Blockade of Transforming Growth Factor β/Smad Signaling in T Cells by Overexpression of Smad7 Enhances Antigen-Induced Airway Inflammation and Airway Reactivity (original) (raw)
Abstract
Transforming growth factor (TGF)-β has been implicated in immunosuppression. However, it remains obscure whether regulation of T cells by TGF-β contributes to the immunosuppression in vivo. To address this issue, we developed transgenic mice expressing Smad7, an intracellular antagonist of TGF-β/Smad signaling, selectively in mature T cells using a plasmid construct coding a promoter element (the distal lck promoter) that directs high expression in peripheral T cells. Peripheral T cells were not growth inhibited by TGF-β in Smad7 transgenic mice. Although Smad7 transgenic mice did not spontaneously show a specific phenotype, antigen-induced airway inflammation and airway reactivity were enhanced in Smad7 transgenic mice associated with high production of both T helper cell type 1 (Th1) and Th2 cytokines. Thus, blockade of TGF-β/Smad signaling in mature T cells by expression of Smad7 enhanced airway inflammation and airway reactivity, suggesting that regulation of T cells by TGF-β was crucial for negative regulation of the inflammatory (immune) response. Our findings also implicated TGF-β/Smad signaling in mature T cells as a regulatory component of allergic asthma.
Keywords: signaling, inhibitor, asthma, mouse, eosinophils
Introduction
TGF-β is a multifunctional cytokine that has diverse effects on a variety of cell types 1. In the immune system, TGF-β regulates growth, differentiation, and function of macrophages, T cells, B cells, and NK cells 2 3 4. Importantly, TGF-β null mice developed extensive inflammation in various organs and died shortly after birth, suggesting that TGF-β played a critical role in suppression of the immune system in vivo 5 6. Downregulation of T cell function by inhibiting T cell proliferation is thought to be an important means for TGF-β–mediated immunosuppression based on in vitro studies. However, since TGF-β acts on multiple targets affecting the immune system 2, it remains obscure whether negative regulation of T cells by TGF-β contributes to the immunosuppression in vivo.
Recent identification of the Smad family of proteins has advanced our understanding of how TGF-β signals from membrane to nucleus 7 8 9 10. The activated TGF-β receptors induce phosphorylation of Smad2 and Smad3, which form heterooligomeric complexes with Smad4. The complexes then translocate to the nucleus and regulate transcriptional responses together with DNA binding cofactors. Recently, Smad7 has been identified as an intracellular antagonist of TGF-β signaling; it inhibits TGF-β–induced transcriptional responses 11 12 13. Smad7 associates with the activated TGF-β receptors and interferes with the activation of Smad2 and Smad3 by preventing their receptor interaction and phosphorylation.
To determine whether negative regulation of T cells by TGF-β contributes to immunosuppression in vivo, we have developed transgenic (Tg) mice expressing Smad7 preferentially in mature T cells using a plasmid construct coding a promoter element (the distal lck promoter) that directs high expression in peripheral T cells 14. This strategy also has the advantage of avoiding complications that may occur by blocking TGF-β/Smad signaling in T cells during early thymocyte maturation 15. Here, we showed that antigen-induced airway inflammation and airway reactivity were enhanced in Smad7 Tg mice, suggesting that TGF-β/Smad signaling in mature T cells was crucial for negative regulation of the inflammatory (immune) response. Since antigen-induced airway inflammation and airway reactivity in mice is a well-established model of allergic asthma 16, our findings also implicated TGF-β/Smad signaling in mature T cells as a regulatory component of allergic asthma.
Materials and Methods
Generation of Tg Mice.
A 1.4-kb fragment including the full-length mouse Flag-tagged Smad7 cDNA was removed from the Smad7–pcDNA3 construct 13 by digestion with HindIII and XhoI. This fragment was subcloned into a pGEM-4Z vector that had been cut with HindIII-SalI. This pGEM-4Z vector containing Smad7 was cut with BamHI, and then ligated into the BamHI site of the pUC19 vector containing the lck distal promoter 14. In the pUC19 vector, the lck distal promoter sequences were fused at the BamHI site to the human growth hormone (hGH) intronic and polyadenylation sequences, the presence of which has been found to improve the expression of cDNA transgenes in a nonspecific fashion. The diagrammatic representation of the lck distal promoter–Smad7 transgene is shown in Fig. 1 A. The SpeI fragment of these lck distal promoter–Smad7 constructions was used for microinjection. The transgenes were injected into the pronuclei of the fertilized eggs of BDF1 mice. The Tg mice were identified by PCR and slot-blot hybridization of genomic DNA to confirm the presence of hGH sequences. C57BL/6 (B6) and BDF1 mice were purchased from Clea, Inc. Transgene-positive founders were backcrossed to B6 mice to establish lines. Animal experiments were approved by the Institutional Review Boards of Chiba and Juntendo Universities.
Figure 1.
