Rapid Tumor Necrosis Factor α-Induced Exocytosis of Glutamate Receptor 2-Lacking AMPA Receptors to Extrasynaptic Plasma Membrane Potentiates Excitotoxicity (original) (raw)
Abstract
The postinjury inflammatory response in the CNS leads to neuronal excitotoxicity. Our previous studies show that a major component of this response, the inflammatory cytokine tumor necrosis factor α (TNFα), causes a rapid increase in AMPA glutamate receptors (AMPARs) on the plasma membrane of cultured hippocampal neurons. This may potentiate neuron death through an increased vulnerability to AMPAR-dependent excitotoxic stress. Here, we test this hypothesis with an in vitro lactose dehydrogenase death assay and examine in detail the AMPAR surface delivery time course, receptor subtype, and synaptic and extrasynaptic distribution after TNFα exposure. These data demonstrate that surface levels of glutamate receptor 2 (GluR2)-lacking Ca2+-permeable AMPARs peak at 15 min after TNFα treatment, and the majority are directed to extrasynaptic sites. TNFα also induces an increase in GluR2-containing surface AMPARs but with a slower time course. We propose that this activity contributes to excitotoxic neuron death because TNFα potentiation of kainate excitotoxicity is blocked by a Ca2+-permeable AMPAR antagonist [NASPM (1-naphthyl acetyl spermine)] and a specific phosphoinositide 3 kinase (PI3 kinase) inhibitor (LY294,002 [2-(4-morpholinyl)-8-phenyl-4_H_-1-benzopyran-4-one]) previously shown to block the TNFα-induced increase in AMPAR surface delivery. This information forms the basis for future in vivo studies examining AMPAR-dependent potentiation of excitotoxic neuron death and dysfunction caused by TNFα after acute injury and during neurodegenerative or neuropsychiatric disorders.
Keywords: TNFα, AMPAR, trafficking, extrasynaptic, excitotoxicity, calcium-permeable
Introduction
Neurodegenerative disease and postinjury environments are characterized by abnormally high levels of the cytokine tumor necrosis factor α (TNFα), a major component of the inflammatory process responsible for neuronal cytotoxicity and dysfunction (Shohami et al., 1999; Perry et al., 2001; Szelenyi, 2001). The specific mechanisms of TNFα-induced neuronal cytotoxicity are unclear. Several studies have searched for the roots of this cytotoxicity by focusing on the protein translation-dependent apoptotic signaling pathways that are part of the complex signaling cascades elicited by TNFα (Zhao et al., 2001; Fontaine et al., 2002; Yang et al., 2002; Zou and Crews, 2005). Recent studies suggest an alternate mechanism for the induction of TNFα-induced neuron death through the rapid TNFα-induced surface expression of AMPA-type glutamate receptors (AMPARs) (Beattie et al., 2002; Ogoshi et al., 2005; Stellwagen et al., 2005). The precise regulation of AMPAR numbers on the postsynaptic plasma membrane is essential for the control of synaptic efficacy (Carroll et al., 2001; Malinow and Malenka, 2002; Song and Huganir, 2002; Bredt and Nicoll, 2003), but disregulation of their trafficking may contribute to excitotoxic vulnerability (Leonoudakis et al., 2004; Pickering et al., 2005).
Our previous work shows that TNFα rapidly increases steady-state glutamate receptor 1 (GluR1)-containing AMPAR total cell surface accumulation 15 min after application to hippocampal neuron cultures (Beattie et al., 2002) through a phosphoinositide 3 kinase (PI3K)-dependent mechanism (Stellwagen et al., 2005). This activity can be observed in cortical neurons (Leonoudakis et al., 2004) as well as in intact spinal cord (Fergusen et al., 2006), suggesting that this is a general response of neurons to TNFα and postinjury inflammation across the CNS. Increased TNFα levels in the CNS contribute to AMPAR-dependent neuron death (Hermann et al., 2001; Noh et al., 2005), and specific blockade of Ca2+-permeable AMPARs (CP-AMPARs) prevents excitotoxicity in intact spinal cord (Corona and Tapia, 2007). These data are consistent with the hypothesis that TNFα induces a rapid rise in CP-AMPARs, which contribute to excitotoxic vulnerability.
Here, we detail the TNFα-induced increase in surface AMPAR levels, concentrating on the spatiotemporal trafficking and subunit composition in cultured hippocampal neurons as well as the functional relevance to excitotoxic vulnerability. We used live fluorescence microscopy of hippocampal neurons to demonstrate that the TNFα-induced increase in surface AMPAR accumulation is attributable to increased exocytosis of internally derived receptors to the neuron plasma membrane. We used biochemical and immunofluorescent microscopy techniques to describe the 1 h time course and synaptic/extrasynaptic distribution of AMPAR surface delivery after TNFα exposure. These data are supportive of GluR2-lacking CP-AMPAR exocytosis to extrasynaptic plasma membrane within 15 min of exogenous TNFα application followed by an increase in GluR2-containing AMPARs after 1 h. Using an in vitro neuron death assay with pharmacological control of AMPAR trafficking and activity, we demonstrated that CP-AMPAR activity is required for TNFα potentiation of neuron death. Compounds that block TNFα-induced AMPAR exocytosis to the plasma membrane block this neuron death. These data reveal a role for TNFα in initiating neuron death by trafficking CP-AMPARs to the extrasynaptic plasma membrane after the neuroinflammation process.
Materials and Methods
Hippocampal cultures.
The primary hippocampal neuron culture protocol is modified from that of Banker and Goslin (1998) and described in detail by Beattie et al. (2002). Briefly, hippocampi of embryonic day 18 (E18) Sprague Dawley rats were removed, digested with papain, and dissociated by trituration. Neurons were plated in Neurobasal medium containing B27 supplement and Glutamax (Invitrogen, Carlsbad, CA) on coverslips or plastic plates coated with poly-d-lysine. After cell attachment (3–4 h), the medium was replaced. Three days later, an equal volume of Neurobasal medium containing N2 supplement and Glutamax was added. Cultures were fed weekly by replacing one-half of the medium with Neurobasal/N2/Glutamax. Astrocyte growth was inhibited by adding 5-fluoro-2′-deoxyuridine on 6 d in vitro (div). Cultures were used for experiments between weeks 3 and 4.
Immunofluorescence AMPAR surface localization.
Before neuron treatment, the medium was changed to prewarmed artificial CSF (ACSF) (25 mm HEPES, pH 7.4, 125 mm NaCl, 2.5 mm KCl, 1 mm MgCl2, 2 mm CaCl2, 33 mm d-glucose with the following receptor antagonists: 0.5 μm tetrodotoxin, 3 μm strychnine, 20 μm bicuculline methiodide, and 20 μm d-APV to eliminate the contributions of spontaneous action potentials, inhibitory signaling, and NMDA receptor activation, respectively) for 30 min. The AMPAR antagonist CNQX had no effect on AMPAR surface expression, and the results presented represent experiments with and without this antagonist. Recombinant rat TNFα (100 ng/ml; R&D Systems, Minneapolis, MN) diluted in ACSF or ACSF alone (control) was added to neurons for the time indicated. After the indicated times, neurons were fixed with 4% paraformaldehyde/4% sucrose in PBS for 10 min, washed with PBS, blocked with 3% BSA, 2% goat serum in PBS (blocking buffer) for 1 h, and incubated with rabbit anti-GluR1 (Calbiochem, La Jolla, CA) antibodies and/or mouse anti-GluR2 antibodies (BD Biosciences, San Jose, CA) (both against extracellular epitopes) overnight at 4°C. Cells were washed three times with PBS and incubated for 45 min at room temperature with goat anti-rabbit Alexa Fluor 555 and/or goat anti-mouse Alexa Fluor 488 (Invitrogen) diluted in blocking buffer. Cells were washed and coverslips were mounted on slides with Fluoromount G (Electron Microscopy Sciences, Hatfield, PA). Fluorescence microscopy was performed on a TE2000 Nikon (Tokyo, Japan) inverted microscope with a Photometrics (Tucson, AZ) Coolsnap HQ CCD camera controlled by MetaMorph software (Molecular Devices, Downingtown, PA). For individual experiments, images for all conditions were analyzed using identical acquisition parameters as described previously (Beattie et al., 2002; Stellwagen et al., 2005). For dendritic analysis, regions of equal size containing dendrites at least 5 μm away from the soma were selected and used for quantitation as above. For postsynaptic density-95 (PSD-95) colocalization, cell surface labeling was performed as above. Cells were then permeabilized with 0.2% Triton X-100 and incubated with a mouse monoclonal anti-PSD-95 antibody (Affinity BioReagents, Golden, CO) for 1 h. Colocalization of PSD-95 and AMPAR subunits was examined by imaging neurons with a single 0.5-μm-thick plane at 1024 × 1024 pixel resolution with a Nikon C1 confocal microscope. Colocalization of PSD-95 and AMPAR subunits was quantified by carefully tracing dendrites using the region measurement tool followed by the use of the “colocalization” feature of MetaMorph software.
AMPAR cell surface biotinylation and detection.
To determine the amount of AMPAR subunit cell surface localization, we used a slight modification of an established protocol (Tsao and von Zastrow, 2000). High-density hippocampal neurons are plated on poly-d-lysine-coated six-well plates (350,000 cells) or 10 cm dishes (2 × 106 cells) and cultured for 3–4 weeks. The medium was changed to prewarmed ACSF with inhibitors as above. TNFα (100 ng/ml) diluted in ACSF or no treatment (control) was added to neurons for the indicated times and incubated at 37°C. Cells were then chilled on ice and washed with cold PBS containing 0.5 mm Ca2+ and 1 mm Mg2+ (PBSCM). Cell surface proteins were biotinylated with the membrane-impermeable reagent NHS-PEO4-biotin [15-([biotinoyl]amino)-4,7,10,13-tetraoxapentadecanoic acid, _N_-hydroxysuccinimidylester] (Pierce, Rockford, IL) at a concentration of 0.25 mg/ml freshly diluted in PBSCM and incubated on ice for 20 min. The reaction was quenched by two 10 min washes with chilled 100 mm glycine in PBSCM. Cells were scraped into 100 μl per well (six-well plates) or 1 ml per 10 cm plate RIPA lysis buffer (1% NP-40, 0.5% deoxycholate, 0.1% SDS, 150 mm NaCl, Tris, pH 8.0, with a protease inhibitor mixture; Roche, Palo Alto, CA), 1 mm EDTA, 1 mm EGTA, 1 mm PMSF, and the phosphatase inhibitors 50 mm NaF, 100 mm Na4P2O7, 10 mm Na β-glycerophosphate, 1 mm Na3VO4, solubilized for 1 h at 4°C on a rotator, and centrifuged for 15 min at 16,000 × g. Supernatant was removed and the protein concentration determined by the Bio-Rad (Hercules, CA) DC protein quantification assay.
