Eukaryotic Acquisition of a Bacterial Operon (original) (raw)
. Author manuscript; available in PMC: 2020 Jun 15.
Published in final edited form as: Cell. 2019 Feb 21;176(6):1356–1366.e10. doi: 10.1016/j.cell.2019.01.034
Summary:
Operons are a hallmark of bacterial genomes, where they allow concerted expression of functionally related genes as single polycistronic transcripts. They are rare in eukaryotes, where each gene usually drives expression of its own independent messenger RNAs. Here we report the horizontal operon transfer of a siderophore biosynthesis pathway from relatives of Escherichia coli into a group of budding yeast taxa. We further show that the co-linearly arranged secondary metabolism genes are expressed, exhibit eukaryotic transcriptional features, and enable the sequestration and uptake of iron. After transfer, several genetic changes occurred during subsequent evolution, including the gain of new transcription start sites that were sometimes within protein-coding sequences, acquisition of polyadenylation sites, structural rearrangements, and integration of eukaryotic genes into the cluster. We conclude that the genes were likely acquired as a unit, modified for eukaryotic gene expression, and maintained by selection to adapt to the highly competitive, iron-limited environment.
Keywords: Central Dogma of Biology, horizontal gene transfer, operon, siderophore biosynthesis, budding yeasts, Wickerhamiella/Starmerella, Saccharomycotina, enterobactin
Introduction:
The core processes of the Central Dogma of Biology, transcription and translation, are broadly conserved across living organisms. Nonetheless, there are seemingly fundamental differences between the domains of life in how these processes are realized. Eukaryotic transcription is spatially and temporally separated from translation and generally operates on individual genes through a complex interplay of transcription factors and chromatin remodeling complexes. Nascent mRNAs are co-transcriptionally processed by adding 3’ polyadenosine (poly(A)) tails and 5’ caps of 7-methyl-guanosine (m7G) before they are trafficked out of the nucleus for translation. In bacteria, transcription is tightly coupled with translation, and both occur inside the cytosol. Furthermore, bacterial transcription often operates on clusters of genes, known as operons, where a single regulatory region controls the expression of physically-linked genes into a polycistronic mRNA that is minimally processed and translated into several polypeptides at similar abundances. In contrast, eukaryotic operons, which are rare in most taxa but are frequently found in nematodes (Blumenthal and Gleason, 2003; Spieth et al., 1993) and tunicates (Ganot et al., 2004; Vandenberghe et al., 2001), are processed by trans-splicing and related mechanisms.
Operon dissemination has been proposed to occur predominantly via horizontal gene transfer (HGT) (Lawrence et al., 1996; Omelchenko et al., 2003), a process where organisms acquire genes from sources other than their parents. HGT is pervasive and richly documented among bacteria, but it is rarer in eukaryotes (Alexander et al., 2016; Keeling and Palmer, 2008; Richards et al., 2011; Slot and Rokas, 2011; Soucy et al., 2015). Recently, several examples of horizontal gene transfer from archaea or bacteria into eukaryotes have been uncovered, and most of these have involved individual genes that are sometimes functionally related, such as genes involved in nucleic acid synthesis and salvage from bacteria into Microsporidia (Alexander et al., 2016), a gene of uncertain origin into Cnidarians (Dana et al., 2012), osmotrophy-related genes from fungi into oomycetes (Richards et al., 2006), various metabolic pathways assembled in several steps from multiple unassociated bacteria into mealybugs (Husnik et al., 2013; Husnik and McCutcheon, 2016), and several examples of bacterial genes into dikaryon fungi (Fitzpatrick, 2012; Marcet-Houben and Gabaldón, 2010).
Only a handful of known cross-domain transfer events have involved multiple genes in a single transfer event. Portions of the genome of an intracellular bacterial endosymbiont of insects have been found in insect genomes, with the transferred chunks ranging in size from roughly 500 bp to nearly the entire endosymbiont genome (Kondo et al., 2002; Nikoh et al., 2007; Hotopp et al., 2007). Furthermore, the plastid genomes of some eustigmatophyte algae harbor an operon of bacterial origin, although the function of the transferred genes in this case is uncertain (Yurchenko et al., 2016; Yurchenko et al., 2018). A two-gene operon of archaeal origin was discovered in the protist Pygsuia nuclear genome, but these genes were subsequently fused into a single open reading frame (ORF) (Stairs et al., 2014). This fused two-gene operon can also be found in the nuclear genome of the anaerobic human protozoan parasite Blastocystis (Tsaousis et al., 2012). It is unclear whether this operon was first transferred to the Pygsuia or the Blastocystis lineage, but both are found in low-oxygen environments: Pygsuia from hypoxic marine sediments and Blastocystis from human gastrointestinal tracts. Additionally, three genes (two of which are now fused into a single ORF in some taxa) from the bacterial peptidoglycan biosynthesis operon can be found in extant Aspergillus spp., although their phylogenetic origin is not fully resolved (Marcet-Houben and Gabaldón, 2010). Despite these tantalizing examples, it remains unclear 1) whether eukaryotes can acquire new traits or capabilities via the horizontal transfer of operons encoding complex multi-gene pathways from free-living bacteria in a single event, 2) whether the expression of such an operon is compatible with the seemingly conflicting characteristics of transcription and translation in a eukaryotic nuclear genome, and 3) how the function of the operon can subsequently be maintained following integration into the eukaryotic genome. Horizontal operon transfer (HOT) events could allow even complex pathways to spread rapidly across domains of life, especially in environments where competition for key nutrients is intense.
One such nutrient is iron, which plays crucial roles in many essential cellular processes (Andrews et al., 2003; Sheftel et al., 2010; Sutak et al., 2008) and is a key determinant of virulence in both animal and plant pathogens (Scharf et al., 2014; Skaar 2010; Toth et al., 2006). Many specialized systems have evolved to sequester iron from the surrounding environment, one of which is the biosynthesis of small-molecule iron chelators called siderophores. Most bacteria synthesize catecholate-class siderophores (Wandersman and Delepelaire, 2004), whereas hydroxamate-class siderophores are commonplace in fungi (Haas et al., 2008). A notable exception is the budding yeast lineage (subphylum Saccharomycotina), which has long been thought to completely lack the ability to synthesize their own siderophores, despite its ability to utilize those produced by other microbes (Haas et al., 2008).
Here we survey a broad range of fungal genomes for known components of iron uptake and storage systems. Although most systems are broadly conserved, we identify a clade of closely related yeast species that contains a bacterial siderophore biosynthesis pathway. Through phylogenetic hypothesis testing, we show that this pathway was acquired through horizontal operon transfer (HOT) from the bacterial order Enterobacteriales, which includes, among others, Escherichia coli, Erwinia carotovora, and Yersinia pestis. Relatives of those species share the insect gut niche with many yeasts of the recipient clade (Gilliam and Valentine, 1974; Gilliam 1997, Moran et al., 2008, Lachance et al., 2001; Rosa et al., 2003). After acquisition, the operon underwent structural changes and successively gained eukaryotic characteristics, while maintaining the clustering of functionally related genes. Transcriptomic experiments show that the transferred siderophore biosynthesis genes are actively expressed in a manner largely consistent with canonical eukaryotic transcription, and in vivo assays demonstrate that the operon is functional in most yeast species that contain it. This remarkable example shows how eukaryotes can acquire a functional bacterial operon, while modifying its transcription to domesticate and maintain expression as a set of linked eukaryotic genes.
Results:
Iron uptake and storage is conserved in fungi
We surveyed the genome sequences of 175 dikaryon fungal species and observed broad conservation of genes involved in low-affinity iron uptake, vacuolar iron storage, reductive iron assimilation, and siderophore import systems (Figure 1, Table S1). In contrast, genes involved in siderophore biosynthesis pathways were more varied in terms of presence and type. Siderophore biosynthesis gene clusters were thought to be completely absent in budding yeasts (Haas et al., 2008), but the genomes of Lipomyces starkeyi and Tortispora caseinolytica contain homologs of the SidA, SidC, SidD, SidF, and SidL genes involved in the biosynthesis of ferricrocin and fusarinine C, which are hydroxamate-class siderophores synthesized from L-ornithine by many filamentous fungi, such as Aspergillus nidulans (Haas et al., 2008). Since these species are highly divergent from all other budding yeast taxa with sequenced genomes, the presence of this pathway in their genomes is likely an ancestral trait inherited from the last common ancestor of the Pezizomycotina and Saccharomycotina, while its absence in most yeasts is likely due to a loss early in budding yeast evolution. A handful of phylogenetically diverse set of budding yeast species contain the newly discovered gene cluster for synthesizing the siderophore pulcherrimin (Krause et al. 2018). Surprisingly, the genomes of three closely related Trichomonascaceae species (Wickerhamiella (Candida) versatilis, Starmerella (Candida) apicola, and Starmerella bombicola) contain multiple homologs of bacterial siderophore biosynthesis genes (entA-F) that are predicted to enable the synthesis of catecholate-class siderophores from chorismate (Adeolu et al., 2016) (Figure S1). These genes are co-linear and predicted to be expressed from the same strand of DNA, features that are both reminiscent of the operons where these genes are found in bacteria.
Figure 1. Distribution of the iron uptake and storage systems among fungi.
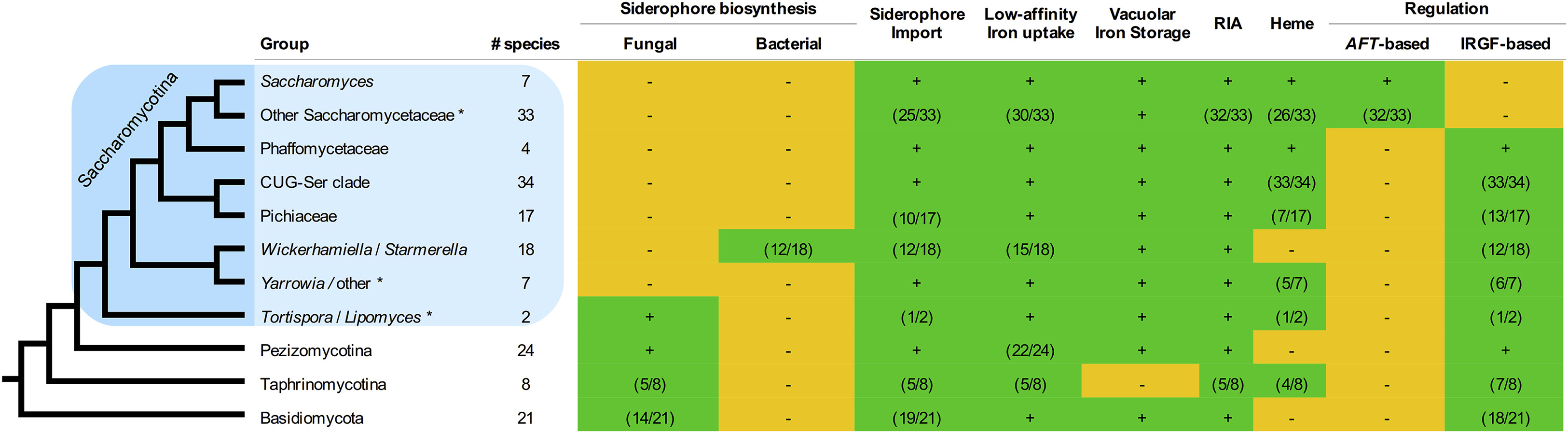
Plus (green) and minus (orange) signs indicate the presence and absence of iron uptake and storage systems in specific taxonomic groups. The numbers in parentheses (green) indicate the number of species in a taxonomic group that possess a specific system, if it is not ubiquitous in that group. The blue box indicates the budding yeasts. RIA - Reductive Iron Assimilation. IRGF - Iron-Responsive GATA Factor. Asterisks (*) mark paraphyletic groups. Note that only Wickerhamiella/Starmerella (W/S) clade fungi contain the bacterial or catecholate-class siderophore biosynthesis pathway, whereas many other dikaryon fungi contain hydroxamate-class siderophore biosynthesis pathways. See also Figure S1 and Table S1.