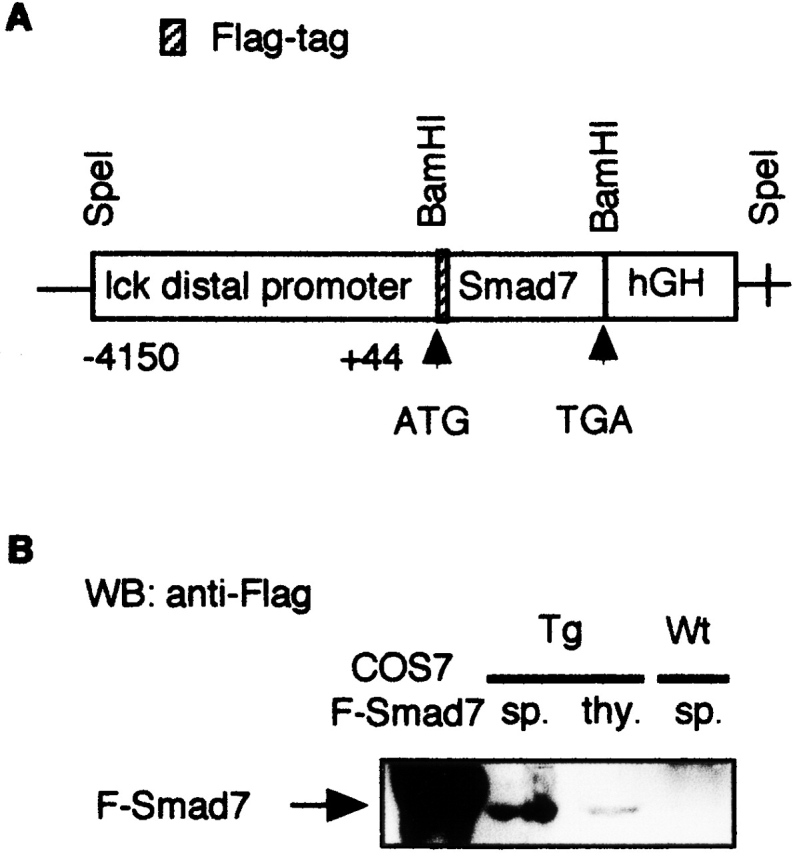
Generation of Smad7 Tg mice. (A) Schematic diagram of constructs used for peripheral T cell–specific expression of the mouse Flag-tagged Smad7 transgene. (B) Expression of the Flag-tagged Smad7 transgene in Smad7 Tg mice. Whole cell lysates were prepared from thymus (thy.) or spleen (sp.) from Smad7 Tg mice or Wt littermates, and subjected to Western blot (WB) analysis using anti-Flag antibody to detect protein expression of the Flag-tagged Smad7 transgene. As a positive control, cell lysate from COS7 cells transfected with Flag-tagged Smad7 (F-Smad7) cDNA was used.
Purification of T Cells.
Mouse CD3+ T cells were purified from spleens harvested from Smad7 Tg mice or wild-type (Wt) littermates by Ficoll-Paque (Amersham Pharmacia Biotech) gradient centrifugation of lymphocytes and magnetic cell sorting using MACS anti-CD3 microbeads (Miltenyi Biotec) following the manufacturer's recommendations. The purity of mouse CD3+ T cells was confirmed by FACScan™ (Becton Dickinson) and was consistently >98%.
In Vitro Stimulation of T Lymphocytes.
For proliferation assays, 96-well microtiter plates were coated with 2 μg/ml of anti-CD3ε mAb (2C11; BD PharMingen). Splenocytes (200 μl/well) were added at a density of 106 cells/ml and incubated in the presence or absence of TGF-β (1 ng/ml) at 37°C in 5% CO2 for 72 h. 1 μCi of [3H]thymidine (Amersham Pharmacia Biotech) was added to the culture for the last 12 h. Thymidine incorporation was assayed by harvesting cells with a PHD cell harvester (Cambridge Technologies). For detection of phosphorylation of Smad2 in T cells, purified CD3+ T cells (106 cells) from mouse spleen were stimulated with TGF-β (5 ng/ml) for 30 min followed by immunoblotting. For cytokine assays, spleen cells (5 × 106 cells) isolated from mice sensitized with OVA at 2-wk intervals were restimulated with OVA (100 μg/ml) 7 d after the second sensitization. 72 h after the culture, supernatants were collected for measurement of cytokines.
Western Blots.
Immunoblotting with anti-Flag antibody (Sigma-Aldrich), anti-Smad2 antibody (Transduction Laboratories), antiphosphorylated Smad2 antibody 17, or anti-Smad7 antibody (KAF) 17 was performed as described previously 18.
Flow Cytometry.
FACS® analysis was performed as described previously 19. In brief, thymus or spleen cells (106) suspended in PBS containing 0.1% NaN3 and 1% FCS were incubated on ice with a mixture of FITC-labeled anti-CD4, PE-labeled anti-CD8, PE-labeled anti-B220, and PE-labeled anti-CD25 antibodies (BD PharMingen). For intracellular cytokine staining, bronchoalveolar lavage (BAL) cells were stained with a mixture of Cy-Chrome–labeled anti-CD4, fixed and permeabilized using a Cytofix/Cytoperm kit (BD PharMingen), and then stained using FITC-labeled anti–IFN-γ antibody and PE-labeled anti–IL-4 antibody (BD PharMingen) according to the manufacturer's recommendations. After gating on CD4 T cells, results were analyzed on a FACSCalibur™ (Becton Dickinson) using CellQUEST™ software (Becton Dickinson).
Immunization and Exposure of Mice.
5–8-wk-old Tg mice or Wt littermates were immunized intraperitoneally with 1 μg of chicken OVA (Sigma-Aldrich) in 4 mg of aluminum hydroxide at 2-wk intervals. 10 d after the second immunization, the sensitized mice inhaled aerosolized OVA (50 mg/ml) dissolved in 0.9% saline by a DeVilbiss 646 nebulizer for 30 min for three consecutive days as described previously 20. As a control, 0.9% saline alone was administered by the nebulizer.
BAL.
BAL was performed as described previously 21. In brief, 48 h after OVA inhalation, the mice were killed by cervical dislocation, the trachea was cannulated, and the airway lumen was washed three times with 0.3 ml of PBS. The BAL fluid (BALF) was centrifuged at 400 g for 10 min at 4°C, and the amount of IL-5, IFN-γ, and TGF-β in the supernatant was measured by enzyme immunoassay as described below. The BAL cells were resuspended in 0.2 ml of PBS and counted using a hemocytometer. For differential cell counts, 2–4 × 103 cells were spun onto glass slides (Cytospin), air dried, fixed with ethanol, and stained with Giemsa solution. The number of eosinophils, neutrophils, lymphocytes, and macrophages in 200 cells was counted based on morphology.
Antigen-induced Eosinophil Infiltration in Mouse Trachea.
Antigen-induced eosinophil infiltration in mouse trachea was evaluated as described previously 20. In brief, at 48 h after the inhalation, the trachea was excised, fixed in 10% formalin, and stained with Luna solution. The number of eosinophils in the submucosal tissue of trachea was counted in Luna-stained sections and expressed as the number of eosinophils per the length of the basement membrane of trachea, which was measured with a digital curvimeter.
Cytokine ELISA.