Biochemical purification of surface AMPARs with streptavidin-agarose.
An equal amount of protein (100 μg) from each experimental condition was precipitated with streptavidin-agarose beads (Pierce) overnight at 4°C with rotation. Beads were washed three times with RIPA buffer, centrifuged through 1 m sucrose/RIPA, and washed once more, and proteins were eluted in SDS sample buffer. Eluted proteins were resolved by SDS-PAGE, transferred to polyvinylidene difluoride (PVDF), and immunoblotted with polyclonal anti-GluR1, polyclonal anti-NR1 (Chemicon, Temecula, CA), monoclonal anti-GluR2 (BD Biosciences), polyclonal anti-actin (Sigma-Aldrich, St. Louis, MO), and anti-PSD95 antibodies (Affinity BioReagents). To detect primary antibodies, blots were incubated goat anti-rabbit or goat anti-mouse antibodies conjugated to horseradish peroxidase (Jackson ImmunoResearch, West Grove, PA), visualized by enhanced chemiluminescence (Super Signal West Femto; Pierce) and digitally imaged with an Alpha Innotech (San Leandro, CA) imager. Before image processing, bands were quantified using Alpha Ease EC analysis software (Alpha Innotech). Figures are inverted images processed with Adobe Photoshop. Surface protein data were normalized to total actin in the lysate of each sample and were expressed as the percentage of untreated controls. Cell membrane integrity during the biotinylation reaction was assessed by immunoblotting the streptavidin-agarose purified protein samples for actin to insure that intracellular proteins were not labeled (data not shown).
For enrichment of synaptic and extrasynaptic surface AMPARs, neurons were biotinylated as above and proteins were solubilized in RIPA buffer containing only 1% NP-40 as the detergent, which does not solubilize postsynaptic densities (Chen et al., 2000; Oh et al., 2006). After incubation for 1 h at 4°C, the homogenate was centrifuged at 100,000 × g for 30 min to pellet the insoluble material. The supernatant (enriched for extrasynaptic AMPARs) was removed, and the insoluble material (enriched for synaptic AMPARs) was solubilized with 1% SDS and diluted 10-fold with 1% NP-40 buffer. These two fractions were then subjected to the streptavidin-agarose pulldowns as above.
Immunoprecipitation and purification of GluR2-lacking GluR1.
Detection of intracellular proteins was accomplished by immunoprecipitation of the supernatants of biotinylated extracts previously cleared of surface protein by streptavidin-agarose precipitation described above. A total of 1 μg of rabbit anti-GluR1 (Chemicon) bound to protein A-agarose (Pierce) beads or 4 μg of goat anti-GluR2 (Santa Cruz Biotechnology, Santa Cruz, CA) bound to protein G-agarose beads (Pierce) were incubated with extract for 2 h and washed as above. Immunoblot detection of precipitated proteins was performed with mouse monoclonal anti-GluR1 (Santa Cruz Biotechnology) and mouse monoclonal anti-GluR2 (BD Biosciences).
For detection of surface-biotinylated GluR2-lacking, GluR1-containing AMPARs, extract was prepared as above. To remove AMPAR complexes containing GluR2 (heteromeric GluR1/GluR2 or GluR2/3 AMPARs) (Oh and Derkach, 2005), equal amounts of protein (100 μg) from each experimental condition were first precipitated with 2 μg per immunoprecipitate (IP) goat anti-GluR2 (Santa Cruz Biotechnology) prebound to protein G-agarose (Pierce) overnight at 4°C. Immunoprecipitates were obtained by centrifugation and the supernatant was immunoprecipitated a second time with fresh 2 μg per IP anti-GluR2/protein G-agarose for 2 h at 4°C to ensure the removal of GluR2-containing AMPARs from the supernatant. The immunoprecipitate was collected by centrifugation and combined with the first immunoprecipitation. The supernatant, cleared of all detectable GluR2-containing AMPARs, was subjected to steptavidin-agarose immunoprecipitation as above followed by immunoblot detection with rabbit anti-GluR1 (Chemicon) detection as above.
Live imaging of superecliptic pHluorin-tagged GluR1 in hippocampal neurons.
A cDNA encoding superecliptic pHluorin (SEP) preceded by a cleavable signal sequence was PCR-amplified using an SEP-tagged adrenergic receptor as template (Yudowski et al., 2006) and fused in-frame between codons 3 and 4 of the cDNA encoding rat GluR1 (Boulter et al., 1990) (flop; accession no. P19490; provided by S. Heinemann, Salk Institute, San Diego, CA), using 5′-_Eco_R1 and 3′-_Bss_HI restriction sites, and inserted into the CMV (cytomegalovirus)-based mammalian expression vector pRK5.
Rat hippocampal neurons were either transfected by AMAXA nucleofection (AMAXA, Gaithersburg, MD) before plating or with Lipofectamine 2000 (Invitrogen) on div 7 with the SEP-GluR1 construct. The 14–17 div neurons were imaged live at 37°C by scanning confocal microscopy (Nikon C1 laser-scanning confocal attached to a Nikon TE2000 microscope) in ACSF with 500 nm TTX, 20 μm bicuculline methiodide, 3 μm strychnine, and 20 μm d-AP5. Neurons were imaged before bleaching, and then surface SEP-GluR1-containing AMPARs were bleached with maximum illumination from the mercury bulb light source for 1 min and allowed to recover in the presence or absence of 100 ng/ml TNFα. This protocol reduces the surface background caused by AMPARs already at the surface and allows for the efficient detection of newly delivered AMPARs derived from internal sources (Yudowski et al., 2007). Internal SEP-GluR1 containing AMPARs are not bleached by this technique because they are not at the appropriate pH and therefore not fluorescent. Images shown are maximum projections of 11 _z_-series images taken at 0.5 μm intervals. Images were then analyzed for changes in fluorescence intensity by setting a low threshold for images immediately after bleaching and comparing to 15 min of recovery with and without TNFα using the same low threshold with MetaMorph software.
Lactose dehydrogenase excitotoxicity assay.
Lactose dehydrogenase (LDH) is a stable cytosolic enzyme released during cell lysis from dead or dying cells and was used as a quantitative measure of neuron toxicity and death (Patel et al., 1993; Glass et al., 2004). Hippocampal neurons were plated at a density of 75,000 cells per well of a 24-well plate and used for assay on div 16–17. Neurons were left untreated (control) or pretreated with 100 ng/ml TNFα, 10 μm CNQX (nonselective AMPAR antagonist), 50 μm 1-naphthyl acetyl spermine (NASPM) (synthetic joro spider toxin analog; Ca2+-permeable AMPAR antagonist), 50 μm 2-(4-morpholinyl)-8-phenyl-4_H_-1-benzopyran-4-one [LY294,002 (LY)] (PI3K inhibitor), or drug plus TNFα for 15 min. Kainate (KA) at 20 μm was then added to the appropriate conditions in a final volume of 250 μl for 18 h at 37°C. Each condition was replicated in quadruplicate for each independent experiment. The medium was removed, centrifuged to remove particulate matter, and the supernatant of each sample was assayed in triplicate for colorimetric measurement of LDH activity using the LDH CytoTox 96 Assay kit following the manufacturer's instructions (Promega, Madison, WI). Each experiment was normalized to control, untreated conditions, and the data from four experiments were compiled.
Statistics.
Populations are described as mean ± SEM and statistical significance was determined by the Student's paired t test (two populations) or z test (immunoblots; two populations). Each experiment was repeated at least three times, and the n values represent the number of individual neurons for immunofluorescence microscopy and number of experiments for immunoblotting.
Results
The surface delivery of GluR1 and GluR2-containing AMPARs over 1 h of TNFα exposure
Our previous immunofluorescence microscopy studies examining the effect of TNFα on GluR1 surface localization in hippocampal neurons used a single time point of 15 min to show that TNFα dramatically increases AMPAR surface delivery (Beattie et al., 2002; Stellwagen et al., 2005). Our present studies use in vitro TNFα treatment of hippocampal neurons to detail the trafficking of AMPARs specifically induced by this singular component of the inflammatory response that neurons experience in vivo after injury. After neurotrauma, TNFα concentrations around CNS neurons rise rapidly above normal levels and these concentrations are maintained for hours to days after the injury (Allan and Rothwell, 2001). We used 100 ng/ml TNFα (used in all of the following experiments) because (1) this concentration is physiologically relevant as shown by studies of TNFα concentration in vivo in the circulation of rats after LPS exposure (Nemeth et al., 1997), and (2) this was the maximum effective concentration for the induction of AMPAR surface localization in our previous in vitro dose–response studies (Beattie et al., 2002). After CNS injury, there is also excessive glutamate release from presynaptic neurons and from astrocytes, which bathes the extrasynaptic as well as synaptic plasma membrane of the postsynaptic neuron (Allan and Rothwell, 2001). Thus, if surface AMPARs (and in particular CP-AMPARs) are maintained above normal levels on the plasma membrane (synaptic, extrasynaptic, or both), the vulnerability of neurons to excitotoxicity should also be increased. To measure the longevity of the TNFα-induced surface localization of AMPARs and to examine the subunit composition of these receptors that arrive at the plasma membrane, we used quantitative immunofluorescence to detect total surface GluR1 (synaptic plus extrasynaptic) or GluR2-containing AMPARs over a 1 h time course of TNFα exposure. Neurons were treated with TNFα for 15, 30, or 60 min or left untreated (control) as described in Materials and Methods. To detect only surface receptors, neurons were fixed without permeabilization and processed for indirect immunofluorescence visualization using extracellular epitope-directed anti-GluR1 and anti-GluR2 antibodies and imaged with wide-field fluorescence microscopy (Fig. 1A,B). We observed a ∼50% increase over control in GluR1 surface localization after just 15 min of TNFα treatment (148 ± 12% of untreated cultures; n = 87; p < 0.01) (Fig. 1A,C). Incubation with TNFα for 30 min maintained a significant increase in GluR1 surface localization but to a lesser extent than at the 15 min time point (131 ± 11%; n = 87; p < 0.05) (Fig. 1C). By 60 min of TNFα treatment, the levels of surface GluR1 had returned to near control levels (109 ± 10%; n = 51) (Fig. 1A,C). Thus, this application of continuously available TNFα caused a rapid and significant increase in surface GluR1 that reached a peak at 15 min of treatment and returned to near baseline levels at 60 min (Fig. 1A,C).
Figure 1.