Horizontal operon transfer (HOT) from bacteria to yeasts
To investigate the evolutionary history of these genes, we sequenced and analyzed 17 additional genomes from the Wickerhamiella/Starmerella clade (W/S clade, Table S1) and identified the catecholate-class siderophore biosynthesis pathway in 12 of these species (Figure 2A, 2C). The genes were located on high-coverage contigs that contained multiple other yeast genes, which assured us that they were not a product of contamination (Table S1). To determine whether the yeast siderophore biosynthesis genes were horizontally acquired from a bacterial operon, we first used the _ent_erobactin genes found in yeasts to perform BLAST queries against the bacterial data present in GenBank and found that the top hits belonged to a range of species from the order Enterobacteriales. Since no single taxon was overrepresented, we surveyed 1,336 publicly available genomes from the class Gammaproteobacteria, to which the order Enterobacteriales belongs, for the presence of entA-entF homologs and extracted them from all 207 genomes where all six genes could be reliably identified (Table S2). We then reconstructed unconstrained maximum-likelihood (ML) phylogenies for each ent gene, as well as for a concatenated super-alignment of all six genes (entABCDEF, Table S2). Since entF contributed nearly two-thirds of the total alignment length, we also evaluated a super-alignment of the remaining five genes (entABCDE, Figure 2A).
Figure 2. Yeast siderophore biosynthesis originated from an Enterobacteriales lineage.
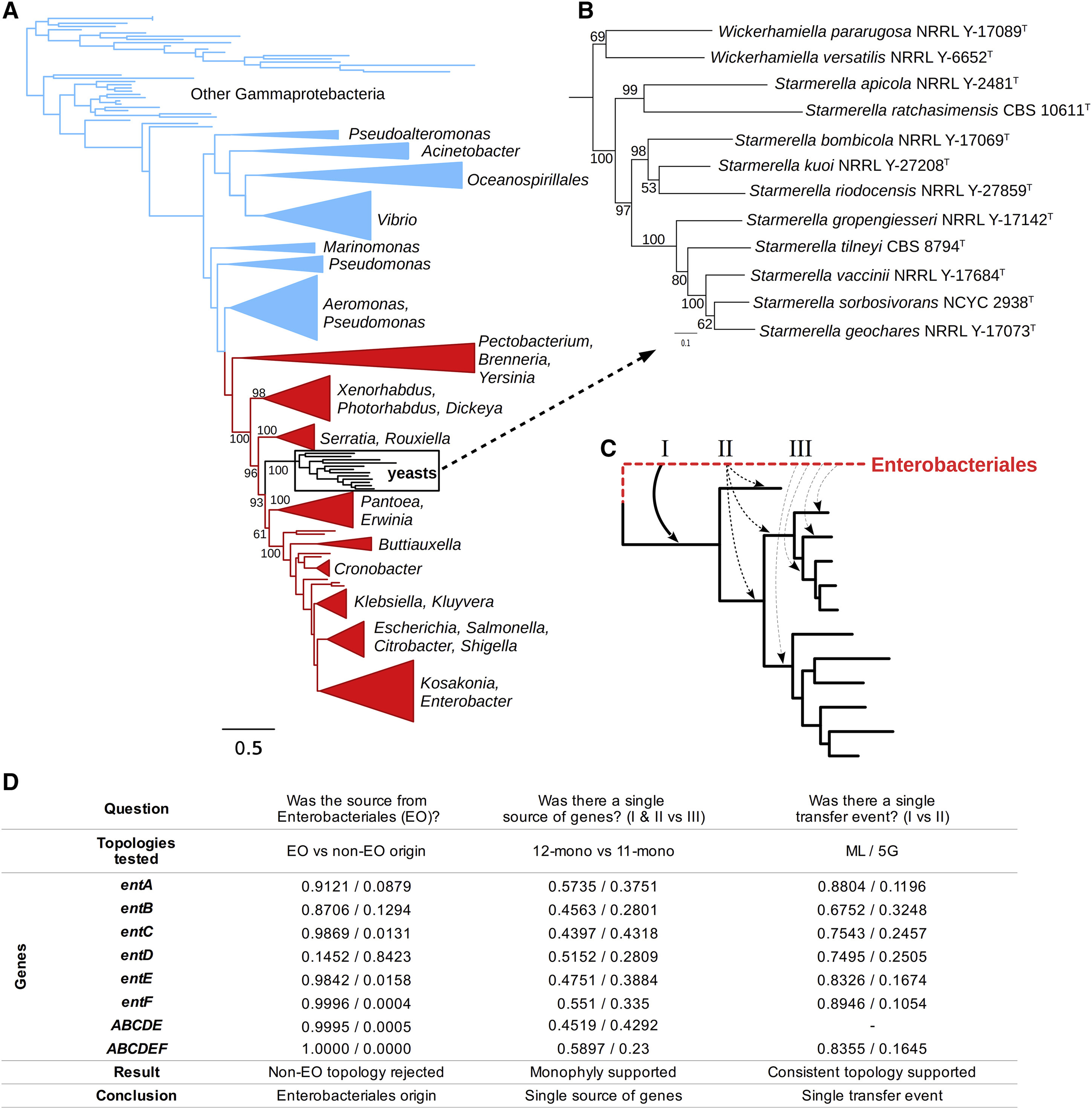
(A) ML phylogeny from the super-alignment of entABCDE genes from 207 Gammaproteobacteria and 12 yeasts, rooted at the midpoint. Bootstrap support values are shown for relevant branches within the Enterobacteriales (red). Other Gammaproteobacteria are blue.(B) Detailed view of the yeast clade from the main phylogeny, with bootstrap support values. (C) Alternative scenarios for the horizontal operon transfer. (D) P-values of the AU tests of different evolutionary hypotheses; EO - Enterobacteriales origin; non-EO - non-Enterobacteriales origin; 12-mono - 12 yeast sequences are monophyletic, 11-mono - 11 yeast sequences monophyletic and one unconstrained (12 alternatives tested, lowest p-value shown, full details in Table S2); 5G - topology of the yeast clade constrained to the one inferred from the super-alignment of entABCDE genes. Shimodaira-Hasegawa (SH) tests had less statistical power but produced fully concordant results. See also Table S1 and Table S2.
Consistent with the BLAST results, the yeast sequences formed a highly supported, monophyletic group nested within the Enterobacteriales lineage. The ent gene phylogenies of the Enterobacteriales were largely congruent with accepted relationships within the order (Adeolu et al., 2016; Baumler et al., 2013), and they placed the ent donor lineage as diverging from its common ancestor with E. coli after its divergence from Serratia and several other genera, but before the divergence of the Pantoea/Erwinia clade and E. coli. To formally test the hypothesis of an Enterobacteriales origin, we reconstructed phylogenies under the constraints that yeast sequences either grouped together with the Enterobacteriales (EO) or outside of that clade (non-EO). We then employed the Approximately Unbiased (AU) tests to determine if the EO phylogenies were a statistically better explanation of the data than the non-EO phylogenies. The EO phylogeny was strongly preferred (p-value < 10−3) for the six- and five-gene concatenation data matrices (Figure 2D). Individual genes carried weak signal due to their short lengths, but the entC, entE, and entF genes nonetheless supported the Enterobacteriales origin (p-value < 0.05); entA and entB had consistent but weaker support; and no individual gene rejected the EO hypothesis. Next, we sought to determine the course of the transfer event and tested a single-source, single-transfer hypothesis against multi-source and multi-transfer alternatives, each of which predicted specific phylogenetic patterns (Figure 2C). AU tests on the reconstructed phylogenies did not support multiple transfer events and, instead, supported the simplest explanation that the HOT event occurred from a single source lineage directly into a single common ancestor of the W/S clade yeasts (Figure 2D).
The bacterial siderophore biosynthesis pathway is functional in yeasts
To determine whether yeasts that contain the ent biosynthesis genes actually produce siderophores, we grew them on a low-iron medium overlaid with an agarose solution containing iron-complexed chromeazurol S (CAS), a colorimetric indicator of iron chelation. In presence of iron chelators, such as siderophores, the indicator changes color from blue to orange in a characteristic halo pattern that tracks the diffusion gradient of siderophores secreted from colonies into the surrounding medium. We tested the 18 yeast species from the W/S clade, together with eight outgroup species spread broadly across the yeast phylogeny (including S. cerevisiae), and E. coli as a positive control. We observed strong signals of siderophore production in six yeast species, all of which contain the siderophore biosynthesis genes (Figure 3B, Figure S4). The lack of signal in other species harboring the siderophore biosynthesis genes could suggest that siderophore production was below the sensitivity of the O-CAS assay under the conditions studied. Conversely, the O-CAS assay could lead to false positives if other mechanisms were sufficiently capable of sequestering iron. Thus, we used HPLC-MS/MS to specifically detect the chemical enterobactin. Under our strict thresholds for detection across multiple experiments and conditions, 9/12 yeast species harboring the siderophore biosynthesis genes were scored as enterobactin producers (Figure 3B). Taken together, these experiments conclusively show that the bacterial siderophore biosynthesis genes are fully functional in at least some W/S clade yeasts.
Figure 3. Evolution of the siderophore biosynthesis genes in yeasts.
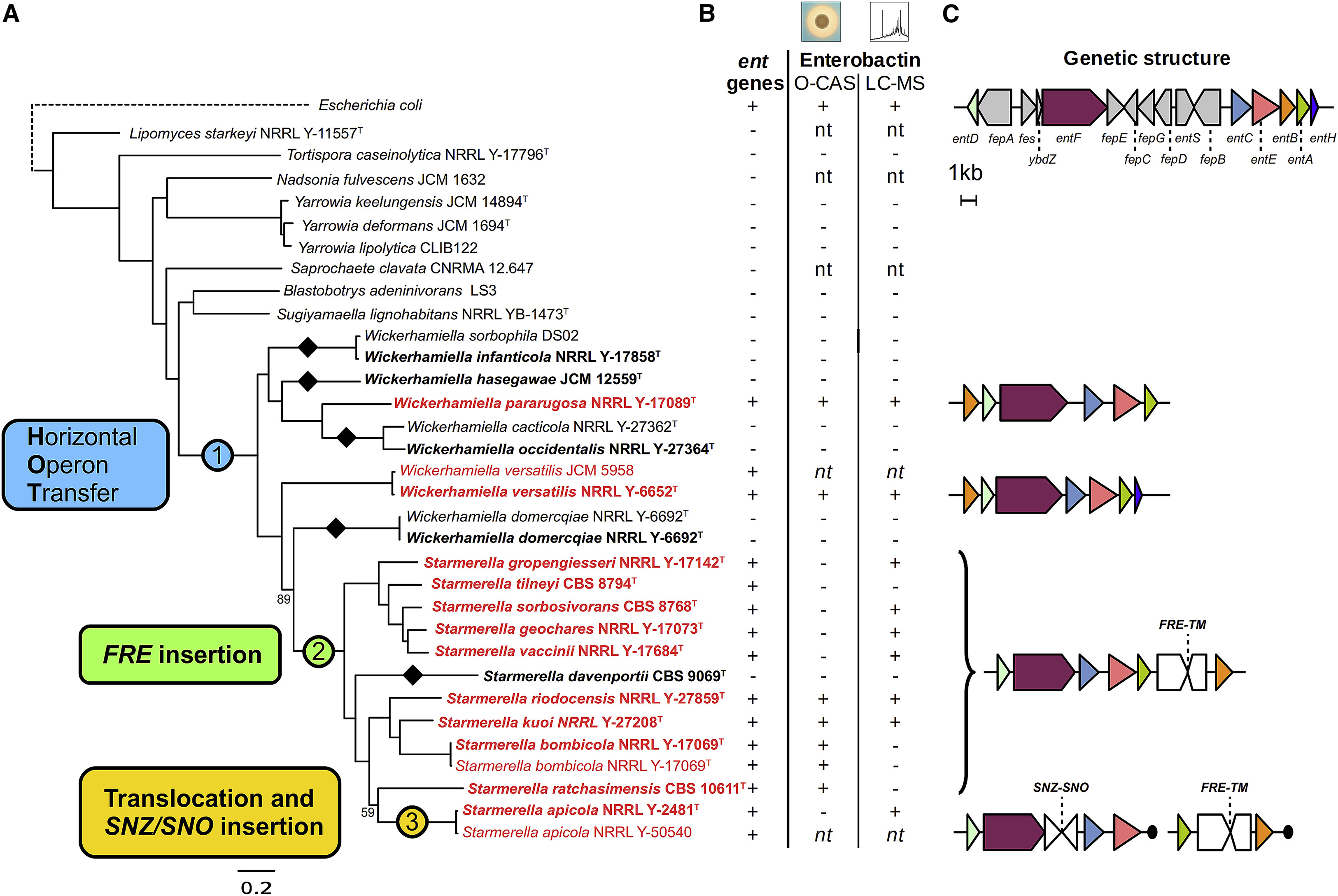
(A) ML phylogeny reconstructed from the concatenated alignment of 661 conserved, single copy genes (834,750 sites), with branch support values below 100 shown. Strains in bold denote genomes sequenced in this study, while strains in red denote genomes containing the siderophore biosynthesis genes. Black diamonds indicate secondary losses in yeast lineages, accompanied by losses of the siderophore importer ARN genes, which are often found in close proximity. (1) Horizontal operon transfer from an Enterobacteriales lineage. (2) Rearrangement and integration of genes encoding ferric reductase (FRE) and an uncharacterized transmembrane protein (TM). (3) Disruption by integration of the SNZ-SNO gene pair and translocation. (B) Species-specific data on presence/absence of the siderophore biosynthesis genes and experimental evidence for the presence of enterobactin in the yeast cultures as determined by an O-CAS assay (not specific to enterobactin) and direct chemical detection by HPLC-MS/MS (enterobactin produced by E. coli was used as the standard; nt - not tested). Note that culture conditions between assays were not identical, and siderophore expression is often condition-dependent (Machuca and Milagres, 2003). (C) Genetic structure of the siderophore biosynthesis operon in E. coli and yeasts, drawn to scale. Individual colors represent homologous genes, and gray marks bacterial genes not found in yeasts. Black circles represent contig termini within 25kb. See also Figure S4 and Table S4.
