Allele-specific copy number analysis of tumors (original) (raw)
Abstract
We present an allele-specific copy number analysis of the in vivo breast cancer genome. We describe a unique bioinformatics approach, ASCAT (allele-specific copy number analysis of tumors), to accurately dissect the allele-specific copy number of solid tumors, simultaneously estimating and adjusting for both tumor ploidy and nonaberrant cell admixture. This allows calculation of “ASCAT profiles” (genome-wide allele-specific copy-number profiles) from which gains, losses, copy number-neutral events, and loss of heterozygosity (LOH) can accurately be determined. In an early-stage breast carcinoma series, we observe aneuploidy (>2.7n) in 45% of the cases and an average nonaberrant cell admixture of 49%. By aggregation of ASCAT profiles across our series, we obtain genomic frequency distributions of gains and losses, as well as genome-wide views of LOH and copy number-neutral events in breast cancer. In addition, the ASCAT profiles reveal differences in aberrant tumor cell fraction, ploidy, gains, losses, LOH, and copy number-neutral events between the five previously identified molecular breast cancer subtypes. Basal-like breast carcinomas have a significantly higher frequency of LOH compared with other subtypes, and their ASCAT profiles show large-scale loss of genomic material during tumor development, followed by a whole-genome duplication, resulting in near-triploid genomes. Finally, from the ASCAT profiles, we construct a genome-wide map of allelic skewness in breast cancer, indicating loci where one allele is preferentially lost, whereas the other allele is preferentially gained. We hypothesize that these alternative alleles have a different influence on breast carcinoma development.
Keywords: breast carcinoma, single-nucleotide polymorphism arrays, bioinformatics, cancer
Genomic changes are key causative events of cancer. Cancer genomes are characterized by numerous sequence changes compared with their normal host counterparts, ranging in size from single base changes (point mutations) to insertions or deletions of large chromosomal fragments and even whole-genome duplications (1, 2). These cancer genomes have been extensively charted by array-comparative genomic hybridization (CGH), SNP arrays (3, 4), and more recently by whole-genome sequencing (5–8). However, correct assembly and interpretation of the data have proven difficult, because tumors often deviate from a diploid state (9, 10), and many contain multiple populations of both tumor and nontumor cells (11, 12). For these reasons, most studies have been limited to reporting gains and losses (array CGH), possibly supplemented by allelic imbalances (SNP arrays), and are unable to assign correct (allele-specific) copy numbers to all loci in the reference genome. Similarly, for correct assembly of complete cancer genomes from sequencing data, the calculation of accurate copy numbers for all loci is a necessary first step for correct interpretation of changes ranging from point mutations to large-scale genomic rearrangements.
We present here an allele-specific copy number analysis of the in vivo breast cancer genome, in which both aneuploidy of the tumor cells and nonaberrant cell infiltration are taken into account. We obtain accurate genome-wide allele-specific copy-number profiles [called “ASCAT (allele-specific copy number analysis of tumors) profiles”] for 91 of 112 breast carcinomas assayed. On the basis of these ASCAT profiles, differences in aberrant tumor cell fraction, ploidy, gains, losses, loss of heterozygosity (LOH), and copy number-neutral events are revealed among the five previously identified molecular breast cancer subtypes. Finally, by evaluating the relative frequency of deletions and duplications of the two possible alleles at each SNP locus, we construct a genome-wide map of allelic skewness, pointing to candidate genes/loci that may drive breast cancer development.
Results
Allele-Specific Copy Number Analysis of Breast Carcinomas.
We performed genotyping of 112 breast carcinoma samples using Illumina 109K SNP arrays and constructed an algorithm (ASCAT) to estimate the fraction of aberrant cells and the tumor ploidy, as well as whole-genome allele-specific copy number profiles taking both properties into account (Fig. 1 and Figs. S1 and S2). Using ASCAT, we obtained genome-wide allele-specific copy number profiles (hereafter called ASCAT profiles) for 91 (81%) of the breast carcinomas. Most of the 21 cases (19%) for which ASCAT indicated that no acceptable solution could be found were characterized by significantly larger residual variance in the Log R profiles (Fig. S3). Hence, ASCAT is able to calculate the allele-specific copy numbers of all assayed SNP loci, taking into account tumor aneuploidy and the fraction of aberrant tumor cells, for most of our breast carcinoma cases, and indicates when the quality of the input data are questionable.
Fig. 1.

ASCAT profiles and their calculation. Two examples are given: (A) a tumor with ploidy close to 2n and (B) a tumor with ploidy close to 4n. (Left) ASCAT first determines the ploidy of the tumor cells ψ_t_ and the fraction of aberrant cells ρ. This procedure evaluates the goodness of fit for a grid of possible values for both parameters (blue, good solution; red, bad solution; detailed in Materials and Methods). On the basis of this goodness of fit, the optimal solution is selected (green cross). Using the resulting tumor ploidy and aberrant cell fraction, an ASCAT profile is calculated (Upper Right), containing the allele-specific copy number of all assayed loci [copy number on the y axis vs. the genomic location on the x axis; green, allele with lowest copy number; red, allele with highest copy number; for illustrative purposes only, both lines are slightly shifted (red, down; green, up) such that they do not overlap; only probes heterozygous in the germline are shown]. Finally, for all aberrations found, an aberration reliability score is calculated (Lower Right).
