Non-self antigens are the cognate specificities of Foxp3+ regulatory T cells (original) (raw)
. Author manuscript; available in PMC: 2008 Sep 1.
Abstract
The majority of regulatory Foxp3+CD4+ T cells naturally arises in the thymus. It has been proposed that T cell receptors (TCRs) on these cells recognize self MHC class II-peptide complexes with high or higher affinity, and that their specificities mirror specificities of autoreactive T cells. Here we analyzed hundreds of TCRs derived from regulatory or non-regulatory T cells and found little evidence that the former population preferably recognizes self-antigens as agonists. Instead, these cells recognized foreign MHC-peptide complexes as often as non-regulatory T cells. Our results show that high affinity, autoreactive TCRs are rare on all CD4+ T cells and suggest that selecting self-peptide is different from the peptide that activates the same regulatory T cells in the periphery.
Introduction
Central tolerance eliminates thymocytes expressing T cell receptors (TCRs) with high affinity to self antigens, but routinely a small fraction of potentially pathogenic T cells escape this process. In the periphery, autoreactive T cells are controlled by active suppression mechanism carried out by regulatory CD25+CD4+ T (Treg) cells that express transcription factor Foxp3 (Hori et al., 2003; Fontenot et al., 2003; Khattri et al., 2003; Shevach et al., 2006). Foxp3 was initially considered as a lineage specification factor that determines the fate of thymic precursors of Treg cells (Fontenot et al., 2005a; Fontenot et al., 2005b). However, as recently shown in different mouse models with a fluorescent reporter knock-in gene, Foxp3 does not act as a master switch for Treg cell lineage commitment (Gavin et al., 2007; Wan and Flavell, 2007). Instead, it stabilizes the phenotype and suppressive function of anergic thymocytes by altering the wide array of Treg cell-specific genes (Gavin et al., 2007; Zheng et al., 2007). Thus, although Foxp3 and the suppressive functions of Treg cells can now be correlated with its late expression in mature CD4+CD8− thymocytes (Fontenot et al., 2005a), the signals inducing the lineage commitment of these cells and Foxp3 expression remain elusive. It has been proposed that some of the thymocytes that recognize self MHC-peptide complexes with high affinity, instead of dying during negative selection or becoming autoreactive T cells, would acquire Foxp3 and differentiate to Treg cells (Coutinho et al., 2005; Fontenot and Rudensky, 2005; Liu, 2006; Cabarrocas et al., 2006).
The direct evidence implying that precursors of Treg cells require high affinity, self-reactive TCRs to become Treg cells came from the TCR transgenic mouse model that co-express cognate peptide in the thymus (Jordan et al., 2001). In this model, interactions between TCR and class II MHC bound with strong but not weak agonist peptide lead to the selection of Foxp3+ Treg cells with regulatory properties (Caton et al., 2004; D'Cruz and Klein, 2005; Kim and Rudensky, 2006). It was also shown in the thymic medulla that expression of agonist peptides induces a non-deletional differentiation to Foxp3+ Treg cells, and that in the periphery suboptimal dose of cognate antigen can convert naive, TCR transgenic cells to Foxp3+ Treg cells (Apostolou et al., 2002; Watanabe et al., 2005; Liu, 2006; Aschenbrenner et al., 2007). However, another study that used different, intrathymically expressed agonist peptide could not reproduce selection of Treg cells, suggesting that factors other than agonist peptides may induce natural differentiation of Treg cells (van Santen et al., 2004).
If higher affinity TCR-MHC-peptide interactions control natural Treg differentiation one should also expect that these cells abundantly express self-reactive TCRs. Indeed, experiments performed in TCRβ transgenic mice, where conventional CD4+ T cells were transduced with TCRs derived from Treg cells, showed that in lymphopenic hosts and in co-cultures with autologous APCs, these transductants proliferate faster than transductants expressing TCRs cloned from naive T cells (Hsieh et al., 2004). Moreover, when these TCRβ transgenic mice were crossed with Foxp3 deficient mice that are prone to autoimmunity, the T and autoreactive Foxp3−CD25+ cells but not the Foxp3−CD25− cells expressed TCRs with overlapping antigenic specificities (Hsieh et al., 2006). These results imply that Treg cells express TCRs with higher affinity to self and that these TCRs overlap with TCRs expressed by pathogenic, self-reactive T cells. However, this proposition remains uncertain, as the contribution of Foxp3-deficient Treg cells in the pathogenesis of scurfy male mice is still unclear and transfer of these cells does not induce autoimmune diseases in lymphopenic hosts (Lin et al., 2007). In addition, the incidence of TCRs specific to foreign antigens on these cells has also been confusing. There are multiple experimental reports that Treg cells recognize exogenous antigens derived from microbes, parasites, neo-antigens or allogeneic tissues, but it is unclear whether these responses are antigen specific or rely on inherent cross-reactivity, dual TCRs or a bystander effect (Belkaid et al., 2002; Suvas et al., 2003; Ochando et al., 2005; Tuovinen et al., 2006; Lerman et al., 2004; Larkin, III et al., 2007). Interestingly, analyses of TCRs in mice expressing a TCRβ transgene or miniature repertoire of TCRs found that dominant TCRs on naive T and regulatory T cells overlap to some extent (Hsieh et al., 2004; Pacholczyk et al., 2006; Wong et al., 2007) and there is an increased diversity of TCRs on Treg cells, suggesting that this repertoire encodes multiple antigen specificities (Pacholczyk et al., 2006). Therefore here we wanted to comprehensively analyze antigenic specificities of Foxp3+ Treg cells and to determine if their TCRs are inherently autoreactive.
For this purpose we characterized hundreds of TCRs originally expressed by CD4+Foxp3+ Treg or CD4+Foxp3− naive T cells and concluded that the frequency of TCRs with high-affinity for self versus non-self antigens is similar within the two CD4+ populations. Our results also demonstrate that Treg cells maintain a highly diverse TCR repertoire that included most of the TCRs dominantly expressed by naive T cells. In contrast, many TCRs expressed by Treg cells were not found on naive CD4+ T cells. Finally, we showed that an outbreak of immune wasting disease in lymphopenic mice relied on the hosts class II MHC ability to present foreign and not self antigens. Therefore, when wasting disease is induced by T cells bearing Treg-derived TCRs (Hsieh et al., 2004), these TCRs recognize non-self antigens at least as efficiently as TCRs expressed on naive T cells.