The amount of IL-4, IL-5, and IFN-γ in the culture supernatant of spleen cells or BALF was determined using murine IL-4 and IL-5 ELISA kits (Endogen) and an IFN-γ ELISA kit (Nalgen). The amount of TGF-β in the supernatant of BALF or plasma was measured using a human TGF-β1 ELISA kit (R&D Systems) that detects mouse TGF-β1 protein because of the high homology of TGF-β1 across species. The assay detects only the active form of TGF-β1. Samples were activated before measuring according to the manufacturer's recommendations. The minimum significant value of IL-4, IL-5, IFN-γ, and TGF-β1 was 5, 5, 1, and 5 pg/ml, respectively.
Measurement of Airway Responsiveness.
Airway responsiveness to acetylcholine challenge was measured as described 21. In brief, at 24 h after OVA or saline inhalation, mice were anesthetized with pentobarbital (50 mg/kg; Abbott Laboratories), intubated with a 20-gauge tracheal cannula, and ventilated at a rate of 120 breaths/min with a constant tidal volume of air (0.2 ml). Airway pressure was measured with a pressure transducer via a port of the tracheal cannula. Muscle paralysis was provided by intravenous administration of pancuronium bromide (0.1 mg/kg; Tocris Cookson). After establishment of a stable airway pressure recording, acetylcholine (500 mg/kg; Wako) was injected intravenously, and the changes in airway pressure were recorded. Airway responsiveness was defined by the time-integrated change in peak airway pressure (airway pressure–time index [APTI]; cmH2O · s).
Data Analysis.
Data are summarized as mean ± SD. The statistical analysis of the results was performed by analysis of variance using Fisher's least significant difference test for multiple comparisons. P < 0.05 was considered significant.
Results
Generation of Smad7 Tg Mice.
To express the Smad7 transgene preferentially in peripheral T cells, we used a plasmid construct expressing Flag-tagged mouse Smad7 13 under the control of a promoter element (the distal lck promoter) that directed high expression in mature thymocytes and peripheral T cells (14; Fig. 1 A). The strategy also has the advantage of avoiding complications that may occur by blocking TGF-β/Smad signaling in T cells during early thymocyte maturation 15. We obtained five transgenic lines, out of which two lines displayed relatively high levels of transgene expression in the spleen (Fig. 1 B). Some level of expression was detected in the thymus (Fig. 1 B), and little expression was observed in the lungs or other organs that were not enriched in T cells (data not shown). We also confirmed expression of the Smad7 transgene in peripheral blood lymphocytes and CD3+ T cells purified from the spleens of Smad7 Tg mice (data not shown). In addition, endogenous Smad7 protein was not detected in CD3+ T cells purified from the spleens of Wt mice and Smad7 Tg mice by Western blotting with anti-Smad7 antibody (data not shown).
Lack of Responsiveness to TGF-β in Splenic T Cells from Smad7 Tg Mice.
To determine whether peripheral T cells expressing the Smad7 transgene were resistant to TGF-β action, we examined the proliferation of splenocytes isolated from Smad7 Tg mice and Wt littermates when activated with mAb to CD3 in the presence and absence of TGF-β. Without stimulation of mAb to CD3, splenocytes from Smad7 Tg mice and Wt littermates did not proliferate (data not shown). The growth-inhibitory effect of TGF-β was largely lost in splenocytes from Smad7 Tg mice when activated with mAb to CD3, although similarly treated Wt splenocytes were growth inhibited ∼60% by TGF-β (Fig. 2 A). The purified population of CD3+ T cells isolated from the spleens of Smad7 Tg mice and activated by anti-CD3 was also insensitive to the inhibitory effect of TGF-β (data not shown). Interestingly, the non–TGF-β–treated levels of proliferation were about threefold higher in splenocytes from Smad7 Tg mice than in those from Wt littermates (Fig. 2 A).
Figure 2.
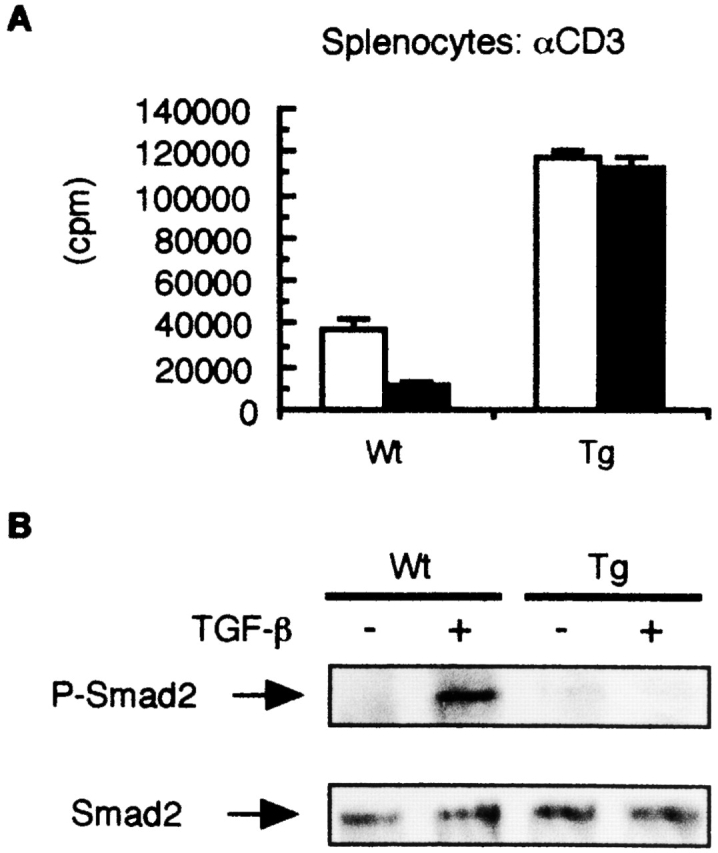
Altered responsiveness of splenic T cells from Smad7 Tg mice to TGF-β. (A) Lack of responsiveness to TGF-β in splenocytes from Smad7 Tg mice. Splenocytes from Smad7 Tg mice or Wt littermates were stimulated with anti-CD3 mAb in the presence or absence (control) of TGF-β (1 ng/ml). Aliquots of cells were pulsed with [3H]thymidine, and incorporated counts (in cpm) were determined. Shown are means ± SD of [3H]thymidine corporation from triplicate cultures. The results are from one representative experiment out of three. (B) Inhibition of TGF-β–mediated phosphorylation of Smad2 in purified T cells from Smad7 Tg mice. Cell lysates were prepared from purified CD3+ T cells from Smad7 Tg mice or Wt littermates, and then immunoblotted with antiphosphorylated Smad2 antibody (top, P-Smad2) or anti-Smad2 antibody (bottom). The results are from one representative experiment out of three.
