Ultrasensitive Detection of Enzymatic Activity Using Single Molecule Arrays (original) (raw)
Abstract

Enzyme assays are important for many applications including clinical diagnostics, functional proteomics, and drug discovery. Current methods for enzymatic activity measurement often suffer from low analytical sensitivity. We developed an ultrasensitive method for the detection of enzymatic activity using Single Molecule Arrays (eSimoa). The eSimoa assay is accomplished by conjugating substrates to paramagnetic beads and measuring the conversion of substrates to products using single molecule analysis. We demonstrated the eSimoa method for the detection of protein kinases, telomerase, histone H3 methyltransferase SET7/9, and polypeptide _N_-acetylgalactosaminyltransferase with unprecedented sensitivity. In addition, we tested enzyme inhibition and performed theoretical calculations for the binding of inhibitor to its target enzyme and show the need for an ultrasensitive enzymatic assay to evaluate the potency of tight binding inhibitors. The eSimoa assay was successfully used to determine inhibition constants of both bosutinib and dasatinib. Due to the ultrasensitivity of this method, we also were able to measure the kinase activities at the single cell level. We show that the eSimoa assay is a simple, fast, and highly sensitive approach, which can be easily extended to detect a variety of other enzymes, providing a promising platform for enzyme-related fundamental research and inhibitor screening.
1. Introduction
Measuring the activity of enzymes, such as protein kinases, proteases, phosphatases, transferases, and telomerase, is important for many applications including drug discovery, clinical diagnostics and prognosis, and physiological function research.1−4 Various methods have been developed for the measurement of enzymatic activity and inhibitor screening, including mass spectrometry, electrochemistry, capillary electrophoresis, radiometric methods, colorimetric analysis, and fluorescence methods.5,6 Colorimetric and fluorescence methods are the most commonly used approaches and enable high-throughput analysis of several thousands of samples per day.7 However, current methods for enzymatic activity measurement often suffer from low analytical sensitivity. In recent years, highly potent small-molecule inhibitors have emerged as efficient drugs for the treatment of many diseases such as cancer.8 The accurate analysis of a tight binding inhibitor with an inhibition constant (Ki) in the subnanomolar or picomolar range requires a low concentration of enzyme near or below the Ki value.9 Since signal generation from subnanomolar concentrations of enzyme is extremely low, an ultrasensitive enzyme assay is necessary. In addition, many studies have demonstrated the heterogeneity of protein concentrations and enzyme activities in cancer cells, and single cell analysis has been shown to provide accurate diagnosis and treatment of cancers,10 which also requires ultrasensitive enzyme assays.
To overcome limitations in analytical sensitivity, our laboratory has developed an ultrasensitive detection platform using Single Molecule Arrays (Simoa).11 Specifically, target analyte molecules are first captured on biofunctionalized paramagnetic beads. A large excess of beads is used relative to the number of target analyte molecules to ensure that there is either zero or one target analyte molecule bound per bead that follows Poisson statistics. A biotinylated detection probe binds to the captured target analyte molecule on the bead, and the formed complex is then labeled with a reporter enzyme streptavidin-β-galactosidase (SβG). The SβG-labeled beads are resuspended in a fluorogenic substrate solution, resorufin-β-D-galactopyranoside (RGP), and loaded onto an array of microwells (50 fL) in which each well is able to hold only one bead. The wells are sealed with oil and the fluorescent product generated by the enzymatic reaction is confined within the microwells, ensuring high local fluorescence intensity that can be easily detected by a digital camera. The Simoa platform has been used for detecting various analytes such as proteins, small molecules, DNA, and mircoRNAs.12−15 However, these assays can only be used for the measurements of analyte concentrations, but not enzymatic activity.
In this work, we describe an enzymatic Simoa method (eSimoa) for ultrasensitive detection of enzymes. Protein kinases and telomerase are used as model enzymes. Protein kinases play a pivotal role in the regulation of numerous intracellular signal transduction pathways and oncogenic transformation.16 Abnormal expression and aberrant phosphorylation of protein kinases are often manifested in a variety of diseases, such as cancer, diabetes, Alzheimer’s disease, and inflammatory diseases.17 Thus, protein kinases are important targets for pharmaceutical intervention, particularly in oncology and inflammation.18 Telomerase is a ribonucleoprotein enzyme that catalyzes the addition of telomeric repeats (TTAGGG)n onto the 3′-end of the human chromosomes.19 Telomerase activity is elevated in more than 85% of cancer cells and absent in a majority of normal cells. As a result, telomerase is used as a biomarker for cancer diagnosis as well as a therapeutic target.20 In the eSimoa assay, substrate-conjugated paramagnetic beads (MBs) were incubated with target enzyme in the presence of cofactors. The substrate is converted to product on the bead and subsequently recognized by a biotinylated detection probe and further labeled by SβG for Simoa analysis. We demonstrate that the analytical sensitivity of these assays is significantly higher than that of conventional enzymatic assays. Due to the high sensitivity, the eSimoa method was applied to evaluate the potency of tight binding inhibitors. Furthermore, this method has been used for determination of protein kinase activity in cell lysates.