We validated the ASCAT profile predictions in three ways. First, we checked ASCAT's consistency and sensitivity to a varying percentage of aberrant tumor cells by applying the algorithm to a dilution series of a tumor sample mixed with different proportions of its germline DNA. Overall, ASCAT profiles were very similar for the different dilutions (Fig. S4). Second, we validated ASCAT's ploidy predictions by experimentally determining the amount of DNA in the tumor cells for 79 of the 91 scored breast cancer cases. We obtained good correspondence with ASCAT's predictions (Fig. 2 and Fig. S5). Finally, FISH experiments were performed on 11 of the breast carcinomas for three loci (Table S1), suggesting a good correspondence with the copy numbers estimated by ASCAT, although FISH seems to consistently underestimate the copy number as compared with ASCAT. Together, these validation experiments confirm that ASCAT accurately predicts ASCAT profiles over a broad range of tumor ploidy and fraction of aberrant tumor cells.
Fig. 2.

Validation of tumor ploidy predicted by ASCAT. ASCAT's ploidy estimates are plotted relative to experimentally measured ploidy. Here we define ploidy as the amount of DNA relative to a haploid genome. For 58 of the 79 assayed samples (73.4%), ASCAT's ploidy predictions correspond well with the experimentally determined ploidy (green triangles close to the diagonal). Three samples (3.8%) have an experimentally determined ploidy larger than 5n (blue triangles), outside of the ploidy range used by ASCAT (1.6n–4.8n, depicted by the blue square). Ten breast carcinomas (12.7%) have a predicted ploidy close to 2n, whereas the experimentally determined ploidy was close to 4n (pink triangles). For most of these cases, manual inspection of the copy number profiles could not reveal any indications (missed by ASCAT) that these samples are in fact close to tetraploid (Fig. S5_A_). Indeed, cases that are tetraploid but only show even-numbered allele-specific copy numbers would be recognized as diploid, because the SNP array data do not provide any information to distinguish such tetraploid samples from diploid samples. Alternatively, the experimental method for ploidy determination, applied to a different part of the tumor as the SNP arrays, could be measuring tumor cells in the S phase of the cell cycle, or a different subclone of the tumor. Finally, eight samples (10.1%) show clearly higher ploidy by ASCAT prediction compared with the experimentally determined ploidy (red triangles). A possible explanation for this is the presence of multiple populations of aberrant tumor cells with (slightly) different aberrations (Fig. S5_B_).
Ploidy and Fraction of Aberrant Tumor Cells in Breast Carcinoma.
To investigate the relevance of aneuploidy and involvement of nonaberrant cells in breast carcinoma, we examined the ploidy and aberrant cell fraction estimates for our breast carcinoma series. Because the tumor samples were macrodissected to remove as much as possible of the surrounding nontumor tissue by a pathologist, the aberrant cell fraction estimates will reflect intratumoral nonaberrant cells and not normal cells surrounding the tumor. We found that the tumors are on average infiltrated with 49% nonaberrant cells and that 45% of them have a ploidy of 2.7n or higher. These results confirm the importance of taking both nonaberrant cell admixture and tumor aneuploidy into account.
Breast carcinomas can be divided into five distinct subtypes, depending on their expression pattern of 561 transcripts (13). These five subgroups, Luminal A, Luminal B, ERBB2, Basal-like, and Normal-like breast carcinoma, are associated with different clinical outcomes (14). We correlated our ploidy and aberrant cell fraction estimates with these gene-expression–based breast cancer subtypes. Stratifying the estimated percentages of aberrant cells by breast cancer subtype revealed considerable differences (Fig. 3_A_), with the highest fraction of aberrant tumor cells for the Luminal A subtype and the lowest fraction for the ERBB2 and Normal-like subtypes. An evaluation of tumor ploidy stratified by molecular subtype showed lowest ploidy for Luminal A, Basal-like, and Normal-like subtypes and highest ploidy for the ERBB2 subtype (Fig. 3_B_). For the Luminal A subtype, the specific ploidy distribution along with its characteristic paucity of aberrations (and preference for aberrations involving whole chromosome arms) implies a common state of diploidy for these tumors, with a minority of Luminal A carcinomas having undergone polyploidization by endoreduplication (with only few additional aberrations), resulting in a tetraploid state.
Fig. 3.

