(34):30879–86. 10.1074/jbc.M203674200 . [DOI] [PubMed] [Google Scholar]
- 106.Nalabothula N, Xi L, Bhattacharyya S, Widom J, Wang JP, Reeve JN, et al. Archaeal nucleosome positioning in vivo and in vitro is directed by primary sequence motifs. BMC Genomics. 2013;14:391 10.1186/1471-2164-14-391 ; PubMed Central PMCID: PMCPMC3691661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wilkinson SP, Ouhammouch M, Geiduschek EP. Transcriptional activation in the context of repression mediated by archaeal histones. Proc Natl Acad Sci U S A. 2010;107(15):6777–81. 10.1073/pnas.1002360107 ; PubMed Central PMCID: PMCPMC2872413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Xie Y, Reeve JN. Transcription by an archaeal RNA polymerase is slowed but not blocked by an archaeal nucleosome. J Bacteriol. 2004;186(11):3492–8. 10.1128/JB.186.11.3492-3498.2004 ; PubMed Central PMCID: PMCPMC415759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ueguchi C, Kakeda M, Mizuno T. Autoregulatory Expression of the Escherichia-Coli-Hns Gene Encoding a Nucleoid Protein—H-Ns Functions as a Repressor of Its Own Transcription. Mol Gen Genet. 1993;236(2–3):171–8. 10.1007/Bf00277109 PubMed PMID: WOS:A1993KK98500003. [DOI] [PubMed] [Google Scholar]
- 110.Dorman CJ. H-NS: A universal regulator for a dynamic genome. Nature Reviews Microbiology. 2004;2(5):391–400. 10.1038/nrmicro883 PubMed PMID: WOS:000221589700013. [DOI] [PubMed] [Google Scholar]
- 111.Baek JH, Rajagopala SV, Chattoraj DK. Chromosome segregation proteins of Vibrio cholerae as transcription regulators. MBio. 2014;5(3):e01061–14. 10.1128/mBio.01061-14 ; PubMed Central PMCID: PMCPMC4010829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ringgaard S, Ebersbach G, Borch J, Gerdes K. Regulatory cross-talk in the double par locus of plasmid pB171. J Biol Chem. 2007;282(5):3134–45. 10.1074/jbc.M609092200 . [DOI] [PubMed] [Google Scholar]
- 113.Breier AM, Grossman AD. Whole-genome analysis of the chromosome partitioning and sporulation protein Spo0J (ParB) reveals spreading and origin-distal sites on the Bacillus subtilis chromosome. Mol Microbiol. 2007;64(3):703–18. 10.1111/j.1365-2958.2007.05690.x . [DOI] [PubMed] [Google Scholar]
- 114.Pratto F, Cicek A, Weihofen WA, Lurz R, Saenger W, Alonso JC. Streptococcus pyogenes pSM19035 requires dynamic assembly of ATP-bound ParA and ParB on parS DNA during plasmid segregation. Nucleic Acids Res. 2008;36(11):3676–89. 10.1093/nar/gkn170 ; PubMed Central PMCID: PMCPMC2441792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.van der Valk RA, Vreede J, Qin L, Moolenaar GF, Hofmann A, Goosen N, et al. Mechanism of environmentally driven conformational changes that modulate H-NS DNA-bridging activity. Elife. 2017;6 10.7554/eLife.27369 ; PubMed Central PMCID: PMCPMC5647153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lang B, Blot N, Bouffartigues E, Buckle M, Geertz M, Gualerzi CO, et al. High-affinity DNA binding sites for H-NS provide a molecular basis for selective silencing within proteobacterial genomes. Nucleic Acids Res. 2007;35(18):6330–7. 10.1093/nar/gkm712 ; PubMed Central PMCID: PMCPMC2094087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ammar R, Torti D, Tsui K, Gebbia M, Durbic T, Bader GD, et al. Chromatin is an ancient innovation conserved between Archaea and Eukarya. Elife. 2012;1. doi: ARTN e00078 10.7554/eLife.00078 PubMed PMID: WOS:000328584600004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tompitak M, Vaillant C, Schiessel H. Genomes of Multicellular Organisms Have Evolved to Attract Nucleosomes to Promoter Regions. Biophys J. 2017;112(3):505–11. 10.1016/j.bpj.2016.12.041 PubMed PMID: WOS:000393734300012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zhao K, Chai X, Marmorstein R. Structure of a Sir2 substrate, Alba, reveals a mechanism for deacetylation-induced enhancement of DNA binding. J Biol Chem. 2003;278(28):26071–7. 10.1074/jbc.M303666200 . [DOI] [PubMed] [Google Scholar]
- 120.Visone V, Vettone A, Serpe M, Valenti A, Perugino G, Rossi M, et al. Chromatin structure and dynamics in hot environments: architectural proteins and DNA topoisomerases of thermophilic archaea. Int J Mol Sci. 2014;15(9):17162–87. 10.3390/ijms150917162 ; PubMed Central PMCID: PMCPMC4200833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Grohmann D, Werner F. Recent advances in the understanding of archaeal transcription. Current Opinion in Microbiology. 2011;14(3):328–34. 10.1016/j.mib.2011.04.012 PubMed PMID: WOS:000292948300016. [DOI] [PubMed] [Google Scholar]
- 122.Sheppard C, Werner F. Structure and mechanisms of viral transcription factors in archaea. Extremophiles. 2017;21(5):829–38. 10.1007/s00792-017-0951-1 PubMed PMID: WOS:000408230000001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Soares DJ, Marc F, Reeve JN. Conserved eukaryotic histone-fold residues substituted into an archaeal histone increase DNA affinity but reduce complex flexibility. J Bacteriol. 2003;185(11):3453–7. 10.1128/JB.185.11.3453-3457.2003 ; PubMed Central PMCID: PMCPMC155370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539 10.1038/msb.2011.75 ; PubMed Central PMCID: PMCPMC3261699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Webb B, Sali A. Comparative Protein Structure Modeling Using MODELLER. Curr Protoc Bioinformatics. 2014;47:5 6 1–32. 10.1002/0471250953.bi0506s47 . [DOI] [PubMed] [Google Scholar]
- 126.Dominguez C, Boelens R, Bonvin AM. HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J Am Chem Soc. 2003;125(7):1731–7. 10.1021/ja026939x . [DOI] [PubMed] [Google Scholar]