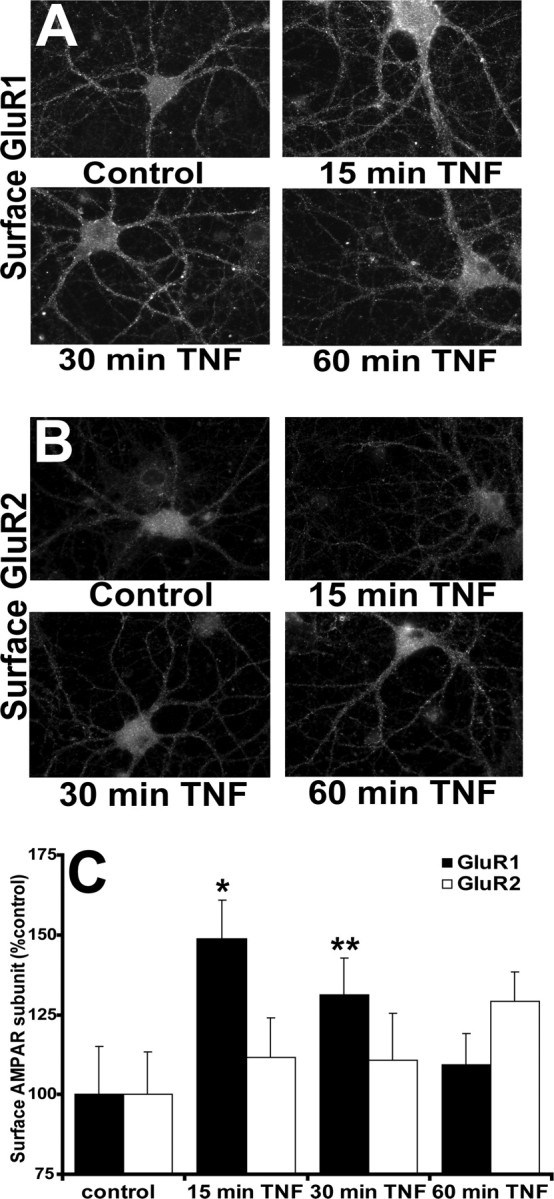
TNFα induces rapid and transient delivery of endogenous GluR1 to the plasma membrane as shown by quantitative immunofluorescence microscopy. A, B, The 17–21 div hippocampal neurons were either treated with TNFα for 15, 30, 60 min or left untreated (control). Neurons were fixed without membrane permeabilization and processed for indirect immunofluorescence using extracellular epitope-directed anti-GluR1 (A) and anti-GluR2 (B) antibodies. Images were acquired using wide-field fluorescence microscopy and were thresholded and quantified using MetaMorph software (see Materials and Methods). Compiled data for GluR1 and GluR2 is expressed in bar graph (C). Error bars indicate SEM. *p < 0.01; **p < 0.05; n = 56–87 cells from eight different experiments.
Our analysis of the surface localization of GluR2 over this time course did not detect a significant increase (15 min, 111 ± 15%, n = 56; 30 min, 110 ± 16%, n = 57; 60 min, 129 ± 15%, n = 52); however, there was a trend of the data toward increased surface GluR2 reaching a maximum at 60 min (Fig. 1B,C) that was confirmed by significant changes seen in the biochemistry below. Future studies using longer time course exposure to TNFα will test our hypothesis that GluR2-containing receptors gradually replace the GluR2-lacking AMPARs that show this dramatic but transient increase in surface localization.
Quantitation of surface and internal pools of GluR1- and GluR2-containing AMPARs over 1 h of TNFα exposure
We used the above immunofluorescence microscopy assays with success to examine increases in discrete aggregations of surface AMPARs or puncta above a given intensity threshold. Much of this analysis then is weighted toward the detection of punctate surface AMPARs, which are likely the aggregation of many individual receptors. We previously showed these AMPAR-containing puncta to be colocalized with both synaptic and extrasynaptic plasma membrane (Beattie et al., 2002). Therefore, to ensure the detection of all surface receptors, even singular AMPARs that might be below the threshold level of our microscopic analysis, we used cell surface biotinylation followed by Western blot analysis to track AMPAR surface localization over a 1 h time course of TNFα exposure. Hippocampal neurons were treated with TNFα for 15, 30, or 60 min, and surface biotinylated (see Materials and Methods). Cells were lysed and the surface biotinylated proteins were purified by streptavidin-agarose precipitation. After SDS-PAGE and electrophoretic membrane transfer, the membrane was probed with either anti-GluR1 or anti-GluR2 antibodies. Initial control experiments demonstrated an increase in surface GluR1 after 15 min TNFα (Fig. 2A) as seen in the immunofluorescence experiments above (Fig. 2B) (127 ± 7% compared with control; n = 7). As a positive control for increased AMPAR surface expression, we used a long-term potentiation chemical induction protocol (chemLTP treatment, 200 μm glycine in ACSF buffer lacking MgCl2 and d-APV) (Lu et al., 2001; Oh and Derkach, 2005). ChemLTP treatment produced a similar increase in total surface GluR1 (Fig. 2B) (128 ± 11% compared with control; n = 7). To ensure that equal concentrations of hippocampal neuron protein extract were used for pulldown experiments, extract before the pulldown was immunoblotted for actin protein levels (Fig. 2A). To ensure that internal proteins were not being labeled during the surface biotinylation procedure, streptavidin-agarose precipitates were routinely immunoblotted for the intracellular protein actin. These controls did not detect actin, suggesting that the plasma membrane was not compromised during surface biotinylation (data not shown).
Figure 2.

TNFα induces a rapid delivery of endogenous GluR1 and a more delayed delivery of GluR2 to the plasma membrane as shown by biochemical analysis. A, Control, untreated neurons or neurons treated with TNFα for 15 min or glycine [long-term potentiation chemical induction protocol (chemLTP); another method for inducing surface AMPAR delivery] and surface biotinylated. A total of 100 μg of soluble protein was processed for streptavidin-agarose pulldowns (see Materials and Methods). A total of 5 μg of total lysate from each condition was immunoblotted for total GluR1 and actin as loading controls (Loading Control). B, Immunoblots in A were quantified by densitometry and normalized to lysate controls, and the compiled data were graphed. *p < 0.01; n = 7. C, Control neurons or neurons were treated with TNFα for 15, 30, or 60 min and biotinylated as above. Streptavidin-agarose was used to precipitate and purify surface biotinylated proteins (left panels, surface receptor); supernatant from these precipitates was immunoprecipitated with either anti-GluR1 or anti-GluR2 antibodies (right panels, intracellular receptor). Precipitates were separated by SDS-PAGE, transferred to PVDF membranes, and immunoblotted with the antibodies to the proteins indicated. A total of 5 μg of lysate (bottom panel) from each condition was immunoblotted as loading controls. D, Immunoblots of surface receptors in C were quantified by densitometry and normalized to actin lysate controls, and the compiled data were graphed. E, Immunoblots of intracellular receptors in C were quantified by densitometry and normalized to actin lysate controls, and the compiled data were graphed. Error bars indicate SEM. *p < 0.05; n = 4 for each condition.
Using this cell surface biotinylation method, we analyzed both the total surface and total intracellular GluR1 and GluR2 AMPARs over a 1 h time course of TNFα treatment (Fig. 2B). Our goal was to confirm our surface immunofluorescence observations and determine possible changes in intracellular and total protein levels of GluR1 and GluR2. The biotinylation/streptavidin-agarose precipitation method described above was used to selectively purify surface AMPAR subunits from lysates of hippocampal cultures with an initial streptavidin pulldown. The GluR1 or GluR2 AMPAR subunits remaining in the supernatant were then immunoprecipitated using antibodies specific to GluR1 or GluR2. This final precipitation yields AMPAR subunits that were intracellularly localized at the time of surface biotinylation.
The surface biotinylation data agreed with the surface immunofluorescence data in that we observed an increase in surface GluR1 at 15 min of TNFα treatment (134 ± 11% of untreated cultures; n = 4; p < 0.005) (Fig. 2C,D). We noted that the significant increase in GluR1 surface expression was reduced to a nonsignificant level over control by 30 min of TNFα treatment (103 ± 14%; n = 3) and remained at near control levels (nonsignificant) even after 60 min of TNFα treatment (109 ± 17%; n = 3), as seen in our microscopy. Biochemical analysis of surface GluR2 accumulation over 60 min of TNFα treatment showed a steady increase to a significant level over untreated controls (15 min, 110 ± 10%; 30 min, 113 ± 16%; 60 min, 143 ± 23%; n = 4; p < 0.05, 60 min compared with control) (Fig. 2C,E). Intracellular levels of GluR1 or GluR2 did not significantly change (GluR1: 15 min, 92 ± 20%; 30 min, 89 ± 12%; 60 min, 96 ± 18%; n = 4; GluR2: 15 min, 104 ± 12%; 30 min, 97 ± 6%; 60 min, 105 ± 11%; n = 4); however, we do observe a trend toward less intracellular GluR1 at 15 min of TNFα treatment, which correlates with the increase in surface GluR1.
The pattern and trend of the data from the microscopy and the biochemistry were similar; however, the biochemical analysis proved less sensitive than the immunofluorescence, possibly because the biochemical analysis measures surface and internal proteins from all cell types in our mixed hippocampal cultures. To eliminate the possibility that astrocytes contributed significantly to this biochemical detection of surface or internal GluR1 or GluR2, we performed the surface biotinylation analysis on pure astrocyte cultures. We detected no surface AMPARs and barely detectable intracellular GluR2 (data not shown). As a control for the specificity of the effect of TNFα on surface AMPARs, we assayed the NR1 subunit of the NMDA receptor (Fig. 2C). Surface NR1 levels did not change significantly (n = 4) (data not shown) in agreement with previous data (Beattie et al., 2002).
These data correlate with our immunofluorescence observations that surface GluR1 levels increase within 15 min of TNFα exposure and then gradually decrease to baseline by 1 h. During this same time frame, surface GluR2 levels have a different temporal profile. Surface GluR2 does not significantly increase until 60 min after TNFα exposure. This suggests that TNFα initially induces a rapid increase in surface GluR2-lacking CP-AMPARs and then causes a slower increase in GluR2-containing (possibly GluR2–3) surface AMPARs. This sequence of initial CP-AMPARs surface delivery followed by replacement with GluR2-containing AMPARs is reminiscent of the trafficking seen during long-term potentiation (LTP) at CA1 hippocampal synapses (Plant et al., 2006). This overall net increase in surface AMPARs seen after TNFα exposure could increase neuronal excitability and sensitivity to excitotoxic cell death.