Percentage of aberrant tumor cells and ploidy across the five breast cancer subtypes. Molecular subtypes used: LumA, Luminal A (n = 45); LumB, Luminal B (n = 10); ERBB2 (n = 12); Basal, Basal-like (n = 12); Normal, Normal-like (n = 8). (A) Distribution of percentage of aberrant tumor cells across the five subtypes. The box plots show the median (thick lines) and the lower and upper quartile (boxes). The whiskers reach up to the most extreme value within 1.5 times the interquartile range from the box. Whereas Luminal A carcinomas harbored the highest levels of aberrant tumor cells (P = 6.9 × 10−6, unpaired t test with unequal variance, testing for differences between the Luminal A subtype and all other carcinomas), tumors of the ERBB2 and Normal-like subtype displayed the lowest fraction of aberrant cells (P = 3.7 × 10−4 and P = 8.4 × 10−3, respectively). (B) Distribution of ploidy across the five subtypes. The vast majority of Luminal A tumors showed a ploidy close to 2n, with a smaller fraction showing a ploidy close to 4n. Carcinomas of the Luminal B subtype were approximately equally divided among 2n and 4n tumors, with two tumors being 3n. On average, the ERBB2 subgroup displayed the highest level of ploidy but also the broadest range. The Basal-like subgroup showed cases with a ploidy 1.6n–2n and cases of 2.8n–3.2n. Normal-like tumors showed a group of cases with ploidy close to 2n and a group of cases with ploidy above 3n.
ASCAT Profiles Allow Accurate Dissection of Gains, Losses, LOH, and Copy Number-Neutral Events and Grant Insight into Tumor Development.
The frequency of gains and losses in a population can be deduced from ASCAT profiles. In our breast carcinoma series, this resulted in similar but slightly more pronounced patterns compared with previous (array-CGH) reports (15–17) (Fig. S6_A_). However, stratification by molecular subtype resulted in considerably higher frequencies of gains and losses specifically for the ERBB2 and the Normal-like subtypes than those directly derived from Log R data (Fig. S6_B_). Hence, contrary to previous reports describing only a limited number of aberrations for these two subtypes (15–17), the use of ASCAT profiles results in clear gains and losses. These aberrations were missed by earlier approaches owing to the high fractions of nonaberrant cells in the ERBB2 and Normal-like subtypes (Fig. 3_A_), a feature taken into account when using ASCAT profiles.
The ASCAT profiles also allow us to investigate LOH and copy number-neutral events. This is not possible using direct evaluation of SNP array data from tumor samples, owing to the admixture with cells not showing these events. A distinct pattern of LOH across the genome emerges (Fig. 4_A_), with LOH being most frequent on chromosome arms 8p, 11q, 16q, and 17p. The q arm of chromosome 16 shows the highest fraction of LOH, including among others SNPs residing in multiple members of the cadherin family (e.g., CDH1, CDH3, CDH15, CDH13, and CDH8). A calculation of the frequency of copy number-neutral events reveals many regions with a frequency above 20%, with some peaks up to 50% (Fig. 4_B_). We define here a copy number-neutral event as an allelic bias for an SNP heterozygous in the germline such that the total copy number does not differ from the tumor ploidy. Following this definition, copy number-neutral events are all genomic aberrations that cannot be detected by array-CGH. We observe that many genomic regions with higher frequency of loss were also more likely to harbor copy number-neutral events. This is particularly apparent for chromosome/chromosome arm 1p, 2, 3, 4q, 9q, 15, and 19p. This suggests that the frequency of actual loss (loss of one allele, possibly combined with gain of the other allele) is considerably higher than what was previously reported (considering only the total amount of DNA, not distinguishing both alleles). Also notable is chromosome 17q, showing both a high frequency of gains and elevated levels of copy number-neutral events. This chromosome arm harbors among others the ERBB2 gene, a gene renowned for its relevance in breast cancer. The overall highest frequencies of copy number-neutral events were seen for chromosome 2, 3, 4, 6, 12, and 15, chromosomes not previously reported as key areas for genomic aberrations in breast cancers, suggesting that copy number-neutral events may represent an as-yet unexplored picture of genomic aberrations in breast cancer.
Fig. 4.

Frequency of LOH and copy number-neutral events. (A) Frequency of LOH across the genome. Probes are shown in genomic order along the x axis, from chromosome 1 to chromosome X, where different chromosomes are delimited by gray lines. (B) Frequency of copy number-neutral events across the genome. For diploid tumors, copy number-neutral events correspond to a subset of LOH (copy number-neutral LOH), but for, for example, tetraploid tumors, a copy number-neutral event can also be three copies of A and one copy of B. (C) Proportion of LOH per case (percentage of probes heterozygous in the germline that have lost this heterozygosity in the tumor), stratified by molecular breast cancer subtypes. Molecular subtypes used and box plot legends are the same as in Fig. 3. The Luminal A subtype shows a significantly lower frequency of LOH compared with the four other subtypes (P = 2.3 × 10−6, unpaired t test with unequal variance). Even more striking is the elevated level of LOH for the Basal-like subtype (P = 1.0 × 10−3). Indeed, two thirds of the Basal-like tumors show LOH at more than 40% of the loci heterozygous in the germline. (D) Proportion of copy number-neutral events per case, stratified by molecular breast cancer subtypes. The Luminal A (P = 4.7 × 10−3, unpaired t test with unequal variance, testing for differences between the Luminal A subtype and all other carcinomas) and Normal-like (P = 0.95) subtype display low levels of copy number-neutral events, the Luminal B subgroup shows intermediate levels (P = 0.99), and the Basal-like (P = 0.043) and ERBB2 subtypes (P = 0.064) show the highest frequencies of copy number-neutral events.