2. Experimental Section
2.1. Materials
Biotinylated antiphosphotyrosine antibody (clone 4G10), adenosine 5′-triphosphate (ATP) disodium salt hydrate, bovine serum albumin (BSA), biotinylated Helix pomatia agglutinin (HPA), anti-H3K4me antibody (M4819), manganese(II) chloride (MnCl2), uridine 5′-diphospho-_N_-acetylgalactosamine disodium salt (UDP-GalNAc), _S_-(5′-adenosyl)-l-methionine chloride dihydrochloride (SAM), phosphatase inhibitor cocktails 2 and 3, magnesium chloride solution (MgCl2), ethylenediaminetetraacetic acid disodium salt solution (EDTA, 0.5 M), 10 mM dNTP mix, Tris buffered saline, Amicon Ultra 0.5 mL centrifugal filters, bosutinib, dasatinib, and Tween 20 were purchased from Sigma-Aldrich (St. Louis, MO). Abltide (KKGEAIYAAPFA-NH2), Srctide (GEEPLYWSFPAKKK-NH2), EA2 (PTTDSTTPAPTTK), and biotinylated Srctide (biotin-GEEPLYWSFPAKKK-NH2) were purchased from Anaspec (Fremont, CA). Telomerase positive control cell pellet (S7701), active Abl protein (14–529), and active Src protein (14–326) were purchased from Millipore (Burlington, MA). Polypeptide _N_-acetylgalactosaminyltransferase 1 (GalNAcT) was purchased from R&D Systems. Histone H3 lysine 4 methyltransferase SET7/9 was purchased from Enzo Life Sciences (Farmingdale, NY). Recombinant histone H3 (C110A) was purchased from Active Motif. 1-Ethyl-3-(3-(dimethylamino)propyl)carbodiimide hydrochloride (EDC), _N_-hydroxysulfosuccinimide (sulfo-NHS), and 20x SSC buffer were purchased from Thermo Fisher (Waltham, MA). Oligonucleotides, the telomerase binding substrate (NH2-T10AATCCGTCGAGCAGAGTT), and detection probe (biotin-CCCTAACCCTAACCCTAACCCTAA) were purchased from IDT. Anti-H3K4me antibodies were biotinylated using NHS-PEG4-biotin according to a previously reported method.21 The Simoa HD-X analyzer and homebrew assay kits were purchased from Quanterix (Lexington, MA).22 Homebrew kits include carboxyl-functionalized paramagnetic beads, 50 nM streptavidin-β-galactosidase (SβG) concentrate, 100 μM resorufin β-d-galactopyranoside (RGP), and diluents (Bead Diluent, Sample Diluent, and SβG Diluent).
2.2. Preparation of Biofunctionalized Beads
Carboxylated 2.7 μm paramagnetic beads (∼4 × 108) were washed three times with 200 μL of bead wash buffer (0.1% Tween 20 in 1x PBS, pH 7.4) and twice with 200 μL of MES buffer (50 mM MES, pH 6.2). EDC and sulfo-NHS were reconstituted in MES buffer to a final concentration of 25 mg/mL. EDC and sulfo-NHS (100 μL each) were added to the beads. The beads were activated on a shaker for 30 min. After activation, the beads were washed three times with bead wash buffer. The activated beads were dispersed in 150 μL of bead wash buffer, and subsequently, 50 μL of 2 mg/mL Abltide or Srctide or EA2 in water were added. For the preparation of telomerase substrate beads, 10 nmol of the substrate DNA was diluted into 200 μL of bead wash buffer and added to the activated beads. The beads were incubated at room temperature with shaking for 3 h, washed three times with bead wash buffer, and incubated in 200 μL of Bead Diluent (Quanterix) with shaking for 1 h. The beads were then washed three times with bead wash buffer and resuspended in 200 μL of Bead Diluent for further use. Histone H3 protein-coated beads and antiphosphotyrosine antibody-coated capture beads were prepared according to a previously published method.23 The beads were counted using a Beckman-Coulter multisizer.
2.3. Preparation of Cell Samples
K-562 leukemia cell lines were purchased from ATCC (CCL-243). All K-562 cells were cultured in ATCC-formulated Iscove’s Modified Dulbecco’s Medium (30–2005) with 10% fetal bovine serum (30–2020, ATCC). HEK-293 kidney cell lines were purchased from ATCC (CRL-1573). All HEK-293 cells were cultured in ATCC formulated Eagle’s Minimum Essential Medium (30–2003) with 10% fetal bovine serum. All cultures were incubated at 37 °C with 5% CO2, and medium was replaced two to three times per week.
Cells were isolated by rinsing culture plates with 5 mL of Dulbecco’s Phosphate Buffered Saline (DPBS, D1283, Millipore Sigma). Cell culture plates were then incubated in 3 mL of trypsin EDTA (30-2101, ATCC) for 3 min. A 7 mL portion of complete growth medium was added to inhibit trypsin activity and resuspend the cells. Cell suspensions were centrifuged for 5 min at 130 g, and the supernatant was aspirated. The resulting cell pellet was resuspended in complete growth medium. An aliquot of cells was stained with Trypan Blue solution (15250-061, Life Technologies) for cell counting. Cells were diluted to a concentration of 1 × 106 cells/mL in DPBS and stained with propidium iodide (P1304MP, Life Technologies) for single cell sorting. Single cells were isolated by fluorescence-activated cell sorting (FACSAria Fusion Cell Sorter). Diluted cells were loaded into the FACSAria instrument, and single cells were sorted into a 96-well plate with 50 μL of lysis buffer already loaded into each well. Lysis buffer was composed of mammalian protein extraction buffer (GE28-9412-79, Millipore Sigma) with 1% phosphatase inhibitor cocktails 2 and 3. After sorting, 96-well plates containing single cell lysates were immediately stored at −80 °C for further analysis. For bulk cell experiments, cells were diluted to a concentration of 1 × 106 to 1 × 107 cells/mL and centrifuged for 5 min at 130 g, and the resulting cell pellet was resuspended in 200 μL of lysis buffer.
The telomerase was extracted from HeLa cells (S7701, Sigma) using the CHAPS method. Hela cell pellets (1 million cells) were first suspended in 200 μL 1× CHAPS lysis buffer and incubated on ice for 30 min. Then, the mixture was spun at 12000 g for 20 min at 4 °C, and 160 μL of the supernatant was transferred to a fresh tube, aliquoted, and stored at −80 °C until use.
