Transforming growth factor-β signaling to T cells inhibits autoimmunity during lymphopenia-driven proliferation (original) (raw)
. Author manuscript; available in PMC: 2013 Jan 1.
Published in final edited form as: Nat Immunol. 2012 May 27;13(7):667–673. doi: 10.1038/ni.2319
Summary
T cell specific deletion of the Transforming growth factor-β (TGF-β) receptor mediated by CD4-cre leads to early onset lethal autoimmune disease that cannot be controlled by regulatory T cells. However, when we delete the receptor using distal Lck (dLck) promoter driven cre, adult mice in which the majority of peripheral CD4+ and CD8+ T cells lacked the TGF-β receptor, showed no signs of autoimmunity. Due to their heightened response to weak T cell receptor stimuli, when transferred into lymphopenic recipients, naive TGF-β unresponsive T cells exhibited dramatically enhanced proliferation, effector differentiation, and induced lymphoproliferative disease. We propose that TGF-β signaling controls self-reactivity of peripheral T cells but in the absence of TGF-β signals, an added trigger, such as lymphopenia, is required to drive overt autoimmune disease.
Transforming growth factor β (TGF-β) is a pleiotropic cytokine that regulates diverse biological processes during fetal development and in adults by binding to TGF-β type I (TGF-βRI) and type II (TGF-βRII) receptors found on the surface of most cells1–3. Upon TGF-β binding, TGF-βRI phosphorylates Smad2 and Smad3 to induce complex formation with Smad4 and nuclear translocation. In addition, several Smad-independent pathways, such as TRAF6-TAK1 dependent p38 and JNK activation, are induced by TGF-β signaling4, 5. In tumorigenesis, TGF-β signaling may initially suppress tumor growth through regulation of cell cycle progression, differentiation and apoptosis. But paradoxically, at later stages, it promotes tumors both by enhancing metastasis and by its immunosuppressive effect on anti-tumor immunity2, 6, 7. In line with its anti-proliferative and immunosuppressive function, mice deficient in TGF-β1 die before or shortly after birth, exhibiting an autoimmune phenotype with massive T cell expansion and organ infiltration8, 9. Depletion of T cells in the knockout mice alleviates the disease10–13, but because TGF-β has regulatory effects on many cell types, whether or not it was having a direct action on T cells in preventing the autoimmune phenotype was unclear. Therefore, in order to dissect the role of TGF-β signaling directly to T cells, researchers have employed mice expressing a dominant negative TGFβRII transgene expressed selectively in T cells14, 15, or have engineered cre-mediated deletion of floxed receptor genes specifically in the T cell lineage (Tgfbr2f/f CD4-cre)16, 17.
In both of these models of T cell specific non-responsiveness to TGF-β, mice develop autoimmune pathology. In particular, mice with CD4-cre-mediated deletion of floxed TGF-βRII die very rapidly (by 3–5 weeks of age) with multiorgan autoimmune lesions, similar to what is observed in mice completely lacking TGF-β16, 17. The severity and rapidity of disease onset in this model of T cell-specific deletion of the receptor for TGF-β resembles that in mice deficient in the transcription factor Foxp3 and therefore lacking regulatory T (Treg) cells18. Because TGF-β signaling is required for the proper development, maintenance and function of Treg cells16, 17, 19–21, the question arose as to what extent the autoimmunity observed in _Tgfbr2_f/f CD4-cre mice was due to the failure of Treg cell function and how much was due to the intrinsic inability of TGF-β unresponsive T cells to control their self reactivity. Certainly the latter was shown to be relevant because neither transfer of wild-type Treg cells into the conditional knockout mice nor the generation of mixed bone marrow chimeras with wildtype plus conditional knockout bone marrow prevented the onset of autoimmunity in the _Tgfbr2_f/f CD4-cre model16, 17.
During their differentiation, T cells are positively selected for low affinity reactivity to self antigens expressed in the thymus. T cells with too high affinity for self are deleted by negative selection, yet the population of peripheral CD4+ and CD8+ T cells retains an affinity for self antigen22. In a fraction of T cells, self reactivity is of sufficient potency to cause clinical signs of autoimmunity under various provocations. Tonic ‘tickling’ of the T cell receptor (TCR) by engagement with self ligands in the periphery is required to maintain the viability of naive T cells. Under normal, lymphoreplete conditions, this weak TCR signaling does not drive T cell division or overt activation. However, in lymphopenic conditions, combined signals from the TCR and the homeostatic γ-chain cytokines, IL-7 and IL-15, drive slow homeostatic proliferation of the majority of T cells23. When transferred into long-term immunodeficient lymphopenic hosts, such as _Rag1_−/− mice, a fraction of T cells undergo rapid IL-7 and IL-15-independent proliferation that is dependent on signals from commensal microbiota24, 25. Finally, lymphopenia has been associated with autoimmunity26-28.
We studied a different model of T cell-specific conditional deletion of TGF-βRII using distal Lck (dLck) promoter driven cre where deletion occurs slowly in the periphery. This contrasts with CD4-cre mediated deletion where all thymus emigrants lack the receptor. Surprisingly, in the dLck deletion model, we observed that adult mice in which the vast majority of peripheral CD4+ and CD8+ T cells lacked the TGF-β receptor, did not show signs of autoimmunity. We hypothesized that CD4-cre mediated early and efficient deletion of TGF-βRII coupled with neonatal lymphopenia were together responsible for the rapid, severe autoimmunity. In a number of in vivo and in vitro experiments we show that TGF-β unresponsive T cells differ from control T cells in their heightened response to weak TCR stimuli. We propose that constitutive TGF-β signaling controls the self reactivity of peripheral T cells but even in the absence of TGF-β responsiveness, an added trigger, such as lymphopenia, is required to drive overt autoimmune disease.
Results
Distal Lck-cre deletion of TGF-βRII in adult mice
To study the function of TGF-β signaling in mature T cells, mice bearing loxp-flanked alleles of the TGF-βRII (_Tgfbr2_f/f) were bred with mice expressing cre under the control of the distal Lck promoter that is only turned on after thymocyte positive selection29, 30. (_Tgfbr2_f/f dLck-cre mice are hereafter referred to as KO and _Tgfbr2_f/+ dLck-cre and Tgfbr2+/+ dLck-cre mice as control mice). In adult mice, dLck-cre mediated deletion of the TGF-β receptor on peripheral CD4+ and CD8+ T cells was almost complete, as shown by surface staining of TGF-βRII, which was indistinguishable from isotype controls (Fig. 1a). CD103 expression on mature CD8+ T cells has been linked with TGF-β signaling31, and the lack of CD103 expression on KO CD8+ T cells (Fig. 1a) reinforces the conclusion that dLck-cre efficiently deletes TGF-βRII on mature T cells in adult mice. CD4+ and CD8+ T cells in adult KO mice were skewed towards an effector or memory phenotype, defined by CD44 and CD62L expression (Fig. 1b) and this was correlated with elevated protein expression of the T helper 1 (TH1)-associated transcription factor T-bet (Fig. 1c). Elevated levels of T-bet were not observed in the T cells bearing a naive phenotype (CD44lo CD62L+) in the KO mice (Fig. 1c). However, no lymphoproliferation or signs of autoimmune diseases were observed in KO mice, all of which remained healthy for up to a year. In contrast, _Tgfbr2_f/f CD4-cre mice bred and maintained in the same animal facility showed dramatic lymphocytic infiltration in many organs early in life (Supplementary Fig. 1), and were moribund by 3–5 weeks of age, as has been reported previously for CD4-cre mediated deletion of the TGF-bRII16, 17. Contrasting with the abundant lymphocytic infiltrates observed in 3 week old _Tgfbr2_f/f CD4-cre mice, the histology of _Tgfbr2_f/f dLck-cre mice was normal even at 18 weeks of age (Supplementary Fig. 1). Therefore, the inability to respond to TGF-β in the majority of peripheral T cells does not lead to autoimmune disease in adult mice.