Stratification of genome-wide LOH and copy number-neutral event profiles by breast cancer subtype reveals hitherto unknown differences (Fig. 4 C and D). A considerably higher frequency of LOH specifically in the Basal-like subtype is immediately apparent (P = 1.0 × 10−3 by an unpaired t test with unequal variance, testing for differences between Basal-like carcinomas and all other carcinomas). This observation, combined with the particular ploidy range of the Basal-like breast carcinomas (Fig. 3_B_), makes us hypothesize that the genomes of Basal-like tumors initially are reduced from a diploid to a partial haploid state (around 1.5n) and subsequently undergo a whole-genome duplication resulting in a ploidy around 3n (Fig. S7).
Alleles Preferentially Gained or Lost in Breast Carcinoma.
The ASCAT profiles of our series of 91 breast carcinomas allow us to create a genome-wide map of alleles preferentially gained or lost (Fig. 5). If for a certain SNP the most commonly lost allele is the B allele, whereas the A allele is preferentially kept (or gained), the A allele likely provides a relative advantage for the breast carcinomas. For example, the SNP rs6575883 within the PPP2R5C gene on 14q32.31 is germline heterozygous in 30 of the cases. Fifteen of those have a loss, all losing the B allele. In addition, there are four gains, all of the A allele, and two copy number-neutral events, both showing gain of A and loss of B. All these observations point to a skewness in the loss/gain of the two alternative alleles and suggest a relative advantage of the A allele and disadvantage of the B allele acting during breast carcinoma development. A statistical evaluation (Fig. S8) resulted in P = 9.5 × 10−7. Hence, despite the relatively limited size of our dataset (further complicated by the fact that for each SNP only heterozygous cases could be evaluated for this analysis), we were able to identify probes with highly significant allelic skewness in a genome-wide statistical evaluation. This confirms that at least part of the allelic skewness shown in Fig. 5 is likely due to selection, suggesting that loci subject to allelic skewness are potential unique markers for breast cancer development.
Fig. 5.

Genome-wide map of allelic skewness. SNPs that show no allelic skewness (no allele is preferentially gained or lost) should show approximately equal frequencies of loss and gain for both alleles. Here, the frequency of the most frequently gained/lost allele is shown. Alleles without allelic skewness should have a frequency of 50% (blue), whereas alleles that are completely skewed have a frequency of 100% (red). For each SNP, we selected the cases from our series that are germline heterozygous. We count how many cases show gains (of A vs. of B), losses (of A vs. of B), and copy number-neutral events (with gain of A and loss of B vs. with gain of B and loss of A). We combined the counts for gain of A, loss of B, and copy number-neutral events with gain of A and loss of B, and the counts for gain of B, loss of A, and copy number-neutral events with gain of B and loss of A, and display the frequency of the most frequently skewed allele. Only probes with a total of at least 10 gains, losses, and copy number-neutral events are shown. Gene symbols shown contain at least one SNP with a most frequently gained/lost allele frequency of 95% or more.
Discussion
Since the initial report in 1992 describing CGH (18), and the later adaptation to array technology (19–21), CGH has established itself as the de facto standard for detection of chromosomal aberrations in tumors. However, more than a decade later, it remains difficult to determine accurate genome-wide copy number profiles of tumors from these high-throughput arrays. Complicating factors are that tumor cells are often aneuploid (9, 10) and that tumor samples contain multiple populations of both tumor and nontumor cells (11, 12). Although some studies aim to bring these effects into the equation (22, 23), these difficulties still remain today. In addition to these limitations, array-CGH provides no information regarding which of the two alternative alleles has been gained or lost, and it overlooks copy number-neutral aberrations.
The introduction of SNP array technology (24, 25) holds the promise to solve these issues, because allele-specific measurements allow estimation of the amount of aberrant and nonaberrant cells in a specimen and clearly show deviations from diploidy. Recently, a number of computational methods have been developed that aim to take into account either tumor aneuploidy or infiltration of nontumoral cells (26–31). However, to calculate correct genome-wide allele-specific copy numbers from SNP array data of nonmicrodissected tumor samples, both these effects need to be modeled simultaneously.
We developed a unique algorithm, ASCAT, to infer ASCAT profiles (accurate genome-wide allele-specific copy number profiles) from SNP array data, estimating and correcting for both tumor cell aneuploidy and nonaberrant cell admixture. We validated ASCAT's copy number predictions by FISH, its sensitivity to increasing nonaberrant cell involvement by application to a dilution series of a tumor sample, and its ploidy predictions by experimental ploidy measurements.
The dissection of cancer genomes is taken to the next step by the recent introduction of cancer genome sequencing (5–8). We believe ASCAT profiles could be useful tools for interpretation of these data, aiding in the assembly of the data and in the identification of changes varying in size from point mutations to complex rearrangements.