Evolution of a bacterial operon inside a eukaryotic host
Given the significant differences in Central Dogma processes between bacteria and eukaryotes, we investigated how the horizontally transferred operon was successfully assimilated into these yeasts. The lengths of intergenic regions were not divisible by three, so we immediately excluded the hypothesis that they were translated as a single fused polypeptide that could be produced from a single transcript by stop codon read-through. Next, we mapped several key changes in gene content, structure, and regulation onto the yeast phylogeny (Figure 3). First, the phylogenetic distribution of the operon genes suggests at least five cases of secondary loss in W/S clade yeasts, a common evolutionary mode for other fungal gene clusters (Campbell et al., 2013; Khaldi et al., 2008; Proctor et al., 2013; Slot and Rokas, 2010). Although all taxa contain the six core genes (entA-F), W. versatilis uniquely harbors a homolog of the entH gene, which encodes a proofreading thioesterase that is not strictly required for siderophore biosynthesis (Leduc et al., 2007). Since no homologs or remnants of other genes from the bacterial operon could be identified, we hypothesize that they were lost due to functional redundancy with genes already present in yeast genomes (e.g. the bacterial ABC transporter genes fepA-G are functionally similar to the yeast major facilitator superfamily transporter genes ARN1–4, while the bacterial esterase gene fes is functionally similar to yeast ferric reductase genes FRE1–8). Second, most extant Enterobacteriales species closely related to the source lineage share an operon structure similar to that of E. coli (Table S2), which is more complex than that of the W/S clade yeasts (Figure 3C). Based on the high amount of sequence divergence, we infer that an ancient bacterial operon, whose structure was somewhere between that of E. coli and W. versatilis, was horizontally transferred into a yeast cell tens of millions of years ago. The operon may have contained fewer genes than extant bacterial operons, or shared gene losses or rearrangements may have occurred to produce a structure similar to that of W. versatilis in the last common ancestor of the W/S clade yeasts. Third, modern yeasts of this clade have evolved at least four different structures through several lineage-specific rearrangements that tended to create derived gene cluster structures with more eukaryotic characteristics, including increasing the size of the intergenic regions, splitting the gene cluster in two in St. apicola, and intercalating at least four eukaryotic genes. The intercalation of a gene encoding a eukaryotic ferric reductase (FRE), which is involved in reductive iron assimilation, between two operon genes in a subset of species offers a particularly telling example. The genetic linkage of these two mechanisms for acquiring iron shows that bacterial and eukaryotic genes can stably co-exist, and perhaps even be selected together as gene clusters for co-inheritance or co-regulation, through eukaryotic mechanisms.
Transferred genes have mainly eukaryotic transcript features
To determine whether and how these yeasts overcame the differences between eukaryotic and bacterial gene expression, we used a strand-specific protocol to sequence mRNA from W. versatilis, St. apicola, and St. bombicola. These species were chosen due to their diverse gene cluster structures and positions on the phylogenetic tree: W. versatilis was chosen because its structure was likely more similar to the ancestral operon, while St. bombicola and St. apicola appeared to represent more derived stages of evolution in the eukaryotic hosts. Each of the three species expressed mRNAs for the siderophore biosynthesis genes, and W. versatilis expression was the highest (Table S3). The W. versatilis genes were expressed at similar levels, whereas St. bombicola and St. apicola genes showed significant diversity in their expression (Table S3, Figure 4, Figures S2–S4). Interestingly, we also observed that the siderophore biosynthesis genes in W. versatilis had much shorter intergenic sequences than their counterparts in St. bombicola and St. apicola, which were each shorter than their respective genome-wide means (within gene cluster means between predicted protein-coding sequences were 158, 484, and 377 bps versus genome-wide means of 370, 549, and 455 bps for W. versatilis, St. bombicola and St. apicola, respectively).
Figure 4. Transcriptomics of the siderophore biosynthesis genes in W. versatilis.
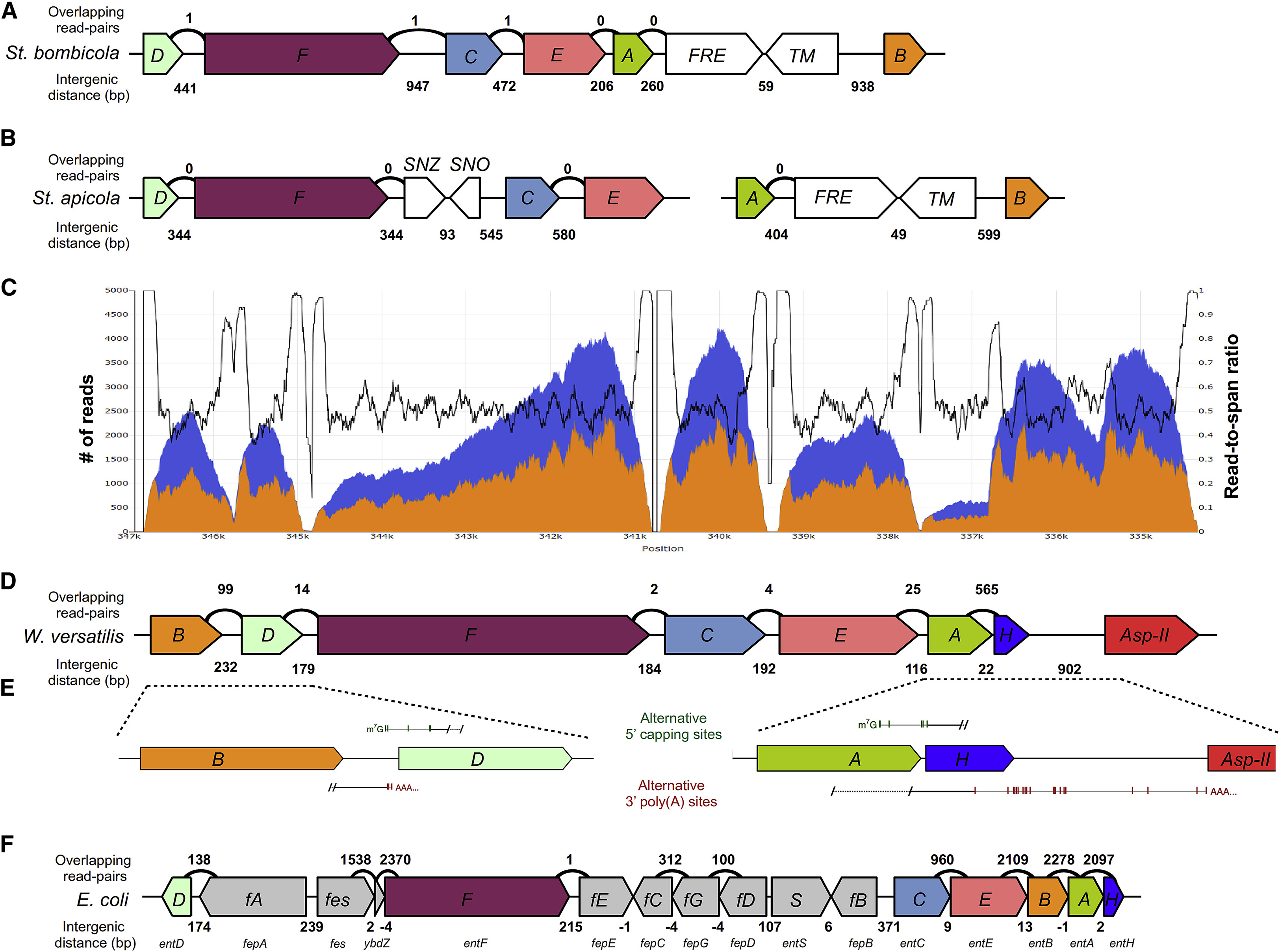
(A, B, D) Diagram of siderophore biosynthesis genes as present in the genomes of St. bombicola (A), St. apicola (B), and W. versatilis (D), drawn to scale. Counts above the diagram indicate read-pairs that map to both predicted protein-coding sequences (low, non-zero read counts are likely DNA contamination). Counts below indicate the size of intergenic regions between adjacent protein-coding sequences, in base pairs. (C) The orange area indicates per-base coverage by RNA-Seq reads (read coverage). The blue area indicates per-base cumulative coverage by RNA-Seq reads and inserts between read-pairs (span coverage). The black line indicates the ratio of the read coverage over the span coverage, which is expected to remain ~50% in the middle of gene transcripts and rise towards 100% at transcript termini. Thus, transcript boundaries are visualized as a coverage trough between two spikes that approach 100% ratios. Ratios below 100% at the putative 5’ or 3’ ends of annotated transcripts, coupled with non-zero coverage of their intergenic regions, suggest overlapping or potentially bicistronic transcripts. The expected 3’ coverage bias can be observed for individual transcripts in the raw coverage data. (E) Results of 5′ and 3′ RACE experiments, depicting the positions of all detected m7G caps (green vertical lines) and poly(A) tails (red vertical lines) in the entB-entD (left) and entA-entH (right) gene pairs in W. versatilis. The outer and inner gene-specific primers are marked by diagonal black lines and were used along with outer and inner primers specific to the 5’ or 3’ RACE adapters provided in the kit (see Materials and Methods), which were adjacent to either the 5’ m7G cap or the 3’ poly(A) tail, respectively. Dotted lines indicate sequences amplified only during the outer nested RACE PCR step, while solid lines indicate the portions of the transcripts that were amplified during the inner nested RACE PCR step, and which were subsequently cloned for sequencing (F) Diagram of siderophore biosynthesis genes as present in the E. coli genome drawn to scale. Counts above the diagram indicate read-pairs crossmapping between genes (based on data from Seo et al., 2014, complete coverage maps shown in Figure S6). Counts below indicate the size of intergenic regions between adjacent protein-coding sequences, in base pairs (negative numbers indicate overlap). The f prefix (fA-fG) indicates the fepA-fepG genes. See also Figure S2, Figure S3 and Table S3.
To further investigate operon-like characteristics that may have been retained, we searched our RNA-Seq data for read-pairs in which each paired read mapped to a different predicted protein-coding sequences. Since both reads of a read-pair originate from the same mRNA molecule, crossmapping of read-pairs could suggest the presence of multiple protein-coding genes on the same transcript. In all three species, most transcripts predicted to be involved in siderophore biosynthesis were clearly monocistronic and included poly(A) tails, as expected from eukaryotic-style gene expression (Figure 4, Figures S2–S4). We did not find any evidence suggesting that 5’ caps were added by trans-splicing (Doren and Hirsh, 1990) or by alternatively cis-splicing a common cassette exon upstream of each protein-coding region (Keren et al., 2010), common eukaryotic transcriptional processing mechanisms that could have produced monocistronic transcripts. Interestingly, W. versatilis produced substantial crossmapping reads for two gene pairs: entB-entD (232 bps apart) and entA-entH (22 bps apart) (Figure 4D). Previously reported yeast bicistronic transcripts have been attributed mainly to inefficiencies in the RNA transcription machinery (David et al., 2006; Pelechano et al., 2013), but given that the yeast ent transcripts encode functionally related steps of a single biosynthesis pathway originally encoded in a bacterial operon, we wondered whether they could have retained some polycistronic characteristics from their ancestry.