Figure 1. Characterization of T cells in adult TGF-βRII KO mice.
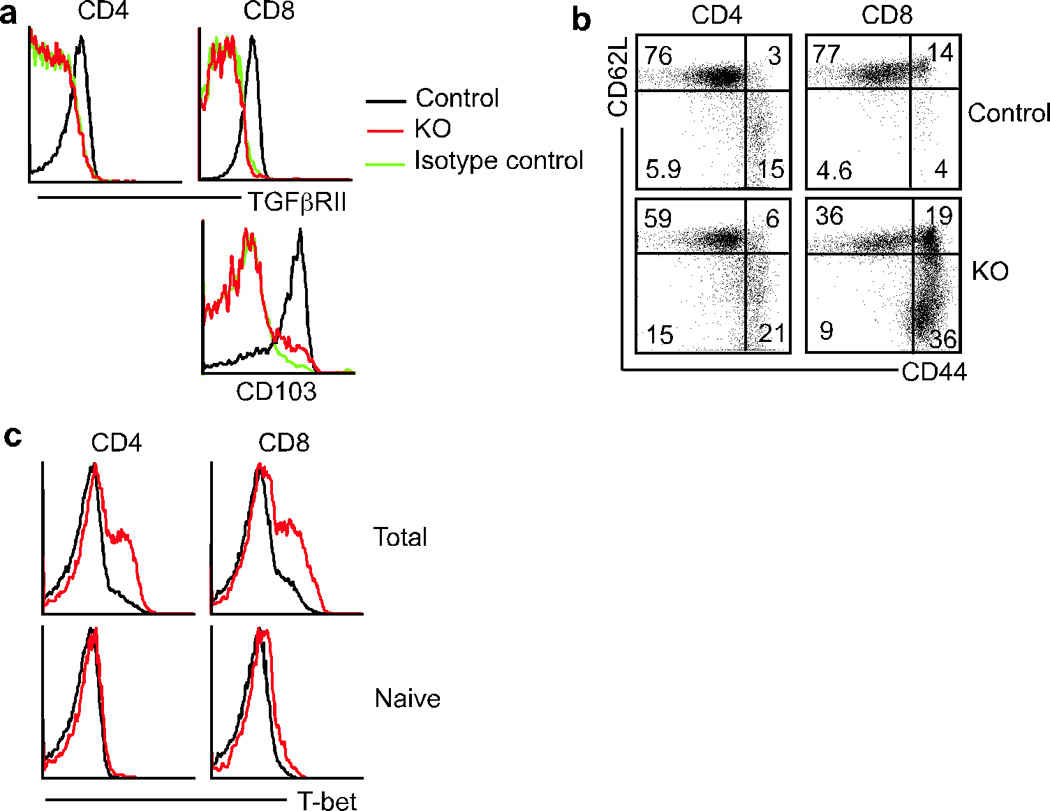
Mice with floxed alleles of TGF-βRII were bred to dLck-cre transgenic mice and examined at 7–9 weeks of age. (a) Surface staining for TGF-βRII and CD103 on gated splenic CD4+ and CD8+ T cells. (b) Splenic CD4+ and CD8+ T cells were stained for CD44 and CD62L. Numbers represent the percentage of cells falling in each quadrant. (c) Intracellular staining for T-bet in total and naive CD44loCD62L+ phenotype CD4+ and CD8+ T cells.
Inefficient deletion of TGF-βRII in neonatal mice
How does one explain the strikingly different outcomes of using dLck-cre versus CD4-cre to delete TGF-βRII on T cells? There are multiple factors that may contribute to the difference. Given that lymphopenia has been linked to autoimmunity26–28, we turned our attention to the importance of lymphopenia during the neonatal period to explain the different phenotypes in the two models of T cell specific TGF-β unresponsiveness. At neonatal day 4, the expression of TGF-βRII on peripheral T cells was indistinguishable between KO (_Tgfbr2_f/f dLck-cre) and control (_Tgfbr2_f/+ dLck-cre and _Tgfbr2_f/+) mice (Supplementary Fig. 2a). Even at 21 days of age, a considerable proportion of thymic and peripheral T cells retained the expression of TGF-βRII in KO mice. In contrast, all T cells emerging from the thymus in mice expressing CD4-cre deleted the TGF-β receptor (Supplementary Fig. 2b). Therefore, during the neonatal lymphopenic period in newborn mice, CD4-cre efficiently deletes TGF-βRII and leads to autoimmunity while dLck-cre is effective later and only mediates efficient deletion in adult mice with no resulting disease.
TGF-β inhibits lymphopenia-induced proliferation
To investigate whether lymphopenia could contribute to the autoimmune phenotype in mice with TGF-β-unresponsive T cells, we turned to mixed cell transfers. Equal numbers of allelically marked naive T cells isolated from control (_Tgfbr2_f/+ dLck-cre) and KO mice were mixed together and transferred into _Rag1_−/− recipients (Fig. 2a and 2b). The cells were labeled prior to transfer with CFSE to monitor their proliferation. A fraction of the control T cells completely diluted their CFSE within a week, corresponding to fast self and/or environmental peptide-major histocompatibility complex (MHC)-dependent proliferation (Fig. 2c) previously reported in these recipient mice24, 25. In addition, a subset of control T cells underwent a few rounds of slow IL-7 or IL-15 dependent homeostatic proliferation (Fig. 2c). In striking contrast to the behavior of the control T cells, slow homeostatic proliferation was almost undetectable in the transferred KO T cells, most of which underwent fast proliferation at both day 5.5 and day 7 time points (Fig. 2c). The enhanced proliferation of the KO T cells resulted in a 3–5 fold increase (day5.5) and a 5-fold increase (day 7) of both CD4+ and CD8+ T cell yield compared to that of control T cells (Fig. 2d). Furthermore, KO CD4+ and CD8+ T cells exhibited heightened effector phenotype and functions, such as KLRG1 expression and Granzyme B and IFN-γ production (Fig. 2e).
Figure 2. Dramatically increased lymphopenia induced proliferation of TGF-βRII KO T cells in _Rag1_−/− mice.