2.4. Preparation of Reagents and Simoa Assay Setup
A three-step assay configuration was chosen for detecting protein kinase activity. Specifically, peptide-modified MBs were diluted in bead buffer (0.05 M Tris-HCl, pH 7.6, 0.1 mg/mL BSA, 15 mM MgCl2 and 3 mM ATP) to a concentration of 10000 beads/μL. Biotinylated antiphosphotyrosine antibodies were diluted in detector buffer (0.05 M Tris-HCl, pH 8.8, 1% BSA and 0.05% Tween 20) to 2 nM. SβG concentrate was diluted to 25 pM in SβG Diluent (Quanterix). Src and Abl standards were serially diluted to desired concentrations in assay buffer (0.05 M Tris-HCl, pH 7.6, and 0.1 mg/mL BSA). The reagents including beads, detector, and SβG were placed in plastic bottles (Quanterix). The samples were loaded onto a 96-well plate (Quanterix). All reagents (beads, detection antibodies, SβG, enzyme substrate RGP, Wash Buffer 1, Wash Buffer 2, and Simoa Sealing Oil) were purchased from Quanterix and loaded onto the Simoa HD-X Analyzer based on the manufacturer’s instructions. A 50 μL portion of bead solution was pipetted into a reaction cuvette. Then 100 μL of sample was added and incubated for 30 min. The beads were pelleted with a magnet, and the supernatant was removed. Following a series of washes, 100 μL of detection antibody was added and incubated for 5 min. The beads were then pelleted again and washed three times. A 100 μL portion of SβG was added and incubated for 5 min. The beads were washed, resuspended in RGP solution, and loaded onto the array. The array was then sealed with oil and imaged. Images of the arrays were analyzed, and AEB (average enzyme per bead) values were calculated by the software in the HD-X Analyzer. For the detection of kinase activity in cell lysates, cell samples were serially diluted in the assay buffer containing 0.5% phosphatase inhibitor cocktails 2 and 3 for each, and 100 μL of sample was measured on the HD-X. For kinase inhibition assay, inhibitors dissolved in DMSO were diluted to different concentrations in the assay buffer. A 60 μL portion of inhibitor solution was mixed with 60 μL of 12 pM Abl or 20 pM Src, and the mixture was incubated at room temperature for 30 and 150 min. A 100 μL portion of the sample was incubated with 50 μL of bead solution (containing 10 μM ATP for Abl and 90 μM ATP for Src) for the Simoa assay.
A two-step assay configuration was chosen when biotin-labeled peptide was used as the substrate. Src stock solution was serially diluted to desired concentrations in the assay buffer. A 60 μL portion of Src sample was mixed with 60 μL of 20 μM biotin-labeled Srctide in the assay buffer containing 2 mM ATP and 10 mM MgCl2, and then the mixture was incubated at room temperature for 60 min. A 6 μL of 0.5 M EDTA was added to stop the reaction. Then 100 μL of the solution was measured on the HD-X. Antiphosphotyrosine antibody coated capture beads were diluted in the assay buffer to 20000 beads/μL. A 100 μL portion of sample and 25 μL of bead solution buffer were incubated for 30 min. The beads were pelleted, and the supernatant was removed. Following a series of washes, 100 μL of SβG (200 pM) was added, and the mixture was incubated for 15 min for the Simoa assay.
For the detection of GalNacT activity, EA2 modified beads were diluted to 10000 beads/μL in the assay buffer containing 15 mM MnCl2 and 0.3 mM UDP-GalNAc. A 50 μL portion of bead solution was incubated with 100 μL of sample for 90 min. The beads were washed, 100 μL of 20 nM biotinylated HPA was added, and the mixture was incubated for 15 min. The beads were washed again. Finally, 100 μL of 200 pM SβG was added and the mixture was incubated for 15 min for the Simoa assay.
For the detection of SET7/9 activity, histone H3 modified beads were diluted to 5000 beads/μL in a Tris buffer (0.05 M Tris-HCl, pH 8.8, 0.1 mg/mL BSA) containing 15 mM MgCl2 and 0.12 mM SAM. A 50 μL portion of bead solution was incubated with 100 μL of sample for 60 min. The beads were washed, 100 μL of 2 nM biotinylated anti-H3K4me antibody in Tris buffer (0.05 M, pH 8.8, 1% BSA and 0.05% Tween 20) was added, and the mixture was incubated for 7.5 min. The beads were washed again. Finally, 100 μL of 50 pM SβG was added and the mixture was incubated for 7.5 min for the Simoa assay.
For the detection of telomerase activity, oligonucleotide-conjugated MBs were suspended in bead buffer (0.05 M Tris-HCl, pH 7.6, 0.15 M sodium chloride, 0.1 mg/mL BSA and 0.2 mM dNTP mix) to 10000 beads/μL. Cell extracts were diluted with an assay buffer (0.05 M Tris-HCl, pH 7.6, 0.15 M sodium chloride, and 0.1 mg/mL BSA) to desired concentrations, and the DNA detection probe was diluted in an SSC buffer (5x SSC, 0.1% Tween, pH 7.0) to 50 nM. A three-step assay configuration was used. A 50 μL portion of bead solution and 50 μL of sample were mixed, and the mixture was incubated for 2 h. After magnetic separation and washing, 100 μL of detection probe was added and the mixture was incubated for 30 min. The beads were pelleted again, and the supernatant was removed. Following a series of washes, 100 μL of 200 pM SβG was added, and the mixture was incubated for 30 min for the Simoa assay.
2.5. Data Analysis
Standard curves were obtained by plotting the signal responses (AEB) against the logarithm of analyte concentrations using Origin software (Origin 9.5). The 4-parameter logistic equation y = A0 + (A1-A0)/[1 + (x/x0)p] was used for curve fitting in the whole concentration range, where A1 is the maximum signal, A0 is the minimum signal, p is the curve slope at the inflection point, and x0 is the concentration at the inflection point. For the inhibition assay, x0 is the IC50 (inhibitor concentration causing a 50% inhibition of the maximum response). The lower the IC50, the higher the potency of inhibitor is. The limit of detection (LOD) was calculated as three standard deviations (SDs) above the background. All measurements were performed in triplicate.
3. Results and Discussion
Enzymatic Simoa Assays
We developed an eSimoa assay that enables ultrasensitive detection of enzymes. For the detection of protein kinase activity (Figure 1), paramagnetic beads (MBs) were modified with specific peptides and incubated with a protein kinase in the presence of the cofactors ATP and MgCl2 to initiate the phosphorylation of tyrosine residues on the peptide. After removal of the protein kinase and cofactors by magnetic separation and washing, the MBs were subsequently incubated with biotinylated antiphosphotyrosine antibodies, which specifically recognize the phosphorylated peptide. Beads with the immunocomplex were then labeled with a reporter enzyme, SβG, via biotin–streptavidin interaction, and detected by SβG enzymatic readout in the Simoa platform. The signal was measured in units of average enzyme (SβG) per bead (AEB), as previously described.24
Figure 1.

