p53 mutations in colorectal cancer- molecular pathogenesis and pharmacological reactivation (original) (raw)
Review Open Access
Copyright ©The Author(s) 2015. Published by Baishideng Publishing Group Inc. All rights reserved.
World J Gastroenterol. Jan 7, 2015; 21(1): 84-93
Published online Jan 7, 2015. doi: 10.3748/wjg.v21.i1.84
p53 mutations in colorectal cancer- molecular pathogenesis and pharmacological reactivation
Xiao-Lan Li, Jianbiao Zhou, Wee-Joo Chng, Cancer Science Institute of Singapore, National University of Singapore, 14 Medical Drive, Centre for Translational Medicine, Singapore 117599, Singapore
Xiao-Lan Li, Zhi-Rong Chen, Department of Gastroenterology, Suzhou Municipal Hospital (Eastern), Suzhou 215001, Jiangsu Province, China
Wee-Joo Chng, Department of Medicine, Yong Loo Lin School of Medicine, National University of Singapore, Singapore 119074, Singapore
Wee-Joo Chng, Department of Hematology-Oncology, National University Hospital, Singapore 119228, Singapore
ORCID number: $[AuthorORCIDs]
Author contributions: Li XL, Zhou J, Chen ZR and Chng WJ all reviewed the literature and wrote the manuscript; all authors approved the final version of the manuscript; and all authors contributed equally to this work and were co-first authors.
Supported by National Research Foundation Singapore and the Singapore Ministry of Education under its Research Centres of Excellence initiative; and NMRC Clinician-Scientist IRG Grant CNIG11nov38 (Zhou J); Chng WJ is also supported by NMRC Clinician Scientist Investigator award
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Correspondence to: Wee-Joo Chng, MD, PhD, Associate Professor, Department of Hematology-Oncology, National University Hospital, 1E, Kent Ridge Road, Singapore 119228, Singapore. mdccwj@nus.edu.sg
Telephone: +65-65161118 Fax: +65-68739664
Received: July 7, 2014
Peer-review started: July 8, 2014
First decision: August 6, 2014
Revised: August 20, 2014
Accepted: October 14, 2014
Article in press: October 15, 2014
Published online: January 7, 2015
Processing time: 184 Days and 0.5 Hours
Abstract
Colorectal cancer (CRC) is one of the most common malignancies with high prevalence and low 5-year survival. CRC is a heterogeneous disease with a complex, genetic and biochemical background. It is now generally accepted that a few important intracellular signaling pathways, including Wnt/β-catenin signaling, Ras signaling, and p53 signaling are frequently dysregulated in CRC. Patients with mutant p53 gene are often resistant to current therapies, conferring poor prognosis. Tumor suppressor p53 protein is a transcription factor inducing cell cycle arrest, senescence, and apoptosis under cellular stress. Emerging evidence from laboratories and clinical trials shows that some small molecule inhibitors exert anti-cancer effect via reactivation and restoration of p53 function. In this review, we summarize the p53 function and characterize its mutations in CRC. The involvement of p53 mutations in pathogenesis of CRC and their clinical impacts will be highlighted. Moreover, we also describe the current achievements of using p53 modulators to reactivate this pathway in CRC, which may have great potential as novel anti-cancer therapy.
Core tip: Dysregulation of p53 tumor suppressor gene is one of the most frequent events contributing to the transformation of colorectal cancer (CRC), as well as the aggressive and metastatic features of CRC. Mutant p53 reactivator, PRIMA-1MET has been tested in Phase I/II clinical trials and shows encouraging benefits. In this review, we systemically and comprehensively summarize the current understanding of p53 mutations in the pathogenesis of CRC and current progress in reactivation of p53 as a novel therapeutic strategy. We hope this review will promote more investigations of reactivation of p53 as a viable treatment option of patients with CRC.