Phosphorylation of Smad2 and Smad3 by activated TGF-β receptors is a key event for the initiation of TGF-β signal transduction 18 22, and Smad7 antagonizes TGF-β signaling by preventing TGF-β receptor–mediated phosphorylation of Smad2 and Smad3 13. Thus, we next determined whether phosphorylation of Smad2 by TGF-β was inhibited in peripheral T cells from Smad7 Tg mice. As shown in Fig. 2 B, we found that TGF-β–mediated phosphorylation of Smad2 was suppressed in CD3+ T cells purified from the spleens of Smad7 Tg mice in contrast to those from Wt littermates (Fig. 2 B). These findings indicated that peripheral T cells from Smad7 Tg mice were insensitive to TGF-β.
Normal T Cell and B Cell Development in Smad7 Tg Mice.
To determine whether any developmental defect in the immune system was observed in Smad7 Tg mice, we examined thymic and splenic T and B cell populations by performing FACS® analysis. As shown in Fig. 3 A, the thymi of Smad7 Tg mice contained normal proportions of CD4 and CD8 single- and double-positive T cells, suggesting that thymic T cell maturation is normal in Smad7 Tg mice. Similarly, the spleens from Smad7 Tg mice contained normal numbers and percentages of B cells and CD4 and CD8 single-positive cells, suggesting that there was not an abnormal expansion of lymphocytes in the spleens of Smad7 Tg mice (Fig. 3 B). These findings indicated that Smad7 Tg mice did not have any developmental defect in their immune system.
Figure 3.
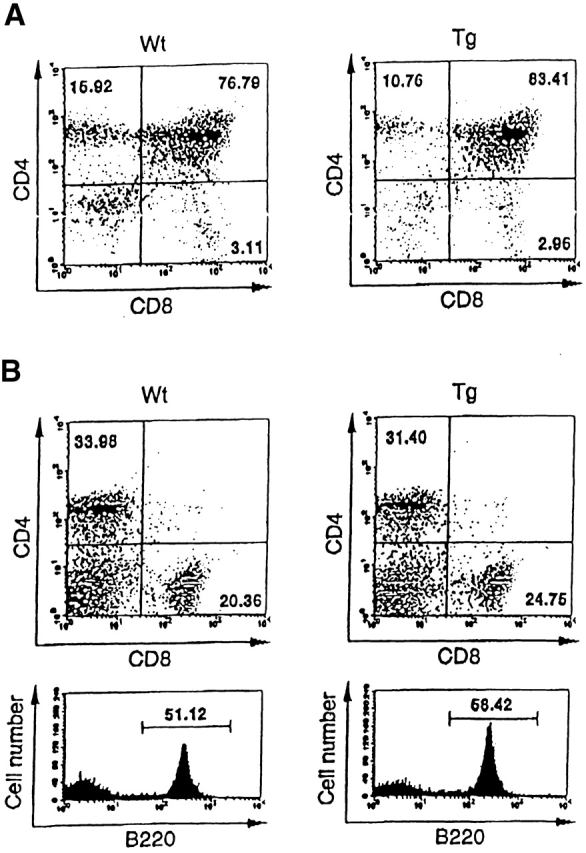
Normal T cell and B cell development in Smad7 Tg mice. FACS® analysis of thymocytes (A) or splenocytes (B) using the indicated conjugated antibodies. All data were gated for viable cells. Percentages represent proportions of viable cells in each region or quadrant. The results are from one representative experiment out of three.
Normal Phenotype of Smad7 Tg Mice.
The Smad7 Tg mice we developed were viable, survived to adulthood, and were phenotypically indistinguishable from their littermate controls at birth and at least until 6 mo of age. Histological examination of lungs or other organs did not show any morphological abnormalities or inflammatory lesions in Smad7 Tg mice (data not shown).
Enhanced Antigen-induced Airway Inflammation in Smad7 Tg Mice.
Since Smad7 Tg mice showed a normal phenotype, contrary to our expectations, we asked whether Smad7 Tg mice were any different in pathological situations, particularly in inflammation. Thus, to determine whether TGF-β/Smad signaling in T cells contributes to regulation of the inflammatory response, we developed airway inflammation in Smad7 Tg mice by inhalation of antigen (OVA) after antigen sensitization as described previously 20. 48 h after the inhaled OVA challenge, BALF was recovered from the animal, and the total cell number in the BALF was counted by cell differentiation analysis. As shown in Fig. 4 A, the total cell count in the BALF recovered from Smad7 Tg mice upon OVA challenge was threefold higher than that in the BALF derived from Wt littermates. Cell differentiation analysis revealed that the number of eosinophils, neutrophils, and lymphocytes in the BALF was significantly increased in Smad7 Tg mice compared with Wt littermates, indicating that OVA-induced airway inflammation was enhanced in Smad7 Tg mice in general (Fig. 4 A). In addition, eosinophil infiltration into the mouse trachea after OVA inhalation was also two- to fourfold higher in Smad7 Tg mice than in Wt littermates (Fig. 4 B). These findings indicated that antigen-induced airway inflammation was enhanced in Smad7 Tg mice, suggesting that TGF-β/Smad signaling in mature T cells had a negative regulatory role in the development of antigen-induced airway inflammation.
Figure 4.