Results
Naïve T and Treg cells express many identical TCRs that are asymmetrically distributed
It has been reported that naive T and Treg cells express largely different repertoires of TCRs, suggesting that these populations are separated based on the affinity of individual TCRs to self MHC-peptide ligands. However, naive T and Treg cells share a portion of TCRs that, depending on the experimental model and evaluation method, varies from 10% to 42% (Hsieh et al., 2006; Pacholczyk et al., 2006; Wong et al., 2007). These analyses included many infrequent TCRs, possibly underestimating the overlap between compared regulatory and non-regulatory populations. To determine the nature of this overlap we took advantage of the recently described TCRmini mouse model, where sequences of CDR3 regions of TCRα chains are utilized to track individual T cell clones. In these animals, T cells express one transgenic TCRβ chain and many TCRα chains derived from a mini-locus that can rearrange one Vα2 segment and two Jα26 or Jα2 segments, during T cell development in the thymus. In the TCRmini mice used here Foxp3+ Treg and Foxp3− naive T cells are naturally selected in the thymus on Ab molecules bound with many endogenous peptides (TCRmini mice) or a single covalently linked Ea(52−68) peptide (TCRminiEp mice). First, we looked at how many of the “most frequent” TCRα transcripts from naive T cell clones were present at least once in the Foxp3+ Treg population. As a cut-off value for determination of frequent TCR clones, we calculated the maximum frequency φmax (with a 95% confidence) of a TCR clone whose sequence could not be found after accumulation of n TCR sequences in a sample (Fig. 1A) (Baron et al., 2003). Based on this evaluation we could assume that we found all TCR clones in the tested repertoire with a frequency above 0.62% (0.91% for TCRminiEp). As shown in Fig. 1B we found 41 different TCRs from TCRmini mice (32 from TCRminiEp) that had a frequency above φmax value, and then compared their frequencies within the naive T and Treg repertoires. 68% of TCRs that were dominantly used by naive T cells were also found on Treg cells, although with very different frequencies. In contrast, 39% of the dominant TCRs for Treg cells were identified on naive T cells. Furthermore, when a similar comparison was performed for TCRs retrieved from TCRminiAbEp mice, 77% of the dominant TCRs from naive T cells were also found on Treg cells, whereas 42% of TCRs from Treg cells were present on naive T cells (Fig. 1B). Thus, when we concentrated TCR analysis on T cell clones with high frequency, the overlap between Treg and naive T populations became substantially greater than initially reported (Pacholczyk et al., 2006). In addition, more TCRs from naive T cells were found on Treg cells, suggesting that TCR repertoire expressed by Treg cells may span over a large portion of the naive T cell repertoire. This result corroborates with our previous finding that Treg cells have higher diversity of TCRs, suggesting that observed variation of dominant TCRs expressed on naive T and Treg cells can arise from asymmetric distribution of the same TCRs not separate selection of different TCRs. The overlap between these two TCR repertoires appeared to be even more significant in TCRminiEp mice, where the diversity of different self class II MHC-peptide complexes is minimal and positive selection of CD4+ T cells proceeds on one peptide covalently bound to Ab molecules. This expression of identical TCRs on Treg and naive T cells argued against the hypothesis that naive T and Treg lineages separate based on the different affinities of TCRs for the selecting MHC-peptide ligands.
Figure 1. Overlap of TCRs between naïve T and Treg populations.

(A) The maximum frequency (ϕmax) of a CDR3α transcript whose sequence did not appear after accumulation of n sequences, with 95% confidence, was computed based on equation 1. Upper and lower limits of the 95% confidence interval for a clone with ϕmax frequency were calculated based on equation 2. Both equations are shown in Experimental Procedures. (B) Observed frequency of most dominant naive and Treg clones found in TCRmini or TCRminiEp mice. Percentage of overlap was calculated by dividing shared clones by a total number of dominant clones of a given population. Shown amino acid sequences represent fragments of CDR3 regions including amino acids beginning with the third amino acid after the invariant C residue in all TCRAV genes (Y-L/F-_C_-A-X-1) and spanning the amino acid immediately preceding the TCRAJ motif (2-F/W-G-X-F-G-T). Clones shared between populations are highlighted by grey box. Table below the graphs shows at what frequency dominant clones of one population were found at least once in second population (for both type of mice).
Homeostatic expansion of T cells in vivo
Naive T cells that express TCRs with higher affinity to self-ligands have improved homeostatic expansion (Ge et. al., 2001). It was also shown that after adoptive transfer to lymphopenic hosts, naive T cells transduced with TCRs cloned from Treg cells expand faster than ones transduced with TCRs derived from naive T cells (Hsieh et al., 2004). This observation has been interpreted in favor of the hypothesis that Treg cells frequently recognize self MHC-peptide complexes with higher affinity and that this feature of Treg-derived TCRs enhances their homeostatic fitness. Because natural Treg cells require self-peptide-MHC complexes for homeostatic expansion and proliferate in vivo in the absence of exogenous antigen, we examined whether the reported higher affinity of Treg cells to self-peptides will enhance the peripheral expansion of these cells in a non-lymphopenic host.
To investigate this issue, we chose the most frequent TCRs for the naive T and Treg clones and followed their expansion ratios between the thymus and the periphery. If increased self-reactivity would help Treg cells expand better, one should expect a higher proportion of Treg than naive T clones expanding after leaving the thymus. The same fraction of both naive T and Treg populations, roughly one third, expanded, contracted, or did not significantly changed their frequency after migration from the thymus to the periphery (Fig. 2). It suggested that the overall fitness required for the survival of naive T and Treg cells in non-lymphopenic mileu is very similar.
Figure 2. Relative extrathymic proliferation of dominant T cell clones from TCRmini mice.

The proportions of peripheral proliferation of most frequent T cell clones (shown in Fig. 1) was calculated by dividing the peripheral frequency of a given clone by its thymic frequency. CDR3α sequences are aligned from the most expanded to the least proliferated clones. Clones shared between populations are highlighted by grey box.
It is also possible that Treg cells, due to their anergic nature, cannot proliferate beyond a particular threshold level. To investigate this possibility, we followed the fate of naive T cell clones expressing TCRs found also on Treg cells (Fig. 2). If Treg cells were selected based on elevated affinity to self MHC/peptide complexes, the naive clones expressing these TCRs should expand better than naive T cell clones bearing TCRs not expressed by Treg cells. We found similar number of naive Tcell clones increased or decreased regardless of whether these cells had “shared” or “non-shared” TCRs, demonstrating that naive T cell clones bearing “regulatory” TCRs do not proliferate faster (Fig. 2). Altogether, these results implied that expression of Treg-like TCRs on naive T or Treg cells does not grant a T cell a homeostatic advantage in a non-lymphopenic environment.
Generation of CD4+ T cells with TCRs derived from Treg cells
The inherent anergy of Treg cells in vitro impedes the study of their antigenic specificities for self and non-self antigens. So far only a few TCRs derived from Treg cells have been analyzed by cloning and individually transducing them into non-regulatory T cells (Hsieh et al., 2004). This elegant approach disconnects the TCR from an anergic parental Treg cells, allowing the TCR specificity to be tested in vivo and in vitro, but it is not suitable to examine many different TCRs on per-cell basis. Furthermore, most of the TCRs analyzed so far were cloned from CD25+ T cells with unknown status of Foxp3, permitting unintentional contamination with TCRs derived from Foxp3−CD25+ autoreactive T cells. To overcome these difficulties, we generated T cell hybridomas from Treg cells, and confirmed their origin by comparing their TCRs with TCRs found on single-cell sorted Foxp3+ Treg cells. CD4+ T cell hybridomas are extensively used to identify low affinity agonists, reveal self antigens responsible for survival and thymic selection or to detect low-abundant, endogenous peptides, demonstrating that it is an established method to study the TCRs specificity (Grubin et al., 1997; Grakoui et al., 1999; Felix et al., 2007).