- Citation: Li XL, Zhou J, Chen ZR, Chng WJ. p53 mutations in colorectal cancer- molecular pathogenesis and pharmacological reactivation. World J Gastroenterol 2015; 21(1): 84-93
- URL: https://www.wjgnet.com/1007-9327/full/v21/i1/84.htm
- DOI: https://dx.doi.org/10.3748/wjg.v21.i1.84
INTRODUCTION
Colorectal cancer (CRC) is the third most common cancer in men and the second most common cancer in women worldwide (http://http://www.wcrf.org). Although diagnosis and therapy have advanced significantly in the last ten years, its prevalence is rising, and the 5-year survival rate is still poor. In 2012, it accounts for nearly 14.1 million cases and 694000 deaths around the world (http://http://www.wcrf.org; http://www.who.int). CRC becomes a serious problem for healthcare in Asian countries too, such as China, Japan, South Korea and Singapore, with a 2-4 fold increase in the incidence during last decades[1]. So more efficacious approaches are urgently needed for CRC patients.
p53 was first discovered and classified as a cellular SV40 large T antigen-binding protein[2,3]. This finding marks the beginning of a brand-new period in cancer research that is expected to have a major impact in the clinic. p53 is a stress-inducible transcription factor, which regulates a large number of diverse downstream genes to exert regulative function in multiple signaling processes. p53 mutation occurs in approximately 40%-50% of sporadic CRC[4]. The status of p53 mutation is closely related to the progression and outcome of sporadic CRC. In recent years, some small molecule compounds have been intensively investigated for reactivation and restoration of p53 via different mechanisms. These promising compounds are being tested in clinical trials and may be approved for the treatment of CRC patients in near future.
P53 FUNCTION: INDUCING CELL CYCLE ARREST AND APOPTOSIS
The human TP53 gene is located on chromosome 17p, and consists of 11 exons and 10 introns[5]. Wild type p53 protein consists of 393 amino acid residues, and several functional domains. In the order from N-terminus to C-terminus, they are: transactivation domain (TAD), proline-rich domain, tetramerization domain and basic domain (Figure 1A). Once activated, p53 upregulates its negative regulator, MDM2 (murine/human double minute 2). MDM2 functions as an E3 ubiquitin-ligase, to regulate the ubiquitination of p53 which leads to its degradation[6]. This forms a negative feedback loop that maintains low levels of p53 in normal cells[7]. Depending on specific context, p53 can induce cell cycle arrest, or apoptosis, or senescence, in the presence of cellular stress, such as DNA damage, hypoxia, oncogene activation, etc. (Figure 1B).
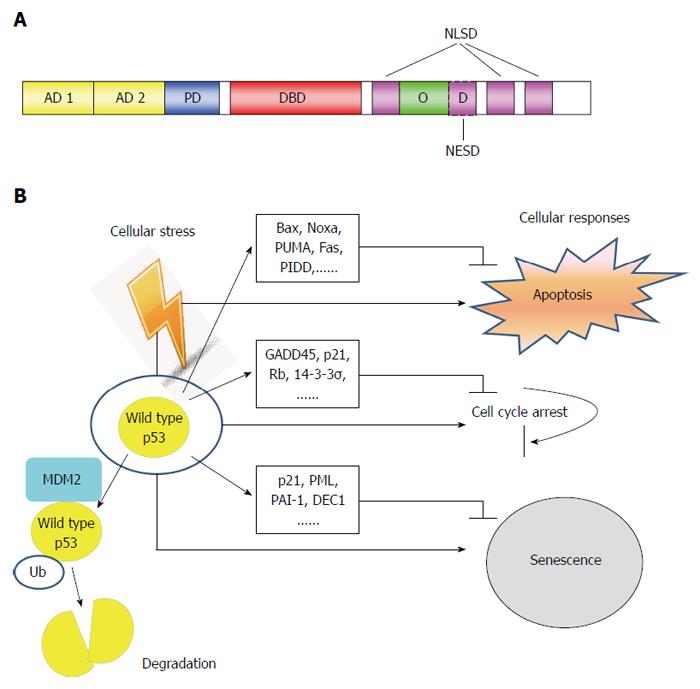
Figure 1 Structure and function of p53 tumor suppressor. A: Schematic of p53 protein structure. The function domains and corresponding amino acid regions are indicated. N-terminus transcription-activation domain (TAD): Residues 1-63; AD1: Residues 1-42 for G1 arrest and apoptotic activity; AD2: Residues 43-63 important for senescence-activity; PD: Residues 64-92 important for apoptotic activity; DBD: Residues 102-292 responsible for binding the p53 co-repressors; NLSD: Residues 316-324, 370-376, 380-386; OD: Residues 325-356; NESD: Residues 340-353; B: In normal cells, p53 activates a plethora of target genes involved in diverse biological processes in response to cellular stress. Ub: Ubiquitin; PD: Poly-proline domain; DBD: DNA binding core domain; OD: Homo-oligomerization domain; NESD: Nuclear export signaling domain; NLSD: Nuclear localization signaling domain.