The TNFα-induced surface AMPAR increase is attributable to plasma membrane insertion of intracellularly derived receptors visualized by live confocal microscopy
The experiments above using fluorescence microscopy of fixed neurons allow us to quantitatively measure relative detectable AMPAR surface localization between groups of cultured hippocampal neurons at discrete time points after TNFα treatment. This technique can also be used to selectively or comparatively measure the surface expression of GluR1- and GluR2-containing AMPARs in relationship to synaptic markers (see Figs. 4, 5). Our present data using this technique along with data from our previous studies suggest that these newly detected receptors are intracellularly derived (Beattie et al., 2002; Stellwagen et al., 2005). However, because the fixed-tissue data has time and spatial resolution limitations, we cannot use them to eliminate the possibility that the newly detectable receptors are the result of aggregation of diffuse surface-localized AMPARs already present on the plasma membrane. Additionally, this technique does not allow us to observe AMPAR localization changes in a single cell after TNFα treatment. To specifically detect any trafficking of intracellularly derived GluR1 to the plasma membrane of individual hippocampal neurons before, during, and after TNFα application, we took advantage of the properties of the SEP form of green fluorescent protein. This is an engineered form of green fluorescent protein whose fluorescence under blue light (488 nm) illumination is quenched at intravesicular pH values of <6 (Miesenbock et al., 1998). Fusion of acidic intracellular vesicles containing SEP with the plasma membrane exposes this marker to the higher pH of the extracellular environment (pH ∼7.4) and results in an instantaneous rise in fluorescence of SEP. Therefore, SEP serves as a useful tool to visualize the delivery of new receptor-SEP fusion proteins to the cell surface from intracellular pools (Ashby et al., 2004; Kopec et al., 2006; Yudowski et al., 2006).
Figure 4.
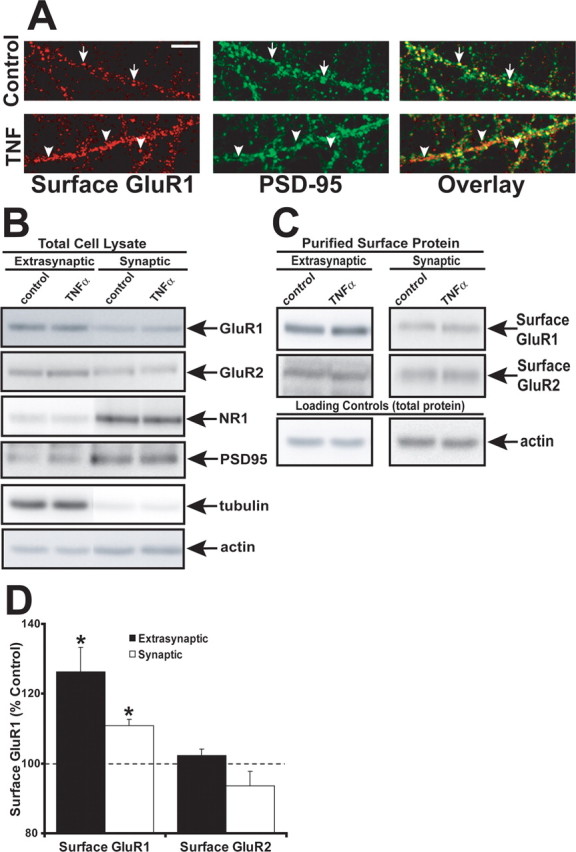
Extrasynaptic surface GluR1 is significantly increased by TNFα treatment. A, Hippocampal neurons were costained with the synaptic marker anti-PSD-95 (green) and anti-GluR1 (red) and imaged by indirect immunofluorescence confocal microscopy. The image at the right displays overlaid images with overlapping fluorescence in yellow. The arrows in the top panels indicate colocalization of GluR1 with PSD-95. The arrowheads in the bottom panels indicate GluR1 not colocalizing with PSD-95. Compiled image quantitation from three experiments demonstrates an increase of 19 ± 7% compared with control (p < 0.001; n = 16). Scale bar, 5 μm. B, To biochemically isolate surface AMPARs, hippocampal cultures were treated with TNFα for 15 min or not treated (control), surface proteins were biotinylated, and neurons were subjected to biochemical synaptic fractionation based on detergent solubility (see Materials and Methods). Total cell lysates from these fractionations (5 μg) were immunoblotted for the synaptic marker NR1 (NMDA receptor subunit) and PSD95, which were enriched in the synaptic fraction, whereas GluR1 is mostly extrasynaptic. C, Surface biotinylated proteins from both extrasynaptic and synaptic fractions were purified by streptavidin-agarose and immunoblotted with anti-GluR1 and anti-GluR2 antibodies. D, Graph of actin-normalized compiled data from immunoblots of TNFα-treated neurons performed as in C. Surface GluR1 is significantly enriched at both extrasynaptic (black bars; *p < 0.001; n = 8) fractions and synaptic fractions (white bars; *p < 0.001; n = 5), whereas GluR2 levels do not change. Error bars indicate SEM.
Figure 5.
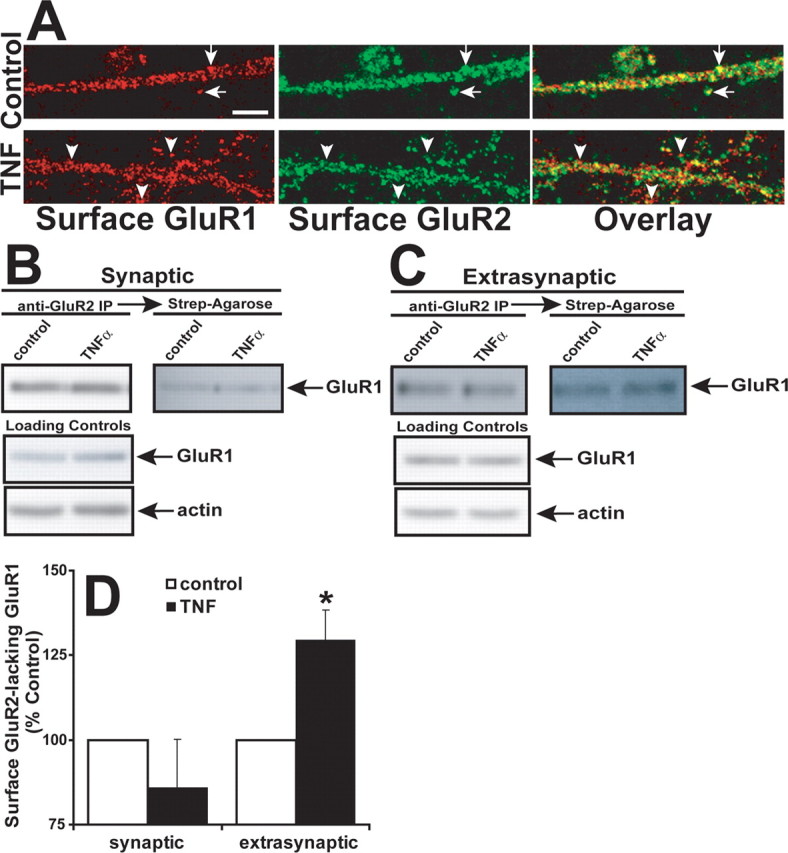
TNFα induces extrasynaptic dendritic accumulation of surface GluR2-lacking GluR1 AMPARs. A, Top, High-magnification, single-plane, confocal images of control, untreated hippocampal dendrites costained for surface GluR1 (red) and GluR2 (green) as in Figure 1. The arrows highlight examples of overlapping colocalization (yellow regions in overlaid image). Bottom, High-magnification images of TNFα-treated neurons stained as above. The arrowheads highlight regions where GluR1 does not overlap with GluR2. Image quantitation demonstrates a decrease of 28 ± 5% compared with control (p < 0.001; n = 15, 3 experiments). Scale bar, 5 μm. B, To biochemically isolate GluR2-lacking AMPARs, hippocampal synaptic lysates were prepared as in Figure 4 and immunoprecipitated with polyclonal anti-GluR2 antibodies to remove GluR2-containing AMPARs. The remaining biotinylated surface proteins in the supernatant were purified by streptavidin-agarose. Isolated proteins were immunoblotted for GluR1. C, Extrasynaptic lysates were immunoprecipitated with anti-GluR2 antibodies followed by streptavidin-agarose purification. Isolated proteins were immunoblotted for GluR1. D, Data from experiments shown in B and C were quantified and compiled. Surface biotinylated extrasynaptic extracts of TNFα-treated cultures showed a significant increase in surface GluR2-lacking GluR1 AMPARs (129 ± 9% of control; *p < 0.001; n = 8), whereas no significant change was detected in surface GluR2-lacking GluR1 AMPARs in synaptic extracts of TNFα-treated neuron cultures (86 ± 14% of control; n = 5). Error bars indicate SEM.
We fused SEP to the extracellular, N terminus of GluR1 (SEP-GluR1) to observe plasma membrane delivery of intracellular vesicles containing GluR1 in response to TNFα with live time-lapse confocal microscopy (Yudowski et al., 2007). To observe only newly membrane inserted SEP-GluR1 AMPARs, we adopted a bleaching protocol to quench the fluorescence of any SEP-GluR1 already on the neuron surface (see Materials and Methods). We then allowed neurons to recover in the presence or absence of TNFα for 15 min. Figure 3, left panels, shows an SEP-GluR1 transfected neuron before bleaching (Pre-Bleach), immediately after bleaching (Post-Bleach), and 15 min of recovery after bleaching (15 min Recov.). In these untreated, control neurons, we observed very little increase in fluorescence intensity. In contrast, after 15 min TNFα treatment, we observed a marked increase in surface fluorescence, often in clusters (Fig. 3, bottom right panel). An analysis of the total surface SEP-GluR1 fluorescence after 15 min of bleach recovery with and without TNFα revealed ∼50% increase in TNFα-treated neurons (percentage change in surface fluorescence, 148 ± 9%) compared with control untreated neurons (percentage change in surface fluorescence, 94 ± 12%) (n = 4 neurons; p < 0.05). These data agree with our immunofluorescence data showing an increase in endogenous surface GluR1 (Fig. 1C). These data indicate that TNFα stimulates the delivery of new GluR1 AMPARs from intracellular stores. Photobleaching could have effects on the immediate health of the neuron and alter normal AMPAR trafficking, but our differential results after photobleaching with control compared with TNFα application supports our contention that the amount of photobleaching we used did not perturb neurons enough to prevent rapid trafficking. Additionally, experiments done without the photobleaching also showed a similar increase of surface SEP-GluR1 over background levels after TNFα treatment (data not shown).
Figure 3.