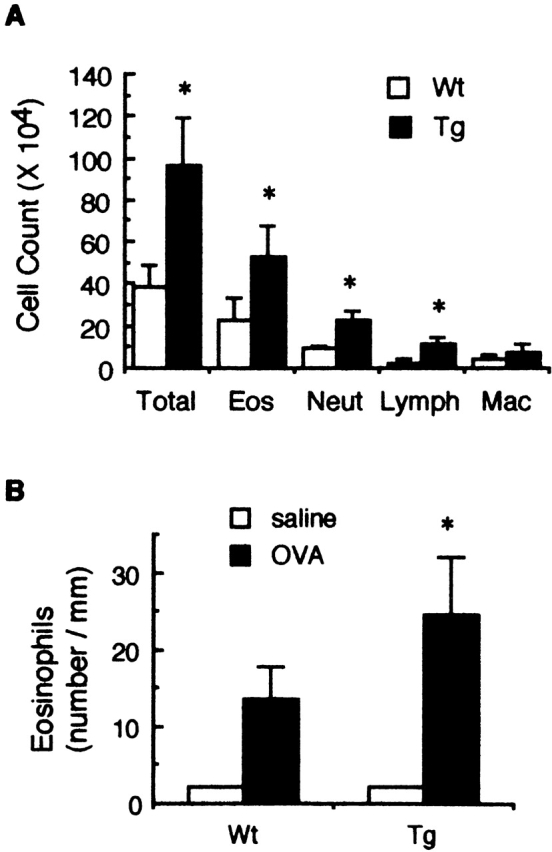
Antigen-induced airway inflammation in Smad7 Tg mice. OVA-sensitized mice were challenged with inhalation of OVA as described in Materials and Methods. Leukocyte infiltration in the BALF (A) and eosinophil infiltration into the trachea (B) of the sensitized mice were then examined at 48 h after OVA inhalation. Data are means ± SD for five mice in each group. The results are from one representative experiment out of three. Eos, eosinophils; Neut, neutrophils; Lymph, lymphocytes; Mac, macrophages. *P < 0.05, significantly different from the mean value of the corresponding control response (Wt).
Cytokine Production in Smad7 Tg Mice.
Since antigen-induced airway inflammation was enhanced in Smad7 Tg mice, we focused on the mechanisms underlying these observations. Because we have previously shown that IL-5 and IFN-γ regulate antigen-induced airway inflammation positively and negatively, respectively 20 23, we examined antigen-induced IL-5 and IFN-γ production in the airways of sensitized mice. IL-5 and IFN-γ levels in the BALF of Smad7 Tg mice were significantly higher at 48 h after OVA inhalation than those of Wt littermates (Fig. 5 A). Little IL-5 or IFN-γ was detected in the BALF of Smad7 Tg mice or Wt littermates after saline inhalation (data not shown). In addition, in vitro OVA-induced IL-4, IL-5, and IFN-γ production was significantly higher in the spleen cells of Smad7 Tg mice than those of Wt littermates (Fig. 5 B). TGF-β levels in the BALF of Smad7 Tg mice at 48 h after OVA inhalation were equivalent to those of Wt littermates (Fig. 5 C). In addition, there was no significant difference in plasma TGF-β levels between Smad7 Tg mice and Wt littermates (Smad7 Tg mice: 1.4 ± 0.4 ng/ml; Wt littermates: 1.2 ± 0.5 ng/ml; n = 5, P = 0.49). These findings indicated that antigen-induced production of Th1 and Th2 cytokines was higher in Smad7 Tg mice than in Wt littermates.
Figure 5.

Cytokine production in Smad7 Tg mice. OVA-sensitized mice were challenged with inhalation of OVA. At 48 h after the inhalation, BALF was collected as described in Materials and Methods. For detection of cytokines in the culture supernatant, spleen cells were isolated from the mice sensitized with OVA/alum and stimulated with OVA (100 μg/ml) in vitro. 72 h after the stimulation, the culture supernatants were collected. IL-5 and IFN-γ levels in the BALF (A), OVA-induced IL-4, IL-5, and IFN-γ production in the supernatant of spleen cells from the sensitized mice (B), and TGF-β1 levels in the BALF (C) were determined by ELISA. Data are means ± SD for five mice in each group. The results are from one representative experiment out of three. *P < 0.05, significantly different from the mean value of the corresponding control response (Wt).
Activated Phenotype of T Lymphocytes in the Airways of Smad7 Tg Mice.
Since we found enhanced IL-5 and IFN-γ levels in the BALF of Smad7 Tg mice upon OVA challenge (Fig. 5 A), we further assessed T cell phenotype in the BALF of Smad7 Tg mice. As shown in Fig. 6, the number of CD4 T cells, CD8 T cells, and CD25+ CD4 T cells (activated phenotype) in the BALF of Smad7 Tg mice was higher than that of Wt littermates upon OVA challenge. Little T cell infiltration into the BALF of Smad7 Tg mice and Wt littermates was observed upon saline challenge (data not shown). In addition, IL-4 and IFN-γ production in CD4 T cells in the BALF was enhanced in Smad7 Tg mice (Fig. 6 C). These findings indicated that T cell infiltration into the airways of Smad7 Tg mice was enhanced and that the infiltrating T cells were more activated in Smad7 Tg mice than in Wt littermates.
Figure 6.