To produce hybridomas from Treg cells, we sorted CD4+CD25+ T cells and cultured them using a protocol optimized to propagate Foxp3+ T cells in vitro. In brief, sorted CD4+CD25+ T cells were cultured in vitro for 7−10 days without loosing expression of Foxp3 and CD25 molecules (Fig. 3A). Importantly, after the expansion phase, the distribution of CDR3 regions of TCRα chains between sorted and expanded Treg cells remained very similar (82% overlap), demonstrating that the expansion did not alter the original distribution of TCRs expressed on Treg cells (Fig. 3B). Expanded Treg cells could be efficiently immortalized by fusion with thymoma BW5147α−β−. After fusion, Treg-derived hybridomas lost Foxp3 expression and other surface markers expressed by dividing Treg blasts but acquired ability to secrete IL-2 upon TCR stimulation - a common feature of conventional CD4+ T cell hybridomas. To further ensure that Treg-derived hybridomas express TCRs from CD25+Foxp3+ Treg cells, we compared their TCR sequences with sequences obtained from a database of single cell-sorted Treg cells. As shown in Fig 3C, after sequencing only forty TCRs, we found the most dominant TCRs originally expressed by Treg cells, which demonstrated that the hybridomas expressed a true representation of TCRs found on Treg cells. Finally, to examine if “regulatory origin” does not limit IL-2 secretion upon TCR stimulation, we directly compared the amounts of IL-2 produced by naive T- and Treg- derived hybridomas. Regardless of origin, all T cell hybridomas produced comparable amounts of IL-2, demonstrating that this assay is appropriate to screen antigenic specificities of TCRs derived from Treg cells (Fig. 3D). To ensure maximum sensitivity, we assayed only CD4hi TCRhi hybridomas and used either bone marrow-derived CD11chi Ab hi DCs or freshly prepared, non-irradiated splenocytes as the APCs. So far, we established over a thousand of T cell hybridomas, of which roughly half was derived from Treg cells and another half was derived from naive T cells. These hybridomas were obtained from three different types of mice: the TCRminiEp, TCRmini and conventional C57BL/6. Use of TCRmini repertoires allowed us to identify the origin of the TCRs sequenced from the T cell hybridomas by comparing them to the database of TCR sequences obtained from single-cell sorted Treg cells (Pacholczyk et al., 2006).
Figure 3. TCRs on T cell hybridomas derived from expanded Treg blasts accurately represent TCRs found on Treg cells.
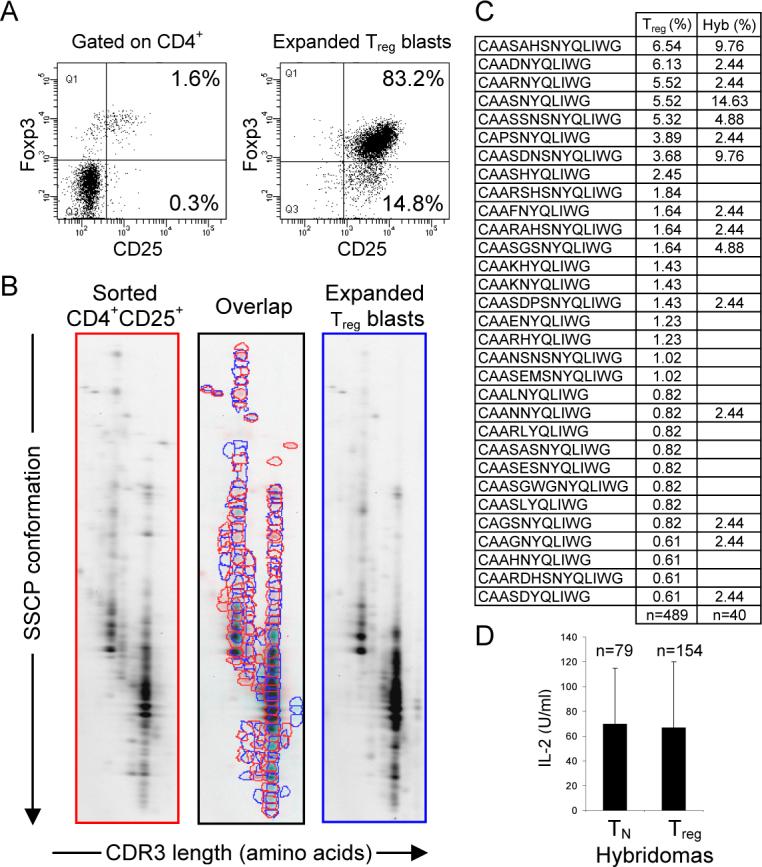
(A) Expression of Foxp3 by Treg cells isolated from lymph nodes of TCRminiEp mice and 7−10 days after in vitro culture of these cells. Cells were surface-stained with antibodies against CD4, CD8, and CD25 molecules, followed by intracellular staining with Foxp3 antibody. (B) 2D-F-SSCP revealed extensive (82%) overlap of CDR3 regions of TCRα chains from freshly sorted and expanded Treg cells. cDNA was synthesized from isolated RNA of both types of cells. Run-off PCR was done with primer specific for TCRVα2, coupled with Cy3 (fluorescent dye). First, fluorescent PCR products were run in the first dimension using capillary electrophoresis in denaturing PAGE. The second dimension was run in a non-denaturing SSCP slab gel. Resulting 2D gels show diversity, distribution, and frequency of dominant CDR3α regions from indicated populations. The intensity of individual dots corresponds to the real frequency of a particular rearrangement in the original sample. Spots migrating with different speed on the second dimension SSCP gel represent different VαJα rearrangements. (C) TCR CDR3α sequences retrieved from Treg-derived hybridomas (Hyb) were compared with the most abundant CDR3α sequences found in Treg cells in TCRminiEp mice (Treg). (D) Hybridomas derived from naïve T or Treg cells of TCRminiEp mice, were co-cultured with splenocytes from C57BL/6 mice. Responding hybridomas were compared for the ability to produce IL-2. Production of IL-2 secreted by activated hybridomas was measured by HT-2 proliferation using MTT assay. Results are shown as arithmetical mean ± SD.
Hybridomas derived from Treg cells of TCRminiEp mice recognize non-self antigens
It has been suggested that TCRs expressed by Treg cells may recognize abundant peripheral self-antigens with an affinity that is above that required for homeostatic expansion (Hsieh et al., 2004; Hsieh et al., 2006). To determine the fraction of Treg cells that recognizes ubiquitously expressed self-antigens, we first examined the antigen specificities of T cell clones derived from naive T and Treg cells from TCRminiEp mice. The advantage of this model is that all self MHC class II molecules in the thymus and periphery remain bound with a single peptide and that the resulting repertoire of TCRs is tolerant to AbEp complex but lacks tolerance to natural, endogenously processed peptides. Also, more than two-thirds of T cell hybridomas obtained from CD4+ T cells produce IL-2 when co-cultured with APCs derived from C57BL/6 mice (Ignatowicz et al., 1996). This feature of TCRs selected by AbEp complex allowed us to precisely test the specificity for self (AbEp) and non-self (AbWT) MHC-peptide complexes. As shown in Fig. 4A, 68% and 87% of hybridomas derived from naive T or Treg cells respectively, become activated after overnight co-culture with APCs expressing non-self AbWT-peptide complexes, but none of these hybridomas produced IL-2 after incubation with self APCs. Furthermore, the same TCRminiAbEp-derived hybridomas were incubated with APCs derived from invariant chain-deficient (AbIi−) mice that express a lower number of Ab molecules loaded with a reduced number of Ii-independent, endogenous peptides. Here, hybridomas derived from naive T and Treg cells also responded to non-self APCs with similar, though expectedly lower, frequency. Importantly, recognition of non-self antigens was observed regardless of whether TCRs on T cell hybridomas represented a pool of “shared” or those only found on Treg cells TCRs (Fig. 4B). In summary, these experiments showed that irrespective of the origin of the antigen(s), the frequency of TCRs directed to non-self antigens is similar between naive T and Treg cells, and that autoreactive TCR specific to abundant self MHC-peptide complexes are rare on all CD4+ T cells.
Figure 4. Treg-derived hybridomas from TCRminiEp mice frequently recognize foreign but not self MHC-peptide complexes.