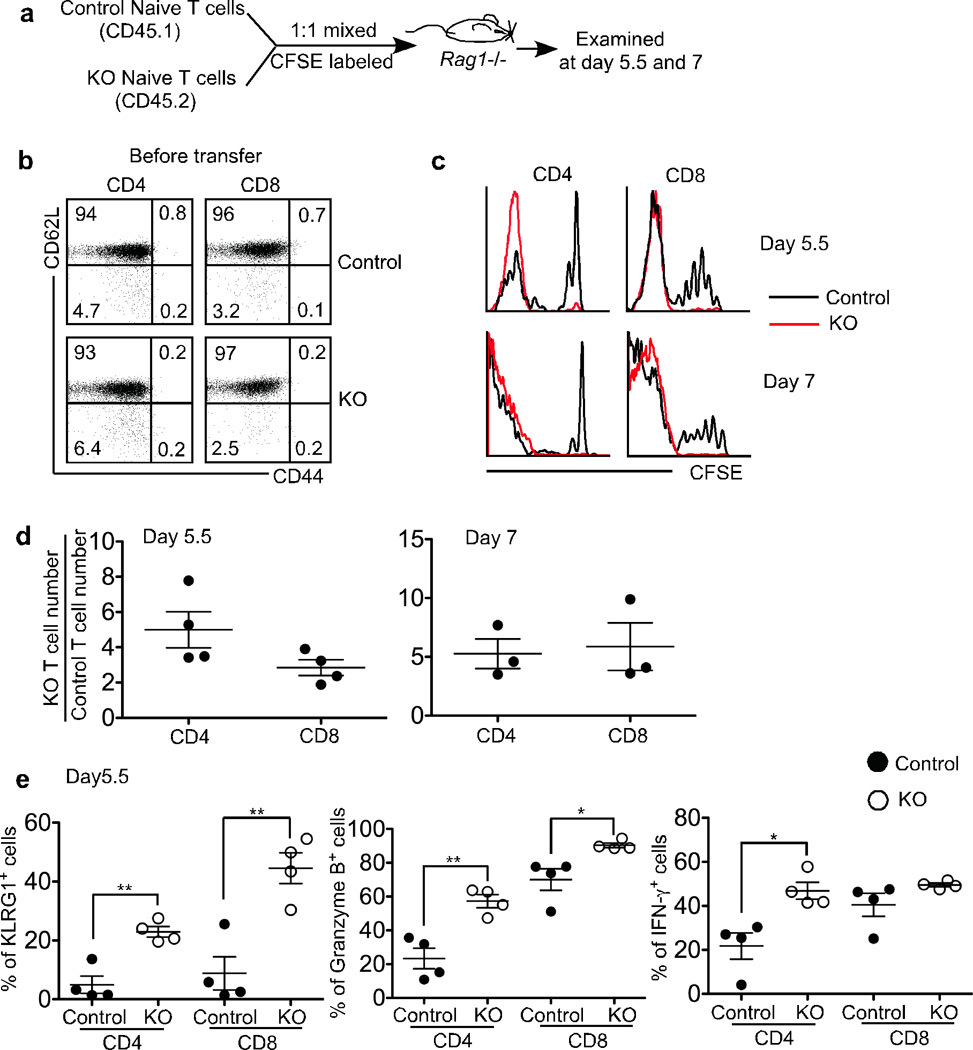
(a) Schematic of the experiment. Naive T cells were purified from control (CD45.1) and KO (CD45.2) mice, mixed at a 1:1 ratio and CFSE labeled. 1×106 cells were transferred into sex-matched _Rag1_−/− recipient mice. 5.5 and 7 days later, recipient mice were sacrificed. (b) Purified naive CD4+ and CD8+ T cells were stained for CD44 and CD62L. (c) Representative CFSE profile of donor T cells in the spleen. (d) Relative KO T cell number was calculated for splenic CD4+ and CD8+ T cells separately. (e) The percentage of KLRG1+, Granzyme B+ and IFN-γ+ cells in the spleen was plotted for donor T cells. Each point represents an individual recipient mouse (* p<0.05 and ** p<0.01). Representative data from two independent experiments are shown.
To study the long-term effects of the enhanced proliferation of KO T cells, naive T cells isolated from control or KO mice were transferred separately into _Rag1_−/− recipients. Starting at 4 weeks after transfer, _Rag1_−/− mice receiving KO T cells exhibited marked weight loss compared with the control group (Fig. 3a). In line with this, we noted that both CD4+ and CD8+ KO T cells accumulated to a much higher level than control cells in all organs examined (a 5–7 fold increase for KO CD4+ T cells and a 9–17 fold increase for KO CD8+ T cells, Fig. 3b). This finding was further confirmed by the greatly increased lymphocytic infiltration in the liver and small intestine of the _Rag1_−/− mice that received KO T cells (Supplementary Fig. 3). Therefore, we conclude that TGF-β signaling directly to naive T cells inhibits rapid proliferation, the acquisition of effector functions and autoimmunity in _Rag1_−/− recipient mice.
Figure 3. TGF-βRII KO T cells induce lymphoproliferative disease in _Rag1_−/− recipient mice.

1.5×106 naive T cells from control or KO mice were transferred separately into _Rag1_−/− recipient mice. (a) Recipient mice were weighed every week after T cell transfer (n=10 for both control and KO groups). (b) 5 weeks post transfer, spleen, lamina propria and liver lymphocytes of the recipient mice were analyzed by flow cytometry. CD4+ and CD8+ T cell number was calculated from the total cellularity and the percentage of T cells in each organ. Each point represents an individual recipient mouse (**p<0.01). Representative data from two independent experiments are shown.
Microbiota-independent rapid proliferation of KO T cells
Because rapid proliferation of T cells transferred into chronically immunodeficient recipients is markedly decreased in germ-free mice, commensal flora are considered a prerequisite for the rapid T cell proliferation24, 32. Both inflammatory signals and antigens from commensal bacteria are required for autoimmunity in these lymphopenic settings. To dissect the underlying mechanisms of the enhanced rapid proliferation of TGF-β unresponsive T cells, experiments similar to those in Fig. 2 were performed except that the recipient _Rag1_−/− mice were pretreated with antibiotics for 7 days before T cell transfer to reduce the level of commensal bacteria and basal inflammation (Supplementary Fig. 4a). As expected, rapid proliferation of T cells from control mice was substantially reduced in antibiotic treated recipients (compare Fig. 2c and Supplementary Fig. 4b). However, rapid proliferation of KO T cells was comparable with or without antibiotic treatment of the _Rag1_−/− recipients. As a result, the relative fold expansion of KO T cells compared to control T cells was increased in the antibiotic treated hosts (compare Fig. 2d and Supplementary Fig. 4c).
In additional experiments, naive T cells from control and KO mice were transferred together into sublethally irradiated wildtype (WT) hosts (Fig. 4a). In contrast to chronic lymphopenic hosts, such acutely lymphopenic hosts did not support rapid proliferation of naive T cells from control mice (Fig. 4b and 24, 25). However, a significant proportion of naive T cells derived from KO mice underwent rapid proliferation in sublethally irradiated B6 mice (Fig. 4b). The enhanced proliferation of KO T cells resulted in a marked increase of both CD4+ and CD8+ T cell yield compared to that of control T cells in all organs examined (Fig 4c). Furthermore, KO T cells exhibited greatly enhanced effector function and phenotype, such as IFN-γ, Granzyme B, and KLRG1 protein expression (Fig. 4d), which was consistent with the elevated levels of T-bet and Eomes observed in KO T cells (Fig. 4d). Similar results were obtained when purified naive OT-1 T cells were transferred into _Rag1_−/− or sublethally irradiated B6 mice (Supplementary Fig. 5). Consistent with previous work24, WT OT-1 T cells did not undergo fast proliferation in either _Rag1_−/− or sublethally irradiated B6 recipients. In contrast, a considerable proportion of KO OT-1 T cells completely diluted their CFSE staining (Supplementary Fig. 5b and 5c). These data suggest that, in contrast to control T cells, KO T cells may not require stimulation from bacterial flora or foreign antigens to undergo rapid proliferation in immunodeficient hosts and this interpretation is supported by our finding that the proliferation of KO OT-1 T cells in response to Listeria-Ova or Vesicular stomatitis virus-Ova in a lymphoreplete host is the same as that of co-transferred control OT-1 T cells. We conclude that weak TCR stimuli from self peptides may be the driving force for the rapid proliferation of TGF-β unresponsive T cells in a lymphopenic environment.