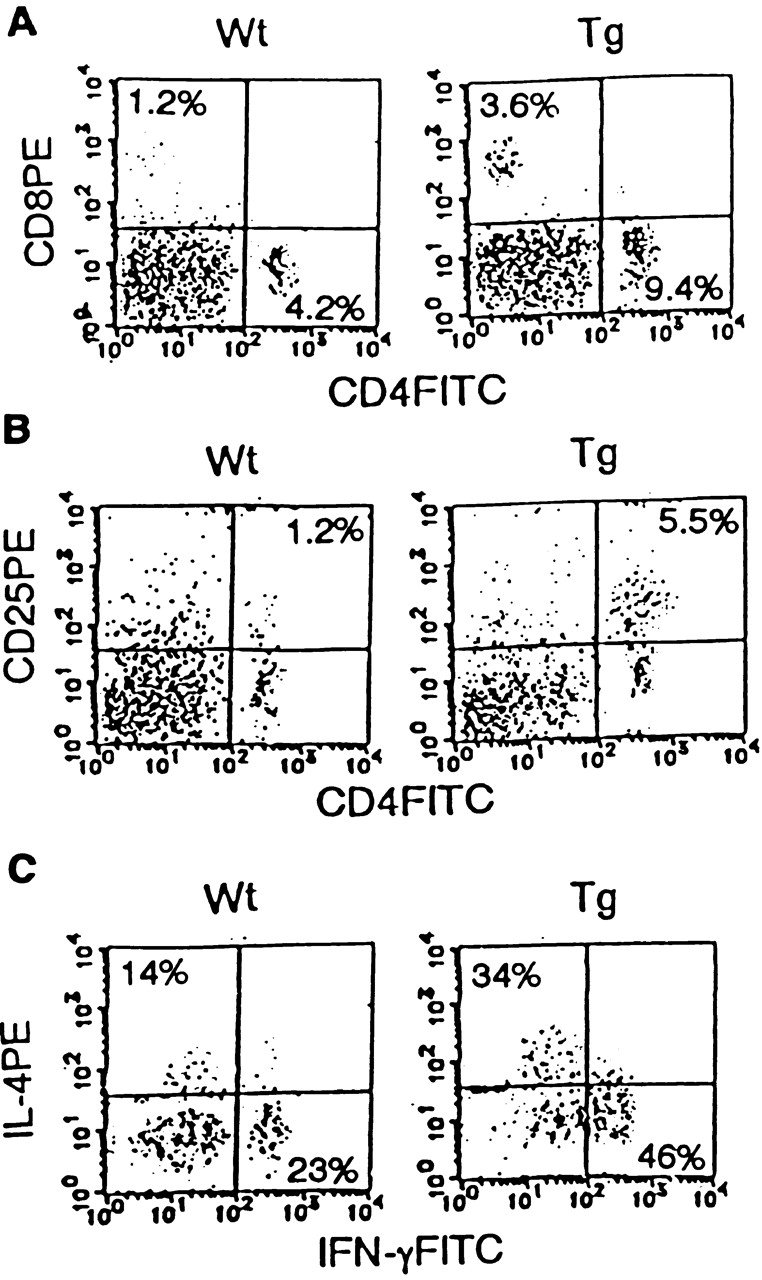
Activated phenotype in T lymphocytes in the airways of Smad7 Tg mice. Representative FACS® profiles of CD4 versus CD8 staining (A), CD4 versus CD25 staining (B), and intracellular staining of IL-4 versus IFN-γ in CD4 T cells (C), on BALF cells using anti-CD4–FITC, anti-CD8–PE, anti-CD25–PE, anti–IFN-γ–FITC, and anti–IL-4–PE antibodies (n = 5 mice in each group). Percentages represent proportions of viable cells in each region or quadrant.
Airway Hyperresponsiveness in Smad7 Tg Mice.
Airway hyperresponsiveness (AHR) is associated with airway inflammation and Th2 cytokines, and is a central feature of asthma 16. Thus, we examined antigen-induced AHR in Smad7 Tg mice. Inhaled OVA challenge of sensitized Wt mice resulted in a significant increase in airway responsiveness to acetylcholine (APTI: control 1,812 ± 64 vs. OVA 2,246 ± 98; n = 5, P < 0.005; Fig. 7) as described previously 21. OVA-induced AHR in sensitized Smad7 Tg mice was significantly higher than that in Wt littermates (APTI: Wt 2,246 ± 98 vs. Tg 2,742 ± 109; n = 5, P < 0.005; Fig. 7). These results indicated that overexpression of Smad7 in mature T cells enhanced antigen-induced AHR, suggesting that TGF-β/Smad signaling in mature T cells played a negative role in antigen-induced AHR in sensitized mice.
Figure 7.

Airway responsiveness to acetylcholine of Smad7 Tg mice. OVA-sensitized mice were challenged with inhalation of OVA (black bars) or saline (white bars). At 24 h after the inhalation, airway responsiveness was assessed by the time-integrated change in peak airway pressure (APTI; cmH2O · s) after intravenous acetylcholine challenge. Data are means ± SD for five mice in each group. The results are from one representative experiment out of three. *P < 0.05, significantly different from the mean value of the corresponding control response (Wt).
Discussion
In this study, we developed Tg mice expressing Smad7 under the control of a distal lck promoter 14, so that we were able to block the TGF-β/Smad signaling pathway preferentially in peripheral T cells. The Tg mice enabled us to determine whether TGF-β/Smad signaling in mature T cells contributed to regulation of the immune system in vivo. We found that Smad7 Tg mice did not spontaneously show a specific phenotype, but showed enhanced antigen-induced airway inflammation and airway reactivity (Fig. 4 and Fig. 7). The findings suggested that TGF-β/Smad signaling in mature T cells contributed to negative regulation of the inflammatory (immune) response, but not to T cell homeostasis under normal circumstances.
Antigen-induced airway inflammation and airway reactivity in mice is a well-established model of allergic asthma 16. Thus, the finding that overexpression of Smad7 in mature T cells enhanced antigen-induced airway inflammation and airway reactivity (Fig. 4 and Fig. 7) also implicated TGF-β/Smad signaling as a regulatory component of allergic asthma. The role of TGF-β/Smad signaling in the pathophysiology of asthma remains unclear. Since our results suggested the importance of TGF-β/Smad signaling in the suppression of allergic airway inflammation, the pathological role of TGF-β/Smad signaling in asthmatic airways should be investigated in the future.
Contrary to our prediction based on the previous finding that TGF-β null mice developed severe inflammation in various organs and an autoimmune phenotype 5 6, Smad7 Tg mice did not spontaneously show a specific phenotype. We also found that development of T cells and B cells was normal in Smad7 Tg mice (Fig. 3). These findings suggested that TGF-β/Smad signaling in mature T cells was not critical for T cell homeostasis under normal circumstances.
The spontaneous phenotype presented here was also distinct from those recently reported by three groups in Smad3 null mice in which splenic T cells were insensitive to TGF-β 19 24 25. Intestinal malignancy 24, a progressive illness associated with opportunistic infection 19, and an early phenotype of forelimb malformation 25 were not observed in our Smad7 Tg mice. In addition, impaired local inflammation in the process of wound healing was recently reported in Smad3 null mice 26. Since TGF-β acts on multiple targets including epithelial cells, neutrophils, and macrophages/monocytes 1, Smad3 null mice should have defects in a variety of TGF-β–responsive cells. For instance, the reduced inflammation during the process of wound healing could be attributed to a blunted chemotactic response of monocytes and neutrophils to TGF-β 26. In our Smad7 Tg mice, those cells are intact in the TGF-β response. Thus, the T cell–selective inhibition of TGF-β/Smad signaling could explain at least in part the phenotypic differences observed between Smad7 Tg mice and Smad3 null mice.