(A) Hybridomas were co-cultured overnight with splenocytes or bone marrow derived dendritic cells (BMDC's) from C57BL/6 (Ab), Ii-, or AbEpIi− (AEp) mice. Production of IL-2 secreted by activated hybridomas was measured by HT-2/MTT assay. In each experiment, all hybridomas were separately tested for response to anti-CD3 stimulation in the presence of autologous APCs, to determine the percentages of hybridomas able to respond to activation. The box beneath the bar graph shows number of hybridomas (n) used in each test. (B) Twelve dominant Treg TCRs identified by single-cell RT-PCR were compared with TCRs found on T cell hybridomas. Based on reactivities of these hybridomas, antigenic specificity was assigned to the particular CDR3α sequence.
Treg cells selected on AbWT complexes rarely recognize self antigens
One could argue that ubiquitous expression of AbEp complexes in TCRminiEp mice enhances deletion of autoreactive TCRs, but in normal animals expressing AbWT complexes, negative selection will be less efficient and incidence of autoreactive TCRs on Treg cells could be higher. Therefore, we made another set of hybridomas from TCRmini and C57BL/6 mice that express AbWT molecules, and tested their ability to respond to APCs expressing self and non-self MHC-peptide complexes. When hybridomas from TCRmini mice were co-cultured with autologous APCs, non of them produced IL-2 indicating that the frequency of Treg cells with potentially autoreactive TCRs is very low (Fig. 5A). To detect the responses of these hybridomas to non-self antigens, we tested their ability to become activated by APCs expressing Abm12 molecules. The Abm12 molecule is similar to autologous Ab molecules, but due to the mutation in antigen binding groove, the Abm12 molecules remain bound with a different set of endogenously processed peptides (Bill et al., 1989). As shown in Fig. 5A, hybridomas derived from Treg and naive T cells recognized the Abm12 APCs, but not other allogeneic APCs expressing Ak and Ek complexes. These results showed that TCRs expressed on Treg cells recognize different non-self peptides bound to MHC, but are not overtly reactive to backbone of class II MHC complexes (Felix et al., 2007). Moreover a similar frequency of Abm12 specific TCRs was observed on hybridomas derived from naive T and Treg cells, confirming that these TCR repertoires have comparable ability to sense non-self APCs.
Figure 5. Hybridomas derived from naïve T and Treg cells respond with the same frequency to stimulation with allogeneic APCs but not autologous APCs.

(A) Hybridomas derived from naive and Treg cells from TCRmini or C57BL/6 mice, were stimulated by splenocytes or BMDC's derived from autologous C57BL/6 (Ab), allogenic CBA Ca/J (AkEk) or bm12 (Abm12) mice. All hybridomas were tested for response to anti-CD3 stimulation in the presence of autologous APCs. Production of IL-2 was measured by HT-2 proliferation using MTT assay. To determine percentages of hybridomas responding to APCs, only hybridomas that responded to the anti-CD3 stimulation were considered. The box beneath the bar graph shows number of hybridomas (n) used in each test. (B) The CDR3α regions of autoreactive TCRs identified in TCRmini repertoire. TCRs on TCRminiEp Treg-derived hybridomas reactive to Ab were compared against database containing CDR3α sequences retrieved by single-cell RT-PCR from T cells in TCRmini mice. Of the six CDR3 sequences identified so far, five were expressed on Foxp3+ Treg cells and two were found on Foxp3− T cells.
Because we have not found autoreactive TCRs in naïve T or Treg populations, we decided to take advantage of the TCRmini repertoire that allows for backtracking receptors to their occurrence in vivo. We sequenced TCRs on hybridomas derived from TCRminiAbEp that responded in vitro to Ab molecules bound with endogenous peptide, and then searched for identical TCRs in our database of almost 1500 TCRs sequenced from CD4+ T cells derived from TCRmini mice. Although there are only a few TCRs shared between these mice, we identified six TCRs from TCRminiEp mice that were highly reactive to AbWT and were found in TCRmini mice as well (Fig. 5B). Interestingly, when we examined on what type of T cells these TCRs have been found in TCRmini mice, two TCRs were expressed on naive T cells and five on Treg cells. Each of these autoreactive TCRs was rare in TCRmini mice suggesting that although some of TCRs expressed by Treg cells are potentially autoreactive, their frequency on naive T and Treg cells was low.
To extrapolate our finding from TCRmini mice to wild-type Treg cells, we also tested antigenic specificities of T cell hybridomas obtained from C57BL/6 mice. As shown in Fig. 5A approximately 10% of the hybridomas responded to allogeneic APCs irrespectively of whether these TCRs originated from naive T or Treg cells. However, none of the hybridomas tested here produced IL-2 following incubation with autologous APCs. This result confirmed our previous findings from TCRmini mice that in general CD4+ T cells rarely express autoreactive TCRs that recognize self class II MHC-peptide complexes as agonists.
Wasting disease induced by adoptive transfer of effector T cells is caused by non-self antigens
Inflamatory bowel disease (IBD) is an immune mediated disease that manifests with inflammation on the colonic and rectal mucosa (ulcerative colitis), and Crohn's disease that affects both the small and large intestines. The genetic and environmental factors responsible for precipitation of the disease are linked with an aberrant inflammatory response toward commensal bacteria. There is an established experimental protocol to induce IBD in mice where transfer of CD4+CD45RBhi T cells into immune-deficient mice, such as Rag2−/− or SCID mice, leads to colitis, causing a wasting disease (reviewed in Coombes et al., 2005). It has also been shown that expansion of non-regulatory T cells engineered to express TCRs cloned from CD25+ T cells causes wasting disease, whereas expansion of T cells expressing TCRs cloned from CD25− T cells does not (Hsieh et al., 2004). These results were interpreted as evidence that repertoire of TCRs expressed by Treg cells is enriched in autoreactive specificities. However, so far there is no direct evidence that self-antigens are responsible for initiation of wasting disease in the transfer model.
To determine whether recognition of self-antigens can cause wasting disease, we sorted naive CD4+CD45RBhi T cells from TCRmini and TCRminiEp mice and transferred them into lymphopenic AbWT TCRα−/− or AbEp TCRα−/− mice, respectively. The host mice that received CD4+CD45RBhi T cells from TCRmini mice started losing weight between 4−5 weeks after adoptive transfer (Fig. 6); this trend continued until mice had to be sacrificed. To ensure that the observed weight loss was a result of colitis, lesions in the intestine were confirmed macroscopically (data not shown). Interestingly, transfer of CD4+CD45RBhi T cells from TCRminiEp mice into AbEp TCRα−/− mice did not result in colitis. Because mice expressing covalent AbEp complexes do not present other antigens our results support previous findings that recognition of bacterial antigens of commensal flora is required for initiation of wasting disease upon transfer of effector CD4+ T cells. Importantly, this experiment also implied that the previously reported precipitation of wasting disease by a transfer of T cells expressing Treg-derived TCRs is caused not by a higher frequency of autoreactive TCRs but rather by sufficient frequency of non-self specificities on Treg cells.
Figure 6. The CD4+CD45RBhi T cells isolated from TCRmini but not TCRminiEp mice cause wasting disease upon adoptive transfer into autologous lymphopenic hosts.

T cells isolated from lymph nodes were FACS sorted using CD4 and CD45RB antibodies. 4×105 of CD4+CD45RBhi T cells (purity above 99%) from TCRmini or TCRminiEp mice were injected intraperitoneally to AbWT TCRα−/− or AbEp TCRα−/− lymphopenic hosts, respectively. The ratio of the starting weight of recipient mice is shown. Data are representative of two independent experiments with 2 or 3 mice per group. Weight loss was associated with colon inflammation, prolapse, bowel wall thickening, and typical hunched-back appearance (data not shown).