To test whether bicistronic transcripts of the siderophore biosynthesis genes were prevalent in W. versatilis, we used the RACE (Rapid Amplification of cDNA Ends) approach to sequence the 5’ and 3’ ends of mRNAs of W. versatilis genes from the two pairs that showed the strongest signal of crossmapping reads (Figure 4E). For the entB-entD gene pair, we found evidence of several overlapping mRNAs (i.e. multiple distinct 3’ ends of mRNAs containing the entB gene overlapped with multiple distinct 5’ ends of mRNAs containing the entD gene), but we did not detect any evidence of bicistronic entB-entD mRNAs. In the case of the entA-entH gene pair, all detected 5’ ends of entH mRNAs overlapped substantially with the detected 3’ ends of entA mRNAs. Several of the 3’ ends of entA mRNAs were also downstream of the predicted entH protein-coding sequence. Although these observations are consistent with the presence of some level of bicistronic entA-entH transcripts, they do not strictly require this interpretation. Indeed, several of the detected 5′ transcription start sites for entH (by both RACE and a large spike in RNA-Seq coverage) occurred within the protein-coding sequence of entA, and we never detected a 5’ transcription start site for entH upstream of entA by RACE. Since both the entA and entH genes express high levels of mRNAs where their own predicted translation start site would be the first one encountered downstream of their respective 5’ cap, the translation of multiple polypeptides from a single transcript is not required to explain the expression of either gene.
Thus, we conclude that, even in W. versatilis, the majority of these transcripts are transcribed and processed through conventional eukaryotic mechanisms that involve distinct promoters and polyadenylation sites for each gene. This densely packed locus produces mRNAs of considerable diversity, including overlapping transcripts, alternative transcription start and polyadenylation sites, transcription start sites within upstream protein-coding sequences, and anti-sense transcripts (Table S3). These observed transcriptional mechanisms suggest that the complexity and noisiness of eukaryotic transcription may have been key to allowing some amount of each gene product to have been expressed by the initially transferred operon.
Discussion:
The horizontal transfer of this siderophore biosynthesis operon has provided an exceptionally illuminating and clearly documented case of the acquisition of a complete multi-gene bacterial operon encoding a complex metabolic pathway by a eukaryotic nuclear genome in a single transfer event. Here we have used phylogenetic hypothesis testing to demonstrate that the ent operon was transferred from the Enterobacteriales lineage into an ancestor of the W/S clade of yeasts in a single event. As the lineage diversified, the operon subsequently underwent structural changes in the recipient eukaryotic genome, including increased intergenic spacing, insertion of eukaryotic genes, separation onto separate contigs, and even outright loss in some cases. We use transcriptomics and RACE to demonstrate that the operon genes are expressed in a manner consistent with canonical eukaryotic transcription, namely that transcripts contain poly(A) tails and are mainly monocistronic. Interestingly, in some cases, the genes evolved new eukaryotic promoters driving the expression of overlapping transcripts. Finally, we used the O-CAS assay, coupled with HPLC-MS/MS, to demonstrate that most species harboring the operon do indeed produce enterobactin, confirming that the operon is fully functional.
The previous scarcity of evidence for HOT into eukaryotes led authors to propose barriers due to pathway complexity (Husnik and McCutcheon, 2018; Wisecaver and Rokas, 2015; Wisecaver et al., 2016) and differences in core Central Dogma processes (Keeling and Palmer, 2008; Richards et al., 2011). Where could the transfer of the siderophore biosynthesis operon between Enterobacteriales and yeasts have occurred, and how could the bacterial operon have been functionally maintained in the yeasts’ genomes? Eukaryotes have been proposed to acquire bacterial genes through several mechanisms, including virus-aided transmission (Routh et al., 2012), environmental stress-induced DNA damage and repair (Flot et al., 2013; Gladyshev et al., 2008), and a phagocytosis-based gene ratchet (Doolittle 1998). The yeast species that harbor the siderophore biosynthesis operon have been isolated predominantly from insects (Lachance et al., 2001; Rosa and Lachance, 1998; Rosa et al., 2003), where stable bacterial and eukaryotic communities coexist inside their guts (Gilliam 1997). Moreover, this niche harbors diverse Enterobacteriales populations in which horizontal gene transfer has been reported (Watanabe and Sato, 1998; Watanabe et al., 1998), and insect guts have recently been described as a “mating nest” for yeasts (Stefanini et al., 2016). Since Enterobacteriales and yeasts can conjugate directly in some cases (Heinemann and Sprague, 1989) and the size of the genomic segment encoding the ent operon in Enterobacteriales (16kb) is within the range of other horizontal transfers reported in the literature, we propose that the last common ancestor of the W/S clade yeasts may have incorporated the operon from a bacterial co-inhabitant of an insect gut. Due to the intense competition for nutrients in this ecosystem, including a constant arms race with the host organism itself (Barber and Elde, 2015), yeasts capable of making their own siderophores and sequestering iron may have had a substantial advantage over those relying on siderophores produced by others. Whether due to ecological or molecular mechanisms, the W/S clade may be particularly prone to HGT since this clade was recently reported to have acquired multiple unlinked bacterial genes (Gonçalves et al., 2018), more than any other clade of known budding yeasts (Shen et al., 2018). Unlike some other HGT reports, the genes transferred into the W/S yeasts did not fuse into multi-domain proteins (Marcet-Houben and Gabaldón, 2010, Stairs et al., 2014, Tsaousis et al., 2012), nor did they gain any introns (Da Lage et al., 2013; Marcet-Houben and Gabaldón, 2010; Tsaousis et al., 2012). Since the donor bacterium lacked introns and budding yeasts are known to be depauperate of introns (Neuveglise et al., 2011; Hooks et al., 2014; Dujon et al., 2017), there seems to have been little evolutionary pressure or insufficient genetic drift to acquire them post-transfer.
Given the fundamental differences between bacterial and eukaryotic gene regulation, how could a bacterial operon have been maintained in a eukaryotic genome upon transfer? If it had not been actively expressed and functional, the genes of the operon would have been rapidly lost from the genome through neutral evolutionary processes. Although eukaryotes do not encode proteins with significant similarity to the negative regulator Fur which controls the expression of the bacterial ent genes, their iron response is similarly governed by transcription factors that also belong to the GATA family, such as Fep1 in S. pombe, SreA in A. nidulans, or Sfu1 in C. albicans. Indeed, the consensus Fur-binding site (5’-_GATAAT_-3’) is remarkably similar to that of the fungal Iron-Responsive GATA Factors (IRGFs, 5’-_WGATAA_-3’) (Chen et al., 2007; Haas et al., 2008). This similarity suggests the intriguing possibility that the siderophore genes could have readily switched from being regulated by a bacterial transcription factor to a eukaryotic transcription factor, at least for the most 5’ promoter. While not a necessary element for successful expression of the ent genes (since both Fur and IRGFs are negative regulators), it could have allowed expression of the transferred genes to be environmentally responsive upon transfer. In addition to containing an enrichment of DNA sequence motifs similar to IRGF binding sites, sequences upstream of the ent genes contain other enriched motifs that could be bound by transcriptional activators (Table S3). Determining whether any of these motifs or any other binding sites regulate the expression of the ent genes in W/S clade yeasts will require more detailed dissection.
Siderophores are highly potent chelators that can efficiently sequester iron at very low concentrations (Boukhalfa and Crumbliss, 2002), so even a low basal expression level of the newly acquired genes, such as from cryptic promoters within upstream protein-coding sequences, could have been enough to convey a considerable selective advantage. This initial eukaryotic expression, perhaps aided by inherently noisy transcriptional and translation processes that include leaky scanning and internal ribosome entry sites (IRESs), could then have been optimized by acquiring more eukaryotic characteristics, such as longer intergenic regions that were gradually refined into improved promoters, distinct polyadenylation sites, and a shift from polycistronic or overlapping transcripts to mainly monocistronic and non-overlapping transcripts. The incorporation of a eukaryotic gene encoding a ferric reductase would have further improved the efficiency of iron acquisition in the highly competitive ecological niche of insect guts, while enhancing the eukaryotic characteristics of the gene cluster. Our HOT finding dramatically expands the boundaries of cross-domain gene flow. The transfer, maintenance, expression, and adaptation of a multi-gene bacterial operon to a eukaryotic host underscore the flexibility of eukaryotic transcriptional and translational systems to produce adaptive changes from novel and unexpected sources of genetic information.
STAR Methods:
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Chris Todd Hittinger (cthittinger@wisc.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Yeast strains were obtained from the USDA Agricultural Research Service (ARS) NRRL Culture Collection in Peoria, Illinois, USA. All sequenced strains have been publicly deposited in the NRRL or CBS. All yeast strains were struck for single colonies from a glycerol freezer stock to YPD (1% w/v yeast extract, 2% w/v peptone, 2% w/v dextrose) agar plates and grown at either room temperature or 30°C until visible colonies formed. Culturing conditions prior to RNA isolation and HPLC sample preparation can be found in the relevant Method Details sections. Strain identifiers and other information for each yeast species can be found in either the STAR Methods Key Resources Table or Table S1.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| E. cloni 10G Chemically Competent Cells | Lucigen | Cat#60107–2 |
| Escherichia coli MG1655 | Blattner et al., 1997 | ATCC: 700926 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Enterobactin | Sigma Aldrich | Cat#E3910 |
| Chromeazurol S | MP Biomedicals, LLC | Cat#154982 |
| Ferric chloride hexahydrate | VWR | Cat#0682–500G |
| Hydrochloric acid | J.T. Baker | Cat#9535–05 |
| Hexadecyltrimethylammonium bromide | Sigma Aldrich | Cat#H6269 |
| Phusion High-Fidelity DNA Polymerase | New England Biolabs | Cat#M0530S |
| Taq DNA Polymerase with Standard Taq Buffer | New England Biolabs | Cat#M0273 |
| Exonuclease I | New England Biolabs | Cat#M0293L |
| Antarctic Phosphatase | New England Biolabs | Cat#M0289L |
| 25:24:1 Phenol:Chloroform:Isoamyl Alcohol | Sigma Aldrich | Cat#P2069 |
| RNase A | VWR Life Science | Cat##97064–064 |
| Critical Commercial Assays | ||
| RNA Clean & Concentrator Kit | Zymo Research | Cat#R1017;Cat#R1018 |
| Qubit RNA Assay Kit | Thermo Fisher | Cat#Q32852 |
| Qubit dsDNA Assay Kit | Thermo Fisher | Cat#Q32853 |
| NEBNext Poly(A) mRNA Magnetic Isolation Module | New England Biolabs | Cat#E7490 |
| NEBNext Ultra Directional RNA Library Prep Kit | New England Biolabs | Cat#E7420 |
| FirstChoice RLM-RACE Kit | Invitrogen | Cat#AM1700 |
| Zymoclean Gel DNA Recovery Kit | Zymo Research | Cat#D4002 |
| pCR-Blunt II-TOPO Kit | Thermo Fisher | Cat#450031 |
| BigDye Terminator Cycle Sequencing Kit | Applied Biosystems | Cat#4337455 |
| ZR Plasmid Miniprep | Zymo Research | Cat#D4016 |
| AxyPrep Mag DyeClean Beads | Fisher Scientific | Cat#14-223-163 |