Activation of p53 can trigger both the mitochondrial (intrinsic) and the death-receptor-induced (extrinsic) apoptotic pathways[8]. p53 induces the expression of pro-apoptotic Bcl-2 (B-cell lymphoma-2) family proteins, mainly Bax, Noxa and PUMA, but downregulates the pro-survival Bcl-2, leading to permeabilization of outer mitochondrial membrane. Then cytochrome c releases from the mitochondria binds to Apaf-1, and induces the activation of the initiator caspase-9, eventually resulting in the activation of executioner caspase-3, -6 and -7[9]. On the other hand, activated p53 also upregulates the expression of some DRs (death receptors), such as Fas (CD95/APO-1), DR5 (TRAIL-R2), and PIDD (p53-induced protein with death domain). Together with caspase-8, they form the death-inducing signaling complex, subsequently activating caspase-3 and inducing apoptosis (Figure 1B). The progression of cell cycle is tightly controlled by cyclins and cyclin-dependent kinases (CDK). p21(WAF1) is one member of CDK inhibitor family, which hinder cell cycle transition from G1 to S phase. p21(WAF1) is a well-characterized p53-downstream gene and its promoter contains consensus p53-binding sequences. It has been shown that p21(WAF1) is one of the major mediator of p53-induced growth arrest. In response to DNA damage, p53 induces not only cell cycle G1 phase arrest, but also G2/M checkpoint arrest. Repression of CDC2, the CDK necessary for initiation of mitosis, by p53 plays an important role in G2/M arrest. Some other p53 target genes, for example, GADD45, p21(WAF1), retinoblastoma protein (Rb), and 14-3-3σ, also cRRIMA-1MET contribute to G2/M arrest. p21(WAF1) and Rb are involved in both G1 to S phase arrest and G2/M arrest induced by p53 (Figure 1B).
Cellular senescence is a specific form of cell cycle arrest, which is prolonged and irreversible[10]. Morphologically, senescence cells significantly increase in size and have prominent nucleoli, as well as abundant cytoplasmic vacuoles[11]. Cellular senescence is an important mechanism for preventing the development of potentially cancerous cells in response to stress-induced DNA damage[12]. Various stress stimuli including DNA-damage response, dysfunctional telomeres, oncogenes, oxidative stress, usually trigger one of the two pivotal routines, either the p53-p21(WAF1) or the p16 (CDKN2A)-Rb pathways to induce senescence[11,13]. In addition to p21(WAF1)[14], genes have been reported as important in p53-induced senescence include tumor suppressor promyelocytic leukemia (PML)[15,16], plasminogen activator inhibitor-1[17], and deleted in esophageal cancer 1 (DEC1)[18] (Figure 1B).
REGULATORS OF P53 ACTIVITY AND THEIR IMPLICATIONS IN CRC
Activating transcription factor 3
Activating transcription factor 3 (ATF3) is one of the p53 target genes and involved in the complicated process of cellular stress response[19,20]. In addition, ATF3 also acts as a co-transcripition factor for p53 achieving maximal induction of DR5 expression upon DNA damage in CRC[21]. DR5 is a trans-membrane TNF (tumor necrosis factor) receptor containing a death domain, which binds to the ligand TRAIL (tumor necrosis factor-related apoptosis-inducing ligand), and triggers cell death by activating the extrinsic apoptotic pathway[22]. Ectopic expression of ATF3 suppresses colon tumor growth and metastasisin mouse xenografts[23]. Post-translational modification of ATF3 by SUMO (small ubiquitin-related modifier) plays a negative role in the regulation of p53 activity[24]. ATF3 was also found to be bound to mutant p53, inactiving its oncogenic potential[25]. Of note, SUMO-1, a member of the SUMO protein family, involves a variety of biologically distinct functions through SUMO attachment of target proteins[26]. Over-expression of SUMO-1 causes the accumulation of sumoylated p53 proteins in colon cancer cells, which leads to more frequent metastasis[27].