Figure 4. Dramatically increased lymphopenia induced proliferation of TGF-βRII KO T cells in sublethally irradiated B6 mice.
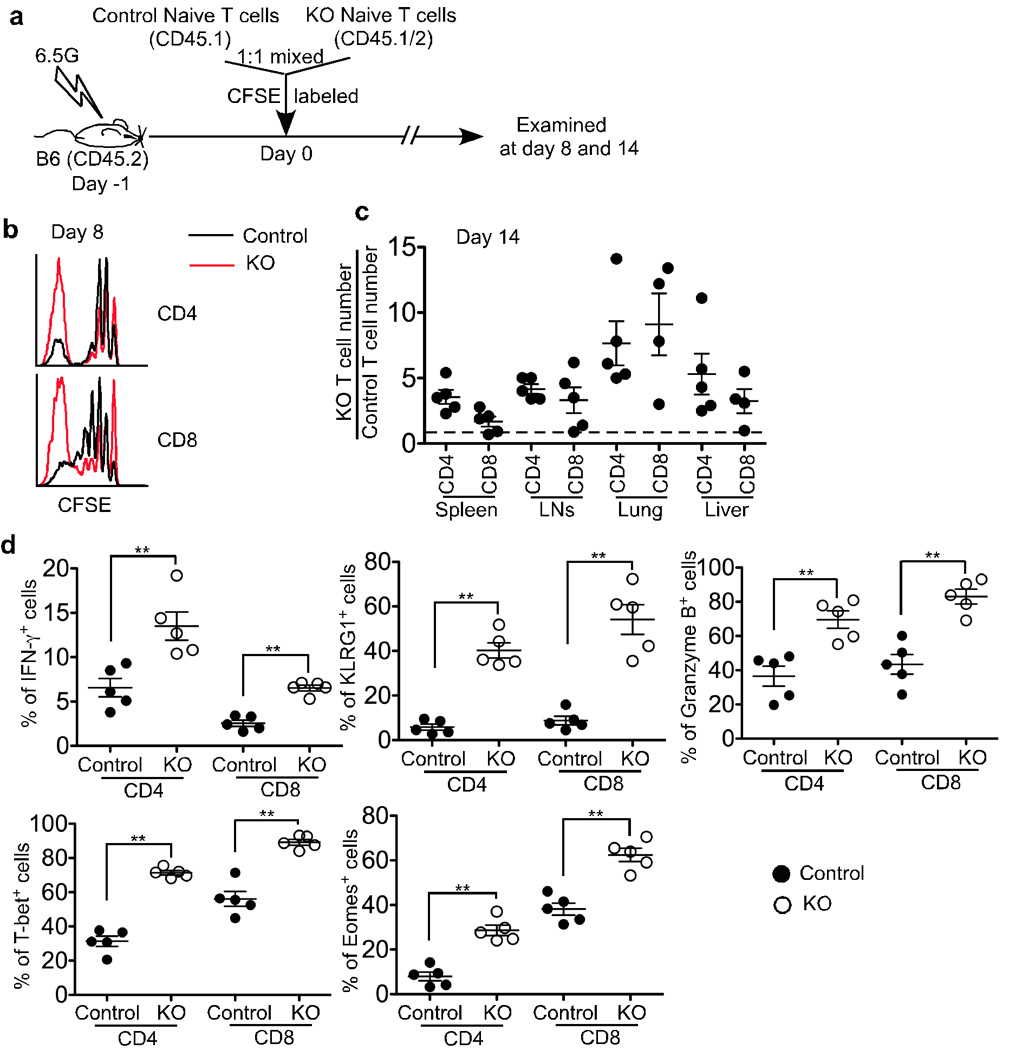
(a) Schematic of the experiment. Sex-matched recipient B6 mice were irradiated at 6.5G. On the following day, naive T cells were purified from control (CD45.1) and KO (CD45.1+CD45.2+) mice, mixed at a 1:1 ratio and CFSE labeled. 1×106 cells were transferred. 8 (b) and 14 (c and d) days later, recipient mice were sacrificed. (b) Representative CFSE profile of donor T cells in the spleen. (c) Relative KO T cell number was calculated for CD4+ and CD8+ T cells separately. (d) The percentage of IFN-γ+, KLRG1+, Granzyme B+, T-bet+ and Eomes+ cells in the spleen was plotted for donor T cells (** p<0.01). Each point represents an individual recipient mouse.
TGF-β inhibits responses to weak TCR stimuli in vitro
To study the effect of TGF-β signaling on T cell proliferation to weak TCR stimuli, OT-1 CD8+ T cells were stimulated with a panel of altered peptide ligands derived from the cognate SIINFEKL peptide of Ova that have been previously characterized for their potency33. Naive CD8+ T cells isolated from control and KO OT-1 mice were mixed together and CFSE labeled (Fig. 5a). T cell and NK cell depleted splenocytes pulsed with the different altered peptide ligands were used as antigen-presenting cells (APCs) in the presence of added TGF-β1. IL-2 was added one day later and the cultures followed for another 4 days. High affinity peptide ligands such as N4 (the cognate ligand) and the variant Y3 induced only slightly enhanced expansion of KO OT-1 cells during the 5-day culture (less than 3 fold, Fig. 5b). Strikingly, as the peptide potency decreased, the relative expansion of KO OT-1 cells increased dramatically, such that by day 4, KO OT-1 T cells outnumbered control OT-1 T cells by 100-fold in cultures stimulated with the lowest affinity ligand, T4 (Fig. 5b and 5c). No expansion was detected in the absence of peptide (Fig. 5b and 5c). The regulatory activity of TGF-β is modulated by the presence of inflammatory cytokines in other cell types1. Indeed, the pro-inflammatory cytokines IFN-α and IL-12 substantially reduced the proliferative advantage of KO OT-1 cells over control OT-1 cells, whereas other pro-inflammatory cytokines, such as IFN-γ, IL-6, TNF, IL-1β and IL-18 had minimal or no effect (Fig. 5c and data not shown). To rule out that the observed effects were unrelated to TGF-β unresponsiveness of KO T cells, TGF-β neutralizing antibody was added to the T4 control cultures which resulted in control OT-1 cells proliferating to the same extent as KO cells (Fig. 5c). Therefore, TGF-β signaling dramatically inhibits CD8+ T cell proliferative response to weak TCR stimuli while it has minimal effects on strong TCR stimulation.
Figure 5. In vitro hyperproliferative response of TGF-βRII KO CD8+ T cells to weak stimulation.