Discussion
We report that contrary to the general belief, non-self antigens constitute a primary source of peptides recognized by TCRs expressed on Treg cells and with an affinity high enough to induce an antigenic response. Moreover these TCRs do not show a major enrichment in high-affinity, autoreactive specificities. This conclusion was obtained based on two approaches: an analysis of TCR usage and homeostatic fitness of naive T and Treg clones in vivo, and an analysis of antigenic specificities of TCRs derived from Foxp3+ Treg cells.
Our comparative analysis showed that 70 percent of the most frequent TCRs on naive T cells were also found on Treg cells, but with very different frequencies. This disparity in the allocation of shared TCRs was already set in the thymus, suggesting that thymic, not peripheral, events enforce this asymmetric distribution. A similar observation of shared TCRs between regulatory and non-regulatory populations was also reported elsewhere, where seven out of ten most frequent TCRs on CD25- and CD25+ CD4+ T cells was shared between both analyzed populations (Hsieh et al., 2006). This substantial fraction of shared TCRs suggests that naive T and Treg cells have, in fact, many shared affinities. In addition, 40 percent of dominant TCRs on Treg cells were also found on naive T cells. Possibly, this overlap could be larger but remained partially hidden due to vastly different frequencies of the same TCRs and likely, a higher diversity of TCRs on Treg cells (Pacholczyk et al., 2006). This could also indicate that TCRs used by naive T cells constitute a subset of TCRs expressed on Treg cells.
The main role of Treg cells is to suppress autoreactive T cells, which suggests that TCRs on Treg cells may recognize self antigens as agonists. However, we found that T cells expressing TCRs derived from a Treg population did not become activated and produced IL-2 upon exposure to self MHC-peptide ligands. Moreover, the majority of these T cell clones responded to non-self antigens that represent endogenously processed peptides and allogenic MHC-peptide complexes. Regardless of whether TCRs were derived from Treg or naive T cells subpopulations, the frequency of TCRs recognizing non-self peptides was similar. Our data corroborate previous findings that partial reversal of the anergic state of Treg cells by co-culture in the presence of IL-2 and/or IL-15 allows them to respond to allogeneic, but not syngeneic, APCs in both mice and humans (Dieckmann et al., 2001; Pacholczyk et al., 2002). Here, the only way we were able to identify potentially autoreactive TCRs specific for AbWT complexes in TCRmini mice was by cross-referencing AbWT-reactive TCRs originally found on hybridomas from TCRminiEp to the database of almost 1500 TCRs previously cloned from TCRmini mice. This result demonstrated that the frequency of high-affinity, self-reactive TCRs in the Treg population is very low, although it may be marginally higher than on naive T cells. One could argue whether T cell hybridomas are an appropriate tool to detect autoreactive TCRs. However, T cell hybridomas made from autoreactive T cells continued to manifest the original self-reactivity in vitro and for years T cell hybridomas were used to detect weak and strong agonists, sometimes at very low levels (Hampl et al., 1997; Grubin et al., 1997). In general, the self-reactive hybridomas are produced either after antigen priming or from autoimmune prone strains, but not directly from naïve TCR repertoire, which is expected to have a very low frequency of autoreactive T cells (Yanoma et al., 1988; Buzas et al., 2003).
It has been hypothesized that TCRs on Treg cells “draw from the same pool of TCRs as autoreactive T cells” and a majority of TCRs on Treg cells have pathogenic potential (Hsieh et al., 2006). This conclusion was based on the observation that CD25+ Treg cells from wild-type mice express TCRs similar to the receptors expressed on pathogenic autoreactive CD4+CD25+ T cells from Foxp3− mice. Indeed, in the recently produced Foxp3-GFP knock-in mice the “disabled” Treg cells expressing GFP left the thymus and partially accumulated in non-lymphatic tissues suggesting direct involvement of these cells in pathogenesis (Gavin et al., 2007; Lin et al., 2007). However, the role of “disabled” Treg cells in the autoimmune disease observed in Foxp3-deficient mice is unclear, since these cells remain anergic, cannot provoke an autoimmune disease upon adoptive transfer to a lymphopenic host, and deteriorate in the absence of other T cells (Lin et al., 2007). Furthermore, when the same questions were addressed in scurfy mice expressing transgenic GFP under the control of Foxp3 regulatory sequences, the proportion of GFP+ in CD4+ T cells in sick scurfy and healthy B6 males were similar and the GFP+ T cells were activated no more than GFP− T cells (Kuczma et al., submitted). Concurrently, the depletion of Foxp3-expressing T cells in adult animals leads to autoimmune disease with a severity similar to the disease observed in the scurfy mice that is entirely driven by autoreactive T cells from the pool of Foxp3− T cells (Kim et al., 2007). Other evidence for pathogenic autoreactive specificities of Treg cells is based on an adoptive transfer of naive T cells that are retrovirally transduced with Treg-derived TCRs and cause autoimmunity manifested by wasting disease (Hsieh et al., 2004). However, onset of colitis depends on an overly aggressive T cell response to a subset of commensal enteric bacteria, and the transfer of naive T cells into germ-free animals does not cause the disease (Gad, 2005; Sartor, 2006). The wasting disease described here was induced by adoptively transferred naive T cells only in animals where class II MHC was able to present foreign antigens, but not in animals where class II MHC was preoccupied with covalently bound self-peptide, confirming that in such an experimental set-up, non-self antigens are a necessity. Hence, the fact that described in other report naive T cells with “regulatory” TCRs cause wasting disease appears to suggest that specificities directed toward non-self (bacterial) antigens are well represented in the Treg repertoire (Hsieh et al., 2004).
In general, conventional T cells require weak interactions with self MHC-peptide complexes to pass thymic selection and to survive in the periphery, and higher affinity interactions with non-self MHC-peptide ligands to induce activation and effector functions. In contrast, it has been proposed that a commitment of Treg cells is driven by higher affinity interactions between TCR and self MHC/peptide complexes (Jordan et al., 2001; Apostolou et al., 2002; Hsieh et al., 2004) and that Treg cells recognize ubiquitous self-ligands with an avidity above that required for selection and survival (Hsieh et al., 2006). These interactions induce Treg phenotype and Foxp3 expression and skew the specificity of TCR repertoire toward self-antigens. Consequently naive T and Treg cells express different sets of TCRs that recognize non-self or self-antigens, respectively. Therefore, under these circumstances Treg cells may use the same self-ligand(s) for positive selection in the thymus and effector function in the periphery, which is fundamentally different from the conventional model of T cell selection and/or activation. Furthermore, if lineage commitment of Treg cells depends on higher TCR affinity for self-ligand(s), the repertoire of TCRs on Treg cells would be less diverse, than the repertoire on naive T cells. This is because for a given antigen there will be fewer TCRs with a higher affinity than TCRs with lower affinity. However, our data show that Treg cells may not only have higher diversity of TCRs than conventional T cells, but also share a substantial portion of TCRs with that population (Fig. 1) (Pacholczyk et al., 2006).