Live confocal imaging of SEP-GluR1-transfected hippocampal neurons treated with TNFα demonstrates exocytosis and rapid surface expression of intracellularly derived SEP-GluR1. The 14 div rat hippocampal neurons AMAXA transfected with SEP-GluR1 were imaged live at 37°C by scanning confocal microscopy (each image is a maximum projection of 11 images taken at 0.5 μm intervals). Fluorescent signal from SEP-GluR1 is visualized only at the neuron plasma membrane. To maximize detection of SEP-GluR1 recently delivered to the neuron surface, 1 min of fluorescent illumination was used to selectively bleach surface SEP-GluR1 already at the plasma membrane (see Materials and Methods). The left three panels show a process from a control live neuron before bleaching (Pre-Bleach), after 1 min of bleaching (Post-Bleach), and 15 min of recovery after bleaching (15 min Recov.). The right three panels depict a neuron imaged before bleaching (Pre-Bleach), after 1 min of bleaching (Post-Bleach), and allowed to recover in presence of TNFα (15 min Recov.). The arrows highlight example areas where AMPARs are either added to existing puncta of surface AMPARs (right arrow) or to areas where new AMPAR puncta appear (left arrow). Image quantitation of four experiments per condition demonstrates ∼50% increase in TNFα-treated neurons (percentage change in surface fluorescence, 148 ± 9%) compared with control, untreated neurons (percentage change in surface fluorescence, 94 ± 12%; n = 4 neurons; p < 0.05). Scale bar, 5 μm.
TNFα increases extrasynaptic surface localization of surface GluR1 in hippocampal neurons
Having established that TNFα induces a maximal surface GluR1 localization at 15 min, we next sought to distinguish AMPARs delivered to synaptic or extrasynaptic regions and measure their distribution at this time point. Extrasynaptic AMPARs are relevant to both synaptic function and excitotoxicity. GluR1-containing AMPARs have been demonstrated to be “primed” in at least two different cases. Activation of the D1 dopamine receptor induces a rapid increase in extrasynaptic surface GluR1, and subsequent NMDA receptor stimulation induces an increase in synaptic GluR1 (Sun et al., 2005; Gao et al., 2006). Furthermore, increased serine-845 phosphorylation of GluR1 was shown to correspond with rapid trafficking of GluR1 AMPARs to extrasynaptic plasma membrane before mobilization to synaptic sites during synaptic activity (Oh et al., 2006). In addition, GluR1-containing AMPARs contribute to inappropriate excitability leading to excitotoxic death during neuronal injury situations in which neurons are exposed to increases in glutamate concentration (Olney and de Gubareff, 1978; Choi, 1994; Mattson and Chan, 2003).
First, we used immunofluorescent microscopy to measure surface GluR1 localized at synapses with and without TNFα treatment. Hippocampal cultures were treated with TNFα for 15 min, fixed, and stained for surface GluR1 and the well established postsynaptic marker PSD-95 (see Materials and Methods). We performed dual-channel confocal microscopy of dendritic surface GluR1 and total PSD-95 of both control neurons and neurons with 15 min of TNFα treatment (Fig. 4A). Merging the two, single-plane, 0.5 μm-thick images revealed overlapping regions of colocalization in yellow (Fig. 4A). We detected surface GluR1 at both synaptic (overlapping with PSD-95) and extrasynaptic (non-PSD-95-overlapping) sites. Quantitation of overlapping dendritic surface GluR1 with PSD-95 revealed a significant increase in the amount of GluR1 not colocalized with PSD-95 (extrasynaptic) in the TNFα-treated samples (increase of 19 ± 7% compared with control; p < 0.001; n = 16–19 neurons, 3 experiments). Because the overall surface localization of GluR1 goes up with TNFα, this increase implies a substantial increase in extrasynaptic GluR1 surface localization.
To confirm these quantitative microscopic results, we used a technique to biochemically enrich surface biotinylated cell lysate fractions for either synaptic or extrasynaptic protein. Hippocampal cultures were either left untreated or treated with TNFα for 15 min, and the surface proteins were biotinylated and cell lysates were processed for biochemical synaptic or extrasynaptic enrichment based on differential detergent solubility (Jo et al., 1999; Chen et al., 2000; Oh et al., 2006) (see Materials and Methods). In total cell lysates from these fractionations, proteins from the 1% NP-40 soluble fraction (“extrasynaptic”) or 1% NP-40 insoluble fraction (“synaptic”) were resolved by SDS-PAGE, transferred to PVDF membrane, and immunoblotted for GluR1, GluR2, and the postsynaptic markers NR1 (NMDA receptor subunit) and PSD-95. NR1 and PSD-95 were greatly enriched in the synaptic fraction of the total cell lysates agreeing with previous measurements of these markers (Jo et al., 1999; Chen et al., 2000), and GluR1 was found to be mostly extrasynaptic (Fig. 4B). In the TNFα-treated samples, quantitation of GluR1 in the surface, extrasynaptic, and synaptic fractions revealed significant enrichment compared with control samples (Fig. 4C,D) (extrasynaptic, 126 ± 7%, n = 8; synaptic, 110 ± 2%, n = 5; p < 0.001 compared with control). Surface extrasynaptic or synaptic GluR2 levels did not change significantly with TNFα treatment (Fig. 4C,D) (extrasynaptic, 102 ± 2%, n = 8; synaptic, 93.5 ± 4%, n = 5). Together with our microscopic synaptic colocalization, these data indicate that a significant portion of TNFα-induced surface accumulation of GluR1 AMPARs is extrasynaptic.
TNFα reduces colocalization of surface GluR1 with GluR2 in hippocampal neurons and increases the surface delivery of extrasynaptic GluR2-lacking AMPARs
The GluR2 AMPAR subunit contains an edited arginine amino acid that lines the ion channel of heteromeric GluR1/2 and GluR2/3 AMPARs that blocks passage of Ca2+ into cells during neuronal depolarization (Hollmann et al., 1991; Burnashev et al., 1992). Without this property of the GluR2 subunit, homomeric GluR1 AMPARs are permeable to Ca2+. Indeed, these GluR2-lacking AMPARs are found on hippocampal neurons (Kumar et al., 2002; Ogoshi and Weiss, 2003; Ju et al., 2004; Thiagarajan et al., 2005) and participate in the normal functioning of neurons by contributing to synaptic strengthening (Gu et al., 1996; Plant et al., 2006). Our previous immunofluorescence and biochemical data demonstrated that TNFα significantly increased surface levels of GluR1, but not GluR2 (Stellwagen et al., 2005). Electrophysiological detection of the increase in miniature EPSC (mEPSC) caused by TNFα was also blocked by an inhibitor specific to GluR1-homomeric AMPARs [_N_-(4-hydroxyphenylpropanoyl) (HPP)-spermine] (Stellwagen et al., 2005). Importantly, another study demonstrated the TNFα-induced surface increase of Ca2+-permeable AMPARs by examining Ca2+ and Co2+ uptake in hippocampal slices (Ogoshi et al., 2005). These data, along with our results above describing increased levels of extrasynaptic GluR1, suggest that TNFα increases the rapid delivery of extrasynaptic surface GluR2-lacking/homomeric GluR1 AMPARs from intracellular locations.
To confirm these findings, we first asked whether surface GluR2-lacking receptors increased compared with GluR2-containing receptors after TNFα treatment. To test this, we used confocal microscopy to detect colocalization of surface GluR1 and surface GluR2 in the presence or absence of exogenous TNFα. Neurons were treated for 15 min with TNFα or left untreated and then fixed and stained with extracellular epitope-directed anti-GluR1 and anti-GluR2 antibodies under nonpermeabilizing conditions (Fig. 5A). Quantification of the amount of dendritic GluR1 colocalizing with GluR2 demonstrates a decrease in colocalization of these two proteins (decrease to 72 ± 5% compared with control; p < 0.001; n = 15, 3 experiments). These results support the idea that new surface delivery of GluR2-lacking AMPARs is induced by TNFα (Stellwagen et al., 2005).
To determine the synaptic and extrasynaptic plasma membrane distribution of these surface-delivered GluR2-lacking AMPARs, we used a biochemical approach based on the detergent solubility of the synapse as in Figure 4. Using biotinylated and detergent-fractionated hippocampal extract derived from control or TNFα-treated cultures, we first immunoprecipitated GluR2 AMPARs using anti-GluR2 antibodies, which effectively removes GluR2-containing complexes from the extract (data not shown) (Oh and Derkach, 2005). The remaining GluR2-depleted supernatant was then incubated with streptavidin-agarose to precipitate surface biotinylated protein and immunoblotted for GluR1. We did not detect a significant change in surface GluR2-lacking GluR1 AMPARs in synaptic extracts of TNFα-treated neuron cultures (Fig. 5B,D) (86 ± 14% of control; n = 5). However, surface biotinylated extrasynaptic extracts of TNFα-treated cultures showed a significant increase in surface GluR2-lacking GluR1 AMPARs (Fig. 5C,D) (129 ± 9% of control; p < 0.001; n = 8). These biochemical data indicate that TNFα causes a significant increase in extrasynaptic surface GluR2-lacking AMPARs.
Hippocampal neurons exposed to TNFα show increased vulnerability to excitotoxicity through Ca2+-permeable AMPAR activation
The functional consequences of TNFα-induced surface localization of CP-AMPARs to extrasynaptic plasma membrane in neurons may include excess Ca2+ influx and subsequent excitotoxicity (Ogoshi and Weiss, 2003; Ogoshi et al., 2005; Kwak and Weiss, 2006). This is especially relevant in traumatic injury situations in which CNS injury or dysfunction may lead to unregulated glutamate release and spillover to extrasynaptic receptors. Thus, our observations of increased extrasynaptic AMPARs after exposure to TNFα suggest that this activity may potentiate excitotoxic cell death.
To test this idea, we used an established LDH neuron excitotoxicity assay to measure neuron death in our hippocampal cultures (Patel et al., 1993; Glass et al., 2004). LDH is a stable cytosolic enzyme released during cell lysis from dead or dying cells and can be easily quantitated by using a colorimetric assay to measure the amount of LDH released into the surrounding medium.
Hippocampal neurons were treated with TNFα for 15 min or left untreated (control). Then, the excitotoxic AMPAR agonist kainate (20 μm) was added and cultures were incubated for 18 h. Culture supernatant was then assayed for LDH activity. Treating neurons with TNFα for 18 h produced no significant increase in toxicity (100 ± 8%) from untreated control samples. Exposure to the excitotoxic drug kainate increased the level of toxicity to 481 ± 21% compared with controls (Fig. 6, black bars). Pretreating the neurons with TNFα for 15 min followed by kainate treatment led to significantly more neuron death (20%) than kainate alone (Fig. 6, black bars) (594 ± 22% from control; significance compared with kainate alone: p > 0.05, n = 4). Experiments were also performed with lower concentrations of KA. KA at 2 μm did not produce an excitotoxic effect after 18 h of exposure and no excitotoxic increase with the addition of TNFα; 10 μm KA plus TNFα compared with 10 μm KA alone showed only a slight, insignificant increase in excitotoxicity (7% increase: 10 μm KA, 144 ± 12%; TNF plus 10 μm KA, 155 ± 8%; n = 3) (data not shown). Neurons pretreated with the AMPAR/KA receptor-specific antagonist CNQX (10 μm) before exposure to kainate were completely protected from excitotoxicity (97 ± 5% compared with control) (data not shown), indicating that the cell death produced by kainate requires AMPAR/KA receptor activity. These results are in agreement with previous in vitro hippocampal studies showing the AMPAR dependence of TNFα-potentiated neuron death (Bernardino et al., 2005).