MicroRNAs
MicroRNA (miRs) are small non-coding RNA molecules consisting of 19-25 nucleotides, with functions in transcriptional and post-transcriptional regulation of gene expression[28]. miRs are believed to be important factors for cell proliferation, apoptosis, senescence and metabolism, which all play crucial role in the carcinogenic process[29]. For example, the high expression of miR-125b which directly targets the 3’UTR of TP53, repressed the endogenous level of p53 proteins, thereby promoting tumor growth and invasion. So, miR-125 acts as oncogene and is associated with the poor prognosis in CRC patients[30]. miR-125b had also been shown to repress both cell cycle-arrest and apoptotic regulators in the p53 network, implicating its role in oncogenesis[31,32] (Figure 2). Conversely, the miR-34 family (miR-34a/b/c) are transcriptional targets of p53[33], and directly suppresses a range of Wnt and epithelial-mesenchymal transition (EMT) genes[34-37]. Thus, part of p53 tumor suppressor function is due to its inhibition of Wnt pathway and EMT transition through miR-34 and loss of this inhibition could trigger the proliferation and tissue-invasion of CRC cells[34,35] (Figure 2).
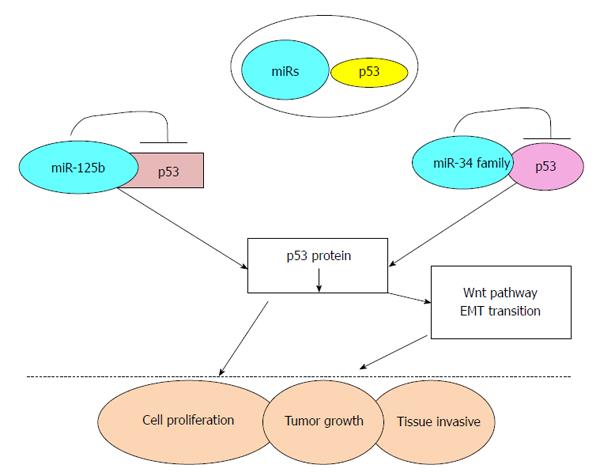
Figure 2 Schematic representation of miRNAs regulating p53 pathway and subsequent tumorigenesis.
P53 MUTATION IN CRC
Development of CRC is a multi-factorial and multi-stage process involving the activation of oncogenes and inactivation of tumor suppressor genes. Confirmed by numerous studies, p53 is a key tumor suppressor gene and is one of the most important elements of our body’s anticancer defense[38]. It is generally known that the progression of CRC follows mutations of the APC, K-Ras, and p53 genes[39]. p53 is the most commonly mutated gene in human cancers[40]. It is thought that p53 mutations play a critical role in the adenoma-carcinoma transition during tumorous pathological process[41]. p53 mutation in CRC occurs in 34% of the proximal colon tumors, and in 45% of the distal colorectal tumors[8,42]. Majority of these mutations occur in exon 5 to 8 (DNA binding doman), and mainly in some hotspot codons, such as 175, 245, 248, 273 and 282, comprising of G to A, C to T transition and leading to the substitution of a single amino acid in p53 protein[41,42] (Table 1). Such substitutions most commonly cluster in the DNA binding domain, causing the disruption of specific DNA binding and sequential transactivation[7,42].
Table 1 Common, high frequency of p53 missense alterations in colorectal cancer.
| Exon | Codon | Codon change | Nucleotide change | Amino acid change |
|---|---|---|---|---|
| 5 | 175 | CGC→CAC | G→A | Arg→His |
| 7 | 245 | GGC→AGC | G→A | Gly→Ser |
| 7 | 245 | GGC→GAC | G→A | Gly→Asp |
| 7 | 248 | CGG→TGG | C→T | Arg→Trp |
| 7 | 248 | CGG→CAG | G→A | Arg→Gln |
| 8 | 273 | CGT→TGT | C→T | Arg→Cys |
| 8 | 273 | CGT→CAT | G→A | Arg→His |
| 8 | 282 | CGG→TGG | C→T | Arg→Trp |
Different types of p53 mutations play a pivotal role in determining the biologic behavior of CRC, such as invasive depth, metastatic site and even the prognosis of patients. p53 mutations are associated with lymphatic invasion in proximal colon cancer, and show significant correlation with both lymphatic and vascular invasion in distal CRC[42] (Table 2). CRC patients with mutant p53 appear more chemo-resistance and have poorer prognosis than those with wild-type p53[43]. In a TP53 colorectal cancer international collaborative study, it was observed that patients with mutant p53 in exon 5 had worse outcome for proximal colon cancer[42] and inactivating mutation of p53 occurred more frequent in advanced stage tumors and were negatively associated with survival[44] (Table 2).