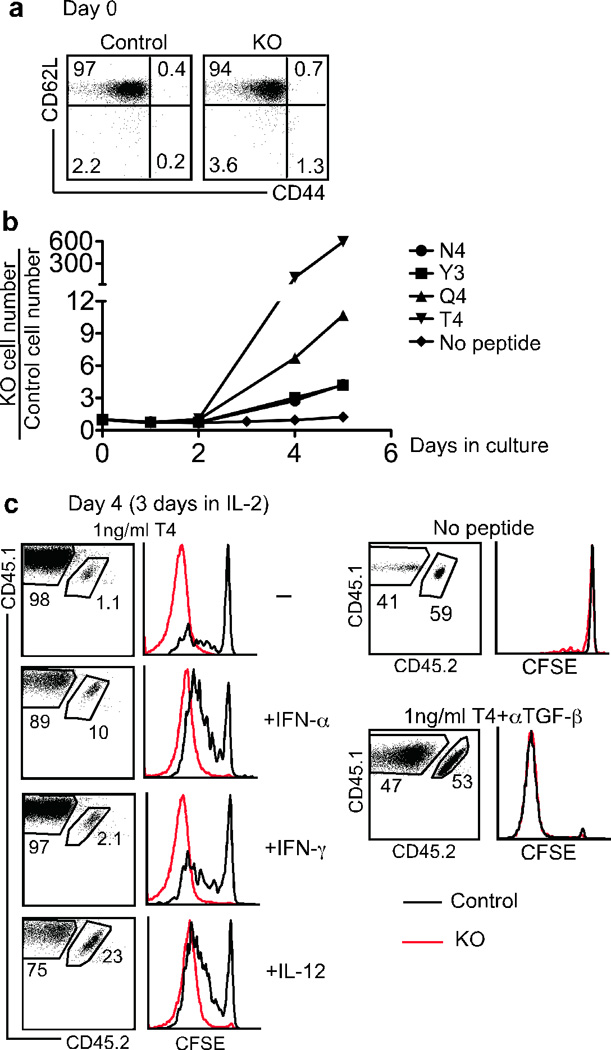
Equal numbers of naive OT-1 cells from control (CD45.1+CD45.2+) and KO (CD45.1) mice were mixed and CFSE labeled. T cell and NK cell depleted splenocytes (as APCs) from B6 (CD45.2) mice were incubated with 1 ng/ml of Ova peptide variants (N4, Y3, Q4, T4) plus 1 µg/ml LPS at 37°C for 1 hour. After extensive washing, an equal number of APCs and OT-1 T cells were mixed in the presence of 2.5 ng/ml hTGF-β1 with or without inflammatory cytokines (1000U/ml IFN-α, 20ng/ml IFN-γ or 20ng/ml IL-12). 50U/ml IL-2 was added one day later. (a) Starting population of mixed control and KO OT-1 T cells. (b) Relative KO OT-1 T cell number in the absence of inflammatory cytokines. (c) Flow cytometry profiles of T4 peptide-stimulated OT-1 T cells after a 4 day in vitro culture. 10 µg/ml αTGF-β neutralizing antibody was added during the culture as shown in the lower right panels. Representative data from three independent experiments are shown.
To address the possibility that naive phenotype T cells from KO mice were already programmed to behave differently to control T cells, we asked if the effect of TGF-β could be reproduced on WT naive cells. WT naive OT-1 cells were purified, CFSE labeled, and mixed with peptide-pulsed APCs at a 1:1 ratio in the presence of TGF-β neutralizing antibody or TGF-β1. IL-2 was added on the following day and the cultures followed for another 4 days. Upon strong TCR stimulation (5ng/ml N4), the presence or absence of TGF-β signaling had minimal effects on WT OT-1 cell proliferation (Fig. 6a and Fig. 6b). However, as the TCR stimulation intensity decreased, the inhibitory effects of TGF-β on WT OT-1 cells increased dramatically. For instance, upon 0.2ng/ml N4 (strong agonist peptide) or 5ng/ml T4 (weak agonist peptide) stimulation, inhibition of TGF-β signaling resulted in a 7-fold increase in OT-1 cell yield during the 5-day culture (Fig. 6a). For 1ng/ml T4 stimulation, OT-1 cell yield was increased over 55 fold by inhibiting TGF-β signaling. Thus we see that in the presence of TGF-β neutralizing antibody, WT OT-1 T cells behaved similarly to KO cells (Fig. 5 and Fig. 6). We conclude that TGF-β signaling specifically inhibits the weak TCR stimulation while largely sparing the response to strong TCR stimulation.
Figure 6. In vitro hyperproliferative response of WT CD8+ T cells to weak stimulation in the absence of TGF-β.
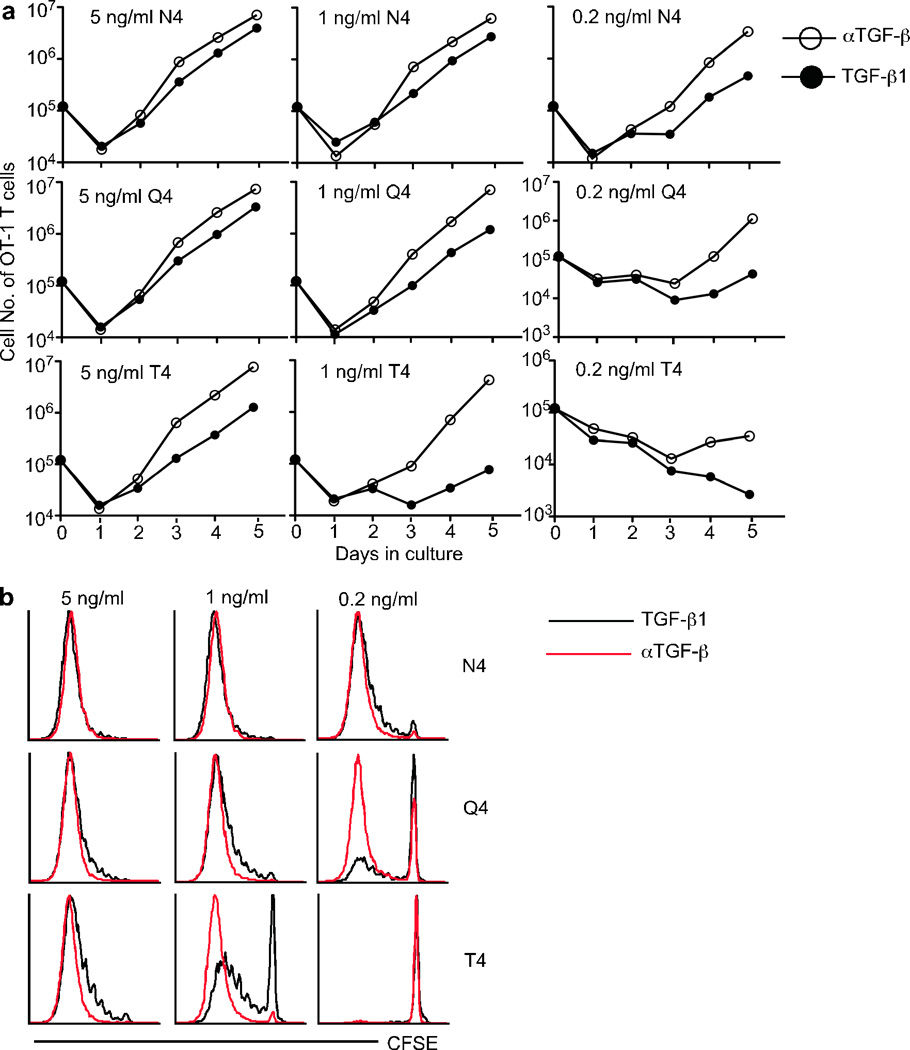
Naive OT-1 cells from WT (CD45.1) mice were purified and CFSE labeled. T cell and NK cell depleted splenocytes (APCs) from B6 (CD45.2) mice were incubated with indicated concentration of Ova peptide variants plus 1 µg/ml LPS at 37°C for 1 hour. After extensive washing, 105 APCs and OT-1 T cells were mixed in the presence of 2.5 ng/ml hTGF-β1 or 10 µg/ml αTGF-β neutralizing antibody. 50U/ml IL-2 was added one day later. (a) Numbers of live OT-1 cells under each condition were determined everyday by flow cytometry with the addition of count standard beads. (b) CFSE profile of OT-1 cells after a 4 day in vitro culture. Representative data from two independent experiments are shown.