| NEBNext Ultra DNA Library Prep Kit | New England Biolabs | Cat#E7370L |
| AxyPrep Mag PCR Beads | Fisher Scientific | Cat#14-223-152 |
| Deposited Data | ||
| Raw DNA reads, raw RNA reads and whole genome assembly data | This paper | BioProject: PRJNA396763 |
| Experimental Models: Organisms/Strains | ||
| Saccharomyces cerevisiae | Hittinger and Carroll, 2007 | FM1282 |
| Kluyveromyces lactis | CBS | CBS 2359 |
| Tortispora caseinolytica | NRRL | NRRL Y-17796T |
| Yarrowia keelungensis | NRRL | NRRL Y-63742T |
| Yarrowia deformans | NRRL | NRRL Y-321T |
| Yarrowia lipolytica | NRRL | NRRL YB-423T |
| Blastobotrys adeninivorans | NRRL | NRRL Y-17592 |
| Sugiyamaella lignohabitans | NRRL | NRRL YB-1473T |
| Wickerhamiella (Candida) infanticola | NRRL | NRRL Y-17858T |
| Wickerhamiella (Candida) hasegawae | JCM | JCM 12559T |
| Wickerhamiella (Candida) pararugosa | NRRL | NRRL Y-17089T |
| Wickerhamiella cacticola | NRRL | NRRL Y-27362T |
| Wickerhamiella occidentalis | NRRL | NRRL Y-27364 |
| Wickerhamiella (Candida) versatilis | NRRL | NRRL Y-6652T |
| Wickerhamiella domercqiae | NRRL | NRRL Y-6692T |
| Starmerella (Candida) gropengiesseri | NRRL | NRRL Y-17142T |
| Starmerella (Candida) tilneyi | CBS | CBS 8794T |
| Starmerella (Candida) sorbosivorans | CBS | CBS 8768T |
| Starmerella (Candida) geochares | NRRL | NRRL Y-17073T |
| Starmerella (Candida) vaccinii | NRRL | NRRL Y-17684T |
| Starmerella (Candida) davenportii | CBS | CBS 9069T |
| Starmerella (Candida) ratchasimensis | CBS | CBS 10611T |
| Starmerella (Candida) apicola | NRRL | NRRL Y-2481T |
| Starmerella (Candida) riodocensis | NRRL | NRRL Y-27859T |
| Starmerella (Candida) kuoi | NRRL | NRRL Y-27208T |
| Starmerella bombicola | NRRL | NRRL Y-17069T |
| Lipomyces starkeyi | NRRL | NRRL Y-11557T |
| Oligonucleotides | ||
| NEBNext Multiplex Oligos for Illumina | New England Biolabs | Cat#E7335, E7500 |
| M13 Forward (−20) and Reverse primers | Included in pCR-Blunt II-TOPO Kit | N/A |
| RACE Primers | This study | See Table S3 |
| Software and Algorithms | ||
| BLAST+ suite v2.2.28 | Altschul et al., 1990 | ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/ |
| iWGS v1.01 | Zhou et al., 2016 | https://github.com/zhouxiaofan1983/iWGS |
| Quast v4.4 | Gurevich et al., 2013 | https://github.com/ablab/quast |
| Maker v2.31.8 | Holt and Yandell, 2011 | http://yandell-lab.org/software/maker.html |
| GeneMark-ES v4.10 | Ter-Hovhannisyan et al., 2008 | http://topaz.gatech.edu/GeneMark/license_download.cgi |
| Augustus v3.2.1 | Stanke et al., 2008 | http://bioinf.unigreifswald.de/augustus/downloads/ |
| SNAP release 2006-07-28 | Korf 2004 | https://github.com/KorfLab/SNAP |
| BUSCO v3 | Simão et al., 2015 | https://busco.ezlab.org/ |
| MAFFT v7 | Katoh and Standley, 2013 | https://mafft.cbrc.jp/alignment/software/ |
| RAxML v8 | Stamatakis 2014 | https://github.com/stamatak/standard-RAxML |
| ExaML v3.0.18 | Kozlov et al., 2015 | https://github.com/stamatak/ExaML |
| IQTREE v1.5.4 | Nguyen et al., 2015 | http://www.iqtree.org/ |
| GSNAP in GMAP release date 2017-05-08 | Wu and Nacu, 2010 | http://research-pub.gene.com/gmap/ |
| Trinity v2.4.0 | Grabherr et al., 2011 | https://github.com/trinityrnaseq/trinityrnaseq/wiki |
| StringTie v1.3.3b | Pertea et al., 2015 | https://github.com/gpertea/stringtie |
| MEME and TOMTOM in MEME suite v5.0.3 | Bailey et al., 2009 | http://meme-suite.org/doc/download.html |
| Other | ||
| 425–600 μη Glass beads, acid-washed | Sigma Aldrich | Cat#G8772–1KG |
| E220evolution Focused-ultrasonicator | Covaris Inc. | N/A |
METHOD DETAILS
Genome sequencing
Genomic DNA (gDNA) isolation and Illumina library prep was done as described previously (Shen et al., 2018). Briefly, cells were grown to saturation in YPD broth, collected by centrifugation with approximately 500 μL 0.5 mm acid-washed beads (Sigma #G8772), and resuspended in DNA lysis buffer (10 mM Tris, 1 mM EDTA, 100 mM NaCl, 1% SDS, 2% Triton X-100 in water). Then, samples were extracted twice with 25:24:1 phenol:chloroform:isoamyl alcohol (Sigma #P2069), precipitated overnight at −80°C in 100% ethanol, collected by centrifugation, washed twice with 70% ethanol, dissolved in 10 mM Tris-Cl (pH 8), and treated with RNase A (VWR #97064–064) for 30 minutes at 37°C. gDNA was then sonicated using a (Covaris E220 Focused-ultrasonicator); followed by end-repair, adapter ligation and size selection with the NEBNext Ultra DNA Library Prep kit (NEB #E7370L), which was performed according to the manufacturer’s protocol except that Axygen AxyPrep Mag PCR beads (Fisher Sci #14-223-152) were used instead of Beckman Coulter AMPure XP beads. Libraries were then submitted for 2×250 bp sequencing on an Illumina HiSeq 2500 instrument.
RNA sequencing
Cells were grown in quadruplicates for either 3 or 6 days on YPD agar, and RNA was extracted using the hot acid phenol protocol (Chomczynski and Sacchi, 1987). Briefly, cells were scraped off of agar plates and flash frozen in a dry ice-ethanol bath. Then, cells were resuspended in TES buffer (10 mM Tris, 10 mM EDTA, 0.5% SDS in water), and added to one volume of 5:1 acid phenol:chloroform. Lysates were then incubated at 70°C for one hour with vortexing every 15 minutes. Then, lysates were extracted twice with one volume of 5:1 phenol:chloroform each and once with chloroform. The aqueous phase of the final chloroform extraction was added to a solution consisiting of 2.5 volumes of 95–100% ethanol and 0.1 volumes of 3 M sodium acetate and was placed at −80°C overnight to precipitate the RNA. RNA pellets were then collected by centrifugation, washed twice with 1 volume of 70% ethanol each, and resuspended in RNase-free water. Purified RNA was then treated with DNase to remove any residual DNA prior to treatment with the RNA Clean & Concentrator kit (Zymo Research #R1017, R1018). Total RNA yields were quantified with the Qubit RNA Assay Kit (Thermo Fisher). Next, mRNA was isolated using the NEBNext Poly(A) mRNA Magnetic Isolation Module (NEB #E7490) and prepared into strand-specific Illumina libraries using the NEBNext Ultra Directional RNA Library Prep Kit (NEB #E7420) and the NEBNext Multiplex Oligos for Illumina (New England Biolabs #E7335, E7500). Library quality was assessed by gel electrophoresis and with the Qubit dsDNA Kit (Thermo Fisher) prior to submission for 2×125 paired-end sequencing with an Illumina HiSeq 2500 instrument.
Mapping transcript ends by 3’ and 5’ RLM-RACE
5’ RNA Ligase-Mediated Rapid Amplification of cDNA Ends (RLM-RACE) and 3’ RACE were carried out using the FirstChoice RLM-RACE Kit (Invitrogen #AM1700), as directed in the kit instructions. Gene-specific outer and inner nested primers used to amplify RACE products were designed to match the annealing temperatures of the RACE outer and inner PCR primers, respectively, with minimal hairpin, self-dimer, and hetero-dimer affinities (Table S3). RACE PCR master mixes were assembled as directed in the kit manual, except we used 1.25 U of Phusion High-Fidelity DNA Polymerase (NEB #M0530S) with HF Buffer. Cycling for nested RACE PCR was carried out under the following conditions: 98°C for 1:00 min; then 35 cycles of 98°C for 10 sec, primer-specific annealing temperature for 30 sec (temperatures indicated in Table S3), and 72°C for 2:30 min; followed by a final extension at 72°C for 5:00 min, and hold at 15°C.
Cloning and confirmation of RACE products
Products from the inner nested RACE PCR were run on a 1% agarose gel and visualized by staining with ethidium bromide. Bands corresponding to distinct RACE PCR products were cut out, extracted with the Zymoclean Gel DNA Recovery Kit (Zymo Research #D4002), and cloned by TOPO blunt-end cloning into pCR-Blunt II-TOPO (Thermo Fisher #450031). The resulting plasmids were transformed into E. coli 10G Chemically Competent Cells (Lucigen #60107–2) and plated onto LB Agar with 50 μg/mL kanamycin. Resulting colonies were inoculated into LB Broth with 50 μg/mL kanamycin sulfate to incubate overnight at 37°C. Their plasmid inserts were then Sanger sequenced by BigDye Terminator Cycle Sequencing (Applied Biosystems) using the M13 forward (−20) or reverse primers, either by miniprep extraction (Zymo Research #D4016) or by colony PCR followed by treatment with Exonuclease I/Antarctic Phosphatase (NEB #M0293L, #M0289L). Excess dideoxy-terminators were removed using AxyPrep Mag DyeClean beads (Fisher Scientific #14-223-163). Colony PCR was conducted with Taq DNA Polymerase with Standard Taq Buffer (NEB #M0273) as follows: 10:00 min at 95°C; then 35 cycles of 95°C for 30 sec, 44°C for 30 sec, and 68°C for 1:00 min; followed by a final extension of 68°C for 5:00 min, and hold at 15°C.
Microbial culturing and O-CAS assays
Low-iron synthetic complete (SC) medium consisted of 5 g/L ammonium sulfate, 1.7 g/L Yeast Nitrogen Base (without amino acids, carbohydrates, ammonium sulfate, ferric chloride, or cupric sulfate), 2 g/L complete dropout mix, 2% dextrose (added after autoclaving), and 200 nM cupric sulfate. M9 minimal medium consisted of 0.4% glucose, 2 mM magnesium sulfate, 100 μM calcium dichloride, and 1x M9 salts (added as a 5x stock solution consisting of 64g/L dibasic sodium phosphate heptahydrate, 15 g/L monobasic potassium phosphate, 2.5 g/L sodium chloride, and 5 g/L ammonium chloride in deionized water).
The chromeazurol S overlay (O-CAS) assay was carried out as previously described (Pérez-Miranda et al., 2007), with some modifications. Specifically, 10X CAS Blue Dye was made by combining the following: 50 mL Solution 1 (60 mg chromeazurol S dissolved in 50 mL deionized H2O), 9 mL Solution 2 (13.5 mg ferric chloride hexahydrate dissolved in 50 mL 10 mM hydrochloric acid), and 40 mL Solution 3 (73 mg hexadecyltrimethylammonium bromide (HDTMA) in 40 mL deionized H2O). Separately, 15.12 g PIPES (free acid) was added to 425 mL deionized water and adjusted to a pH of approximately 6.8 with 2.46 g sodium hydroxide. 4.5 g agarose was added as a solidifying agent, and the resulting solution was brought up to 450 mL with deionized water in a 1-L Erlenmeyer flask. To make the CAS overlay, the agarose-PIPES solution was heated to melt the agarose and added in a ratio of 9:1 to 10X CAS Blue Dye, and 6 mL of the resulting O-CAS solution were overlaid onto low-iron SC plates.
Yeast strains were grown to saturation in 3 mL YPD medium at 30°C on a rotating culture wheel, centrifuged at 3000 rpm for 5 minutes to collect the cells, and resuspended in 3 mL deionized water. A volume of 5 μL of the resulting cell suspension was spotted onto 60 mm diameter plates containing low-iron SC medium using agarose (1% w/v) as a gelling agent and incubated at 30°C for 7 days before adding 6 mL of O-CAS solution. E. coli cells were grown overnight in M9 minimal medium at 37°C, and 5 μL of culture was spotted onto low-iron SC plates that had already been overlaid with 6 mL of O-CAS solution and allowed to dry for at least 1 hour. Pictures of yeast colonies were taken 2 days after the O-CAS was poured, while E. coli colonies were photographed 5 days after the O-CAS was poured. With exposure and focus lock enabled, pictures were taken of the plates set on top of a miniature white light trans-illuminator placed under a gel-imaging dark box.
HPLC analysis of yeast culture extracts
Yeasts were grown on YPD plates until colonies formed; a colony was inoculated into low-iron SC medium with either 2% dextrose or 2% glycerol as a carbon source, as indicated (Table S4), and the resulting culture was incubated at 30°C for either 3 or 6 days, as indicated. The spent medium was then centrifuged, and the supernatant was filtered through a 0.2 μm nylon filter directly into a 2 mL LC vial. The filtered samples were analyzed for the presence of enterobactin on a Shimadzu LCMS8040 using 50 μL injections of the filtered yeast culture. Sample to sample carryover was minimized by rinsing the needle assembly and using two blank injections of 10 μL isopropanol between samples. The mobile phase was a binary gradient of acetonitrile and water, pumped at 0.7 mL/min through a Phemonenex Kinetex 5μ XB-C18 column (P/N: 00G-4605-E0, 100 Å, 250′4.6 mm) and heated at 60°C.