Table 2 Summary of major conclusions on the importance of p53 in colorectal cancer development.
| Ref. | Major conclusions |
|---|---|
| Taketani _et al_[21] | p53 partners with ATF3 in maximal induction of DR5 upon |
| DNA damage | |
| Wei _et al_[25] | ATF3 binds mutant p53 and inhibits its oncogenic function |
| Nishida _et al_[30] | High expression of miRNA-125b predicts poor survival in CRC. miRNA-125 decreases p53 expression |
| Kim _et al_[34,35] | Loss of p53 de-represses Wnt pathway and EMT transition through miRNA-34 |
| López _et al_[41] | p53 mutations occur in 54% of sporadic CRC |
| Russo _et al_[42] | p53 mutations correlate with the site, biologic behaviour and outcome of CRC |
| Iacopetta _et al_[44] | p53 mutations that lose transactivational ability are more common in advanced CRC and associated with poor survival |
PHARMACOLOGICAL REACTIVATION OF P53 AS CANCER THERAPY
Results from a large number of studies have unequivocally evidence demonstrated that mutant p53 not only plays a pivotal role in the transformation of CRC, but also contributes to the aggressiveness and invasiveness of CRC. It is not surprising that manipulation of the p53 pathway has attracted interest soon after the discovery of p53 gene. Although reintroduction of wild type p53 by gene therapy appears a straightforward and logical choice, this approached is impeded by the technical challenge of efficient gene delivery and safety issues inherent in the use of viral vectors[45]. In recent years, we witness an array of small molecule inhibitors modulating the p53 pathway being developed (Figure 3). Some of these compounds have been tested as potential therapeutic agents in CRC.

Figure 3 Small molecule compounds pharmacologically reactivating of p53 function. MI43 and Nutlin-3 bound to MDM2 blocking MDM2-p53 interaction. RITA bound to p53 interfering MDM2-p53 interaction. α-Lipoic acid increased p53 protein stability and its apoptotic effect. Quinacrine induced the autophagy-associated cell death in a p53-dependent manner. NSC17632 activated p53-like activatity dependent on p73. PRIMA-1/PRIMA-1MET restored mutant p53 to exert apoptotic effect. Maslinic acid and Epicatechin gallate as plant extraction modulated the expression of p53 and its target genes in p53-dependent apoptotic and cell cycle arrest pathway.
Modulation of wild-type p53 activity via inhibiting MDM2-p53 interaction
MDM2 protein, the E3 ubiquitin protein ligase, binds to the amino-terminal of p53, and ubiquitylates p53, leading to its proteasomal degradiaiton; this inhibits its suppressive function in cancer cells[46]. Pharmacological inhibitors of MDM2 have already been extensively researched for their anti-cancer activities through stabilization of p53 protein[47-49]. Activation of p53 without DNA damage should be a great advantage, compared to many traditional chemotherapeutic agents[47].
MIs (MDM2 Inhibitors): In recent years, a number of MIs that disrupt the MDM2-p53 interaction have been discovered. The spiro oxindole MI-43 is one of these specific MDM2 antagonists that cause p53 accumulation and lead to the induction of target genes, e.g., p21, Puma, and Noxa[50]. In colon cancer cells, cell cycle arrest and apoptosis were induced by MI-43 in a p53-dependent manner[51]. MI-219 is an improved MDM2-p53 inhibitor with improved pharmacokinetic profile and higher binding affinity to MDM2. MI-219 showed potent efficacy as a single agent in inducing apoptosis in HCT-116 colon cancer cell line. Furthermore, the combination of MI-219 with chemotherapeutic drug, Oxaliplatin, achieved high synergism in p53-mediated apoptotic response[52].