Schematic of eSimoa assay for the detection of protein kinase using peptide-modified MBs as the substrate.
Proto-oncogene tyrosine-protein kinase Src was selected as a model enzyme, since it is found to be overexpressed and highly activated in various human cancers, and it may have an influence on the development of the metastatic phenotype.25 Numerous radiometric and fluorescent assays have been developed for detecting Src activity, but the sensitivity is relatively low.26,27 Thus, ultrasensitive methods are highly desirable. For the detection of Src activity, Srctide was covalently linked to MBs through a reaction between activated carboxyl groups on the MBs and amino groups on Srctide. The Srctide-modified MBs were used as the substrate, and the corresponding phosphorylated peptide was recognized by antiphosphotyrosine antibodies. Figure 2A shows the response curve for the detection of Src. The signal (AEB) increases proportionally with increasing Src concentration, as more peptides are phosphorylated at higher enzyme concentrations. The background value is 0.0241 ± 0.0015, and the signal to background ratio (S/B) is 27.4 for 1 pM Src. The LOD is 4.9 fM, which is much lower than that of current state-of-the-art techniques (typically in the subnanomolar concentrations, Table S1), demonstrating that the eSimoa assay is highly sensitive. Additionally, we explored a second Simoa method for Src detection using a biotinylated Srctide as the substrate (Figures S1 and S2). The sensitivity of this approach is much lower (with LOD of 49.2 pM), which might be because the peptide chain is too short to allow for efficient capture by antiphosphotyrosine antibody modified MBs and SβG.
Figure 2.

Response curves for the detection of (A) Src and (B) Abl activity.
To evaluate the ability of the eSimoa assay to detect other protein kinases, an activity assay for Abelson murine leukemia viral oncogene homologue 1 (Abl) was developed. Abl is a cytoplasmic and nuclear protein tyrosine kinase that influences many cellular processes including cell differentiation, cell division, cell adhesion, and the oxidative stress response.28,29 For example, the Abl-Bcr (breakpoint cluster region protein) fusion protein can promote the occurrence and progression of chronic myeloid leukemia (CML) by allowing CML cells to rapidly grow. Therefore, Abl is an important molecular target for the pharmacological therapy of CML. It is therefore important to develop a simple and sensitive method for determining Abl activity and screening of inhibitors. Many methods such as electrochemical and fluorescent assays have been developed for detecting Abl activity, but these assays suffer from low analytical sensitivity.30,31 We demonstrate that the eSimoa assay using Abltide-conjugated MBs can quantify Abl concentrations (Figure 2B) with an LOD of 4.2 fM. The eSimoa assay for Abl is more sensitive than all conventional protein kinase assays: many orders of magnitude more sensitive than any other methods (Table S1). The background value is 0.0305 ± 0.0022, and the S/B is 63.6 for 1 pM Abl. These results indicate that the eSimoa assay is a powerful method with unprecedented sensitivity that could be used to detect the activity of other protein kinases, such as epidermal growth factor receptor (EGFR) and serine/threonine-protein kinase B-Raf (BRAF).
To examine the capability of the eSimoa assay for detecting other classes of enzymes, an activity assay for telomerase, an RNA-dependent polymerase, was established. The most commonly used telomerase detection assay is the polymerase chain reaction (PCR)-based telomeric repeat amplification protocol (TRAP).32 However, TRAP requires the use of DNA polymerases and is therefore susceptible to PCR-derived artifacts.20 PCR-free assays for telomerase activity are highly desirable. As shown in Figure 3, the substrate oligonucleotides were preconjugated to the surface of MBs. Telomerase extracted from HeLa cells was first complexed to its substrate oligonucleotide. The bound telomerase catalyzes the addition of telomeric repeats (TTAGGG)n onto the 3′-end of the substrate in the presence of dNTP mix using the telomerase’s intrinsic RNA as a template. After incubation and magnetic separation, the bound telomerase was washed away and a biotin-labeled detection probe comprised of a complementary sequence was added. The elongated oligonucleotide sequences specifically bound with the detection probes, and excess detection probes were removed after magnetic separation. Finally, SβG was added to label the MBs for Simoa analysis. As shown in Figure 4, the signal increased proportionally with the number of cells analyzed by Simoa as increasing levels of telomerase were extracted. The detection limit was 70 cells, comparable to the PCR-based assay for telomerase (typically with detection limits of tens of cells),20 demonstrating the high sensitivity of the eSimoa assay.
Figure 3.

Schematic of eSimoa assay for the detection of telomerase.
Figure 4.
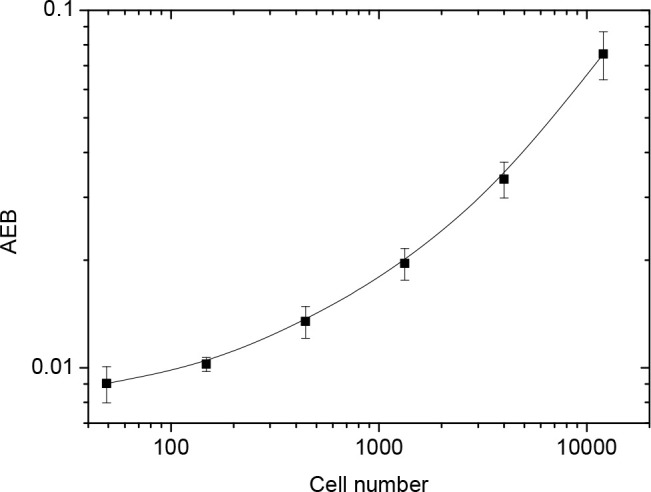
Response curve for the detection of telomerase activity using Simoa.