Another possible explanation for the increased proliferation of KO T cells in a lymphopenic environment is that KO T cells exhibit an enhanced response to common γ chain cytokines, such as IL-7, IL-15 or IL-2. However, IL-2, IL-7 or IL-15 alone could not induce KO OT-1 T cell proliferation in vitro (Fig. 5 and Supplementary Fig. 6). Furthermore, when sublethally irradiated _Il15_−/− mice were used as recipient mice, similar results as presented in Fig. 4 were obtained and, in a lymphoreplete environment, KO T cells did not respond better to super-agonist IL-2/anti-IL-2 complexes (data not shown). Therefore, weak TCR stimulation is required to induce hyperproliferative response of KO T cells.
CD4+ T cells promote KO CD8+ T cell expansion
To dissect the contribution of CD4+ versus CD8+ T cells in provoking autoimmunity in lymphopenic recipients of TGF-β unresponsive T cells, naive CD4+ and CD8+ T cells isolated from control and KO mice were transferred separately into _Rag1_−/− recipients (Fig. 7a). KO CD4+ T cells exhibited more than a 10-fold increase in numbers over control CD4+ T cells, while KO CD8+ T cells expanded to a similar level as control CD8+ T cells (Fig. 7b). In line with this, KO CD4+ T cells rapidly induced weight loss in _Rag1_−/− recipients whereas KO CD8+ T cells did not (Fig. 7c and Fig. 7d). However, even CD4+ T cells from control mice promoted KO CD8+ T cell expansion in _Rag1_−/− recipients (Supplementary Fig. 7), and, as a consequence, the co-transfer of control CD4+ T cells with KO CD8+ T cells precipitated the development of autoimmunity (Fig. 7d). In summary, KO CD4+ T cells alone and KO CD8+ T cells with CD4+ help induce autoimmunity under lymphopenic conditions.
Figure 7. TGF-βRII KO CD4+ T cells induce autoimmune disease in _Rag1_−/− mice whereas KO CD8+ T cells only do so in the presence of CD4+ T cells.
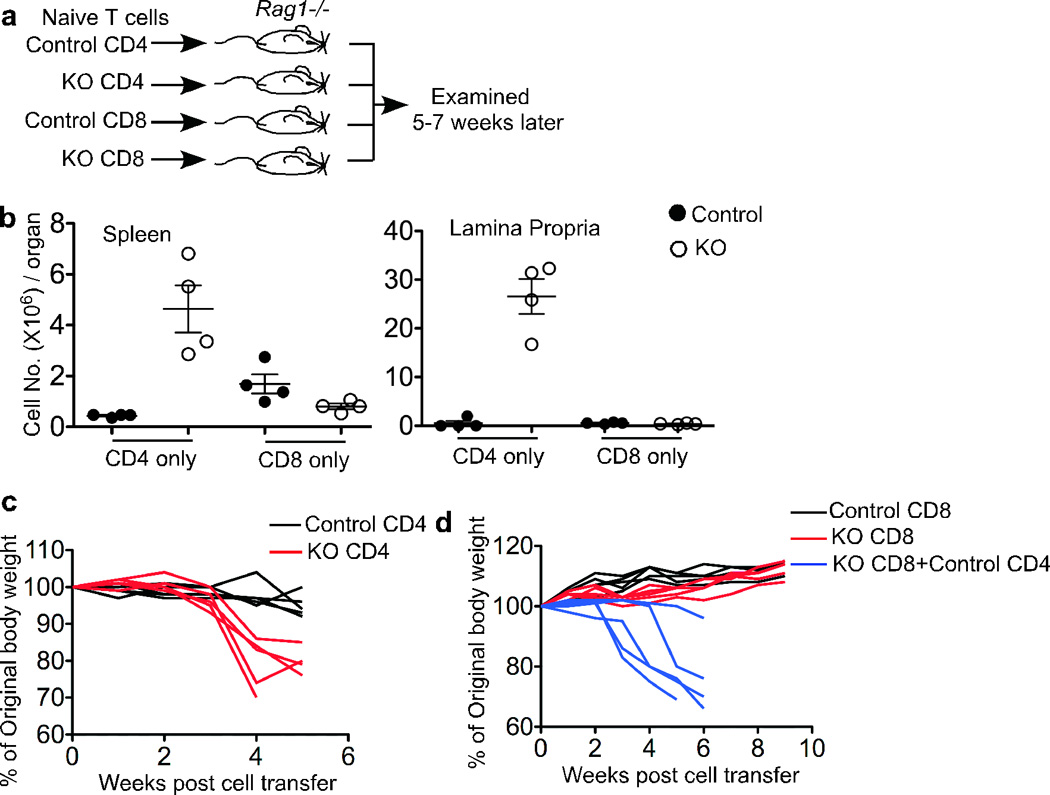
(a) Schematic of the experiment. 1×106 naive CD4+ or CD8+ T cells from control or KO mice were transferred into _Rag1_−/− recipient mice that were examined 5–7 weeks later. (b) Donor T cell number was calculated from the total cellularity and the percentage of each population in spleen and lamina propria. (c) and (d) Body weight was examined every week for the _Rag1_−/− mice receiving CD4+ T cells (c) and CD8+ T cells with or without CD4+ T cells (d). Each line represents an individual recipient mouse.
Discussion
We show that CD4+ and CD8+ T cells that are unresponsive to TGF-β signals because they lack the TGF-βRII may remain quiescent in the lymphoid periphery of conventionally housed, unimmunized mice. Thus, adult mice in which the majority of peripheral T cells lack the TGF-β receptor nonetheless appear healthy, showing no signs of lymphoproliferation, weight loss or autoimmune disease for the duration of the analysis (18 weeks). This result, obtained by the T cell specific deletion of the TGF-β receptor by cre expressed under the control of the distal Lck promoter was surprising because it differed sharply from the outcome when the receptor is deleted by cre driven by the CD4 promoter. Others have reported, and we have confirmed, that such _Tgfbr2_f/f CD4-cre mice show signs of disease shortly after birth and die by around 3–5 weeks of age with massive lymphoproliferation and multiorgan lymphocytic infiltrates16, 17. We sought to explain the strikingly different outcomes of using dLck-cre versus CD4-cre to delete the TGF-β receptor. There are many differences between the two cre deletion models. For instance, CD4-cre is turned on at the early double-negative (DN)-to-double positive (DP) stage of thymopoiesis. As a result, NKT cell development is abrogated34 and essentially all developing T cells, including CD4 Treg cells, lack the receptor. In addition, the absence of the TGF-β receptor on thymocytes during DP-to-single positive (SP) development may result in the selection of an altered TCR repertoire35. In contrast, in _Tgfbr2_f/f dLck-cre mice, cre-mediated deletion starts at the SP stage and continues in peripheral T cells. In this model, NKT development is normal and most peripheral Treg cells retain TGF-β receptor expression (data not shown). With regard to the possibility that it is the difference in the TGF-β responsiveness of Treg cells in the two models that explains the different outcomes, it is notable that others have reported that neonatal injection of wildtype Treg cells into _Tgfbr2_f/f CD4-cre mice does not prevent disease, and mixed bone marrow chimeras, made with a 50:50 mixture of wildtype and _Tgfbr2_f/f CD4-cre stem cells, also succumb to autoimmunity16, 17. Thus it appears that WT Treg cells cannot prevent autoimmunity in the CD4-cre model. Furthermore, the different response to Treg cells cannot explain all the phenomena we observed. For instance when purified OT-1 T cells were transferred into _Rag1_−/− mice, there were no Treg cell present. In addition, purified naive OT-1 T cells were used in our in vitro experiments and Treg cells were not present in the culture. However, under these settings, clear differences were observed between KO and control T cells. These data strongly suggest that intrinsic defects other than Treg cell responsiveness contribute to the striking phenotype in KO T cells.