The mobile phase was held at 0% acetonitrile for 0.2 minutes, then ramped to 100% acetonitrile over 3.05 minutes, and held there for 2.25 minutes to wash the column. The mobile phase was then ramped back to 0% acetonitrile over 0.5 minutes and held at 0% acetonitrile for 4 minutes to reset the column’s initial condition. The eluent was diverted to waste for the first 2 minutes to minimize salt buildup on the ion source. After 2 minutes, the eluent flowed through a PDA detector scanning from 190–400 nm and into the MS ionization source operating in DUIS (ESI/APCI) mode with 2.5 L/min nebulizing gas, 20 L/min drying gas, 300°C DL temperature, and 400°C heat block. The MS scanned the ions in negative mode from 200–1000 m/z, and monitored three MRM transitions (668→178, 668→222, and 668→445, Table S4) with argon collision gas at 230 kPa. The retention time and relative intensities of the three MRM transitions were determined using an enterobactin standard (Sigma Aldrich #E3910, Figure S4). The results of the yeast strain screening are presented in Figure 3 and Table S4.
QUANTIFICATION AND STATISTICAL ANALYSIS
Identification of iron metabolism genes
Amino acid sequences of proteins known to be involved in iron uptake and storage were used as BLASTP and TBLASTN v2.2.28+ (Altschul et al., 1990) queries against genomes and proteomes of a broad range of dikaryon fungal species (see Table S1). The genomic data was obtained from GenBank, as well as from draft genome assemblies generated for 20 strains by the RIKEN BioResource Center and RIKEN Center for Life Science Technologies through the Genome Information Upgrading Program of the National Bio-Resource Project of the MEXT. S. cerevisiae homologs were used, except for: the fungal hydroxamate-class siderophore biosynthesis proteins, which came from A. nidulans; the bacterial catecholate-class siderophore biosynthesis proteins, which came from E. coli; and the iron-responsive GATA factor sequences, which came from A. nidulans (SreA), Ustilago maydis (Urbs1), Phanerochate chrysosporium (SreP), Neurospora crassa (Sre), Candida albicans (Sfu1), and Schizosaccharomyces pombe (Fep1). Identification of entA-entF genes in bacterial genomes was performed using E. coli protein sequences as queries for BLASTP and TBLASTN to search 1,382 Enterobacteriales genomes and proteomes downloaded from GenBank. Only genes from the 207 genomes where all six genes could be identified at E-value cutoff of 1E-10 were considered for further phylogenetic analyses.
Genome assembly and annotation
To generate whole-genome assemblies, Illumina reads were used as input to the meta-assembler pipeline iWGS v1.01 (Zhou et al., 2016). Briefly, this pipeline performed quality-based read trimming, followed by k-mer length optimization, and used a range of state-of-the-art assemblers to generate multiple genome assemblies. Assembly quality was assessed using QUAST v4.4 (Gurevich et al., 2013), and the best assembly for each species was chosen based on the N50 statistic. ORFs were annotated in genomes using the MAKER pipeline v2 (Holt and Yandell, 2011) and the GeneMark-ES v4.10 (Ter-Hovhannisyan et al., 2008), Augustus v3.2.1 (Stanke et al., 2008), and SNAP (release 2006-07-28) (Korf 2004) gene predictors.
Phylogenetic reconstruction and tests
The species phylogeny was obtained by analyzing conserved single-copy fungal orthologs by using a previously described phylogenomic approach (Shen et al., 2016). Briefly, sequences of conserved, single-copy orthologous genes were identified in the genome assemblies using the BUSCO v3 software (Simão et al., 2015), single-copy BUSCO genes shared by at least 80% of species were aligned using MAFFT v7 (Katoh and Standley, 2013), and these orthologs were used for maximum-likelihood phylogenetic reconstruction with RAxML v8 (Stamatakis 2014). The reconstruction was performed under the LG model of amino acid substitution (Le and Gascuel, 2008) with empirical amino acid frequencies, four gamma distribution rate categories to estimate rate heterogeneity, and 100 rapid bootstrap pseudoreplicates. A concatenated super-alignment of all genes was also used for phylogenetic reconstruction by running ExaML v3.0.18 (Kozlov et al., 2015) under the JTT substitution matrix (chosen by the built-in maximum-likelihood model selection), per-site rate heterogeneity model with median approximation of the GAMMA rates, and with memory saving option for gappy alignments turned on. Constrained phylogeny reconstructions were conducted in RAxML through the “-g” option, and the AU and SH topology tests were performed with IQ-TREE v1.5.4 (Nguyen et al., 2015) using 10,000 bootstrap pseudoreplicates.
Three evolutionary scenarios were considered to explain the course of the horizontal transfer event: (I) single-source, single-target; (II) single-source, multiple-targets; and (III) multiple-sources. Each of them predicted specific phylogenetic patterns. Scenario I predicted that the yeast sequences would form a strongly supported monophyletic group with a consistent internal topology. Scenario II predicted that yeast sequences would form a strongly supported monophyletic group but not follow a consistent internal topology. Scenario III predicted that yeast sequences would not form a monophyletic group. To first establish whether there was a single (I & II) or multiple donor species (III), we determined whether the yeast sequences created a strongly supported monophyletic group within the Enterobacteriales phylogeny. The monophyletic base of the yeast clade had full bootstrap support, but we used the AU test again to compare the phylogeny constrained so that all 12 yeast taxa formed a separate monophyletic group versus 12 alternative phylogenies constrained so that 11 yeast taxa were monophyletic, and the remaining single taxon could be placed freely on the tree. For all six genes, every phylogeny tested was within the 95% confidence interval of the best-supported phylogeny and thus showed no statistical difference, strongly supporting the single origin scenario (Figure 2D, Table S2). To determine whether the transfer happened into an ancestor of the W/S clade species studied (scenario I) or into various yeast lineages independently (scenario II), we looked at the topology of the yeast clade. Under the first scenario, yeast sequences would recapitulate a consistent, well-supported phylogeny, whereas under the second scenario, the phylogenetic signal would be weak, and the topology would be poorly supported. Bootstrap supports at the internal nodes in the five- and six-gene phylogenies were high (Figure 2A, Figure 2B), which indicated consistent phylogenetic signal in the alignments. We next compared the unconstrained ML phylogenies versus phylogenies constrained so that the yeast sequences follow the topology of the five-gene concatenation (5G), and the AU test again showed that there was no statistically significant difference between them, which we took as indication that the transfer most likely happened from a single ancestral Enterobacteriales lineage into the ancestor of the 12 yeast taxa (Figure 2D, Table S2). SH tests had less statistical power but produced fully concordant results with the same constraints (Table S2).
RNA-Seq analysis
Reads were mapped to their respective genome assemblies using GSNAP (Wu and Nacu, 2010) from the GMAP package (release date 2017-05-08) with the novel splicing site search option enabled. De novo transcriptome assembly was performed using the Trinity pipeline v2.4.0 (Grabherr et al., 2011), which was run in the RF strand-specific mode with the jaccard-clip option enabled. Transcript abundances of siderophore biosynthesis genes were estimated using StringTie v1.3.3b (Pertea et al., 2015).
Evidence of transcriptional processing was evaluated by inspecting parts of the RNA-Seq reads that were soft-clipped from the ends of reads during the mapping step. 3’ ends were inspected for evidence of poly(A) tails of at least three consecutive As or Ts, which were not encoded in the genome. The power of such analysis is limited by the fact that only small fraction of reads (~0.05%) are expected to be initiated using the (A)6 or (T)6 primers, which increases the rate of false negative results, but true positive results remain unaffected. With this caveat, we note that evidence of poly(A) tails was not detected from the W. versatilis entE, entA, and entH genes. 5’ ends were inspected for presence of common sequences, encoded elsewhere in the genome, which could have been indicative of splicing leaders (in case of trans-splicing) or cassette exons (in case of alternative cis-splicing).
Identification of enriched motifs in promoters
Motifs enriched in the promoter regions of the 12 species that harbor the siderophore biosynthesis genes were identified and analyzed using the MEME-suite v5.0.3 (Bailey et al., 2009). Sequences of 100, 200, 300, 400, and 500 nucleotides upstream from the start codon of each ent gene were extracted and searched for statistically enriched DNA motifs of 10, 15, or 20 bases in length using the program MEME. The program was run on both the sense and antisense strands (option “-revcomp”), looking for any number of motif repetitions (option “-mod anr”), extracting up to 20 motifs (option “-nmotifs 20”) or up to the E-value of 1e-3 (option “-evt 1e3”). Identified motifs were then queried against the YEASTRACT database of experimentally determined transcription factor binding sites using the program TOMTOM, run with default options.
HPLC analysis of yeast culture extracts
Yeast strains were determined to be positive for the production of enterobactin by HPLC-MS/MS if the chromatograms from at least four of the six biological cultures (see Table S4) were found to have an enterobactin peak (correct retention time, all three MRM transitions with relative intensities of 30% the authentic standard, and > 5:1 signal-to-noise in the MRM chromatogram of the 668→178 transition).
DATA AND SOFTWARE AVAILABILITY
Raw DNA and RNA sequencing data were deposited in GenBank under Bioproject ID PRJNA396763 (www.ncbi.nlm.nih.gov/bioproject/PRJNA396763). Whole Genome Shotgun assemblies have been deposited at DDBJ/ENA/GenBank under the accessions NRDR00000000-NREI00000000. Genome-specific accessions are listed in Table S1.
KEY RESOURCES TABLE
The table highlights the genetically modified organisms and strains, cell lines, reagents, software, and source data essential to reproduce results presented in the manuscript. Depending on the nature of the study, this may include standard laboratory materials (i.e., food chow for metabolism studies), but the Table is not meant to be comprehensive list of all materials and resources used (e.g., essential chemicals such as SDS, sucrose, or standard culture media don’t need to be listed in the Table). Items in the Table must also be reported in the Method Details section within the context of their use.The number of primers and RNA sequences that may be listed in the Table is restricted to no more than ten each. If there are more than ten primers or RNA sequences to report, please provide this information as a supplementary document and reference this file (e.g., See Table S1 for XX) in the Key Resources Table.
Please note that ALL references cited in the Key Resources Table must be included in the References list. Please report the information as follows:
- REAGENT or RESOURCE: Provide full descriptive name of the item so that it can be identified and linked with its description in the manuscript (e.g., provide version number for software, host source for antibody, strain name). In the Experimental Models section, please include all models used in the paper and describe each line/strain as: model organism: name used for strain/line in paper: genotype. (i.e., Mouse: OXTRfl/fl: B6.129(SJL)-Oxtrtm1.1Wsy/J). In the Biological Samples section, please list all samples obtained from commercial sources or biological repositories. Please note that software mentioned in the Methods Details or Data and Software Availability section needs to be also included in the table. See the sample Table at the end of this document for examples of how to report reagents.
- SOURCE: Report the company, manufacturer, or individual that provided the item or where the item can obtained (e.g., stock center or repository). For materials distributed by Addgene, please cite the article describing the plasmid and include “Addgene” as part of the identifier. If an item is from another lab, please include the name of the principal investigator and a citation if it has been previously published. If the material is being reported for the first time in the current paper, please indicate as “this paper.” For software, please provide the company name if it is commercially available or cite the paper in which it has been initially described.
- IDENTIFIER: Include catalog numbers (entered in the column as “Cat#” followed by the number, e.g., Cat#3879S). Where available, please include unique entities such as RRIDs, Model Organism Database numbers, accession numbers, and PDB or CAS IDs. For antibodies, if applicable and available, please also include the lot number or clone identity. For software or data resources, please include the URL where the resource can be downloaded. Please ensure accuracy of the identifiers, as they are essential for generation of hyperlinks to external sources when available. Please see the Elsevier list of Data Repositories with automated bidirectional linking for details. When listing more than one identifier for the same item, use semicolons to separate them (e.g. Cat#3879S; RRID: AB_2255011). If an identifier is not available, please enter “N/A” in the column.
- A NOTE ABOUT RRIDs: We highly recommend using RRIDs as the identifier (in particular for antibodies and organisms, but also for software tools and databases). For more details on how to obtain or generate an RRID for existing or newly generated resources, please visit the RII or search for RRIDs.