In the eSimoa assay, an enzyme molecule first binds with a substrate-conjugated bead and then converts the substrate into product. After the formation of product on the bead, the enzyme molecule dissociates from the bead and a new reaction cycle starts. One enzyme molecule can catalyze the formation of many product molecules. The formed product molecules are recognized by specific detection probes and further labeled with SβG for Simoa analysis. The beads are trapped in femtoliter-sized wells, where only one bead can fit per well, allowing for a digital readout of each individual bead to determine if it contains a product molecule or not. Because enzymes catalyze multiple reaction cycles, they have an intrinsic amplification effect. Coupled with the ability of Simoa to analyze single molecules, the eSimoa assay shows ultrahigh sensitivity. Furthermore, as shown in Figures S3–S6, this method has been successfully used for the detection of histone methyltransferase SET7/9 and polypeptide _N_-acetylgalactosaminyl-transferase (GalNAcT). These results indicate that the eSimoa approach is a general method, which provides a promising platform for enzyme-related research and clinical diagnostics.
Evaluating Potency of Inhibitors
An important application of enzymatic activity assay is to screen inhibitors. Small-molecule inhibitors are an important class of pharmacological agents that exhibit competitive inhibition of substrate binding. To examine whether the eSimoa method is suitable for small-molecule drug screening, we first performed theoretical calculations to determine the factors that influence the analytical performance of an enzyme inhibition test. The theoretical calculations were derived from simple equations based on bimolecular interactions described in the Supporting Information. Specifically, half maximal inhibitory concentration (IC50) is used to evaluate the potency of an inhibitor. When an enzyme is incubated with a competitive inhibitor and substrate, the inhibitor directly competes with the substrate in binding to the enzyme. In this case, IC50 is determined by a multivariable eq (eq 1) that depends on Ki, Km, [S], and [E0].33
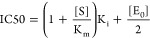 |
1 |
|---|
where Ki is the inhibition constant, Km is the Michaelis constant, [E0] is the total concentration of enzyme, and [S] is the substrate concentration. In many cases, a high substrate concentration [S] can be used to make sure that [E0] is much smaller than (1+ [S]/Km)*Ki(i.e., Kiapp), and IC50 is determined by Kiapp where it can reflect the potency of an inhibitor.
When an inhibitor binds to its target enzyme with very high affinity, such as with Ki values in the nanomolar range or even lower, the binding is tight and typically a long incubation time is required to reach equilibrium. Tight binding inhibitors are ideal drug candidates due to high binding affinity and long residence time of the compound on the target enzyme, which allows more efficient inhibition of a target enzyme at small doses. To evaluate the potency of tight binding inhibitors, the enzyme is typically preincubated with the inhibitor and then the substrate is added to initiate the reaction. In this case, IC50 is calculated using eq 2
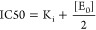 |
2 |
|---|
The IC50 is determined by Ki and [E0], independent of the substrate concentration. Accurate analysis of tight binding inhibitors should be performed at enzyme concentrations near or below the Ki value. Otherwise, the enzyme concentration will have a major effect on the IC50 and small experimental errors can have significant effects on the determination of Ki.9 Challenges of evaluating the potency of tight binding inhibitors with subnanomolar affinities can be overcome by using the ultrasensitive platform described here to measure picomolar enzyme concentrations.
We first determined the Km values for Src and Abl. Different concentrations of ATP were used in the phosphorylation reaction, while the concentrations of peptide-modified beads and enzymes were kept constant. As shown in Figure S7, with increasing ATP concentration, the signal increased proportionally because more tyrosine groups were phosphorylated at higher ATP concentrations. The Km values calculated from the half maximal effective concentration (EC50) are 3.69 μM for Abl and 29.2 μM for Src, which are consistent with reported values in the literature.26
We next used the eSimoa assay to test two inhibitors, bosutinib and dasatinib, against Abl and Src. Bosutinib is a dual inhibitor of Abl and Src kinases, with inhibition constants in the nanomolar range. Dasatinib is more potent than bosutinib, inhibiting both Abl and Src with dissociation constants in the picomolar range. Each enzyme (12 pM for Abl and 20 pM for Src) was incubated with the inhibitors for different times and then detected using the eSimoa assay. As shown in Figure 5, both enzymes exhibit a decrease in enzymatic activity as the inhibitor concentration increases. When bosutinib directly competed with ATP, IC50s were 2.33 nM for Src and 2.91 nM for Abl, while the IC50s slightly decreased to 2.24 nM for Src and 2.58 nM for Abl when bosutinib was preincubated with the enzymes for 30 min (Figure 5A,B). This result demonstrates that preincubation has a small effect on the assays. The Ki values calculated from eq 1 were 1.16 nM for Src and 1.45 nM for Abl, consistent with the reported results (IC50:1.0 nM for Src and 1.2 nM for Bcr-Abl).34
Figure 5.

Dose response curves at different preincubation times of bosutinib inhibition of Src (A) and Abl (B), and dasatinib inhibition of Src (C) and Abl (D).
In contrast, when the enzymes were preincubated with dasatinib, the dose response curves were left-shifted to lower concentrations (Figure 5C,D). Preincubation is required to accurately determine the potency of dasatinib. IC50s were 466.7 pM for Src and 735.1 pM for Abl when dasatinib directly competed with ATP. The IC50s decreased to 39.2 pM for Src and 29.8 pM for Abl when the enzymes were preincubated with dasatinib for 150 min. The apparent Ki values calculated from eq 2 were 14.6 pM for Src and 11.9 pM for Abl, which are in good agreement with the reported values determined from protein–ligand binding experiments (Ki: 16 ± 1.0 pM for Src and 30 ± 22 pM for Bcr-Abl).35 These results demonstrate that the eSimoa assay can be used to evaluate the potency of inhibitors, especially for the screening of tight binding inhibitors. In contrast to kinetic assays for enzymes, the eSimoa assay is an end-point method. Preincubation of the inhibitor with its target enzyme is usually required, and the preincubation time needs to be optimized to ensure that binding between the inhibitor and the enzyme reaches equilibrium.