Antigen-induced airway inflammation was enhanced in Smad7 Tg mice (Fig. 4). Since CD4 T cells and IL-5 play a central role in regulating antigen-induced airway inflammation 20 and antigen-induced IL-5 production was enhanced in Smad7 Tg mice (Fig. 5), the enhanced antigen-induced airway inflammation could be attributed to the enhanced IL-5 production in Smad7 Tg mice. CD4 T cells were thought to be a major source of IL-5 production in the airways because we had preliminary data that IL-5 (and also INF-γ) production in the BALF of sensitized mice after OVA challenge was not detected after depletion of CD4 T cells with anti-CD4 antibody immediately before the OVA challenge (data not shown).
Recently, a cooperative role of Th1 cells with Th2 was reported 27 28, indicating that IFN-γ at physiologically relevant levels was insufficient to neutralize the effects of IL-5 in vivo. Thus, although we observed enhanced IFN-γ production in Smad7 Tg mice (Fig. 5), relative contributions of IFN-γ to the airway inflammation might be small.
We found that T cells infiltrating into the airways were increased and more activated in Smad7 Tg mice (Fig. 6). In addition, production of TGF-β, which was shown to inhibit the production of and response to cytokines in T cells 4, in the airways of Smad7 Tg mice was equivalent in Wt littermates (Fig. 5 C). Taken together, these findings suggested that enhanced IL-5 and IFN-γ production in the airways of Smad7 Tg mice might be due to the enhanced activation of T cells that could have escaped from the inhibitory action of TGF-β.
Another reason for the enhanced cytokine production in Smad7 Tg mice might be that blockade of TGF-β/Smad signaling by Smad7 in T cells augmented differentiation of naive T cells into both Th1 and Th2 cells, resulting in enhanced cytokine production. The role of TGF-β/Smad signaling in T cell differentiation towards Th1 and/or Th2 subsets is still unclear 4. Our findings of higher production of IL-4, IL-5, and IFN-γ in Smad7 Tg mice than in Wt littermates (Fig. 5 and Fig. 6) suggested that generation of Th1 and Th2 cells was enhanced under the conditions in which differentiation of T cells was not influenced by TGF-β. However, more defined experiments using a purified population of CD4+ T cells are clearly needed to claim that generation of Th1 and Th2 cells was enhanced in Smad7 Tg mice.
Furthermore, most recently Gorelik et al. showed that abrogation of TGF-β signaling in T cells led to a spontaneous autoimmune phenotype using Tg mice expressing the dominant-negative TGF-β type II receptor selectively in T cells 29. Their Tg mice developed inflammatory infiltration in several organs and circulating autoimmune antibodies after 3–4 mo of age, which were not seen in our Smad7 Tg mice. The different mouse backgrounds may explain this difference, since their Tg mice were backcrossed onto a B10.BR background whereas our Tg mice were backcrossed onto a B6 background. Another explanation is that there may be TGF-β signals that are not blocked by overexpression of Smad7 in T cells, which would lead to the largely normal phenotype in our Smad7 Tg mice. The latter possibility should be investigated in the future.
In summary, we showed that blockade of TGF-β/Smad signaling in mature T cells by overexpression of Smad7 enhanced antigen-induced airway inflammation and airway reactivity. Our findings suggested that TGF-β/Smad signaling in mature T cells was crucial for negative regulation of the inflammatory (immune) response. Our findings also implicated TGF-β/Smad signaling in mature T cells as a regulatory component of allergic asthma.
Acknowledgments
We thank M. Mamura, H. Ushio, and K. Maeda for helpful discussion and technical assistance, T. Nakayama for the distal lck promoter plasmid construct, S. Itoh and P. ten Dijke for anti-Smad antibodies, and E. Kamijima for excellent secretarial assistance.
This work was supported in part by grants from the Ministry of Education, Science, and Culture, Japan, the Mochida Foundation (to A. Nakao), and the Naito Foundation (to A. Nakao).
Footnotes
Abbreviations used in this paper: AHR, airway hyperresponsiveness; APTI, airway pressure–time index; B6, CB57L/6; BAL, bronchoalveolar lavage; BALF, BAL fluid; hGH, human growth hormone; Tg, transgenic; Wt, wild-type.
A. Nakao and S. Miike contributed equally to this work.
References
- Roberts A.B., Sporn M.B. The transforming growth factors-β. In: Sporn M.B., Roberts A.B., editors. Handbook of Experimental Pharmacology. Springer-Verlag; Heidelberg: 1990. pp. 418–472. [Google Scholar]
- Wahl S.M. Transforming growth factor-β in inflammationa cause and a cure. J. Clin. Immunol. 1992;12:61–74. doi: 10.1007/BF00918135. [DOI] [PubMed] [Google Scholar]
- Miyazono K., ten Dijke P., Heldin C.-H. Receptors for transforming growth factor-β. Adv. Immunol. 1994;55:181–220. [PubMed] [Google Scholar]
- Letterio J.J., Roberts A.B. Regulation of immune responses by TGF-β. Annu. Rev. Immunol. 1998;16:137–161. doi: 10.1146/annurev.immunol.16.1.137. [DOI] [PubMed] [Google Scholar]
- Shull M.M., Ormsby I., Kier A.B., Pawlowski S., Diebold R.J., Yin M., Allen R., Sidman C., Proetzel G., Calvin D. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature. 1992;359:693–699. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni A.B., Huh C.G., Becker D., Geiser A., Lyght M., Flanders K.C., Roberts A.B., Sporn M.B., Ward J.M., Karlsson S. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc. Natl. Acad. Sci. USA. 1993;90:770–774. doi: 10.1073/pnas.90.2.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derynck R., Feng X.H. TGF-β receptor signaling. Biochem. Biophys. Acta. 1997;1333:F105–F150. doi: 10.1016/s0304-419x(97)00017-6. [DOI] [PubMed] [Google Scholar]