Nutlins: Nutlins are cis-imidazoline analogs, which occupy the binding pocket of MDM2, thus disrupting MDM2-p53 interaction. Nutlins were first discovered using biochemical screening strategy by Vassilev and colleagues in Roche in 2004[53]. Among them, Nutlin-3 (R1772) has been widely tested in a variety of cancers in vitro, in mouse xenografts bearing human tumors, as well as clinical trials in human subjects[54]. Nutlin-3 was observed to act as MDM2 antagonist, stabilize p53 and activate p53 target genes in CRC cells expressing wild-type p53. MDMX, another member of MDM protein family, shares a similar amino acid sequence and structural organization with MDM2. Although both MDM2 and MDMX negatively regulate p53, the relative abundance of MDM2 and MDMX level influences cancer cells response to Nutlin-3. Cancer cells overexpressing MDM2 are sensitive to Nutlin-3, in contrast, cancer cells overexpressing MDMX are resistant to Nutlin-3 due to its inability to block p53-MDMX interaction[55]. Nutlin-3a, but not the aftermentioned RIAT (reactivation of p53 and induction of tumor cell apoptosis), has been shown to specifically downregulate α5 integrin in p53 wild type colon cancer[56]. These findings are useful in patient selection in a clinical trial aiming to evaluate Nutlin-3 against CRC. Nutlin may offer clinical benefits for CRC bearing high expression MDM2 or α5 integrin.
Cancer cells often acquire secondary resistance after a prolonged exposure of single agent, so it is clinically desirable to treatment the cancer patients with combination therapy. Nutlin has been tested in combination with other drugs in CRC. Tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) is one of the DNA damage-inducible p53 target gene[57]. Notably, TRAIL induces cell death mainly through the induction of extrinsic apoptosis pathway, while Nutlin works predominately through inducing the intrinsic apoptosis pathway. Combination of Nutlin-3 and TRAIL synergistically enhances cell death in human p53 wild type sarcoma HOS cells and colon cancer HCT116 cells owing to the simultaneous engagement of intrinsic and extrinsic apoptosis pathways[58]. Furthermore, Nutlin-3 treatment increases DR5 expression on both mRNA and protein levels[58,59]. Controlled, concomitant release of Nutlin-3 and Doxil, the liposomal preparation of doxorubicin, by novel drug engineering, leads to synergistic anti-proliferative effect and induction of cell death in CRC cells carrying both wild-type and mutant p53[60]. Combination treatment with Nutlin-3 and Inauhzin, a SIRT1 (Sirtuin 1) inhibitor in colon and lung cancer cell lines, is able to enhances their apoptotic effect in a p53-dependent manner[61]. It is also noteworthy that Nutlin-3 can mediate the phosphorylation of p53 at key DNA-damage-specific serine residues (Ser15, 20 and 37) and initiate the DNA damage signaling pathway which resulted in cell cycle arrest in p53-independent manner[62]. Currently, Nutlin-3 has already been evaluated in phase I clinical trial to treat patients suffering from hematologic neoplasms[63]. Taken together, Nutlin-3 may be a helpful addition to our armamentarium combating CRC, particularly used in conjunction with other drugs.
RITA: RITA was identified from National Cancer Institute library compound Challenge set for its ability to inhibit the proliferation of HCT-116 (p53 wild type) much more than its p53 null counterpart[64]. RITA has been shown to suppress colon cancer growth in a mouse xenofgraft model. Mechanically, this compound directly binds to p53 rather than MDM2, and induces a conformational change in p53, which interfered with the p53-MDM2 interaction, and p53 ubiquitination, resulting in p53 accumulation and cellular apoptosis[64,65]. The study carried out by Di Marzo _et al_[66] implicated that RITA also reactivated mutant p53 function in malignant mesothelioma. Whether RITA is also effective in CRC cells harboring mutant p53 would merit further investigation.