Our findings question the direct role of TCR signaling in the linage commitment of Treg cells, but support the view that recognition of self MHC-peptide complexes is required for these cells to survive in the thymus and periphery. Subsequently, we would like to offer an alternative model where commitment of Treg precursors occurs prior to thymic selection by the TCR (Pacholczyk et al., 2002; van Santen et al., 2004; Pennington et al., 2006) and even weak interactions with self-ligands can induce survival of thymic Treg cells. In this model precursors of Treg cells (pre-Treg) possess a unique physiological status and exist as a fraction of late CD4−CD8− (DN) or cortical CD4+CD8+ (DP) thymocytes. This condition may be triggered by a “near-death experience,” different proliferation propensity prior to the DP stage, or a currently unknown signal(s) delivered upon the contacts of thymocyte with other thymocytes or thymic stromal cells. Different physiological status of pre-Treg cells, not signaling through the TCR, will determine their lineage commitment and TCR distribution. Positive selection of both lineages proceeds on the same pool of self ligands generating unbiased TCRs distribution. Both naive T and Treg repertoires are primarily directed to recognize non-self antigens with small fractions of potentially autoreactive TCRs. After positive selection, the TCR repertoires on Treg cells is wider because physiological status of pre-Treg thymocytes can make positive selection of Treg cells more promiscuous. Some Treg cells can express autoreactive TCRs, but in contrast to the current dogma, the self peptide(s) involved in positive selection of Treg cells is not responsible for activation of their suppressor functions in the periphery. In support of a “pre-comittment” hypothesis, it has been recently reported that Foxp3 expression can occur in pre-selected CD4−CD8− thymocytes, when these cells develop in the absence of trans-conditioning by CD4+CD8+ thymocytes (Pennington et al., 2006).
Experimental Procedures
Mice
TCRminiAbWT mice were obtained as previously described (Pacholczyk 2006). TCRminiAbEp mice were generated by crossing VαJα/βTg AbWT mice with mice deficient for the invariant chain and endogenous Aβb chain, but expressing a single covalent AbEp complex (Ignatowicz et al., 1996). To eliminate expression of endogenous TCRα chains, all TCRmini mice used in this study were crossed with mice deficient in endogenous TCRα genes. All TCRmini mice were 6 to 8 weeks old and were heterozygous for the TCRα mini locus to ensure expression of a single TCR.
All mice were housed under specific pathogen-free conditions in the animal care facility at the Medical College of Georgia. All work involving animals was conducted under protocols approved by the Animal Care and Use Committee at Medical College of Georgia.
Cells preparation and flow cytometry analysis
Single-cell suspensions were prepared from lymph nodes by mechanical disruption through nylon mesh. The following antibodies were used for flow cytometry analyses: anti-CD4-APC, anti-CD8-PerCP, anti-CD25-FITC, anti-CD45RB-PE, anti-Vα2-PE, anti-Vβ14-FITC, anti-TCRβ-APC and anti-Foxp3-APC (all from BD PharMingen or eBioscience). Intracellular staining for Foxp3 expression was performed using intracellular staining kit, according to the manufacturer's protocol (eBioscience). FACS analysis was performed using BD FACSCanto and Diva software (BD PharMingen). Dead cells were excluded by gating of forward and side scatter.
Generation of T cell hybridomas
To generate conventional T cell hybridomas, CD4+ T cells sorted from lymph nodes were cultured for 3 days in culture medium in the presence of plate-bound aCD3 mAb and autologus irradiated splenocytes. On day 3, dead cells were removed by gradient centrifugation on Lymphocyte Separation Media (Cellgro, Herndon, VA). The recovered live cells were expanded in the presence of IL-2 for 4 days, and T cell blasts were converted into T cell hybridomas as previously described (Ignatowicz et al., 1996). To generate T cell hybridomas derived from regulatory T cells, we sorted CD4+CD25+ lymphocytes and induced their proliferation by aCD3 and aCD28 antibodies fixed on the solid surface in the presence of exogenous IL-2 for 10 days (Singh et al, manuscript in preparation). Expanded CD25+ T cell blasts were also converted into T cell hybridomas.
HT-2 assay
T cell hybridomas were tested for their responsiveness toward self and non-self APCs by HT-2 assay (Ignatowicz et al., 1996). Briefly, 105 hybridoma cells were incubated with 5×105 splenocytes or 105 bone marrow dendritic cells, from different mice, as indicated. After 24 h, the amount of secreted IL-2 was measured using the detector HT-2 cell line. The proliferation of HT-2 cells in response to IL-2 was measured using 3-(4,5-dimethythiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma) assay.
MoFlo sorting of CD4+CD45hi T cells and adoptive transfer to lymphopenic hosts
Lymphocytes were stained with appropriate antibodies and were subjected to double sorting on MoFlo sorter (Cytomation) (Pacholczyk et al., 2006). Purity of the CD4+CD25+ or CD4+CD45RBhi populations was >99% _TCRα_−/− mice were injected intraperitoneally with sorted CD4+ T cell subpopulations in PBS. Mice received 4 × 105 CD45RBhi CD4+ cells. Following adoptive transfer mice weight was monitored on a weakly basis.
RT-PCR and two-dimensional, fluorescent, single-stranded conformational polymorphism (2D-F-SSCP)
RNA was isolated from sorted populations using Trizol according to the manufacturer's protocol (Invitrogen). cDNA synthesis was done using M-MLV reverse transcriptase and Random Hexamers (all from Promega). cDNA was amplified in a standard PCR reaction with Vα2-specific primers (Va2_287s, CA_29r). 2 μl of PCR product was used as a template for a run-off reaction with a fluorescent primer Va2_287s labeled on the 5' end with Cy3 (synthesized by IDT Inc.). The denatured fluorescent products were then subjected to 2D electrophoresis as previously described (Pacholczyk et al., 2006) Fluorescent images were acquired by scanning the slab gel in a Typhoon 9410 imager (Amersham-Pharmacia). Images were analyzed using Image Master 5.0 Platinum software (Amersham-Pharmacia).
Estimation of most frequent TCRs
Maximum frequency _ϕ_max of possibly yet unseen TCR clone was calculated based on equation published elsewhere (Baron et al., 2003):
| n(ϕmax−1.65ϕmax(1−ϕmax)n−1)=1, | (1) |
|---|
where n is a total number of accumulated sequences. To estimate boundaries of a real frequency (_ϕ_clon) of a TCR clone with observed frequency _f_clon = _ϕ_max we used equation:
| ϕclon∈[fclon−1.96fclon(1−fclon)n−1,fclon+1.96fclon(1−fclon)n−1], | (2) |
|---|
Table 1.
| Treg-derived CDR3α sequences from TCRminiEp mice | Used by cells: | Hybridomas reactive to: | |
|---|---|---|---|
| AbWT (Non-self) | AbEp (Self) | ||
| CAASAHSNYQLIWG | TN / Treg | + | − |
| CAADNYQLIWG | Treg | + | − |
| CAARNYQLIWG | TN / Treg | + | − |
| CAASNYQLIWG | TN / Treg | + | − |
| CAASSNSNYQLIWG | Treg | + | − |
| CAPSNYQLIWG | Treg | + | − |
| CAASDNSNYQLIWG | Treg | + | − |
| CAASHYQLIWG | TN / Treg | N/A | N/A |
| CAARSHSNYQLIWG | Treg | N/A | N/A |
| CAAFNYQLIWG | TN / Treg | + | − |
| CAARAHSNYQLIWG | TN / Treg | + | − |
| CAASGSNYQLIWG | TN / Treg | − | − |
Table 2.