- Heldin C.-H., Miyazono K., ten Dijke P. TGF-β signalling from cell membrane to nucleus through SMAD proteins. Nature. 1997;390:465–471. doi: 10.1038/37284. [DOI] [PubMed] [Google Scholar]
- Attisano L., Wrana J. Mads and Smads in TGF-β signaling. Curr. Opin. Cell Biol. 1998;10:188–194. doi: 10.1016/s0955-0674(98)80141-5. [DOI] [PubMed] [Google Scholar]
- Massague J. TGF-β signal transduction. Annu. Rev. Biochem. 1998;67:753–791. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- Hayashi H., Abdollah S., Qiu Y., Cai J., Xu Y.Y., Grinnell B.W., Richardson M.A., Topper J.N., Gimbrone M.A., Jr., Wrana J., Falb D. The MAD-related protein Smad7 associates with the TGF-β receptor and functions as an antagonist of TGF-β signaling. Cell. 1997;89:1165–1173. doi: 10.1016/s0092-8674(00)80303-7. [DOI] [PubMed] [Google Scholar]
- Topper J.N., Cai J., Qiu Y., Anderson K.R., Xu Y.Y., Deeds J.D., Feeley R., Gimeno C.J., Woolf E.A., Tayber O. Vascular MADstwo novel MAD-related genes selectively inducible by flow in human vascular endothelium. Proc. Natl. Acad. Sci. USA. 1997;94:9314–9319. doi: 10.1073/pnas.94.17.9314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakao A., Afrakhte M., Moren A., Nakayama T., Christian J.L., Heuchel R., Itoh S., Kawabata M., Heldin N.E., Heldin C.-H., ten Dijke P. Identification of Smad7, a TGF-β-inducible antagonist of TGF-β signaling. Nature. 1997;389:631–635. doi: 10.1038/39369. [DOI] [PubMed] [Google Scholar]
- Wildin R.S., Wang H.U., Forbush K.A., Perlmutter R.M. Functional dissection of the murine lck distal promoter. J. Immunol. 1995;155:1286–1295. [PubMed] [Google Scholar]
- Plum J., De Smedt M., Leclercq G., Vandekerckhove B. Influence of TGF-β on murine thymocyte development in fetal thymus organ culture. J. Immunol. 1995;154:5789–5798. [PubMed] [Google Scholar]
- Wills-Karp M. Immunological basis of antigen-induced airway hyperresponsiveness. Annu. Rev. Immunol. 1999;17:255–281. doi: 10.1146/annurev.immunol.17.1.255. [DOI] [PubMed] [Google Scholar]
- Brodin G., ten Dijke P., Funa K., Heldin C.-H., Landstrom M. Increased smad expression and activation are associated with apoptosis in normal and malignant prostate after castration. Cancer Res. 1999;59:2731–2738. [PubMed] [Google Scholar]
- Nakao A., Imamura T., Souchenlnytskyi S., Kawabata M., Ishisaki A., Oeda E., Tamaki K., Hanai J., Heldin C.-H., Miyazono K., ten Dijke P. TGF-β receptor-mediated signaling through Smad2, Smad3, and Smad4. EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:5353–5362. doi: 10.1093/emboj/16.17.5353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datto M.B., Frederick J.P., Pan L., Borton A.J., Zhang Y., Wang W.-F. Targeted disruption of Smad3 reveals an essential role in transforming growth factor β-mediated signal transduction. Mol. Cell. Biol. 1999;19:2495–2504. doi: 10.1128/mcb.19.4.2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima H., Iwamoto I., Tomoe R., Matsumura R., Tomioka H., Takatsu K., Yoshida S. CD4 positive T lymphocytes and interleukin-5 mediate antigen-induced eosinophil infiltration into the mouse trachea. Am. Rev. Respir. Dis. 1992;144:374–377. doi: 10.1164/ajrccm/146.2.374. [DOI] [PubMed] [Google Scholar]
- Kumano K., Nakao A., Nakajima H., Hayashi F., Kurimoto M., Okamura H., Saito Y., Iwamoto I. Interleukin-18 enhances antigen-induced eosinophil recruitment into the mouse airways. Am. J. Respir. Crit. Care Med. 1999;160:873–878. doi: 10.1164/ajrccm.160.3.9805026. [DOI] [PubMed] [Google Scholar]
- Macias-Silva M., Abdollah S., Hoodless P.A., Pirone R., Attisano L., Wrana J. MADR2 is a substrate of the TGF-β receptor and its phosphorylation is required for nuclear accumulation and signaling. Cell. 1996;87:1215–1224. doi: 10.1016/s0092-8674(00)81817-6. [DOI] [PubMed] [Google Scholar]
- Iwamoto I., Nakajima H., Endo H., Yoshida S. Interferon-γ regulates antigen-induced eosinophil recruitment into the mouse airways by inhibiting the infiltration of CD4 T cells. J. Exp. Med. 1993;177:573–576. doi: 10.1084/jem.177.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y., Richardson J.A., Parada L.F., Graff J.M. Smad3 mutant mice develop metastatic colorectal cancer. Cell. 1998;94:703–714. doi: 10.1016/s0092-8674(00)81730-4. [DOI] [PubMed] [Google Scholar]
- Yang X., Letterio J.L., Lechleider R.J., Chen L., Hayman R., Gu H., Roberts A.B., Deng C. Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T cell responsiveness to TGF-β. EMBO (Eur. Mol. Biol. Organ.) J. 1999;18:1280–1291. doi: 10.1093/emboj/18.5.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft G.S., Yang X., Glick A.B., Weinstein M., Letterio J.L., Mizel D.E., Anzano M., Greenwell-Wild T., Wahl S.M., Deng C., Roberts A.B. Mice lacking Smad3 show accelerated wound healing and an impaired local inflammatory response. Nat. Cell Biol. 1999;1:260–266. doi: 10.1038/12971. [DOI] [PubMed] [Google Scholar]
- Hansen G., Berry G., DeKruyff R.H., Umetsu D.T. Allergen-specific Th1 cells fail to counterbalance Th2 cell–induced airway hyperreactivity but cause severe airway inflammation. J. Clin. Invest. 1999;103:175–183. doi: 10.1172/JCI5155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randolf D.A., Stephens R., Carruthers C.J.L., Chaplin D.D. Cooperation between Th1 and Th2 cells in a murine model of eosinophilic airway inflammation. J. Clin. Invest. 1999;104:1021–1029. doi: 10.1172/JCI7631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorelik L., Flavell R.A. Abrogation of TGF-β signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity. 2000;12:171–181. doi: 10.1016/s1074-7613(00)80170-3. [DOI] [PubMed] [Google Scholar]