The distribution of ploidy and aberrant cell fraction across our early-stage breast carcinoma series (Fig. 3) suggests that analysis methods not taking both ploidy and nonaberrant cell infiltration into account will misinterpret at least 50% of the cases. This may lead to underestimation of the number of aberrations in tumors showing high nonaberrant cell admixture (as observed in ERBB2 and Normal-like breast carcinomas) or to misinterpretation of nearly all aberrations in aneuploid tumors.
ASCAT profiles allow identification of LOH and copy number-neutral events, invisible to array-CGH (and misinterpreted by SNP array methods not correcting for both aneuploidy and nonaberrant cell infiltration). The genomic distribution of LOH (Fig. 4_A_) corresponds very well with that of losses (Fig. S6_A_). This is in large part because many losses also result in loss of heterozygosity. Unexpectedly, however, there were also correspondences between losses and copy number-neutral events (two entirely distinct classes of aberrations), suggesting that the frequency of loss of one allele (possibly combined with gain of the other allele) is considerably higher than previously reported.
We identify characteristic differences in tumor ploidy, nonaberrant cell admixture, and frequency of gains, losses, LOH, and copy number-neutral events among the five molecularly defined breast cancer subtypes, many of which were previously unknown. These findings confirm the added value of our approach and at the same time support the hypothesis that these molecular subtypes are distinct biological entities. For example, we find a high fraction of nonaberrant cells in the ERBB2 and Normal-like subtypes (Fig. 3_A_), two tumor subtypes for which previous reports described only a limited number of aberrations (15–17). The correction for nonaberrant cell involvement shows that these tumors in fact do not harbor fewer aberrations than the other subtypes (Fig. S6_B_) but rather that these aberrations were missed by earlier approaches not adjusting for nonaberrant cell involvement. The ERBB2 gene is an important tumor antigen for the induction of CD8+-mediated T-cell responses in breast carcinomas, and patients carrying tumors overexpressing this growth factor receptor are often mounting immune responses to ERBB2-derived peptides (32). Hence, the attraction of T cells may at least in part explain the higher fraction of nonaberrant cells in tumors of the ERBB2 subtype.
Our findings also allow insight into tumor development. Luminal A breast carcinomas are typically diploid, showing only a limited number of aberrations (mostly affecting entire chromosome arms). A minority of them have become tetraploid by endoreduplication, with very few additional aberrations. Breast carcinomas of the Basal-like subtype in contrast show numerous aberrations, with most chromosomes affected. There are considerably more losses than gains, resulting in a ploidy of 1.6n to 2n. In a later stage, some of these tumors undergo a whole-genome duplication. We hypothesize that these partially haploid genomes become unstable (at around 1.5n), resulting in a selection for more stable triploid genomes. The ASCAT profiles of these triploid basal-like carcinomas, displaying extensive LOH, confirm that this whole-genome duplication occurs late in tumor development, after the tumor genome has acquired a large number of aberrations (Fig. S7).
Finally, we construct a genome-wide map of allelic skewness, indicating loci where one allele is preferentially lost whereas the other allele is preferentially gained. We hypothesize that these alternative alleles have a different effect on breast carcinoma development, with the allele preferentially gained showing a beneficial effect on the breast carcinoma, compared with the allele preferentially lost. Interestingly, the gene containing the SNP showing the most extreme allelic skewness (21 aberrations, all pointing to preferential gain of A and preferential loss of B), PPP2R5C (PP2A, B subunit, B56γ isoform), has been shown to mediate DNA-damage induced dephosphorylation of p53 (33), and as part of the heterotrimeric complex PP2A may play an important tumor suppressive role in multiple cancers, including breast cancer (34, 35).
Materials and Methods
Breast Carcinoma Series.
The study population of early-stage breast carcinomas has been described previously (16, 36). It consists of 112 blood-tumor pairs. A part of each of the surgically removed tumor specimens was frozen directly at −80 °C and stored. The frozen tumors were then macrodissected by a pathologist. Two frozen sections from each were examined by microscope to secure representative tumor tissue before DNA extraction. The blood DNA was isolated from the lymphocyte fraction of peripheral blood. Both were analyzed using the Human-1 109K BeadChip SNP array platform (Illumina). A more detailed description can be found in our earlier study (16). Because these were all breast cancers from female patients, the SNP array data for chromosome Y was not used, leaving 109,302 SNPs in total.
Although multiple methods have been developed to perform molecular breast cancer subtyping (37, 38), in this study we opted to use te same subtypes as in our previous study (16, 36), for easier comparison.
ASCAT Algorithm.
Illumina SNP arrays deliver two output tracks: Log R, a measure of total signal intensity, and B allele frequency (BAF), a measure of allelic contrast (25). The Log R track is similar to the output given by common array-CGH platforms and quantifies the (total) copy number of each genomic locus. The BAF track shows the relative presence of each of the two alternative nucleotides (called “A” and “B”) at each SNP locus profiled.