Activation of p53-like activity via other p53 family members, p67 and p73
In addition to p53, the p53 family includes two other members, p63 and p73[67]. They encode proteins with significant sequence homology and functional similarity with p53. A derivative of the cytotoxic plant alkaloid ellipticine, NSC176327 induced potent killing in CRC cells regardless of p53 status. Further experiments revealed that NSC176327 treatment increased the expression of p73, p21 and DR5, while knockdown of p73 in p53 null cells rendered these cells resistant to this drug treatment[68]. The notion that p73 is also a drug target in CRC is reinforced by other studies. Ray _et al_[69] reported that MDM2 inhibitors, like Nutlin-3 , could also disrupt the MDM2-p73 binding, and induce the expression of apoptotic proteins such as Noxa, PUMA and cell cycle arrest protein p21 in CRC cells lacking of functional p53[70]. Securinine, a widely used alkaloid, was identified to promote p73-dependent apoptosis in p53-deficent CRC cells[71]. In conclusion, these results shed new light on the induction of p73 as a therapeutic option in CRC patients with either mutant p53 or p53 null.
Reactivation of mutant p53
It has been long recognized that mutant p53 protein not only abrogates the tumor suppressor function, but also gain novel oncogenic function, which promotes a more aggressive, metastatic cancer phenotype. However, it is until recently that promising compounds that specifically targeting this type of mutant oncogenic p53 proteins have been developed. Aiming to screen compounds that specifically targeting mutant p53, Bykov _et al_[72] discovered one compound 2,2-bis(hydroxymethyl)-1-azabicyclo[2,2,2]octan -3-one, which inhibited the growth of Saos-2-His-273 cells, a Tet-off mutant p53 cell line. This compound was named PRIMA-1 (p53-reactivation and induction of massive apoptosis-1, APR-017)[72]. Late, its methylated form, RRIMA-1MET (APR-246) which is more efficient, was developed by the same group[73]. PRIMA-1 restores the sequence-specific DNA binding region via forming adducts with thiols in mutant p53 and activating several p53 target genes, promoting apoptosis in human cancer cells with mutant p53[74]. The initial consideration was that these two compounds had potent effects on p53-mutant cells, compared to cells with wild-type p53. However, emerging evidence demonstrated that unfolded mutant p53 and unfolded wild-type p53 could also be refolded by PRIMA-1 and PRIMA-1MET[74,75]. So PRIMA-1 and PRIMA-1MET may induce apoptosis in cancer cells carrying either wild-type p53 or mutant p53. Among the class of small molecules that can selectively induce apoptosis in cancer cells with mutant p53, PRIMA-1MET is the first drug which has already advanced to a phase I/II clinical trial for hematologic malignancies and prostate cancer[76,77]. However, there is little investigation about the ability of PRIMA-1MET to induce apoptosis and inhibit tumor growth in different CRC cell lines with different p53 status, thus, more studies are necessary to intensively explore RRIMA-1MET as a novel therapeutic strategy in CRC.
Natural agents extracted from plants
Recently, the anticancer function of agents extracted from nature plants is attracting some attention. The mechanisms implicated have been uncovered constantly.
Maslinic acid: Maslinic acid (MA) is a natural triterpene from Olea europaea, and possesses potent anticancer property aganist CRC cells. Exposure to MA induced the expression of JNK (c-Jun NH2-terminal kinase), p53, and increased the mitochondrial apoptotic signaling molecules, resulting in cell cycle arrest and apoptosis[78,79]. In p53-deficient CRC cells, apoptosis could also be induced by MA without requiring the mitochondrial pathway[80].
Epicatechin gallate: Experimental and epidemiological evidences reveal that dietary polyphenolic plant-derived compounds have anti-proliferative and anti-invasive activity in cancers of gastrointestinal tract, lung, skin, prostate and breast[81-83]. Epicatechin gallate (ECG) is one of the most important compounds of polyphenols found in green tea, which stimulated the expression of p53, p21, and MAPKs (mitogen-activated protein kinases) in CRC cells, leading to cell cycle arrest at G0/G1-S phase in a time-dependent manner[82]. Furthermore, ECG could inhibit the degradation of p53 protein and RNA that contributed to the stabilization of p53.
Other compounds
p53 proteins can be targeted for proteasomal degradation in both normal and cancer cells. α - Lipoic acid (α-LA) is the most common drug worldwide to treat diabetic polyneuropathy. Yoo and colleagues had shown α-LA inhibited proliferation and induced apoptosis in colon cancer cells via preventing p53 degradation. Specifically, α-LA treatment downregulated ribosomal protein p90S6K (RPS6KA4) which was confirmed to inhibit p53 function. Furthermore, α-LA exerted an inhibitory effect on the nuclear translocation of nuclear factor-κB (NF-κB), which played an important role in regulating RPS6KA4 gene expression[84].