Detection of Protein Kinase Activity in Cell Samples
In the previous sections, purified recombinant protein kinases were used in the eSimoa assays. However, a recombinant protein may not reflect the native conformation and activity of the target in its physiological context, because of the absence of interacting regulatory proteins, expression of alternative splice variants, incorrect protein folding, or post-translational modifications.36 As protein kinases have crucial and multifaceted effects on a majority of cellular pathways, especially those involved in signal transduction,37 it is essential to measure the activity of native protein kinases in cell samples to truly validate the methodology. The standard experimental approach is to immunoprecipitate the kinase of interest and measure the phosphorylation of a purified protein or peptide substrate using radiolabeled ATP.38 A loss of enzymatic activity may occur during the isolation process, and usually a large amount of cells is required due to the relatively low sensitivity of such assays. We examined whether the eSimoa method could be applied to detect protein kinase in cell lysates. We selected two cell lines, K562 and HEK293, because Bcr-Abl fusion proteins are overexpressed in K562 cells, while the Abl activity in HEK293 cells is usually low. Cell samples were lysed and phosphatase inhibitor cocktails were added to avoid interference from endogenous phosphatases. The cell lysates were serially diluted to desired concentrations and then incubated with Abltide-modified MBs for Simoa analysis. As shown in Figure 6, the signal increased with extracts from increasing numbers of cells, with the lowest concentrations of kinase detected corresponding to the concentration that would be extracted from one cell for K562 and from 30 cells for HEK293. We found the signal intensity (AEB) of K562 cells to be 21.4 times as high as that of HEK 293 cells (100 cells for each type).
Figure 6.

Bar graphs of Abl activity measurements in cell lysates of (A) K562 and (B) HEK293.
Since Bcr-Abl is overexpressed in K562 cells, we tested the inhibition efficiency of bosutinib and dasatinib in the cell lysates. As shown in Figure S8, the shape of the dose response curves for the cell-based assays was similar to those of assays using purified recombinant enzymes. The response curves were left-shifted to lower concentrations when dasatinib was preincubated with the cell lysates. Specifically, IC50 was 433.7 pM when dasatinib directly competed with ATP. When dasatinib was preincubated with the cell lysates for 30 min, the IC50 decreased to 82.2 pM. For bosutinib, there is only a small decrease in the IC50 values when the inhibitor was preincubated with the cell lysates (from 3.19 nM to 2.57 nM). These results are consistent with the assays using purified recombinant enzymes, indicating the eSimoa assays can be used to reliably determine the potency of inhibitors in cell samples.
We further tested whether it is possible to detect Abl activity in single cells. Single K562 cells were isolated by fluorescence-activated cell sorting and lysed. Bosutinib and dasatinib were added to the single cell lysates to final concentrations of 50 and 10 nM, respectively. The samples were then analyzed by Simoa. As shown in Figure 7, the signal has a relatively broad distribution when only buffer was added to the single cell lysates, demonstrating a heterogeneous Abl activity from cell to cell. In the presence of dasatinib and bosutinib, the signal distribution moved to lower AEB signals and the average signal decreased due to inhibition. These results indicate that the eSimoa assay can be used to measure kinase activity in single cells.
Figure 7.

Detection of Bcr-Abl activity in individual single cells (A) and average signal changes (B).
Conclusions
eSimoa assays have been developed for measuring the activity of Src, Abl, telomerase, SET7/9, and GalNAcT (Figures 1–4 and S1–S6). Although these enzymes operate by very different mechanisms and on different types of substrates, all the tested assays worked well, indicating the eSimoa method is a general approach that is both robust and flexible. Single molecule analysis is much more sensitive than conventional enzymatic assays, enabling measurements of enzymes at concentrations previously unachievable and opens a window into biological phenomena that were previously inaccessible, such as the screening of tight binding inhibitors and measurements of enzymatic activity in single cells.
According to theoretical calculations, it is difficult to screen an inhibitor with Ki in the picomolar range when nanomolar enzyme is used in the screening test. Due to the ultrasensitivity of the eSimoa method, we can use a very low concentration of enzyme to evaluate the potency of tight binding inhibitors. The eSimoa method has been successfully used to determine inhibition constants of both bosutinib and dasatinib (Figure 5). To the best of our knowledge, this is the first time that the inhibition constants of dasatinib against Abl and Src have been determined by enzyme assays. The obtained Ki values are consistent with those reported in the literature determined from protein–ligand binding experiments. The interference due to denatured enzymes in conventional protein-binding experiments is effectively avoided by the enzymatic assay. The eSimoa inhibition tests are performed in 96-well plates and run in an automated way, enabling medium-throughput screening of enzyme inhibitors. Future efforts are focused on screening of tight binding inhibitors against some disease-related therapeutic targets such as EGFR and BRAF.
In addition, the ultrasensitive eSimoa assay provides an unprecedented opportunity to study enzymatic activity in cell samples. Recent studies have demonstrated that many anticancer drugs kill most but not all the cells in a tumor, often resulting in relapse of cancer.39,40 It has been proposed that cell-to-cell variability in protein activity contributes to this different response to drugs. As most conventional techniques only provide a population-averaged measurement, they do not reflect an accurate picture of a heterogeneous population of cells that are in different signaling states. We demonstrated that the eSimoa assay can be applied to determine kinase activity in cell lysates, and kinase activity in single cells can be detected (Figures 6 and 7). Combined with fluorescence-activated cell sorting, the eSimoa method allows the analysis of enzymatic activity in hundreds of single cells within 3 h. The ability to measure protein kinase activity at the single cell level enables us to study cellular heterogeneity in drug response, which has relevance to the treatment of cancers. In this work, we studied the effects of inhibitors, but the influence of other factors that regulate signaling pathways can be also investigated using this method.
The developed eSimoa method could be expanded to the detection of other enzymes because: (1) the preparation of substrate-conjugated MBs is simple and general; (2) the use of MBs enables the enzymatic reaction to be more controllable. The formed product can be easily purified, owing to efficient magnetic separation, and then detected using Simoa as long as there is a suitable detection probe; (3) the approach shows ultrahigh sensitivity. eSimoa is a simple and robust approach, which should be applicable to a diverse range of areas, such as drug discovery, physiological function research, and medical diagnostics.
Acknowledgments
We thank Limor Cohen at Wyss Institute for Biologically Inspired Engineering, Harvard University, for taking part in discussion and revising the manuscript. This work was supported in part by a grant (award #: OPP1157033) from the Bill and Melinda Gates Foundation.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c06599.