Given that lymphopenia has been linked to autoimmunity26–28, we focused on the importance of the neonatal lymphopenic period to explain the different phenotypes in the two models of T cell specific TGF-βRII deletion. This was sparked by our observation that deletion of the receptor was rather inefficient in the periphery and thymus of newborn and young _Tgfbr2_f/f dLck-cre mice. In contrast, all T cells emerging from the thymus in mice expressing CD4-cre have deleted the TGF-β receptor, including during the neonatal period. While not ruling out a contribution of other explanations (such as differences in Treg cells, presence or absence of NKT, or an altered TCR repertoire) for the different phenotypes observed in the two models, our data show clearly that naive CD4+ and CD8+ T cells are regulated by TGF-β signals in a lymphopenic environment. In particular, we show that, compared to transfer of TGF-β responsive T cells, TGF-β unresponsive T cells with a naive phenotype show enhanced proliferation and acquisition of effector phenotype and rapid disease induction in lymphopenic recipients.
Because naive T cell proliferation in a lymphopenic environment is partly dependent on the weak TCR stimulation derived from self MHC ligands23, we modeled this by comparing the response of control and receptor knockout CD8+ T cells to potent and weak TCR stimuli in vitro. In mixed cultures of control-plus-knockout T cells, we again observed the dramatic proliferative advantage of the knockout T cells to weak TCR stimulation. Previous work from this laboratory has shown that the weakest ligand used, T4, is about 70-fold less potent than the cognate SIINFEKL peptide in activating OT-I cells33 and is reported to be close to the ‘threshold’ affinity for thymocyte positive versus negative selection36. Importantly, neither TCR repertoire differences nor the action of Treg cells can explain this in vitro result obtained with purified OT-1 CD8+ T cell responders. To rule out that KO T cell defects other than acute unresponsiveness to TGF-β were related to the phenotype, WT OT-1 cells were cultured with or without TGF-β signaling. Indeed, in the absence of TGF-β signaling, WT OT-1 behaved similarly to KO cells.
Combined, these results allow us to hypothesize that under non-inflammatory conditions, but with enhanced exposure to self antigen such as occurs in a lymphopenic environment, TGF-β signaling directly to T cells is necessary to control T cell activation and proliferation. Our results and those of others affirm the dominant and persistent role of TGF-β in maintaining T cell homeostasis. The dampening role on T cell activation and resulting autoimmunity is especially important when other factors, such as enhanced exposure to self antigen and greater access to homeostatic cytokines, impinge on peripheral T cells.
Methods
Mice
_Tgfbr2_f/f mice came from Stefan Karlsson (Lund University, Sweden 37). Distal Lck-cre transgenic mice were provided by Pamela J. Fink (University of Washington, Seattle, WA) and originally came from Nigel Killeen30 (University of California, San Francisco, CA). C57BL/6 (stock no. 000664) mice and _Rag1_−/− mice (stock no. 002216) were obtained from The Jackson Laboratory and housed in specific pathogen-free conditions in the animal facilities at the University of Washington (Seattle, WA). All recipient mice were used at 6 to 12 wk of age. All experiments were done in accordance with the University of Washington Institutional Animal Care and Use Committee guidelines.
Adoptive transfer and antibiotic treatment
Total naive T cells, CD4+, CD8+ and OT-1 T cells were isolated from spleen and lymph nodes using a Pan T cell isolation kit, a CD4 isolation kit or a CD8 isolation kit (Miltenyi) following the manufacturer’s instruction but with the addition of biotin-αCD44 antibody during the biotin antibody cocktail incubation step. The indicated number of T cells was transferred intravenously into appropriate recipient mice. As indicated, some recipient mice had been treated with antibiotic water (100U/ml polymixin B, 1mg/ml neomycin and 1mg/ml ampicillin) for 7 days before T cell transfer.
In vitro proliferation
Naive OT-1 T cells were purified using a CD8 isolation kit (Miltenyi) with supplemental anti-CD44, and labeled with 5 μM CFSE (Invitrogen). T cell and NK cell depleted splenocytes were purified by a similar procedure as OT-1 T cells with an antibody mixture containing biotin-αTCRβ (H57-597), biotin-αTCRγδ (eBioGL3) and biotin-αNK1.1 (pk136) and used as APCs. APCs were pulsed with 0.2–5ng/ml peptide (SIINFEKL and all variant peptides were from Genemed Synthesis) and 1µg/ml LPS (Sigma) at 37°C for 1 hour followed by extensive washing. OT-1 T cells and APCs were mixed and cultured in the presence of 2.5ng/ml hTGF-β1 (R&D systems). As indicated, 1000U/ml IFN-αA (PBL InterferonSource), 20ng/ml IFN-γ and 20ng/ml IL-12 (Peprotech) were added to the culture. 50U/ml IL-2 (eBioscience) was added one day later. 10µg/ml αTGF-β neutralizing antibody (clone#1D11, R&D systems) was added as indicated in the absence exogenous hTGF-β1. To calculate the T cell number during in vitro cultures, 20µl of Flow Cytometry Absolute Count Standard beads (Bangs Lab) was added to each sample before flow cytometric analysis.
Antibodies and flow cytometry
Single-cell suspensions were prepared from the spleen, small intestine and liver after perfusion of the animal at the indicated time-points post cell transfer. Cells were typically stained with antibodies specific for CD8 (53-6.7), CD4 (RM4-5), CD103 (2E7), CD90.2 (53-2.1), CD62L (MEL-14), CD44 (IM7), TCRβ (H57-597), CD45.1 (A20), CD45.2 (104), KLRG1 (2F1), IFN-γ (XMG1.2), Eomes (Dan11mag) and T-bet (eBio4B10) (eBioscience, Biolegend and BD), TGF-βRII (R&D Systems) and Granzyme B (clone#GB11, Invitrogen). For intracellular cytokine staining, cells were stimulated ex vivo with 5µg/ml αCD3 + 5µg/ml αCD28 for 4 hours at 37°C in the presence of brefeldin A followed by surface staining and treatment with the Cytofix/Cytoperm kit (BD). Cells were analyzed using a FACSCanto (BD) and analyzed using FLOWJO (TreeStar) software.
Supplementary Material
1
ACKNOWLEDGMENTS
We thank Andy G. Farr for help with the histology, Tessa Bergsbaken for help with the immunofluorescent microscopy and Pamela J. Fink for comments on the manuscript.