Figure 6.

Hippocampal neurons preexposed to TNFα display increased vulnerability to excitotoxicity. Neurons were left untreated (control) or pretreated with TNFα (TNF) alone or in combination with 50 μm LY294,002 (LY), 50 μm NASPM (NASPM) for 15 min. Kainate at 20 μm was then added to untreated (KA), TNFα-pretreated (TNF+KA), drug pretreated (LY+KA; NASPM+KA), or neurons pretreated with drug and TNFα (LY+KA+TNF; NASPM+KA+TNF) and incubated for 18 h. The graph is the compiled data of lactose dehydrogenase activity released from dead or dying cells into the incubation media normalized to untreated controls (see Materials and Methods). n = 4 experiments, 4 samples per experiment; *p < 0.05 compared with kainate alone. Error bars indicate SEM.
To further support the hypothesis that TNFα potentiates kainate excitotoxicity through increasing the number of surface AMPARs, we blocked the PI3K pathway with the inhibitor LY (50 μm). LY was shown previously to inhibit the TNFα-induced surface AMPAR delivery (Stellwagen et al., 2005), and we tested whether it could reduce or eliminate the TNFα excitotoxic potentiation effect. LY alone had no effect on excitotoxicity during this treatment protocol, showing little difference from controls (Fig. 6, gray bars) (151 ± 45% of control). LY pretreatment reduced the excitotoxic effect of kainate to 315 ± 29% of control, a 34.5% reduction compared with kainate alone (Fig. 6) (p < 0.005). When TNFα was added to the pretreatment conditions with LY, no additional potentiation of toxicity was observed (Fig. 6) (326 ± 12%; significance compared with LY plus kainate, p = 0.75). These data suggest that blocking the PI3 kinase pathway could eliminate TNFα-mediated potentiation of kainate excitotoxicity.
The immunofluorescence and biochemical data presented above as well as in previous studies (Ogoshi et al., 2005; Stellwagen et al., 2005) suggest that TNFα increases the surface delivery of Ca2+-permeable, GluR2-lacking AMPARs within 15 min of TNFα exposure. Ca2+-permeable AMPARs have been demonstrated to be major mediators of excitotoxic cell death (Carriedo et al., 1996; Oguro et al., 1999; Kwak and Weiss, 2006). If these Ca2+-permeable AMPARs are a significant component of the TNFα-induced surface AMPAR accumulation, then specifically blocking their activity, and thus Ca2+ influx through these channels, should reduce the extent of kainate-induced toxicity. Neurons were pretreated with the Ca2+-permeable AMPAR antagonist NASPM (50 μm; synthetic joro spider toxin analog) with and without TNFα for 15 min before kainate exposure. NASPM alone had no effect on excitotoxicity during this treatment protocol (Fig. 6) (89 ± 4% of control). Pretreatment with NASPM reduced the amount of kainate-induced excitotoxicity to 313 ± 54% of control, a 35% reduction from kainate treatment alone (Fig. 6) (p < 0.05). This reduction in toxicity from kainate treatment may reflect the block of existing surface GluR2-lacking AMPARs, and the remaining cell death induced through activation of GluR2-containing AMPARs and kainate receptors. Unlike the potentiation observed with TNFα alone (Fig. 6, black bars), pretreatment of cultures with NASPM followed by KA plus TNFα produced no additional potentiation (Fig. 6, white bars) (317 ± 53%; p = 0.95). These functional data suggest that blocking CP-AMPARs eliminates the TNFα-mediated potentiation of kainate excitotoxicity and support the data above indicating that TNFα increases surface GluR2-lacking CP-AMPARs. Together, these results show that TNFα-mediated surface delivery of CP-AMPARs contributes to the excitotoxic vulnerability of neurons and potentiates neuron injury and death.
Discussion
We used live-cell microscopy, fixed-cell microscopy, and biochemical analysis to detail the pattern, distribution, and subunit composition of TNFα-induced AMPAR surface expression. Our experiments measuring the potentiation of kainate-induced excitotoxicity by TNFα show the specific relevance of CP-AMPARs to neuronal death. This work adds new detail to our previous studies supporting the rapid (after 15 min of TNFα exposure) surface delivery of GluR2-lacking CP-AMPARs through a PI3 kinase-dependent mechanism (Beattie et al., 2002; Stellwagen et al., 2005).
Our live imaging data of TNFα-induced surface delivery of SEP-GluR1 in hippocampal neurons confirms that the source of the increased AMPAR surface accumulation seen in our original observations is intracellular and is not simply the aggregation of receptors already at the plasma membrane. Future studies will examine the nature of these AMPAR intracellular source compartments, yet it is likely that at least some of these will include recycling endosomes because induction of LTP increases surface AMPARs from these compartments (Park et al., 2004, 2006). Another possibility is the rapid synthesis and translocation of GluR1-containing AMPARs from dendritic shaft-localized endoplasmic reticulum (Job and Eberwine, 2001; Steward and Schuman, 2001; Ju et al., 2004).
Our biochemical and microscopic data examining total surface AMPAR levels over a 1 h time course of specific TNFα receptor activation is supportive of a maximum increase in total surface accumulation of CP-AMPARs at 15 min, which returns to baseline by 1 h. The amount of increased total surface CP-AMPARs (∼50% above control levels) after 15 min of TNFα is in close agreement with both our previous results (Beattie et al., 2002; Stellwagen et al., 2005) and other studies examining the TNFα effect on AMPARs in hippocampal neuron cultures (Ogoshi et al., 2005). Interestingly, rapid surface delivery of CP-AMPARs has also been observed after LTP induction of CA1 hippocampal neurons in slices with maximum surface increases and a time course profile comparable with our present results (Plant et al., 2006). CP-AMPARs are important functionally because they have stronger current passing capabilities compared with GluR2-containing AMPARs and are Ca2+-permeable (Hollmann et al., 1991; Burnashev et al., 1992). When neurons are exposed to excess glutamate after neuronal damage, excessive surface expression of CP-AMPARs induced by increased TNFα levels may lead to the abnormal influx of Ca2+ and overactivation of AMPARs resulting in potentiated excitotoxicity (Gelbard et al., 1993; Chao and Hu, 1994; Hermann et al., 2001; Bernardino et al., 2005).
Our LDH excitotoxicity data agree with studies showing an increase in GluR1 levels in NT2 cells after TNFα treatment and potentiation of kainate-induced cell death (Yu et al., 2002). Importantly, our current data show that specifically blocking the activity of CP-AMPARs with NASPM eliminates the potentiation of excitotoxicity by TNFα. Additionally, our functional kainate-induced excitotoxicity assay demonstrated that blocking TNFα-induced surface delivery AMPARs with the PI3 kinase inhibitor LY294,002 also eliminates potentiation of excitotoxic neuron death. The application of TNFα in our in vitro assay for 18 h did not itself increase neuron death, suggesting that programmed apoptotic death did not contribute to the kainate plus TNFα potentiation of neuron death. These results are in agreement with previous in vitro hippocampal slice studies showing that AMPA-induced excitotoxicity was dependent on the TNFα receptor TNFR1 but not the TNFα receptor TNFR2 (Bernardino et al., 2005). These results in combination with our data demonstrating TNFα-induced trafficking through TNFR1 activation (Stellwagen et al., 2005) fit the hypothesis that TNFR1 activation may contribute to excitotoxic death, whereas TNFR2 may signal for neuroprotection (Fontaine et al., 2002).
Increases in total surface levels of GluR2-containing AMPARs as seen by our biochemical analysis follow a more delayed time course than GluR2-lacking CP-AMPARs with a significant increase after 1 h of TNFα exposure. These results agree with other findings that GluR1-containing AMPARs are rapidly trafficked to synapses after LTP induction and then are gradually replaced by GluR2-containing AMPARs after LTP induction (Hayashi et al., 2000; Shi et al., 2001). This suggests that CP-AMPARs and GluR2-containing AMPARs are recruited to or maintained on the surface by different mechanisms. Much of the research into differential trafficking of GluR1 and GluR2 has focused on protein–protein interactions in the PSD (Malinow and Malenka, 2002; Bredt and Nicoll, 2003), yet our biochemical data show that most of the increase in surface GluR2-lacking AMPARs elicited by TNFα is extrasynaptic. How TNFα induces differential surface AMPAR subunit composition is unclear, but we established that glial-derived TNFα acts specifically through the high-affinity TNFR1 subtype of TNF receptor to increase surface AMPARs; inhibiting the PI3 kinase pathway to block TNFα-induced surface trafficking of AMPARs (Stellwagen et al., 2005). Future studies will be aimed at further identifying components of this signaling cascade in the hopes that specifically inhibiting key steps may prevent the damaging effects of excessive surface CP-AMPARs.
Our cell surface biotinylation of GluR1-containing AMPARs (which include GluR1/1 and GluR1/2) detected significant increases in both synaptic and extrasynaptic sites (Fig. 4D). However, the cell surface biotinylation and purification of GluR2-lacking/GluR1-containing CP-AMPARs (which likely are GluR1 homomeric) showed a significant increase in extrasynaptic but not synaptic localization (Fig. 5C). A previous electrophysiological experiment has demonstrated that CP-AMPARs can be increased by TNFα at synapses by measuring mEPSCs while blocking CP-AMPARs with the antagonist HPP-spermine (in addition to synaptic activity blockade) (Stellwagen et al., 2005). This block was somewhat small (82% of control) and was measured in single pyramidal neurons. It is possible that our biochemical assay, which pools data from hippocampal pyramidal neurons and interneurons, is unable to detect the small changes measurable by whole-cell electrophysiology of individual pyramidal neurons.