Please use the empty table that follows to organize the information in the sections defined by the subheading, skipping sections not relevant to your study. Please do not add subheadings. To add a row, place the cursor at the end of the row above where you would like to add the row, just outside the right border of the table. Then press the ENTER key to add the row. Please delete empty rows. Each entry must be on a separate row; do not list multiple items in a single table cell. Please see the sample table at the end of this document for examples of how reagents should be cited.
TABLE FOR AUTHOR TO COMPLETE
Please upload the completed table as a separate document. Please do not add subheadings to the Key Resources Table. If you wish to make an entry that does not fall into one of the subheadings below, please contact your handling editor. (NOTE: For authors publishing in Current Biology, please note that references within the KRT should be in numbered style, rather than Harvard.)
Supplementary Material
Supplemental Table 3
Supplemental Table 1
Supplemental Table 2
Supplemental Table 4
1
Acknowledgments:
We thank David J. Eide and Michael D. Bucci for advice on low-iron media; Nicole T. Perna and Jeremy D. Glasner for E. coli strain MG1655; Michael G. Thomas, John Ralph and the Eide, Perna, Rokas, and Hittinger labs for comments and discussions; RIKEN for publicly releasing 20 genome sequences prior to publication; Lucigen Corporation (Middleton, WI) for use of their Covaris for gDNA sonication; and the University of Wisconsin Biotechnology Center DNA Sequencing Facility for providing Illumina sequencing facilities and services. This work was conducted in part using the resources of the Wisconsin Energy Institute, the Center for High-Throughput Computing at the University of Wisconsin-Madison, and the UW Biotechnology Center DNA Sequencing Facility. This material is based upon work supported by the National Science Foundation under Grant Nos. DEB-1442113 (to A.R.) and DEB-1442148 (to C.T.H. and C.P.K.), in part by the DOE Great Lakes Bioenergy Research Center (DOE BER Office of Science DE-SC0018409 and DE-FC0207ER64494 to Timothy J. Donohue), the USDA National Institute of Food and Agriculture (Hatch Project 1003258 to C.T.H.), and National Institutes of Health (NIAID AI105619 to A.R.), and a Guggenheim Fellowship (to A.R). C.T.H. is a Pew Scholar in the Biomedical Sciences and a Vilas Early Career Investigator, supported by the Pew Charitable Trusts and Vilas Trust Estate, respectively. D.T.D. was supported by a NHGRI training grant to the Genomic Sciences Training Program (5T32HG002760). Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture. USDA is an equal opportunity provider and employer.
Footnotes
Declaration of Interests:
The authors declare no competing interests.
References:
- Adeolu M, Alnajar S, Naushad S, Gupta RS (2016). Genome-based phylogeny and taxonomy of the “_Enterobacteriales_”: proposal for Enterobacterales ord. nov. divided into the families Enterobacteriaceae, Erwiniaceae fam. nov., Pectobacteriaceae fam. nov., Yersiniaceae fam. nov., Hafniaceae fam. nov., Morganellaceae fam. nov., and Budviciaceae fam. nov. Int. J. Syst. Evol. Microbiol 66, 5575–5599. [DOI] [PubMed] [Google Scholar]
- Alexander WG, Wisecaver JH, Rokas A, Hittinger CT (2016). Horizontally acquired genes in early-diverging pathogenic fungi enable the use of host nucleosides and nucleotides. Proc. Natl. Acad. Sci. U. S. A 113, 4116–4121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990). Basic local alignment search tool. J. Mol. Biol 215, 403–410. [DOI] [PubMed] [Google Scholar]
- Andrews SC, Robinson AK, Rodríguez-Quiñones F (2003). Bacterial iron homeostasis. FEMS Microbiol. Rev 27, 215–237. [DOI] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS (2009). MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res, 37(suppl_2), W202–W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber MF, Elde NC (2015). Buried Treasure: Evolutionary Perspectives on Microbial Iron Piracy. Trends Genet. 31, 627–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumler DJ, Ma B, Reed JL, Perna NT (2013). Inferring ancient metabolism using ancestral core metabolic models of enterobacteria. BMC Syst. Biol 7, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blattner FR, Plunkett G, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, et al. (1997). The Complete Genome Sequence of Escherichia coli K-12. Science 277, 1453–1462. [DOI] [PubMed] [Google Scholar]
- Blumenthal T, Gleason KS (2003). Caenorhabditis elegans operons: form and function. Nat. Rev. Genet 4, 112–120. [DOI] [PubMed] [Google Scholar]
- Boukhalfa H, Crumbliss AL (2002). Chemical aspects of siderophore mediated iron transport. Biometals. 15, 325–339. [DOI] [PubMed] [Google Scholar]
- Campbell MA, Staats M, van Kan JAL, Rokas A, Slot JC (2013). Repeated loss of an anciently horizontally transferred gene cluster in Botrytis. Mycologia. 105, 1126–1134. [DOI] [PubMed] [Google Scholar]
- Chen Z, Lewis KA, Shultzaberger RK, Lyakhov IG, Zheng M, Doan B, Storz G, Schneider TD (2007). Discovery of Fur binding site clusters in Escherichia coli by information theory models. Nucleic Acids Res. 35, 6762–6777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N (1987). Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem 162, 156–159. [DOI] [PubMed] [Google Scholar]
- Da Lage JL, Binder M, Hua-Van A, Janeček Š, & Casane D (2013). Gene make-up: rapid and massive intron gains after horizontal transfer of a bacterial α-amylase gene to Basidiomycetes. BMC Evol, 13(1), 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dana CE, Glauber KM, Chan TA, Bridge DM, Steele RE (2012). Incorporation of a Horizontally Transferred Gene into an Operon during Cnidarian Evolution. PLOS ONE. 7, e31643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David L, Huber W, Granovskaia M, Toedling J, Palm CJ, Bofkin L, Jones T, Davis RW, Steinmetz LM (2006). A high-resolution map of transcription in the yeast genome. Proc. Natl. Acad. Sci. U. S. A 103, 5320–5325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doolittle WF (1998). You are what you eat: a gene transfer ratchet could account for bacterial genes in eukaryotic nuclear genomes. Trends Genet. 14, 307–311. [DOI] [PubMed] [Google Scholar]
- Doren KV, Hirsh D (1990). mRNAs that mature through trans-splicing in Caenorhabditis elegans have a trimethylguanosine cap at their 5’ termini. Mol. Cell. Biol 10, 1769–1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dujon BA, Louis EJ (2017). Genome diversity and evolution in the budding yeasts (Saccharomycotina). Genetics, 206(2), 717–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick DA (2012). Horizontal gene transfer in fungi. FEMS Microbiol. Lett 329, 1–8. [DOI] [PubMed] [Google Scholar]
- Flot JF, Hespeels B, Li X, Noel B, Arkhipova I, Danchin EGJ, Hejnol A, Henrissat B, Koszul R, Aury JM, et al. (2013). Genomic evidence for ameiotic evolution in the bdelloid rotifer Adineta vaga. Nature. 500, 453–457. [DOI] [PubMed] [Google Scholar]
- Ganot P, Kallesøe T, Reinhardt R, Chourrout D, Thompson EM (2004). Spliced-Leader RNA trans Splicing in a Chordate, Oikopleura dioica, with a Compact Genome. Mol. Cell. Biol 24, 7795–7805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilliam M, & Valentine DK (1974). Enterobacteriaceae isolated from foraging worker honey bees, Apis mellifera. Journal of Invertebrate Pathology, 23(1), 38–41. [Google Scholar]
- Gilliam M (1997). Identification and roles of non-pathogenic microflora associated with honey bees. FEMS Microbiol. Lett 155, 1–10. [Google Scholar]
- Gladyshev EA, Meselson M, Arkhipova IR (2008). Massive Horizontal Gene Transfer in Bdelloid Rotifers. Science. 320, 1210–1213. [DOI] [PubMed] [Google Scholar]
- Gonçalves C, Wisecaver JH, Kominek J, Oom MS, Leandro MJ, Shen XX Opulente DA, Zhou X, Peris D, Kurtzman CP, et al. (2018). Evidence for loss and reacquisition of alcoholic fermentation in a fructophilic yeast lineage. Elife,7, e33034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol 29, 644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich A, Saveliev V, Vyahhi N, Tesler G (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics. 29, 1072–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas H, Eisendle M, Turgeon BG (2008). Siderophores in fungal physiology and virulence. Annu. Rev. Phytopathol 46, 149–187. [DOI] [PubMed] [Google Scholar]
- Heinemann JA, Sprague GF (1989). Bacterial conjugative plasmids mobilize DNA transfer between bacteria and yeast. Nature. 340, 205–209. [DOI] [PubMed] [Google Scholar]
- Hittinger CT, Carroll SB (2007). Gene duplication and the adaptive evolution of a classic genetic switch. Nature 449, 677–681. [DOI] [PubMed] [Google Scholar]
- Hittinger CT, Gonçalves P, Sampaio JP, Dover J, Johnston M, Rokas A (2010). Remarkably ancient balanced polymorphisms in a multi-locus gene network. Nature. 464, 54–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt C, Yandell M (2011). MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics. 12, 491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooks KB, Delneri D, & Griffiths-Jones S (2014). Intron evolution in Saccharomycetaceae. Gen Biol Evol, 6(9), 2543–2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotopp JCD, Clark ME, Oliveira DCSG, Foster JM, Fischer P, Torres MCM, Giebel JD, Kumar N, Ishmael N, Wang S, et al. (2007). Widespread Lateral Gene Transfer from Intracellular Bacteria to Multicellular Eukaryotes. Science. 317, 1753–1756. [DOI] [PubMed] [Google Scholar]
- Husnik F, Nikoh N, Koga R, Ross L, Duncan RP, Fujie M, Tanaka M, Satoh N, Bachtrog D, Wilson ACC, et al. (2013). Horizontal gene transfer from diverse bacteria to an insect genome enables a tripartite nested mealybug symbiosis. Cell, 153(7), 1567–1578. [DOI] [PubMed] [Google Scholar]
- Husnik F, McCutcheon JP (2016). Repeated replacement of an intrabacterial symbiont in the tripartite nested mealybug symbiosis. Proceedings of the National Academy of Sciences, 113(37), E5416–E5424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husnik F, McCutcheon JP (2018). Functional horizontal gene transfer from bacteria to eukaryotes. Nat. Rev. Microbiol 16, 67–79. [DOI] [PubMed] [Google Scholar]
- Katoh K, Standley DM (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol 30, 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeling PJ, Palmer JD (2008). Horizontal gene transfer in eukaryotic evolution. Nat. Rev. Genet 9, 605–618. [DOI] [PubMed] [Google Scholar]
- Keren H, Maor LG, Ast G (2010). Alternative splicing and evolution: diversification, exon definition and function. Nat. Rev. Genet 11, 345–355. [DOI] [PubMed] [Google Scholar]
- Khaldi N, Collemare J Lebrun MH, Wolfe KH (2008). Evidence for horizontal transfer of a secondary metabolite gene cluster between fungi. Genome Biol. 9, R18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo N, Nikoh N, Ijichi N, Shimada M, Fukatsu T (2002). Genome fragment of Wolbachia endosymbiont transferred to X chromosome of host insect. Proc Natl. Acad. Sci. U. S. A 99, 14280–14285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korf I (2004). Gene finding in novel genomes. BMC Bioinformatics. 5, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlov AM, Aberer AJ, Stamatakis A (2015). ExaML version 3: a tool for phylogenomic analyses on supercomputers. Bioinformatics. 31, 2577–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause DJ, Kominek J, Opulente DA, Shen XX, Zhou X, Langdon QK, DeVirgilio J, Hulfachor AB, Kurtzman CP, Rokas A, Hittinger CT (2018). Functional and evolutionary characterization of a secondary metabolite gene cluster in budding yeasts. Proc. Natl. Acad. Sci. U. S. A, 115(43), 11030–11035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurland CG, Andersson SG (2000). Origin and evolution of the mitochondrial proteome. Microbiol. Mol. Bio. Rev 64, 786–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachance MA, Starmer WT, Rosa CA, Bowles JM, Barker JSF, Janzen DH (2001). Biogeography of the yeasts of ephemeral flowers and their insects. FEMS Yeast Res. 1, 1–8. [DOI] [PubMed] [Google Scholar]