Two recurrent phenomena complicate the analysis of genotype profiles of cancer samples and occurred frequently in our series as well: infiltration of nonaberrant cells and aneuploidy of tumor cells (Fig. S1). We express the Log R and BAF data (r and b, respectively) as a function of the allele-specific copy number (nA,i and nB,i), accounting for nonaberrant cell infiltration and tumor aneuploidy (SI Materials and Methods for details):
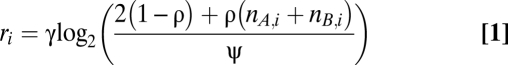
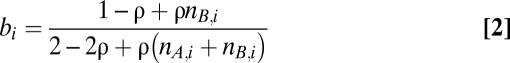
In Eqs. 1 and 2, i represents the genomic location, and γ is a constant depending on the SNP array technology used. The average ploidy of the sample is modeled by ψ = 2(1 − ρ) + ρψ_t_, with ψ_t_ the tumor ploidy (ranging from 1.6 to 4.8, corresponding with a tumor ploidy range of 1.6n to 4.8n). The aberrant cell fraction of a sample is modeled by ρ, a value between 0 and 1. The parameter γ can be obtained from the literature (25) (it is the drop in Log R in case of a deletion in a 100% pure sample), whereas ρ and ψ_t_ need to be estimated from the data for each tumor sample separately. On the basis of these equations, we can express the allele-specific copy number estimates as a function of the data and the parameters (SI Materials and Methods).
To make our method less sensitive to noise in the input data, both Log R and BAF are preprocessed by a specially designed segmentation and filtering algorithm, Allele-Specific Piecewise Constant Fitting (ASPCF) (SI Materials and Methods for details). First, probes for which the germline DNA is homozygous (i.e., probes in the BAF bands at heights 0 and 1) are removed from the BAF track, because they are uninformative for determination of the total copy number. Because our breast carcinoma series consisted of blood and tumor pairs, we used the genotypes generated from the blood samples to eliminate the probes homozygous in the germline (Fig. S2_A_). ASPCF then fits piecewise constant functions simultaneously to the Log R and BAF data, requiring change points to appear at the same genomic locations in the two fitted functions (Fig. S2_B_). As a result, a segmentation of the genome is obtained, each segment corresponding to a genomic region between two adjacent change points (or between a change point and the start/end of a chromosome arm). For Log R, a single fitted value is obtained for each segment, whereas for BAF the output from ASPCF may consist of either one or two values per segment. These values are symmetric around 0.5. If the aberrant cells are found to be balanced (equal number of As and Bs), only one value, 0.5, is returned. If the aberrant cells show an allelic bias, it will be present in both directions (e.g., SNPs with ABB and AAB genotype will both be present), resulting in two values symmetric around 0.5 being output from ASPCF (Fig. S2_B_).
These ASPCF-smoothed data are subsequently used as input of our ASCAT algorithm (implemented in R), to estimate the parameters ρ (aberrant cell fraction) and ψ_t_ (tumor ploidy), as well as the absolute allele-specific copy number calls (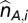 and
and 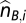 ). Using the fact that true copy numbers are nonnegative whole numbers, we seek values for ρ and ψ_t_ such that the allele-specific copy number estimates are as close as possible to nonnegative whole numbers for germline heterozygous SNPs. Optimal values for ρ and ψ_t_ were estimated as follows:
). Using the fact that true copy numbers are nonnegative whole numbers, we seek values for ρ and ψ_t_ such that the allele-specific copy number estimates are as close as possible to nonnegative whole numbers for germline heterozygous SNPs. Optimal values for ρ and ψ_t_ were estimated as follows:
- (i) Genome-wide allele-specific copy number profiles were calculated for a grid of ρ (0.10, 0.11, …, 1.05) and ψ_t_ values (1.00, 1.05, …, 5.40)
- (ii) for each parameter value combination, the total distance to a nonnegative whole-number solution for the genome-wide allele-specific copy number profiles was calculated and summed over all SNPs (Eq. 3).
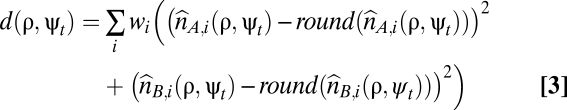
- Here, the round() function rounds to the nearest nonnegative whole number. The weight wi = 1 for probes in segments with allelic bias (BAF ≠ 0.5), and wi = 0.05 for probes in segments without allelic bias (BAF = 0.5), because the former were deemed more likely aberrant segments.
- (iii) All local minima were determined and were considered as possible interpretations of the data. For each possible interpretation, a goodness-of-fit score is calculated. This goodness-of-fit g is calculated as a linear rescaling of the total distance to nonnegative whole numbers to a percentage: g = 100% when d = 0 and g = 0 when d = the distance obtained when the allele-specific copy numbers for each SNP differ 0.25 from nonnegative whole numbers (
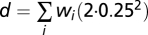 ). The value 0.25 was selected as a reasonable maximum distance (averaged over all probes), taking into consideration the fact that this goodness-of-fit is calculated only for local minima.