The postinjury milieu in the CNS is a complex mix of activity-driven responses and inflammatory mediators like the inflammatory cytokine TNFα. Our intent in this study as in our previous work was to isolate the effect of TNFα on the trafficking of AMPARs in an in vitro model by the addition of inhibitors including TTX and APV (Beattie et al., 2002; Stellwagen et al., 2005). The data presented in this study suggests that specific TNFα receptor activation increases delivery of GluR2-lacking CP-AMPARs to extrasynaptic plasma membrane. The location of initial AMPAR surface delivery is being debated in the literature. Our current results add to the mounting evidence that AMPARs are first exocytosed to extrasynaptic locations. Our recent publication showing that AMPARs are exclusively exocytosed to extrasynaptic plasma membrane after NMDAR activation via chemLTP treatment also supports this hypothesis (Yudowski et al., 2007). Some of the first researchers to support this idea have shown that D1 receptor activation increases the extrasynaptic surface pool of AMPAR (step 1) and concurrent NMDAR activity allows for synaptic localization (step 2) (Sun et al., 2005; Gao et al., 2006). Another group also supports this two-step delivery of AMPARs first to extrasynaptic and then to synaptic plasma membrane after LTP induction (Oh et al., 2006). Studies have also demonstrated the surface membrane delivery of AMPARs to extrasynaptic sites during development (King et al., 2006). Other studies show that surface AMPARs can move laterally across the membrane to synapses where they are trapped and stabilized by synaptic activity (Groc et al., 2004; Adesnik et al., 2005; Bats et al., 2007; Ehlers et al., 2007). We believe the TTX and APV activity block we use to isolate TNFα-specific components of AMPAR mobilization provides the analytical benefit of capturing AMPARs in extrasynaptic plasma membrane immediately after exocytosis. Future studies in our laboratory will examine the concurrent effects of activity and TNFα signaling on synaptic localization of AMPARs.
We provide detailed data about trafficking of AMPARs through 1 h of TNF application. At the end of this hour, we show that the levels of surface GluR2-lacking CP-AMPARs are back to baseline, whereas calcium-impermeant GluR2-containing AMPARs may be moderately increased. This trafficking data in combination with our observation that the specific CP-AMPAR blocker NASPM blocks the potentiation of KA-induced death by TNFα suggests that the potentiation is effected through the initial, rapid increase in GluR2-lacking CP-AMPARs within 15 min of TNFα exposure. These data reveal a previously unexamined window of excitotoxic vulnerability caused by TNFα. Inhibiting this activity through modulation of CP-AMPAR trafficking may reduce excitotoxicity after acute trauma or during neurological disorders.
Footnotes
This work was supported by National Institutes of Health Grants MH067931 and NS038079 as well as by The Forbes Norris ALS Center, the California Pacific Medical Center Research Institute, and ALS Association Starter Grant 766. We thank Ruth Chu and Jae-Young Seo for expert technical assistance with hippocampal dissection. We thank Steven Braithwaite, Dave Stellwagen, Michael Beattie, Jacqueline Bresnahan, Adam Ferguson, and Brandon Miller for critical discussions and suggestions.
References
- Adesnik H, Nicoll RA, England PM. Photoinactivation of native AMPA receptors reveals their real-time trafficking. Neuron. 2005;48:977–985. doi: 10.1016/j.neuron.2005.11.030. [DOI] [PubMed] [Google Scholar]
- Allan SM, Rothwell NJ. Cytokines and acute neurodegeneration. Nat Rev Neurosci. 2001;2:734–744. doi: 10.1038/35094583. [DOI] [PubMed] [Google Scholar]
- Ashby MC, De La Rue SA, Ralph GS, Uney J, Collingridge GL, Henley JM. Removal of AMPA receptors (AMPARs) from synapses is preceded by transient endocytosis of extrasynaptic AMPARs. J Neurosci. 2004;24:5172–5176. doi: 10.1523/JNEUROSCI.1042-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banker G, Goslin K. Culturing nerve cells. Ed 2. Cambridge, MA: MIT; 1998. [Google Scholar]
- Bats C, Groc L, Choquet D. The interaction between Stargazin and PSD-95 regulates AMPA receptor surface trafficking. Neuron. 2007;53:719–734. doi: 10.1016/j.neuron.2007.01.030. [DOI] [PubMed] [Google Scholar]
- Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, Beattie MS, Malenka RC. Control of synaptic strength by glial TNFalpha. Science. 2002;295:2282–2285. doi: 10.1126/science.1067859. [DOI] [PubMed] [Google Scholar]
- Bernardino L, Xapelli S, Silva AP, Jakobsen B, Poulsen FR, Oliveira CR, Vezzani A, Malva JO, Zimmer J. Modulator effects of interleukin-1β and tumor necrosis factor-α on AMPA-induced excitotoxicity in mouse organotypic hippocampal slice cultures. J Neurosci. 2005;25:6734–6744. doi: 10.1523/JNEUROSCI.1510-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulter J, Hollmann M, O'Shea-Greenfield A, Hartley M, Deneris E, Maron C, Heinemann S. Molecular cloning and functional expression of glutamate receptor subunit genes. Science. 1990;249:1033–1037. doi: 10.1126/science.2168579. [DOI] [PubMed] [Google Scholar]
- Bredt DS, Nicoll RA. AMPA receptor trafficking at excitatory synapses. Neuron. 2003;40:361–379. doi: 10.1016/s0896-6273(03)00640-8. [DOI] [PubMed] [Google Scholar]
- Burnashev N, Monyer H, Seeburg PH, Sakmann B. Divalent ion permeability of AMPA receptor channels is dominated by the edited form of a single subunit. Neuron. 1992;8:189–198. doi: 10.1016/0896-6273(92)90120-3. [DOI] [PubMed] [Google Scholar]
- Carriedo S, Yin H, Weiss J. Motor neurons are selectively vulnerable to AMPA/kainate receptor-mediated injury in vitro. J Neurosci. 1996;16:4069–4079. doi: 10.1523/JNEUROSCI.16-13-04069.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll RC, Beattie EC, von Zastrow M, Malenka RC. Role of AMPA receptor endocytosis in synaptic plasticity. Nat Rev Neurosci. 2001;2:315–324. doi: 10.1038/35072500. [DOI] [PubMed] [Google Scholar]
- Chao CC, Hu S. Tumor necrosis factor-alpha potentiates glutamate neurotoxicity in human fetal brain cell cultures. Dev Neurosci. 1994;16:172–179. doi: 10.1159/000112104. [DOI] [PubMed] [Google Scholar]
- Chen L, Chetkovich DM, Petralia RS, Sweeney NT, Kawasaki Y, Wenthold RJ, Bredt DS, Nicoll RA. Stargazin regulates synaptic targeting of AMPA receptors by two distinct mechanisms. Nature. 2000;408:936–943. doi: 10.1038/35050030. [DOI] [PubMed] [Google Scholar]
- Choi DW. Glutamate receptors and the induction of excitotoxic neuronal death. Prog Brain Res. 1994;100:47–51. doi: 10.1016/s0079-6123(08)60767-0. [DOI] [PubMed] [Google Scholar]
- Corona JC, Tapia R. Ca2+-permeable AMPA receptors and intracellular Ca2+ determine motoneuron vulnerability in rat spinal cord in vivo. Neuropharmacology. 2007;52:1219–1228. doi: 10.1016/j.neuropharm.2006.12.008. [DOI] [PubMed] [Google Scholar]
- Ehlers MD, Heine M, Groc L, Lee MC, Choquet D. Diffusional trapping of GluR1 AMPA receptors by input-specific synaptic activity. Neuron. 2007;54:447–460. doi: 10.1016/j.neuron.2007.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fergusen AR, Christensen RN, Gensel JC, Miller BA, Beattie EC, Bresnahan JC, Beattie MS. Effect of tumor necrosis factor alpha on AMPA-type glutamate receptor trafficking in spinal cord neurons in vivo. Soc Neurosci Abstr. 2006;32:475–11. [Google Scholar]
- Fontaine V, Mohand-Said S, Hanoteau N, Fuchs C, Pfizenmaier K, Eisel U. Neurodegenerative and neuroprotective effects of tumor necrosis factor. 2002 doi: 10.1523/JNEUROSCI.22-07-j0001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao C, Sun X, Wolf ME. Activation of D1 dopamine receptors increases surface expression of AMPA receptors and facilitates their synaptic incorporation in cultured hippocampal neurons. J Neurochem. 2006;98:1664–1677. doi: 10.1111/j.1471-4159.2006.03999.x. [DOI] [PubMed] [Google Scholar]
- Gelbard HA, Dzenko KA, DiLoreto D, del Cerro C, del Cerro M, Epstein LG. Neurotoxic effects of tumor necrosis factor alpha in primary human neuronal cultures are mediated by activation of the glutamate AMPA receptor subtype: implications for AIDS neuropathogenesis. Dev Neurosci. 1993;15:417–422. doi: 10.1159/000111367. [DOI] [PubMed] [Google Scholar]
- Glass TF, Reeves B, Sharp FR. The impact of excitotoxic blockade on the evolution of injury following combined mechanical and hypoxic insults in primary rat neuronal culture. Neurobiol Dis. 2004;17:378–384. doi: 10.1016/j.nbd.2004.07.017. [DOI] [PubMed] [Google Scholar]
- Groc L, Heine M, Cognet L, Brickley K, Stephenson FA, Lounis B, Choquet D. Differential activity-dependent regulation of the lateral mobilities of AMPA and NMDA receptors. Nat Neurosci. 2004;7:695–696. doi: 10.1038/nn1270. [DOI] [PubMed] [Google Scholar]
- Gu JG, Albuquerque C, Lee CJ, MacDermott AB. Synaptic strengthening through activation of Ca2+-permeable AMPA receptors. Nature. 1996;381:793–796. doi: 10.1038/381793a0. [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Shi SH, Esteban JA, Piccini A, Poncer JC, Malinow R. Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science. 2000;287:2262–2267. doi: 10.1126/science.287.5461.2262. [DOI] [PubMed] [Google Scholar]
- Hermann GE, Rogers RC, Bresnahan JC, Beattie MS. Tumor necrosis factor-alpha induces cFOS and strongly potentiates glutamate-mediated cell death in the rat spinal cord. Neurobiol Dis. 2001;8:590–599. doi: 10.1006/nbdi.2001.0414. [DOI] [PubMed] [Google Scholar]
- Hollmann M, Hartley M, Heinemann S. Ca2+ permeability of KA-AMPA-gated glutamate receptor channels depends on subunit composition. Science. 1991;252:851–853. doi: 10.1126/science.1709304. [DOI] [PubMed] [Google Scholar]