- Schematics and response curves of eSimoa assays, effects of ATP on eSimoa assays, dose response curves of bosutinib and dasatinib inhibition, analytical performances of various methods for Src and Abl activity detection, theoretical considerations (PDF)
The authors declare the following competing financial interest(s): David R. Walt is a board member and equity holder of Quanterix Corporation. All other authors declare no competing financial interest.
Supplementary Material
References
- Kwong G. A.; Von Maltzahn G.; Murugappan G.; Abudayyeh O.; Mo S.; Papayannopoulos I. A.; Sverdlov D. Y.; Liu S. B.; Warren A. D.; Popov Y. Mass-encoded synthetic biomarkers for multiplexed urinary monitoring of disease. Nat. Biotechnol. 2013, 31 (1), 63. 10.1038/nbt.2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shults M. D.; Janes K. A.; Lauffenburger D. A.; Imperiali B. A multiplexed homogeneous fluorescence-based assay for protein kinase activity in cell lysates. Nat. Methods 2005, 2 (4), 277. 10.1038/nmeth747. [DOI] [PubMed] [Google Scholar]
- Courtney T. M.; Deiters A. Optical control of protein phosphatase function. Nat. Commun. 2019, 10 (1), 1–10. 10.1038/s41467-019-12260-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W.; Fu P.; Sun M.; Xu L.; Kuang H.; Xu C. Dual quantification of microRNAs and telomerase in living cells. J. Am. Chem. Soc. 2017, 139 (34), 11752–11759. 10.1021/jacs.7b03617. [DOI] [PubMed] [Google Scholar]
- Yi F.; Huang X.; Ren J. Simple and sensitive method for determination of protein kinase activity based on surface charge change of peptide-modified gold nanoparticles as substrates. Anal. Chem. 2018, 90 (6), 3871–3877. 10.1021/acs.analchem.7b04569. [DOI] [PubMed] [Google Scholar]
- Kovarik M. L.; Allbritton N. L. Measuring enzyme activity in single cells. Trends Biotechnol. 2011, 29 (5), 222–230. 10.1016/j.tibtech.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore B. D.; Stevenson L.; Watt A.; Flitsch S.; Turner N. J.; Cassidy C.; Graham D. Rapid and ultra-sensitive determination of enzyme activities using surface-enhanced resonance Raman scattering. Nat. Biotechnol. 2004, 22 (9), 1133. 10.1038/nbt1003. [DOI] [PubMed] [Google Scholar]
- Hoelder S.; Clarke P. A.; Workman P. Discovery of small molecule cancer drugs: successes, challenges and opportunities. Mol. Oncol. 2012, 6 (2), 155–176. 10.1016/j.molonc.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy D. J. Determination of accurate KI values for tight-binding enzyme inhibitors: an in silico study of experimental error and assay design. Anal. Biochem. 2004, 327 (1), 61–67. 10.1016/j.ab.2003.12.018. [DOI] [PubMed] [Google Scholar]
- Lawson D. A.; Kessenbrock K.; Davis R. T.; Pervolarakis N.; Werb Z. Tumour heterogeneity and metastasis at single-cell resolution. Nat. Cell Biol. 2018, 20 (12), 1349–1360. 10.1038/s41556-018-0236-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rissin D. M.; Kan C. W.; Campbell T. G.; Howes S. C.; Fournier D. R.; Song L.; Piech T.; Patel P. P.; Chang L.; Rivnak A. J. Single-molecule enzyme-linked immunosorbent assay detects serum proteins at subfemtomolar concentrations. Nat. Biotechnol. 2010, 28 (6), 595. 10.1038/nbt.1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen L.; Walt D. R. Single-molecule arrays for protein and nucleic acid analysis. Annu. Rev. Anal. Chem. 2017, 10, 345–363. 10.1146/annurev-anchem-061516-045340. [DOI] [PubMed] [Google Scholar]
- Wang X.; Cohen L.; Wang J.; Walt D. R. Competitive immunoassays for the detection of small molecules using single molecule arrays. J. Am. Chem. Soc. 2018, 140 (51), 18132–18139. 10.1021/jacs.8b11185. [DOI] [PubMed] [Google Scholar]
- Cohen L.; Hartman M. R.; Amardey-Wellington A.; Walt D. R. Digital direct detection of microRNAs using single molecule arrays. Nucleic Acids Res. 2017, 45 (14), e137-e137 10.1093/nar/gkx542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L.; Shan D.; Zhao M.; Pink B. A.; Minnehan K. A.; York L.; Gardel M.; Sullivan S.; Phillips A. F.; Hayman R. B. Direct detection of bacterial genomic DNA at sub-femtomolar concentrations using single molecule arrays. Anal. Chem. 2013, 85 (3), 1932–1939. 10.1021/ac303426b. [DOI] [PubMed] [Google Scholar]
- Pawson T.; Scott J. D. Protein phosphorylation in signaling-50 years and counting. Trends Biochem. Sci. 2005, 30 (6), 286–290. 10.1016/j.tibs.2005.04.013. [DOI] [PubMed] [Google Scholar]
- Lahiry P.; Torkamani A.; Schork N. J.; Hegele R. A. Kinase mutations in human disease: interpreting genotype-phenotype relationships. Nat. Rev. Genet. 2010, 11 (1), 60. 10.1038/nrg2707. [DOI] [PubMed] [Google Scholar]
- Anastassiadis T.; Deacon S. W.; Devarajan K.; Ma H.; Peterson J. R. Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29 (11), 1039. 10.1038/nbt.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shay J. W.; Wright W. E. Telomeres and telomerase: three decades of progress. Nat. Rev. Genet. 2019, 20, 299–309. 10.1038/s41576-019-0099-1. [DOI] [PubMed] [Google Scholar]