). The value 0.25 was selected as a reasonable maximum distance (averaged over all probes), taking into consideration the fact that this goodness-of-fit is calculated only for local minima. - (iv) Local minima corresponding to unlikely interpretations are automatically excluded by ASCAT: (1) solutions with ploidy (calculated as the average total copy number) outside a user-defined range (1.6n–4.8n), (2) solutions with a too-low percentage of aberrant tumor cells (ρ < 0.20), (3) “floating” solutions—solutions that show genomic aberrations but that do not show any SNPs with copy number 0 of either allele (by this criterion, ASCAT avoids interpretations with higher ploidy when there is no evidence of higher ploidy), and (4) solutions with a goodness-of-fit below 80%.
- (v) If one candidate solution remains, then this solution is reported. If multiple solutions remain, these are ranked according to their goodness of fit, and the highest ranking solution is reported. For the reported solution, ASCAT returns the percentage of aberrant tumor cells, the tumor ploidy (calculated as the average total copy number), the goodness of fit, and the whole-genome allele-specific copy number profile of the tumor (ASCAT profile), as well as an aberration reliability score for each aberration found (SI Materials and Methods for details).
Software and Data Availability.
The ASCAT and ASPCF software, the SNP array data, and the allelic skewness data are available at http://www.ifi.uio.no/bioinf/Projects/ASCAT.
Supplementary Material
Supporting Information
Acknowledgments
We thank Elmar Bucher and Tuuli Lappalainen for bioinformatical assistance; Therese Sørlie for sharing the correlation values to the molecular breast cancer subtypes; Grethe I. G. Alnaes and Fredrik E. Johansen for performing part of the Illumina genotyping; and Trond Stokke for valuable discussion. Operating costs for genotyping were provided by Norwegian Research Council Grants 155218/V40 and 175240/S10 (to A.-L.B.-D.), Functional Genomics-Norsk Forskningsråd (Norwegian Research Council) (FUGE-NFR) FUGE-NFR 181600/V11 (to V.N.K.), and a Swizz Bridge Award (A.-L.B.-D.). Laboratory assistance was funded by Norwegian Cancer Society Grants D99061 (to A.-L.B.-D.) and D03067 (to V.N.K.). P.V.L. is a postdoctoral researcher of the Research Foundation–Flanders (FWO) and is as a visiting scientist at the Institute for Cancer Research supported by travel grants from the FWO and from the European Association for Cancer Research. S.H.N. is a postdoctoral fellow of the Norwegian Cancer Association (PK01-2007-0356) and is supported by a travel grant from Lillemor Grobstoks Legacy for Cancer Research. C.M.P. was supported by National Cancer Institute Breast Specialized Program of Research Excellence Grant P50-CA58223-09A1, the Breast Cancer Research Foundation, and the V Foundation for Cancer Research.
Footnotes
*This Direct Submission article had a prearranged editor.
The authors declare no conflict of interest.
References
- 1.Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009;458:719–724. doi: 10.1038/nature07943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Balmain A, Gray J, Ponder B. The genetics and genomics of cancer. Nat Genet. 2003;33(Suppl):238–244. doi: 10.1038/ng1107. [DOI] [PubMed] [Google Scholar]
- 3.Mullighan CG, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007;446:758–764. doi: 10.1038/nature05690. [DOI] [PubMed] [Google Scholar]
- 4.Cancer Genome Atlas Research Network Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mardis ER, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361:1058–1066. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stephens PJ, et al. Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature. 2009;462:1005–1010. doi: 10.1038/nature08645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pleasance ED, et al. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature. 2010;463:184–190. doi: 10.1038/nature08629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pleasance ED, et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature. 2010;463:191–196. doi: 10.1038/nature08658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Holland AJ, Cleveland DW. Boveri revisited: Chromosomal instability, aneuploidy and tumorigenesis. Nat Rev Mol Cell Biol. 2009;10:478–487. doi: 10.1038/nrm2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rajagopalan H, Lengauer C. Aneuploidy and cancer. Nature. 2004;432:338–341. doi: 10.1038/nature03099. [DOI] [PubMed] [Google Scholar]
- 11.Campbell LL, Polyak K. Breast tumor heterogeneity: Cancer stem cells or clonal evolution? Cell Cycle. 2007;6:2332–2338. doi: 10.4161/cc.6.19.4914. [DOI] [PubMed] [Google Scholar]
- 12.Witz IP, Levy-Nissenbaum O. The tumor microenvironment in the post-PAGET era. Cancer Lett. 2006;242:1–10. doi: 10.1016/j.canlet.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 13.Perou CM, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 14.Sørlie T, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA. 2001;98:10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bergamaschi A, et al. Distinct patterns of DNA copy number alteration are associated with different clinicopathological features and gene-expression subtypes of breast cancer. Genes Chromosomes Cancer. 2006;45:1033–1040. doi: 10.1002/gcc.20366. [DOI] [PubMed] [Google Scholar]