- Lawrence JG, Roth JR (1996). Selfish Operons: Horizontal Transfer May Drive the Evolution of Gene Clusters. Genetics. 143, 1843–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le SQ, Gascuel O (2008). An improved general amino acid replacement matrix. Mol. Biol. Evol 25, 1307–1320. [DOI] [PubMed] [Google Scholar]
- Leduc D, Battesti A, Bouveret E (2007). The hotdog thioesterase EntH (YbdB) plays a role in vivo in optimal enterobactin biosynthesis by interacting with the ArCP domain of EntB. J. Bacteriol 189, 7112–7126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machuca A, Milagres AMF (2003). Use of CAS-agar plate modified to study the effect of different variables on the siderophore production by Aspergillus. Lett. Appl. Microbiol 36, 177–181. [DOI] [PubMed] [Google Scholar]
- Marcet-Houben M, Gabaldón T (2010). Acquisition of prokaryotic genes by fungal genomes. Trends Genet. 26, 5–8. [DOI] [PubMed] [Google Scholar]
- Martin W, Rujan T, Richly E, Hansen A, Cornelsen S, Lins T, Leister D, Stoebe B, Hasegawa M, Penny D (2002) Evolutionary analysis of Arabidopsis, cyanobacterial, and chloroplast genomes reveals plastid phylogeny and thousands of cyanobacterial genes in the nucleus. Proc. Natl. Acad. Sci. U. S. A 99, 12246–12251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran NA, McCutcheon JP, & Nakabachi A (2008). Genomics and evolution of heritable bacterial symbionts. Annu. Rev. Genet, 42, 165–190. [DOI] [PubMed] [Google Scholar]
- Neuvéglise C, Marck C, & Gaillardin C (2011). The intronome of budding yeasts. C. R. Biol, 334(8–9), 662–670. [DOI] [PubMed] [Google Scholar]
- Nikoh N, Tanaka K, Shibata F, Kondo N, Hizume M, Shimada M, Fukatsu T (2007). Wolbachia genome integrated in an insect chromosome: Evolution and fate of laterally transferred endosymbiont genes. Genome Res. 18, 272–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol 32, 268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omelchenko MV, Makarova KS, Wolf YI, Rogozin IB, Koonin EV (2003). Evolution of mosaic operons by horizontal gene transfer and gene displacement in situ. Genome Biol. 4, R55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelechano V, Wei W, Steinmetz LM (2013). Extensive transcriptional heterogeneity revealed by isoform profiling. Nature. 497, 127–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Miranda S, Cabirol N (2007). R. George-Téllez, L. S. Zamudio-Rivera, F. J. Fernández, O-CAS, a fast and universal method for siderophore detection. J. Microbiol. Methods 70, 127–131. [DOI] [PubMed] [Google Scholar]
- Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL (2015). StringTie enables improved reconstruction of a transcriptome from RNA-Seq reads. Nat. Biotechnol 33, 290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proctor RH, Van Hove F, Susca A, Stea G, Busman M, van der Lee T, Waalwijk C, Moretti A, Ward TJ (2013). Birth, death and horizontal transfer of the fumonisin biosynthetic gene cluster during the evolutionary diversification of Fusarium. Mol. Microbiol 90, 290–306. [DOI] [PubMed] [Google Scholar]
- Richards TA, Dacks JB, Jenkinson JM, Thornton CR, Talbot NJ (2006). Evolution of Filamentous Plant Pathogens: Gene Exchange across Eukaryotic Kingdoms. Curr. Biol 16, 1857–1864. [DOI] [PubMed] [Google Scholar]
- Richards TA, Leonard G, Soanes DM, Talbot NJ (2011). Gene transfer into the fungi. Fungal Biol. Rev 25, 98–110. [Google Scholar]
- Richards TA, Soanes DM, Jones MDM, Vasieva O, Leonard G, Paszkiewicz K, Foster PG, Hall N, Talbot NJ (2011). Horizontal gene transfer facilitated the evolution of plant parasitic mechanisms in the oomycetes. Proc. Natl. Acad. Sci. U. S. A 108, 15258–15263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosa CA, Lachance MA, Silva JOC, Teixeira ACP, Marini MM, Antonini Y, Martins RP (2003). Yeast communities associated with stingless bees. FEMS Yeast Res. 4, 271–275. [DOI] [PubMed] [Google Scholar]
- Rosa CA, Lachance MA, The yeast genus Starmerella gen. nov. and Starmerella bombicola sp. nov., the teleomorph of Candida bombicola (Spencer, Gorin & Tullock) Meyer & Yarrow. Int. J. Syst. Evol. Microbiol 48, 1413–1417. [DOI] [PubMed] [Google Scholar]
- Routh A, Domitrovic T, Johnson JE (2012). Host RNAs, including transposons, are encapsidated by a eukaryotic single-stranded RNA virus. Proc. Natl. Acad. Sci. U. S. A 109, 1907–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharf DH, Heinekamp T, Brakhage AA (2014). Human and Plant Fungal Pathogens: The Role of Secondary Metabolites. PLOS Pathog 10, e1003859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo SW, Kim D, Latif H, O’Brien EJ, Szubin R, Palsson BO (2014). Deciphering Fur transcriptional regulatory network highlights its complex role beyond iron metabolism in Escherichia coli. Nat. Commun 5, 4910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheftel A, Stehling O, Lill R (2010). Iron–sulfur proteins in health and disease. Trends Endocrinol. Metab 21, 302–314. [DOI] [PubMed] [Google Scholar]
- Shen XX, Zhou X, Kominek J, Kurtzman CP, Hittinger CT, Rokas A (2016). Reconstructing the Backbone of the Saccharomycotina Yeast Phylogeny Using Genome-Scale Data. G3, 6, 3927–3939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen XX, Opulente DA, Kominek J, Zhou X, Steenwyk JL, Buh KV, Haase MAB, Wisecaver JH, Wang M, Doering DT, et al. (2018). Tempo and Mode of Genome Evolution in the Budding Yeast Subphylum. Cell, 175(6), 1533–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 31, 3210–3212. [DOI] [PubMed] [Google Scholar]
- Skaar EP (2010). The battle for iron between bacterial pathogens and their vertebrate hosts. PLOS Pathog. 6, e1000949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slot JC, Rokas A (2011). Horizontal Transfer of a Large and Highly Toxic Secondary Metabolic Gene Cluster between Fungi. Curr. Biol 21, 134–139. [DOI] [PubMed] [Google Scholar]
- Slot JC, Rokas A (2010). Multiple GAL pathway gene clusters evolved independently and by different mechanisms in fungi. Proc. Natl. Acad. Sci. U. S. A 107, 10136–10141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soucy SM, Huang J, Gogarten JP (2015). Horizontal gene transfer: building the web of life. Nat. Rev. Genet 16, 472–482. [DOI] [PubMed] [Google Scholar]
- Spieth J, Brooke G, Kuersten S, Lea K, Blumenthal T (1993). Operons in C. elegans: polycistronic mRNA precursors are processed by trans-splicing of SL2 to downstream coding regions. Cell. 73, 521–532. [DOI] [PubMed] [Google Scholar]
- Stairs CW, Eme L, Brown MW, Mutsaers C, Susko E, Dellaire G, Soanes DM, van der Giezen M, Roger AJ (2014). A SUF Fe-S Cluster Biogenesis System in the Mitochondrion-Related Organelles of the Anaerobic Protist Pygsuia. Curr. Biol 24, 1176–1186. [DOI] [PubMed] [Google Scholar]
- Stamatakis A (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30, 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M, Diekhans M, Baertsch R, Haussler D (2008). Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics. 24, 637–644. [DOI] [PubMed] [Google Scholar]
- Stefanini I, Dapporto L, Berná L, Polsinelli M, Turillazzi S, Cavalieri D (2016). Social wasps are a Saccharomyces mating nest. Proc. Natl. Acad. Sci. U. S. A 113, 2247–2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutak R, Lesuisse E, Tachezy J, Richardson DR (2008). Crusade for iron: iron uptake in unicellular eukaryotes and its significance for virulence. Trends Microbiol. 16, 261–268. [DOI] [PubMed] [Google Scholar]
- Ter-Hovhannisyan V, Lomsadze A, Chernoff YO, Borodovsky M (2008). Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res. 18, 1979–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmis JN, Ayliffe MA, Huang CY, Martin W (2004). Endosymbiotic gene transfer: organelle genomes forge eukaryotic chromosomes. Nat. Rev. Genet 5, 123–135. [DOI] [PubMed] [Google Scholar]
- Toth IK, Pritchard L, Birch PRJ (2006). Comparative Genomics Reveals What Makes An Enterobacterial Plant Pathogen. Annu. Rev. Phytopathol 44, 305–336. [DOI] [PubMed] [Google Scholar]
- Tsaousis AD, Ollagnier de Choudens SO, Gentekaki E, Long S, Gaston D, Stechmann A, Vinella D, Py B, Fontecave M, Barras F, Lukeš J, Roger AJ (2012). Evolution of Fe/S cluster biogenesis in the anaerobic parasite Blastocystis. Proc. Natl. Acad. Sci. U. S. A, 109(26), 10426–10431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberghe AE, Meedel TH, Hastings KEM (2001). mRNA 5′-leader trans-splicing in the chordates. Genes Dev. 15, 294–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh CT, Liu J, Rusnak F, Sakaitani M (1990). Molecular studies on enzymes in chorismate metabolism and the enterobactin biosynthetic pathway. Chem. Rev 90, 1105–1129. [Google Scholar]
- Wandersman C, Delepelaire P (2004). Bacterial Iron Sources: From Siderophores to Hemophores. Annu. Rev. Microbiol 58, 611–647. [DOI] [PubMed] [Google Scholar]
- Watanabe K, Hara W, Sato M (1998). Evidence for Growth of Strains of the Plant Epiphytic Bacterium Erwinia herbicola and Transconjugation among the Bacterial Strains in Guts of the Silkworm Bombyx mori. J Invertebr Pathol. 72, 104–111. [DOI] [PubMed] [Google Scholar]
- Watanabe K, Sato M (1998). Plasmid-Mediated Gene Transfer Between Insect-Resident Bacteria, Enterobacter cloacae, and Plant-Epiphytic Bacteria, Erwinia herbicola, in Guts of Silkworm Larvae. Curr. Microbiol 37, 352–355. [DOI] [PubMed] [Google Scholar]
- Weigel BJ, Burgett SG, Chen VJ, Skatrud PL, Frolik CA, Queener SW, Ingolia TD (1988). Cloning and expression in Escherichia coli of isopenicillin N synthetase genes from Streptomyces lipmanii and Aspergillus nidulans. J. Bacteriol 170, 3817–3826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisecaver JH, Rokas A (2015). Fungal metabolic gene clusters-caravans traveling across genomes and environments. Front. Microbiol 6, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisecaver JH, Alexander WG, King SB, Hittinger CT, Rokas A (2016). Dynamic evolution of nitric oxide detoxifying flavohemoglobins, a family of single-protein metabolic modules in bacteria and eukaryotes. Mol. Biol. Evol 33(8), 1979–1987. [DOI] [PubMed] [Google Scholar]
- Wu TD, Nacu S (2010). Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics. 26, 873–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yurchenko T, Ševčíková T, Strnad H, Butenko A, Eliás M (2016). The plastid genome of some eustigmatophyte algae harbours a bacteria-derived six-gene cluster for biosynthesis of a novel secondary metabolite. Open Biol. 6, 160249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yurchenko T, Ševčíková T, Přibyl P, El Karkouri K, Klimeš V, Amaral R, Zbránková V, Kim E, Raoult D, Santos LMA (2018). A gene transfer event suggests a long-term partnership between eustigmatophyte algae and a novel lineage of endosymbiotic bacteria. ISME J. 2018 September;12(9):2163–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Peris D, Kominek J, Kurtzman CP, Hittinger CT, Rokas A (2016). in silico Whole Genome Sequencer & Analyzer (iWGS): A Computational Pipeline to Guide the Design and Analysis of de novo Genome Sequencing Studies. G3, 6, 3655–3662. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 3
Supplemental Table 1
Supplemental Table 2
Supplemental Table 4
1
Data Availability Statement
Raw DNA and RNA sequencing data were deposited in GenBank under Bioproject ID PRJNA396763 (www.ncbi.nlm.nih.gov/bioproject/PRJNA396763). Whole Genome Shotgun assemblies have been deposited at DDBJ/ENA/GenBank under the accessions NRDR00000000-NREI00000000. Genome-specific accessions are listed in Table S1.