FUTURE PERSPECTIVES
There is no doubt that reactivation and restoration of p53 function have great potential as a novel therapeutic strategy in CRC. However, the majority of molecules that lead to cell cycle arrest and apoptosis in CRC cells, has only been tested in cell lines and animal models, and has yet to enter in clinical trials. In addition, it is clear that mutant p53 promotes various oncogenic events. Nevertheless, the critical mechanisms are still not completely understood. The issue that different mutations might affect p53 function differently makes small molecule inhibitors targeting mutant p53 more complicated to assess in a clinical trial. This theme needs to be explored further. Importantly, resistance to treatments and poor prognosis for CRC patients with new p53 mutations will require the continuing development of new agent targeting these novel mutations. Riding on the last 30 years of intensive research in p53 area, this is now the time to harvest the fruits from this body of work and translate our knowledge of p53 into clinical practice for CRC patients.
Footnotes
P- Reviewer: Gao CM, Lakatos PL, Moussata D S- Editor: Ma YJ L- Editor: A E- Editor: Wang CH
References
| 3. | Tan TH, Wallis J, Levine AJ. Identification of the p53 protein domain involved in formation of the simian virus 40 large T-antigen-p53 protein complex. J Virol. 1986;59:574-583. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 8. | Ryan KM, Phillips AC, Vousden KH. Regulation and function of the p53 tumor suppressor protein. Curr Opin Cell Biol. 2001;13:332-337. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 12. | Mallette FA, Ferbeyre G. The DNA damage signaling pathway connects oncogenic stress to cellular senescence. Cell Cycle. 2007;6:1831-1836. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 13. | Shay JW, Pereira-Smith OM, Wright WE. A role for both RB and p53 in the regulation of human cellular senescence. Exp Cell Res. 1991;196:33-39. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 15. | Ferbeyre G, de Stanchina E, Querido E, Baptiste N, Prives C, Lowe SW. PML is induced by oncogenic ras and promotes premature senescence. Genes Dev. 2000;14:2015-2027. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 19. | Zhang C, Gao C, Kawauchi J, Hashimoto Y, Tsuchida N, Kitajima S. Transcriptional activation of the human stress-inducible transcriptional repressor ATF3 gene promoter by p53. Biochem Biophys Res Commun. 2002;297:1302-1310. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 22. | MacFarlane M, Ahmad M, Srinivasula SM, Fernandes-Alnemri T, Cohen GM, Alnemri ES. Identification and molecular cloning of two novel receptors for the cytotoxic ligand TRAIL. J Biol Chem. 1997;272:25417-25420. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 39. | Cottu PH, Muzeau F, Estreicher A, Fléjou JF, Iggo R, Thomas G, Hamelin R. Inverse correlation between RER+ status and p53 mutation in colorectal cancer cell lines. Oncogene. 1996;13:2727-2730. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 50. | Sun SH, Zheng M, Ding K, Wang S, Sun Y. A small molecule that disrupts Mdm2-p53 binding activates p53, induces apoptosis and sensitizes lung cancer cells to chemotherapy. Cancer Biol Ther. 2008;7:845-852. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 52. | Azmi AS, Banerjee S, Ali S, Wang Z, Bao B, Beck FW, Maitah M, Choi M, Shields TF, Philip PA. Network modeling of MDM2 inhibitor-oxaliplatin combination reveals biological synergy in wt-p53 solid tumors. Oncotarget. 2011;2:378-392. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 54. | Shen H, Maki CG. Pharmacologic activation of p53 by small-molecule MDM2 antagonists. Curr Pharm Des. 2011;17:560-568. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 67. | Damia G, Broggini M. Cell cycle checkpoint proteins and cellular response to treatment by anticancer agents. Cell Cycle. 2004;3:46-50. [PubMed] [DOI] [Cited in This Article: ] |
|---|
| 68. | Lu C, Wang W, El-Deiry WS. Non-genotoxic anti-neoplastic effects of ellipticine derivative NSC176327 in p53-deficient human colon carcinoma cells involve stimulation of p73. Cancer Biol Ther. 2008;7:2039-2046. [PubMed] [DOI] [Cited in This Article: ] |
|---|