- Zheng G.; Daniel W. L.; Mirkin C. A. A new approach to amplified telomerase detection with polyvalent oligonucleotide nanoparticle conjugates. J. Am. Chem. Soc. 2008, 130 (30), 9644–9645. 10.1021/ja803035p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen L.; Walt D. R. Evaluation of antibody biotinylation approaches for enhanced sensitivity of single molecule array (Simoa) immunoassays. Bioconjugate Chem. 2018, 29 (10), 3452–3458. 10.1021/acs.bioconjchem.8b00601. [DOI] [PubMed] [Google Scholar]
- Wilson D. H.; Rissin D. M.; Kan C. W.; Fournier D. R.; Piech T.; Campbell T. G.; Meyer R. E.; Fishburn M. W.; Cabrera C.; Patel P. P. The Simoa HD-1 analyzer: a novel fully automated digital immunoassay analyzer with single-molecule sensitivity and multiplexing. J. Lab. Autom. 2016, 21 (4), 533–547. 10.1177/2211068215589580. [DOI] [PubMed] [Google Scholar]
- Cohen L.; Xie L.; Xylas M. E.; Walt D. R. Single molecule arrays for ultra-sensitive detection of rat cytokines in serum. J. Immunol. Methods 2018, 452, 20–25. 10.1016/j.jim.2017.10.002. [DOI] [PubMed] [Google Scholar]
- Rissin D. M.; Fournier D. R.; Piech T.; Kan C. W.; Campbell T. G.; Song L.; Chang L.; Rivnak A. J.; Patel P. P.; Provuncher G. K. Simultaneous detection of single molecules and singulated ensembles of molecules enables immunoassays with broad dynamic range. Anal. Chem. 2011, 83 (6), 2279–2285. 10.1021/ac103161b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irby R. B.; Yeatman T. J. Role of Src expression and activation in human cancer. Oncogene 2000, 19 (49), 5636. 10.1038/sj.onc.1203912. [DOI] [PubMed] [Google Scholar]
- Card A.; Caldwell C.; Min H.; Lokchander B.; Xi H.; Sciabola S.; Kamath A. V.; Clugston S. L.; Tschantz W. R.; Wang L. High-throughput biochemical kinase selectivity assays: panel development and screening applications. J. Biomol. Screening 2009, 14 (1), 31–42. 10.1177/1087057108326663. [DOI] [PubMed] [Google Scholar]
- Huang Y.; Huang H.; Qin J.; Liu X.; Zhao S.; Chen Z.-F.; Liang H. A graphene oxide-based multiplexed fluorescence assay for the detection of protein kinase activity in cell lysates and the evaluation of protein kinase inhibition. Sens. Actuators, B 2017, 238, 908–916. 10.1016/j.snb.2016.07.140. [DOI] [Google Scholar]
- Shaul Y. c-Abl: activation and nuclear targets. Cell Death Differ. 2000, 7 (1), 10. 10.1038/sj.cdd.4400626. [DOI] [PubMed] [Google Scholar]
- Shaul Y.; Merav B.-Y. Role of c-Abl in the DNA damage stress response. Cell Res. 2005, 15 (1), 33. 10.1038/sj.cr.7290261. [DOI] [PubMed] [Google Scholar]
- Diculescu V. C.; Enache T. A. Electrochemical evaluation of Abelson tyrosine-protein kinase 1 activity and inhibition by imatinib mesylate and danusertib. Anal. Chim. Acta 2014, 845, 23–29. 10.1016/j.aca.2014.06.025. [DOI] [PubMed] [Google Scholar]
- Ghadiali J. E.; Cohen B. E.; Stevens M. M. Protein kinase-actuated resonance energy transfer in quantum dot- peptide conjugates. ACS Nano 2010, 4 (8), 4915–4919. 10.1021/nn101293s. [DOI] [PubMed] [Google Scholar]
- Herbert B.-S.; Hochreiter A. E.; Wright W. E.; Shay J. W. Nonradioactive detection of telomerase activity using the telomeric repeat amplification protocol. Nat. Protoc. 2006, 1 (3), 1583. 10.1038/nprot.2006.239. [DOI] [PubMed] [Google Scholar]
- Copeland R. A.Evaluation of enzyme inhibitors in drug discovery: a guide for medicinal chemists and pharmacologists. John Wiley & Sons: pp 245–285, 2013. [PubMed] [Google Scholar]
- Golas J. M.; Arndt K.; Etienne C.; Lucas J.; Nardin D.; Gibbons J.; Frost P.; Ye F.; Boschelli D. H.; Boschelli F. SKI-606, a 4-anilino-3-quinolinecarbonitrile dual inhibitor of Src and Abl kinases, is a potent antiproliferative agent against chronic myelogenous leukemia cells in culture and causes regression of K562 xenografts in nude mice. Cancer Res. 2003, 63 (2), 375–381. [PubMed] [Google Scholar]
- Lombardo L. J.; Lee F. Y.; Chen P.; Norris D.; Barrish J. C.; Behnia K.; Castaneda S.; Cornelius L. A.; Das J.; Doweyko A. M. Discovery of N-(2-chloro-6-methyl-phenyl)-2-(6-(4-(2-hydroxyethyl)-piperazin-1-yl)-2-methylpyrimidin-4-ylamino) thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J. Med. Chem. 2004, 47 (27), 6658–6661. 10.1021/jm049486a. [DOI] [PubMed] [Google Scholar]
- Bantscheff M.; Eberhard D.; Abraham Y.; Bastuck S.; Boesche M.; Hobson S.; Mathieson T.; Perrin J.; Raida M.; Rau C. Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nat. Biotechnol. 2007, 25 (9), 1035–1044. 10.1038/nbt1328. [DOI] [PubMed] [Google Scholar]
- Regot S.; Hughey J. J.; Bajar B. T.; Carrasco S.; Covert M. W. High-sensitivity measurements of multiple kinase activities in live single cells. Cell 2014, 157 (7), 1724–1734. 10.1016/j.cell.2014.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastie C. J.; McLauchlan H. J.; Cohen P. Assay of protein kinases using radiolabeled ATP: a protocol. Nat. Protoc. 2006, 1 (2), 968. 10.1038/nprot.2006.149. [DOI] [PubMed] [Google Scholar]
- Xia X.; Owen M.; Lee R.; Gaudet S. Cell-to-cell variability in cell death: can systems biology help us make sense of it all?. Cell Death Dis. 2014, 5 (5), e1261-e1261 10.1038/cddis.2014.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer S. L.; Gaudet S.; Albeck J. G.; Burke J. M.; Sorger P. K. Non-genetic origins of cell-to-cell variability in TRAIL-induced apoptosis. Nature 2009, 459 (7245), 428–432. 10.1038/nature08012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.