- Jo K, Derin R, Li M, Bredt DS. Characterization of MALS/Velis-1, -2, and -3: a family of mammalian LIN-7 homologs enriched at brain synapses in association with the postsynaptic density-95/NMDA receptor postsynaptic complex. J Neurosci. 1999;19:4189–4199. doi: 10.1523/JNEUROSCI.19-11-04189.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Job C, Eberwine J. Localization and translation of mRNA in dendrites and axons. Nat Rev Neurosci. 2001;2:889–898. doi: 10.1038/35104069. [DOI] [PubMed] [Google Scholar]
- Ju W, Morishita W, Tsui J, Gaietta G, Deerinck TJ, Adams SR, Garner CC, Tsien RY, Ellisman MH, Malenka RC. Activity-dependent regulation of dendritic synthesis and trafficking of AMPA receptors. Nat Neurosci. 2004;7:244–253. doi: 10.1038/nn1189. [DOI] [PubMed] [Google Scholar]
- King AE, Chung RS, Vickers JC, Dickson TC. Localization of glutamate receptors in developing cortical neurons in culture and relationship to susceptibility to excitotoxicity. J Comp Neurol. 2006;498:277–294. doi: 10.1002/cne.21053. [DOI] [PubMed] [Google Scholar]
- Kopec CD, Li B, Wei W, Boehm J, Malinow R. Glutamate receptor exocytosis and spine enlargement during chemically induced long-term potentiation. J Neurosci. 2006;26:2000–2009. doi: 10.1523/JNEUROSCI.3918-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar SS, Bacci A, Kharazia V, Huguenard JR. A developmental switch of AMPA receptor subunits in neocortical pyramidal neurons. J Neurosci. 2002;22:3005–3015. doi: 10.1523/JNEUROSCI.22-08-03005.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak S, Weiss JH. Calcium-permeable AMPA channels in neurodegenerative disease and ischemia. Curr Opin Neurobiol. 2006;16:281–287. doi: 10.1016/j.conb.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Leonoudakis D, Braithwaite SP, Beattie MS, Beattie EC. TNFalpha-induced AMPA-receptor trafficking in CNS neurons; relevance to excitotoxicity? Neuron Glia Biol. 2004;1:263–273. doi: 10.1017/S1740925X05000608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Man H, Ju W, Trimble WS, MacDonald JF, Wang YT. Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron. 2001;29:243–254. doi: 10.1016/s0896-6273(01)00194-5. [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Chan SL. Neuronal and glial calcium signaling in Alzheimer's disease. Cell Calcium. 2003;34:385–397. doi: 10.1016/s0143-4160(03)00128-3. [DOI] [PubMed] [Google Scholar]
- Miesenbock G, De Angelis DA, Rothman JE. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 1998;394:192–195. doi: 10.1038/28190. [DOI] [PubMed] [Google Scholar]
- Nemeth ZH, Hasko G, Szabo C, Vizi ES. Amrinone and theophylline differentially regulate cytokine and nitric oxide production in endotoxemic mice. Shock. 1997;7:371–375. doi: 10.1097/00024382-199705000-00010. [DOI] [PubMed] [Google Scholar]
- Noh KM, Yokota H, Mashiko T, Castillo PE, Zukin RS, Bennett MV. Blockade of calcium-permeable AMPA receptors protects hippocampal neurons against global ischemia-induced death. Proc Natl Acad Sci USA. 2005;102:12230–12235. doi: 10.1073/pnas.0505408102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogoshi F, Weiss JH. Heterogeneity of Ca2+-permeable AMPA/kainate channel expression in hippocampal pyramidal neurons: fluo-rescence imaging and immunocytochemical assessment. J Neurosci. 2003;23:10521–10530. doi: 10.1523/JNEUROSCI.23-33-10521.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogoshi F, Yin HZ, Kuppumbatti Y, Song B, Amindari S, Weiss JH. Tumor necrosis-factor-alpha (TNF-alpha) induces rapid insertion of Ca2+-permeable alpha-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA)/kainate (Ca-A/K) channels in a subset of hippocampal pyramidal neurons. Exp Neurol. 2005;193:384–393. doi: 10.1016/j.expneurol.2004.12.026. [DOI] [PubMed] [Google Scholar]
- Oguro K, Oguro N, Kojima T, Grooms SY, Calderone A, Zheng X, Bennett MV, Zukin RS. Knockdown of AMPA receptor GluR2 expression causes delayed neurodegeneration and increases damage by sublethal ischemia in hippocampal CA1 and CA3 neurons. J Neurosci. 1999;19:9218–9227. doi: 10.1523/JNEUROSCI.19-21-09218.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh MC, Derkach VA. Dominant role of the GluR2 subunit in regulation of AMPA receptors by CaMKII. Nat Neurosci. 2005;8:853–854. doi: 10.1038/nn1476. [DOI] [PubMed] [Google Scholar]
- Oh MC, Derkach VA, Guire ES, Soderling TR. Extrasynaptic membrane trafficking regulated by GluR1 serine 845 phosphorylation primes AMPA receptors for long-term potentiation. J Biol Chem. 2006;281:752–758. doi: 10.1074/jbc.M509677200. [DOI] [PubMed] [Google Scholar]
- Olney JW, de Gubareff T. Glutamate neurotoxicity and Huntington's chorea. Nature. 1978;271:557–559. doi: 10.1038/271557a0. [DOI] [PubMed] [Google Scholar]
- Park M, Penick EC, Edwards JG, Kauer JA, Ehlers MD. Recycling endosomes supply AMPA receptors for LTP. Science. 2004;305:1972–1975. doi: 10.1126/science.1102026. [DOI] [PubMed] [Google Scholar]
- Park M, Salgado JM, Ostroff L, Helton TD, Robinson CG, Harris KM, Ehlers MD. Plasticity-induced growth of dendritic spines by exocytic trafficking from recycling endosomes. Neuron. 2006;52:817–830. doi: 10.1016/j.neuron.2006.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel MN, Yim GK, Isom GE. N-Methyl-d-aspartate receptors mediate cyanide-induced cytotoxicity in hippocampal cultures. Neurotoxicology. 1993;14:35–40. [PubMed] [Google Scholar]
- Perry RT, Collins JS, Wiener H, Acton R, Go RC. The role of TNF and its receptors in Alzheimer's disease. Neurobiol Aging. 2001;22:873–883. doi: 10.1016/s0197-4580(01)00291-3. [DOI] [PubMed] [Google Scholar]
- Pickering M, Cumiskey D, O'Connor JJ. Actions of TNF-alpha on glutamatergic synaptic transmission in the central nervous system. Exp Physiol. 2005;90:663–670. doi: 10.1113/expphysiol.2005.030734. [DOI] [PubMed] [Google Scholar]
- Plant K, Pelkey KA, Bortolotto ZA, Morita D, Terashima A, McBain CJ, Collingridge GL, Isaac JT. Transient incorporation of native GluR2-lacking AMPA receptors during hippocampal long-term potentiation. Nat Neurosci. 2006;9:602–604. doi: 10.1038/nn1678. [DOI] [PubMed] [Google Scholar]
- Shi S, Hayashi Y, Esteban JA, Malinow R. Subunit-specific rules governing AMPA receptor trafficking to synapses in hippocampal pyramidal neurons. Cell. 2001;105:331–343. doi: 10.1016/s0092-8674(01)00321-x. [DOI] [PubMed] [Google Scholar]
- Shohami E, Ginis I, Hallenbeck JM. Dual role of tumor necrosis factor alpha in brain injury. Cytokine Growth Factor Rev. 1999;10:119–130. doi: 10.1016/s1359-6101(99)00008-8. [DOI] [PubMed] [Google Scholar]
- Song I, Huganir RL. Regulation of AMPA receptors during synaptic plasticity. Trends Neurosci. 2002;25:578–588. doi: 10.1016/s0166-2236(02)02270-1. [DOI] [PubMed] [Google Scholar]
- Stellwagen D, Beattie EC, Seo JY, Malenka RC. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-α. J Neurosci. 2005;25:3219–3228. doi: 10.1523/JNEUROSCI.4486-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steward O, Schuman EM. Protein synthesis at synaptic sites on dendrites. Annu Rev Neurosci. 2001;24:299–325. doi: 10.1146/annurev.neuro.24.1.299. [DOI] [PubMed] [Google Scholar]
- Sun X, Zhao Y, Wolf ME. Dopamine receptor stimulation modulates AMPA receptor synaptic insertion in prefrontal cortex neurons. J Neurosci. 2005;25:7342–7351. doi: 10.1523/JNEUROSCI.4603-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szelenyi J. Cytokines and the central nervous system. Brain Res Bull. 2001;54:329–338. doi: 10.1016/s0361-9230(01)00428-2. [DOI] [PubMed] [Google Scholar]
- Thiagarajan TC, Lindskog M, Tsien RW. Adaptation to synaptic inactivity in hippocampal neurons. Neuron. 2005;47:725–737. doi: 10.1016/j.neuron.2005.06.037. [DOI] [PubMed] [Google Scholar]
- Tsao PI, von Zastrow M. Type-specific sorting of G protein-coupled receptors after endocytosis. J Biol Chem. 2000;275:11130–11140. doi: 10.1074/jbc.275.15.11130. [DOI] [PubMed] [Google Scholar]
- Yang L, Lindholm K, Konishi Y, Li R, Shen Y. Target depletion of distinct tumor necrosis factor receptor subtypes reveals hippocampal neuron death and survival through different signal transduction pathways. J Neurosci. 2002;22:3025–3032. doi: 10.1523/JNEUROSCI.22-08-03025.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z, Cheng G, Wen X, Wu GD, Lee WT, Pleasure D. Tumor necrosis factor alpha increases neuronal vulnerability to excitotoxic necrosis by inducing expression of the AMPA-glutamate receptor subunit GluR1 via an acid sphingomyelinase- and NF-kappaB-dependent mechanism. Neurobiol Dis. 2002;11:199–213. doi: 10.1006/nbdi.2002.0530. [DOI] [PubMed] [Google Scholar]
- Yudowski GA, Puthenveedu MA, von Zastrow M. Distinct modes of regulated receptor insertion to the somatodendritic plasma membrane. Nat Neurosci. 2006;9:622–627. doi: 10.1038/nn1679. [DOI] [PubMed] [Google Scholar]
- Yudowski GA, Puthenveedu MA, Leonoudakis D, Panicker S, Thorn KS, Beattie EC, von Zastrow M. Real-time imaging of discrete exocytic events mediating surface delivery of AMPA receptors. J Neurosci. 2007;27:11112–11121. doi: 10.1523/JNEUROSCI.2465-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Bausano B, Pike BR, Newcomb-Fernandez JK, Wang KK, Shohami E, Ringger NC, DeFord SM, Anderson DK, Hayes RL. TNF-alpha stimulates caspase-3 activation and apoptotic cell death in primary septo-hippocampal cultures. J Neurosci Res. 2001;64:121–131. doi: 10.1002/jnr.1059. [DOI] [PubMed] [Google Scholar]
- Zou JY, Crews FT. TNF alpha potentiates glutamate neurotoxicity by inhibiting glutamate uptake in organotypic brain slice cultures: neuroprotection by NF kappa B inhibition. Brain Res. 2005;1034:11–24. doi: 10.1016/j.brainres.2004.11.014. [DOI] [PubMed] [Google Scholar]