| Sequences of TCRs reactive to AbWT found in TCRminiEp and TCRmini mice | Distribution on CD4+ T cells in TCRmini mice | |
|---|---|---|
| Treg | TN | |
| CAASNYQLIWG | + | |
| CAASAHSNYQLIWG | + | |
| CAGSNYQLIWG | + | |
| CAARSHSNYQLIWG | + | + |
| CAASDYQLIWG | + | |
| CAASGWDSNYQLIWG | + |
Acknowledgements
We thank D. Daniely and R. Markowitz for editing the manuscript and H. Ignatowicz and P. Merai for technical assistance. All work involving animals was conducted under protocols approved by the Animal Care and Use Committee at Medical College of Georgia. This work was supported by basic research grants from NIH (AI041145-07A1) and ROTR Foundation. R.P. is supported by intramural STP grant from MCG. P.K. is supported by grants from the Georgia Cancer Coalition and NCI (CA107349-01A1).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Apostolou I, Sarukhan A, Klein L, von Boehmer H. Origin of regulatory T cells with known specificity for antigen. Nat. Immunol. 2002;3:756–763. doi: 10.1038/ni816. [DOI] [PubMed] [Google Scholar]
- Aschenbrenner K, D'Cruz LM, Vollmann EH, Hinterberger M, Emmerich J, Swee LK, Rolink A, Klein L. Selection of Foxp3(+) regulatory T cells specific for self antigen expressed and presented by Aire(+) medullary thymic epithelial cells. Nat. Immunol. 2007;8:351–358. doi: 10.1038/ni1444. [DOI] [PubMed] [Google Scholar]
- Baron V, Bouneaud C, Cumano A, Lim A, Arstila TP, Kourilsky P, Ferradini L, Pannetier C. The repertoires of circulating human CD8(+) central and effector memory T cell subsets are largely distinct. Immunity. 2003;18:193–204. doi: 10.1016/s1074-7613(03)00020-7. [DOI] [PubMed] [Google Scholar]
- Belkaid Y, Piccirillo CA, Mendez S, Shevach EM, Sacks DL. CD4+CD25+ regulatory T cells control Leishmania major persistence and immunity. Nature. 2002;420:502–507. doi: 10.1038/nature01152. [DOI] [PubMed] [Google Scholar]
- Bill J, Ronchese F, Germain RN, Palmer E. The contribution of mutant amino acids to alloantigenicity. J Exp. Med. 1989;170:739–750. doi: 10.1084/jem.170.3.739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzas EI, Hanyecz A, Murad Y, Hudecz F, Rajnavolgyi E, Mikecz K, Glant TT. Differential recognition of altered peptide ligands distinguishes two functionally discordant (arthritogenic and nonarthritogenic) autoreactive T cell hybridoma clones. J Immunol. 2003;171:3025–3033. doi: 10.4049/jimmunol.171.6.3025. [DOI] [PubMed] [Google Scholar]
- Cabarrocas J, Cassan C, Magnusson F, Piaggio E, Mars L, Derbinski J, Kyewski B, Gross DA, Salomon BL, Khazaie K, Saoudi A, Liblau RS. Foxp3+ CD25+ regulatory T cells specific for a neo-self-antigen develop at the double-positive thymic stage. Proc. Natl. Acad. Sci. U. S. A. 2006;103:8453–8458. doi: 10.1073/pnas.0603086103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caton AJ, Cozzo C, Larkin J,, III, Lerman MA, Boesteanu A, Jordan MS. CD4(+) CD25(+) regulatory T cell selection. Ann. N. Y. Acad. Sci. 2004;1029:101–114. doi: 10.1196/annals.1309.028. [DOI] [PubMed] [Google Scholar]
- Coombes JL, Robinson NJ, Maloy KJ, Uhlig HH, Powrie F. Regulatory T cells and intestinal homeostasis. Immunol Rev. 2005;204:184–194. doi: 10.1111/j.0105-2896.2005.00250.x. [DOI] [PubMed] [Google Scholar]
- Coutinho A, Caramalho I, Seixas E, Demengeot J. Thymic commitment of regulatory T cells is a pathway of TCR-dependent selection that isolates repertoires undergoing positive or negative selection. Curr. Top. Microbiol. Immunol. 2005;293:43–71. doi: 10.1007/3-540-27702-1_3. [DOI] [PubMed] [Google Scholar]
- D'Cruz LM, Klein L. Development and function of agonist-induced CD25+Foxp3+ regulatory T cells in the absence of interleukin 2 signaling. Nat. Immunol. 2005;6:1152–1159. doi: 10.1038/ni1264. [DOI] [PubMed] [Google Scholar]
- Dieckmann D, Plottner H, Berchtold S, Berger T, Schuler G. Ex vivo isolation and characterization of CD4(+)CD25(+) T cells with regulatory properties from human blood. J Exp. Med. 2001;193:1303–1310. doi: 10.1084/jem.193.11.1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felix NJ, Donermeyer DL, Horvath S, Walters JJ, Gross ML, Suri A, Allen PM. Alloreactive T cells respond specifically to multiple distinct peptide-MHC complexes. Nat. Immunol. 2007;8:388–397. doi: 10.1038/ni1446. [DOI] [PubMed] [Google Scholar]
- Fontenot JD, Dooley JL, Farr AG, Rudensky AY. Developmental regulation of Foxp3 expression during ontogeny. J Exp. Med. 2005a;202:901–906. doi: 10.1084/jem.20050784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 2003;4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity. 2005b;22:329–341. doi: 10.1016/j.immuni.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Fontenot JD, Rudensky AY. A well adapted regulatory contrivance: regulatory T cell development and the forkhead family transcription factor Foxp3. Nat. Immunol. 2005;6:331–337. doi: 10.1038/ni1179. [DOI] [PubMed] [Google Scholar]
- Gad M. Regulatory T cells in experimental colitis. Curr. Top. Microbiol. Immunol. 2005;293:179–208. doi: 10.1007/3-540-27702-1_9. [DOI] [PubMed] [Google Scholar]
- Gavin MA, Rasmussen JP, Fontenot JD, Vasta V, Manganiello VC, Beavo JA, Rudensky AY. Foxp3-dependent programme of regulatory T-cell differentiation. Nature. 2007;445:771–775. doi: 10.1038/nature05543. [DOI] [PubMed] [Google Scholar]
- Ge Q, Rao VP, Cho BK, Eisen HN, Chen J. Dependence of lymphopenia-induced T cell proliferation on the abundance of peptide/MHC epitopes and strength of their interaction with T cell receptors. Proc. Natl. Acad. Sci. USA. 2001;98:1728–1733. doi: 10.1073/pnas.98.4.1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grakoui A, Donermeyer DL, Kanagawa O, Murphy KM, Allen PM. TCR-independent pathways mediate the effects of antigen dose and altered peptide ligands on Th cell polarization. J Immunol. 1999;162:1923–1930. [PubMed] [Google Scholar]
- Grubin CE, Kovats S, deRoos P, Rudensky AY. Deficient positive selection of CD4 T cells in mice displaying altered repertoires of MHC class II-bound self-peptides. Immunity. 1997;7:197–208. doi: 10.1016/s1074-7613(00)80523-3. [DOI] [PubMed] [Google Scholar]
- Hampl J, Chien YH, Davis MM. CD4 augments the response of a T cell to agonist but not to antagonist ligands. Immunity. 1997;7:379–385. doi: 10.1016/s1074-7613(00)80359-3. [DOI] [PubMed] [Google Scholar]
- Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–1061. [PubMed] [Google Scholar]