References
- 1.Li MO, Wan YY, Sanjabi S, Robertson AK, Flavell RA. Transforming growth factor-beta regulation of immune responses. Annu Rev Immunol. 2006;24:99–146. doi: 10.1146/annurev.immunol.24.021605.090737. [DOI] [PubMed] [Google Scholar]
- 2.Massague J. TGFbeta in Cancer. Cell. 2008;134:215–230. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blobe GC, Schiemann WP, Lodish HF. Role of transforming growth factor beta in human disease. N Engl J Med. 2000;342:1350–1358. doi: 10.1056/NEJM200005043421807. [DOI] [PubMed] [Google Scholar]
- 4.Hill CS. Nucleocytoplasmic shuttling of Smad proteins. Cell Res. 2009;19:36–46. doi: 10.1038/cr.2008.325. [DOI] [PubMed] [Google Scholar]
- 5.Zhang YE. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009;19:128–139. doi: 10.1038/cr.2008.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ikushima H, Miyazono K. TGFbeta signalling: a complex web in cancer progression. Nat Rev Cancer. 2010;10:415–424. doi: 10.1038/nrc2853. [DOI] [PubMed] [Google Scholar]
- 7.Yang L, Pang Y, Moses HL. TGF-beta and immune cells: an important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010;31:220–227. doi: 10.1016/j.it.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kulkarni AB, et al. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci U S A. 1993;90:770–774. doi: 10.1073/pnas.90.2.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shull MM, et al. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature. 1992;359:693–699. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Diebold RJ, et al. Early-onset multifocal inflammation in the transforming growth factor beta 1-null mouse is lymphocyte mediated. Proc Natl Acad Sci U S A. 1995;92:12215–12219. doi: 10.1073/pnas.92.26.12215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bommireddy R, et al. Elimination of both CD4+ and CD8+ T cells but not B cells eliminates inflammation and prolongs the survival of TGFbeta1-deficient mice. Cell Immunol. 2004;232:96–104. doi: 10.1016/j.cellimm.2005.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rudner LA, et al. Necroinflammatory liver disease in BALB/c background, TGF-beta 1-deficient mice requires CD4+ T cells. J Immunol. 2003;170:4785–4792. doi: 10.4049/jimmunol.170.9.4785. [DOI] [PubMed] [Google Scholar]
- 13.Letterio J, et al. Autoimmunity associated with TGF-beta1-deficiency in mice is dependent on MHC class II antigen expression. J Clin Invest. 1996;98:2109–2119. doi: 10.1172/JCI119017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gorelik L, Flavell RA. Abrogation of TGFbeta signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity. 2000;12:171–181. doi: 10.1016/s1074-7613(00)80170-3. [DOI] [PubMed] [Google Scholar]
- 15.Lucas PJ, Kim SJ, Melby SJ, Gress RE. Disruption of T cell homeostasis in mice expressing a T cell-specific dominant negative transforming growth factor beta II receptor. J Exp Med. 2000;191:1187–1196. doi: 10.1084/jem.191.7.1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li MO, Sanjabi S, Flavell RA. Transforming growth factor-beta controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity. 2006;25:455–471. doi: 10.1016/j.immuni.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 17.Marie JC, Liggitt D, Rudensky AY. Cellular mechanisms of fatal early-onset autoimmunity in mice with the T cell-specific targeting of transforming growth factor-beta receptor. Immunity. 2006;25:441–454. doi: 10.1016/j.immuni.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 18.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 19.Li MO, Wan YY, Flavell RA. T cell-produced transforming growth factor-beta1 controls T cell tolerance and regulates Th1-and Th17-cell differentiation. Immunity. 2007;26:579–591. doi: 10.1016/j.immuni.2007.03.014. [DOI] [PubMed] [Google Scholar]
- 20.Liu Y, et al. A critical function for TGF-beta signaling in the development of natural CD4+CD25+Foxp3+ regulatory T cells. Nat Immunol. 2008;9:632–640. doi: 10.1038/ni.1607. [DOI] [PubMed] [Google Scholar]
- 21.Marie JC, Letterio JJ, Gavin M, Rudensky AY. TGF-beta1 maintains suppressor function and Foxp3 expression in CD4+CD25+ regulatory T cells. J Exp Med. 2005;201:1061–1067. doi: 10.1084/jem.20042276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Starr TK, Jameson SC, Hogquist KA. Positive and negative selection of T cells. Annu Rev Immunol. 2003;21:139–176. doi: 10.1146/annurev.immunol.21.120601.141107. [DOI] [PubMed] [Google Scholar]
- 23.Sprent J, Surh CD. Normal T cell homeostasis: the conversion of naive cells into memory-phenotype cells. Nat Immunol. 2011;12:478–484. doi: 10.1038/ni.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kieper WC, et al. Recent immune status determines the source of antigens that drive homeostatic T cell expansion. J Immunol. 2005;174:3158–3163. doi: 10.4049/jimmunol.174.6.3158. [DOI] [PubMed] [Google Scholar]
- 25.Min B, Yamane H, Hu-Li J, Paul WE. Spontaneous and homeostatic proliferation of CD4 T cells are regulated by different mechanisms. J Immunol. 2005;174:6039–6044. doi: 10.4049/jimmunol.174.10.6039. [DOI] [PubMed] [Google Scholar]
- 26.King C, Ilic A, Koelsch K, Sarvetnick N. Homeostatic expansion of T cells during immune insufficiency generates autoimmunity. Cell. 2004;117:265–277. doi: 10.1016/s0092-8674(04)00335-6. [DOI] [PubMed] [Google Scholar]
- 27.Krupica T, Jr, Fry TJ, Mackall CL. Autoimmunity during lymphopenia: a two-hit model. Clin Immunol. 2006;120:121–128. doi: 10.1016/j.clim.2006.04.569. [DOI] [PubMed] [Google Scholar]
- 28.Le Campion A, et al. Lymphopenia-induced spontaneous T-cell proliferation as a cofactor for autoimmune disease development. Blood. 2009;114:1784–1793. doi: 10.1182/blood-2008-12-192120. [DOI] [PubMed] [Google Scholar]
- 29.Hale JS, Ames KT, Boursalian TE, Fink PJ. Cutting Edge: Rag deletion in peripheral T cells blocks TCR revision. J Immunol. 2010;184:5964–5968. doi: 10.4049/jimmunol.1000876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang DJ, et al. Selective expression of the Cre recombinase in late-stage thymocytes using the distal promoter of the Lck gene. J Immunol. 2005;174:6725–6731. doi: 10.4049/jimmunol.174.11.6725. [DOI] [PubMed] [Google Scholar]
- 31.El-Asady R, et al. TGF-{beta}-dependent CD103 expression by CD8(+) T cells promotes selective destruction of the host intestinal epithelium during graft-versus-host disease. J Exp Med. 2005;201:1647–1657. doi: 10.1084/jem.20041044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Feng T, Wang L, Schoeb TR, Elson CO, Cong Y. Microbiota innate stimulation is a prerequisite for T cell spontaneous proliferation and induction of experimental colitis. J Exp Med. 2010;207:1321–1332. doi: 10.1084/jem.20092253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zehn D, Lee SY, Bevan MJ. Complete but curtailed T-cell response to very low-affinity antigen. Nature. 2009;458:211–214. doi: 10.1038/nature07657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Doisne JM, et al. iNKT cell development is orchestrated by different branches of TGF-beta signaling. J Exp Med. 2009;206:1365–1378. doi: 10.1084/jem.20090127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ouyang W, Beckett O, Ma Q, Li MO. Transforming growth factor-beta signaling curbs thymic negative selection promoting regulatory T cell development. Immunity. 2010;32:642–653. doi: 10.1016/j.immuni.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Daniels MA, et al. Thymic selection threshold defined by compartmentalization of Ras/MAPK signalling. Nature. 2006;444:724–729. doi: 10.1038/nature05269. [DOI] [PubMed] [Google Scholar]
- 37.Leveen P, et al. TGF-beta type II receptor-deficient thymocytes develop normally but demonstrate increased CD8+ proliferation in vivo. Blood. 2005;106:4234–4240. doi: 10.1182/blood-2005-05-1871. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1