- 16.Nordgard SH, et al. Genome-wide analysis identifies 16q deletion associated with survival, molecular subtypes, mRNA expression, and germline haplotypes in breast cancer patients. Genes Chromosomes Cancer. 2008;47:680–696. doi: 10.1002/gcc.20569. [DOI] [PubMed] [Google Scholar]
- 17.Chin K, et al. Genomic and transcriptional aberrations linked to breast cancer pathophysiologies. Cancer Cell. 2006;10:529–541. doi: 10.1016/j.ccr.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 18.Kallioniemi A, et al. Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science. 1992;258:818–821. doi: 10.1126/science.1359641. [DOI] [PubMed] [Google Scholar]
- 19.Solinas-Toldo S, et al. Matrix-based comparative genomic hybridization: Biochips to screen for genomic imbalances. Genes Chromosomes Cancer. 1997;20:399–407. [PubMed] [Google Scholar]
- 20.Pinkel D, et al. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat Genet. 1998;20:207–211. doi: 10.1038/2524. [DOI] [PubMed] [Google Scholar]
- 21.Pollack JR, et al. Genome-wide analysis of DNA copy-number changes using cDNA microarrays. Nat Genet. 1999;23:41–46. doi: 10.1038/12640. [DOI] [PubMed] [Google Scholar]
- 22.Lyng H, et al. GeneCount: Genome-wide calculation of absolute tumor DNA copy numbers from array comparative genomic hybridization data. Genome Biol. 2008;9:R86. doi: 10.1186/gb-2008-9-5-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang K, Li J, Li S, Bolund L, Wiuf C. Estimation of tumor heterogeneity using CGH array data. BMC Bioinformatics. 2009;10:12. doi: 10.1186/1471-2105-10-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McCarroll SA, et al. Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat Genet. 2008;40:1166–1174. doi: 10.1038/ng.238. [DOI] [PubMed] [Google Scholar]
- 25.Peiffer DA, et al. High-resolution genomic profiling of chromosomal aberrations using Infinium whole-genome genotyping. Genome Res. 2006;16:1136–1148. doi: 10.1101/gr.5402306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Attiyeh EF, et al. Genomic copy number determination in cancer cells from single nucleotide polymorphism microarrays based on quantitative genotyping corrected for aneuploidy. Genome Res. 2009;19:276–283. doi: 10.1101/gr.075671.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Staaf J, et al. Segmentation-based detection of allelic imbalance and loss-of-heterozygosity in cancer cells using whole genome SNP arrays. Genome Biol. 2008;9:R136. doi: 10.1186/gb-2008-9-9-r136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gardina PJ, Lo KC, Lee W, Cowell JK, Turpaz Y. Ploidy status and copy number aberrations in primary glioblastomas defined by integrated analysis of allelic ratios, signal ratios and loss of heterozygosity using 500K SNP Mapping Arrays. BMC Genomics. 2008;9:489. doi: 10.1186/1471-2164-9-489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pounds S, et al. Reference alignment of SNP microarray signals for copy number analysis of tumors. Bioinformatics. 2009;25:315–321. doi: 10.1093/bioinformatics/btn624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Greenman CD, et al. PICNIC: An algorithm to predict absolute allelic copy number variation with microarray cancer data. Biostatistics. 2010;11:164–175. doi: 10.1093/biostatistics/kxp045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun W, et al. Integrated study of copy number states and genotype calls using high-density SNP arrays. Nucleic Acids Res. 2009;37:5365–5377. doi: 10.1093/nar/gkp493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pupa SM, Tagliabue E, Ménard S, Anichini A. HER-2: A biomarker at the crossroads of breast cancer immunotherapy and molecular medicine. J Cell Physiol. 2005;205:10–18. doi: 10.1002/jcp.20387. [DOI] [PubMed] [Google Scholar]
- 33.Li HH, Cai X, Shouse GP, Piluso LG, Liu X. A specific PP2A regulatory subunit, B56gamma, mediates DNA damage-induced dephosphorylation of p53 at Thr55. EMBO J. 2007;26:402–411. doi: 10.1038/sj.emboj.7601519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sablina AA, et al. The tumor suppressor PP2A Abeta regulates the RalA GTPase. Cell. 2007;129:969–982. doi: 10.1016/j.cell.2007.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Esplin ED, et al. The glycine 90 to aspartate alteration in the Abeta subunit of PP2A (PPP2R1B) associates with breast cancer and causes a deficit in protein function. Genes Chromosomes Cancer. 2006;45:182–190. doi: 10.1002/gcc.20284. [DOI] [PubMed] [Google Scholar]
- 36.Naume B, et al. Presence of bone marrow micrometastasis is associated with different recurrence risk within molecular subtypes of breast cancer. Mol Oncol. 2007;1:160–171. doi: 10.1016/j.molonc.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sørlie T, et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci USA. 2003;100:8418–8423. doi: 10.1073/pnas.0932692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parker JS, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol. 2009;27:1160–1167. doi: 10.1200/JCO.2008.18.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information