- Hsieh CS, Liang Y, Tyznik AJ, Self SG, Liggitt D, Rudensky AY. Recognition of the peripheral self by naturally arising CD25+ CD4+ T cell receptors. Immunity. 2004;21:267–277. doi: 10.1016/j.immuni.2004.07.009. [DOI] [PubMed] [Google Scholar]
- Hsieh CS, Zheng Y, Liang Y, Fontenot JD, Rudensky AY. An intersection between the self-reactive regulatory and nonregulatory T cell receptor repertoires. Nat. Immunol. 2006;7:401–410. doi: 10.1038/ni1318. [DOI] [PubMed] [Google Scholar]
- Ignatowicz L, Kappler J, Marrack P. The repertoire of T cells shaped by a single MHC/peptide ligand. Cell. 1996;84:521–529. doi: 10.1016/s0092-8674(00)81028-4. [DOI] [PubMed] [Google Scholar]
- Jordan MS, Boesteanu A, Reed AJ, Petrone AL, Holenbeck AE, Lerman MA, Naji A, Caton AJ. Thymic selection of CD4+CD25+ regulatory T cells induced by an agonist self-peptide. Nat. Immunol. 2001;2:301–306. doi: 10.1038/86302. [DOI] [PubMed] [Google Scholar]
- Khattri R, Cox T, Yasayko SA, Ramsdell F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat. Immunol. 2003;4:337–342. doi: 10.1038/ni909. [DOI] [PubMed] [Google Scholar]
- Kim JM, Rasmussen JP, Rudensky AY. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat. Immunol. 2007;8:191–197. doi: 10.1038/ni1428. [DOI] [PubMed] [Google Scholar]
- Kim JM, Rudensky A. The role of the transcription factor Foxp3 in the development of regulatory T cells. Immunol Rev. 2006;212:86–98. doi: 10.1111/j.0105-2896.2006.00426.x. [DOI] [PubMed] [Google Scholar]
- Larkin J,, III, Picca CC, Caton AJ. Activation of CD4+ CD25+ regulatory T cell suppressor function by analogs of the selecting peptide. Eur. J Immunol. 2007;37:139–146. doi: 10.1002/eji.200636577. [DOI] [PubMed] [Google Scholar]
- Lerman MA, Larkin J,, III, Cozzo C, Jordan MS, Caton AJ. CD4+ CD25+ regulatory T cell repertoire formation in response to varying expression of a neo-self-antigen. J Immunol. 2004;173:236–244. doi: 10.4049/jimmunol.173.1.236. [DOI] [PubMed] [Google Scholar]
- Lin W, Haribhai D, Relland L, Truong N, Carlson M, Williams C, Chatila T. Regulatory T cell development in the absence of functional Foxp3. Nat. Immunol. 2007;8:359–368. doi: 10.1038/ni1445. [DOI] [PubMed] [Google Scholar]
- Liu YJ. A unified theory of central tolerance in the thymus. Trends in Immunology. 2006;27:215–221. doi: 10.1016/j.it.2006.03.004. [DOI] [PubMed] [Google Scholar]
- Morrissey PJ, Charrier K, Braddy S, Liggitt D, Watson JD. CD4+ T cells that express high levels of CD45RB induce wasting disease when transferred into congenic severe combined immunodeficient mice. Disease development is prevented by cotransfer of purified CD4+ T cells. J Exp. Med. 1993;178:237–244. doi: 10.1084/jem.178.1.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochando JC, Yopp AC, Yang Y, Garin A, Li Y, Boros P, Llodra J, Ding Y, Lira SA, Krieger NR, Bromberg JS. Lymph node occupancy is required for the peripheral development of alloantigen-specific Foxp3+ regulatory T cells. J Immunol. 2005;174:6993–7005. doi: 10.4049/jimmunol.174.11.6993. [DOI] [PubMed] [Google Scholar]
- Pacholczyk R, Ignatowicz H, Kraj P, Ignatowicz L. Origin and T Cell Receptor Diversity of Foxp3(+)CD4(+)CD25(+) T Cells. Immunity. 2006;25:249–259. doi: 10.1016/j.immuni.2006.05.016. [DOI] [PubMed] [Google Scholar]
- Pacholczyk R, Kraj P, Ignatowicz L. Peptide specificity of thymic selection of CD4+CD25+ T cells. J Immunol. 2002;168:613–620. doi: 10.4049/jimmunol.168.2.613. [DOI] [PubMed] [Google Scholar]
- Pennington DJ, Silva-Santos B, Silberzahn T, Escorcio-Correia M, Woodward MJ, Roberts SJ, Smith AL, Dyson PJ, Hayday AC. Early events in the thymus affect the balance of effector and regulatory T cells. Nature. 2006;444:1073–1077. doi: 10.1038/nature06051. [DOI] [PubMed] [Google Scholar]
- Powrie F, Leach MW, Mauze S, Caddle LB, Coffman RL. Phenotypically distinct subsets of CD4+ T cells induce or protect from chronic intestinal inflammation in C. B-17 scid mice. Int. Immunol. 1993;5:1461–1471. doi: 10.1093/intimm/5.11.1461. [DOI] [PubMed] [Google Scholar]
- Sartor RB. Mechanisms of disease: pathogenesis of Crohn's disease and ulcerative colitis. Nat. Clin. Pract. Gastroenterol. Hepatol. 2006;3:390–407. doi: 10.1038/ncpgasthep0528. [DOI] [PubMed] [Google Scholar]
- Shevach EM, DiPaolo RA, Andersson J, Zhao DM, Stephens GL, Thornton AM. The lifestyle of naturally occurring CD4+ CD25+ Foxp3+ regulatory T cells. Immunol Rev. 2006;212:60–73. doi: 10.1111/j.0105-2896.2006.00415.x. [DOI] [PubMed] [Google Scholar]
- Suvas S, Kumaraguru U, Pack CD, Lee S, Rouse BT. CD4+CD25+ T cells regulate virus-specific primary and memory CD8+ T cell responses. J Exp. Med. 2003;198:889–901. doi: 10.1084/jem.20030171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuovinen H, Salminen JT, Arstila TP. Most human thymic and peripheral-blood CD4+ CD25+ regulatory T cells express 2 T-cell receptors. Blood. 2006;108:4063–4070. doi: 10.1182/blood-2006-04-016105. [DOI] [PubMed] [Google Scholar]
- van Santen HM, Benoist C, Mathis D. Number of T reg cells that differentiate does not increase upon encounter of agonist ligand on thymic epithelial cells. J Exp. Med. 2004;200:1221–1230. doi: 10.1084/jem.20041022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan YY, Flavell RA. Regulatory T-cell functions are subverted and converted owing to attenuated Foxp3 expression. Nature. 2007;445:766–770. doi: 10.1038/nature05479. [DOI] [PubMed] [Google Scholar]
- Watanabe N, Wang YH, Lee HK, Ito T, Wang YH, Cao W, Liu YJ. Hassall's corpuscles instruct dendritic cells to induce CD4+CD25+ regulatory T cells in human thymus. Nature. 2005;436:1181–1185. doi: 10.1038/nature03886. [DOI] [PubMed] [Google Scholar]
- Wong J, Obst R, Correia-Neves M, Losyev G, Mathis D, Benoist C. Adaptation of TCR repertoires to self-peptides in regulatory and nonregulatory CD4+ T cells. J. Immunol. 2007;178:7032–7041. doi: 10.4049/jimmunol.178.11.7032. [DOI] [PubMed] [Google Scholar]
- Yanoma S, Aoki I, Ishii N, Tani K, David CS, Okuda K. The role of autoreactive T-cell hybridomas from autoimmune model mice. Immunology. 1988;64:113–119. [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Josefowicz SZ, Kas A, Chu TT, Gavin MA, Rudensky AY. Genome-wide analysis of Foxp3 target genes in developing and mature regulatory T cells. Nature. 2007;445:936–940. doi: 10.1038/nature05563. [DOI] [PubMed] [Google Scholar]